
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiosynthesis of a new skyllamycin in Streptomyces nodosus: a cytochrome P450 forms an epoxide in the cinnamoyl chain†
Yuhao
Song
 a,
Jose A.
Amaya
b,
Vidhi C.
Murarka
b,
Hugo
Mendez
b,
Mark
Hogan
a,
Jimmy
Muldoon
c,
Paul
Evans
c,
Yannick
Ortin
c,
Steven L.
Kelly
d,
David C.
Lamb
d,
Thomas L.
Poulos
b and
Patrick
Caffrey
*a
a,
Jose A.
Amaya
b,
Vidhi C.
Murarka
b,
Hugo
Mendez
b,
Mark
Hogan
a,
Jimmy
Muldoon
c,
Paul
Evans
c,
Yannick
Ortin
c,
Steven L.
Kelly
d,
David C.
Lamb
d,
Thomas L.
Poulos
b and
Patrick
Caffrey
*a
aCentre for Synthesis and Chemical Biology and School of Biomolecular and Biomedical Science, University College Dublin, Ireland. E-mail: patrick.caffrey@ucd.ie
bDepartments of Molecular Biology and Biochemistry, Pharmaceutical Sciences and Chemistry, University of California, Irvine, California, USA
cCentre for Synthesis and Chemical Biology and School of Chemistry, University College Dublin, Ireland
dFaculty of Medicine, Health and Life Science, Institute of Life Science, Swansea University, Singleton Park, Swansea, SA2 8PP, UK
First published on 18th March 2024
Abstract
Activation of a silent gene cluster in Streptomyces nodosus leads to synthesis of a cinnamoyl-containing non-ribosomal peptide (CCNP) that is related to skyllamycins. This novel CCNP was isolated and its structure was interrogated using mass spectrometry and nuclear magnetic resonance spectroscopy. The isolated compound is an oxidised skyllamycin A in which an additional oxygen atom is incorporated in the cinnamoyl side-chain in the form of an epoxide. The gene for the epoxide-forming cytochrome P450 was identified by targeted disruption. The enzyme was overproduced in Escherichia coli and a 1.43 Å high-resolution crystal structure was determined. This is the first crystal structure for a P450 that forms an epoxide in a substituted cinnamoyl chain of a lipopeptide. These results confirm the proposed functions of P450s encoded by biosynthetic gene clusters for other epoxidized CCNPs and will assist investigation of how epoxide stereochemistry is determined in these natural products.
Introduction
There is growing pharmaceutical interest in non-ribosomal lipopeptides that contain substituted cinnamoyl acyl chains.1–9 These cinnamoyl-containing peptides (CCNPs) display diverse biological activities (Table S1†) and have potential applications as anticancer drugs, antibiotics and biofilm-dispersing agents. One of the best-studied CCNPs is skyllamycin A (Fig. 1). This complex propenyl-cinnamoyl-containing depsipeptide has anti-cancer activity that results from inhibition of the platelet-derived growth factor (PGDF) signalling pathway in tumor cells.10 The structure of skyllamycin A has been determined and a total synthesis has been achieved.11,12 The biosynthetic pathway has been extensively investigated.13 The propenyl-cinnamoyl unit is assembled by a highly reducing type II polyketide synthase (PKS) (Fig. S1†). The chain is completed on a discrete acyl carrier protein (ACP) that interacts with the first module of a non-ribosomal peptide synthetase (NRPS). The cinnamoyl chain is attached to the first activated amino acid and the NRPS assembly line synthesizes a lipopeptidyl thioester that is cyclized to give the final product (Fig. S2 and S3†). | ||
| Fig. 1 Structure of skyllamycin A.10 The carbon atoms of the propenyl-cinnamoyl chain are numbered 1 to 12. Non-standard amino acid residues are labelled. Known skyllamycin analogues (skyllamycins B–E) result from incorporation of aspartate rather than β-methylaspartate, and differences in the C2–C3 region of the cinnamoyl chain.13–15 The structures of skyllamycins B, C, D and E are shown in Fig. S3.† | ||
The sequence of the peptidyl thioester prior to cyclization is: propenylcinnamoyl-L-Thr-L-Ala-β-methyl-L-Asp-Gly-β-hydroxy-L-Phe-L-Pro-O-methyl-β-hydroxy-L-Tyr-D-Trp-α-hydroxy-Gly-D-Leu-β-hydroxy-L-Leu. The β-methyl-L-Asp is formed by a glutamate mutase encoded within the gene cluster. The β-hydroxy-L-Phe, O-methyl-β-hydroxy-L-Tyr, and β-hydroxy-Leu residues are formed by a single P450 enzyme that catalyzes stereospecific β-hydroxylation of aminoacyl building blocks, thioester-linked to peptidyl carrier protein domains within the NRPS.16,17 The α-(S)-hydroxyglycine residue is formed by a flavin-dependent monooxygenase.18 The D-Trp and D-Leu residues are generated by standard epimerase domains embedded within NRPS modules. The chain-terminating thioesterase (TE) has an additional epimerase activity that forms the final β-hydroxy-D-leucine residue from its L-isomer prior to macrocycle formation.19
While all CCNPs feature ortho-substituted cinnamates, slight differences between PKS systems give variations in chain length, functionalization, and double bond geometry.20,21 In a few cases, the substituted cinnamoyl chain is modified by a cytochrome P450.1,22 Biological activities of CCNPs are determined by the peptide macrocycle and by the precise structure of the cinnamoyl chain.23
The amphotericin B producer Streptomyces nodosus has a chromosomal region that is almost identical to biosynthetic gene cluster for skyllamycin A. However, the S. nodosus cluster also includes a gene for P450Sky2, a unique cytochrome P450 that has no counterpart in the well-characterized skyllamycin producer Streptomyces sp Acta 2897.13 Activation of the silent S. nodosus cluster led to production of a lipopeptide with a molecular mass of 1498.6 g mol−1, which is 16 mass units greater than that of skyllamycin A.24 These observations suggested that P450Sky2 inserts an extra oxygen atom during biosynthesis of this lipopeptide analogue. The position of this modification was unknown.
To gain further insights into biosynthesis of skyllamycin lipopeptides, we investigated the new analogue synthesised by S. nodosus, which we term oxy-skyllamycin. Purification and structural analysis revealed an epoxide in the cinnamoyl chain. Targeted gene disruption established that P450Sky2 catalyzes epoxide formation. The P450Sky2 enzyme was overproduced and a high-resolution crystal structure was obtained. Our results pave the way for future P450Sky2 mutagenesis and combinatorial chemistry approaches to generate new classes of bioactive compounds including much needed anticancer drugs and antibiotics.
Experimental
Purification of oxy-skyllamycin analogue
The skyllamycin analogue was extracted from cultures of amphotericin-deficient S. nodosus ΔamphI pIAGO-LuxR, as described previously.24 Purification of the directly extracted material by HPLC was carried out using a Varian ProStar system with a semi-preparative Agilent Zorbax SB-C18 column (9.4 × 150 mm, 5 mm). For initial purification, gradients were generated by mixing solvent A, 0.1% (v/v) formic acid in H2O, and solvent B, 0.1% (v/v) formic acid in methanol. The column was equilibrated with 50% B and loaded with 500 μl of crude lipopeptide extract in methanol. Components were separated by applying a gradient of 60% to 90% B over 35 min with a flow rate of 4 mL per min. Fractions were collected and those containing oxy-skyllamycin were concentrated in a SpeedVac vacuum centrifuge. Several runs were required to obtain sufficient material. The peptide was purified further by a second HPLC process using a different solvent system: 0.1% (v/v) formic acid in H2O for solvent A and 0.1% (v/v) formic acid in acetonitrile for solvent B. The gradient was 50% to 70% B over 35 min at a 4 mL per min flow rate. In this manner, approximately 3 mg of oxy-skyllamycin was obtained per litre of S. nodosus production culture.Mass spectrometry
Mass spectrometry was carried out using an Agilent 6546 series Q-TOF LC/MS system equipped with an Agilent Jetstream electrospray ionisation source, coupled to an Agilent 1260 infinity prime II LC system. Chromatography was carried out using an Agilent InfinityLab Poroshell 120 SB-C18 column, 2.1 × 50 mm. Solvent A was 0.1% (v/v) formic acid in H2O, solvent B was 0.1% (v/v) formic acid in acetonitrile. These were used to form a linear gradient in which the acetonitrile concentration increased from 10% to 90% over 5 minutes at a flow rate of 0.6 mL min−1. The sample injection volume was 1 μL.A small sample of authentic skyllamycin A was kindly provided by Professor Roderich Süssmuth. Cleavage of skyllamycin A and isolated oxy-skyllamycin was carried out as described by Pohle and co-workers.13 Approximately 0.1 mg of each peptide was dissolved in 500 μl of a mixture of acetonitrile, water and concentrated NH4OH (49![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :49:2). Each suspension was left at room temperature for 4 hours then dried in a SpeedVac vacuum centrifuge. For MS–MS analysis of acyl-octapeptide products were fragmented at collision energies of 20 eV and 40 eV.
:49:2). Each suspension was left at room temperature for 4 hours then dried in a SpeedVac vacuum centrifuge. For MS–MS analysis of acyl-octapeptide products were fragmented at collision energies of 20 eV and 40 eV.
To display the difference in skyllamycins produced by pIAGO-LuxR transformants of S. nodosus NM and S. nodosus NM ΔP450Sky2, extracted ion chromatograms were generated for skyllamycin A and oxy-skyllamycin. These show elution of skyllamycin A ions [(M + H)+ = 1483.6780; (M + Na)+ = 1505.6600] or oxy-skyllamycin A ions [(M + H)+ = 1499.6729; (M + Na)+ = 1521.6549] over time.
NMR spectroscopy
Approximately 2 mg of the purified oxy-skyllamycin was dissolved in 0.65 mL deuterated methanol (CD3OD) and filtered. One-dimensional (1H), and two-dimensional (1H–1H-COSY, 1H–13C-HSQC and TOCSY) spectra were collected using an Agilent DD2 500 MHz console NMR spectrometer equipped with a OneNMR probe. Spectra were recorded at 25 °C.Inactivation of the S. nodosus NM P450Sky2 gene
The gene for P450Sky2 includes two internal Bgl II sites. A chromosomal region of 3211 bp including this gene was amplified by PCR with primers SkyF1 5′ GAGTTCGTCGACATCCAGGTTGCG 3′ and SkyR1 5′ GATCAAGCTTCTGCAGGGCATGCACGTCCGCAAGGGCATTC 3′. The PCR product was digested with Bam HI and Hin dIII and cloned into pUC118. The resulting plasmid pUC118-P450Sky2 was digested with Bgl II and religated to generate pUC118-P450Sky2ΔBgl II. This plasmid contained a P450Sky2 gene with a 591 bp in-frame internal deletion, along with upstream and downstream flanking sequences. The 2620 bp insert from this plasmid was excised with Bam HI and Pst I and cloned into KC-UCD1 digested with the same enzymes. Positive phage clones were identified by PCR with primers SChk1 5′ CTGGATGTTCAGCGACGAGTACAC 3′ and SChk2 5′ GACGTCCGGGAAACGTTCGAGGAC 3′. The KC-P4502Sky2ΔBgl II phage was used to replace the P450Sky2 gene with an inactive copy. The details of the gene replacement method have been described previously.25P450Sky2 cloning, heterologous expression, protein purification and spectral characterization
The NCBI accession number for the P450Sky2 protein is WP_052454353. The gene was amplified from S. nodosus genomic DNA by PCR with primers P450F3 5′ AGAGCACATATGGCAGCCGGCGGAGCACTTTC 3′ and P450R 5′ GTACAAGCTTCTGCGGCTGAGGAATGTCAG 3′. In the amplified gene, the P450F3 primer creates an Nde I site (5′ CATATG 3′) overlapping the start codon. The P450R primer is complementary to the sequence just downstream from the stop codon of the P450 gene; it introduces a Hin dIII restriction site (5′ AAGCTT 3′). The amplified DNA was purified using a QIAEx kit, digested with Nde I and Hin dIII, and cloned between the Nde I and Hin dIII sites of the expression vector pET28. The resulting construct gave a recombinant form of the protein with an N-terminal hexahistidine tag.The plasmid containing the gene for P450Sky2 was transformed into E. coli BL21 (DE3) and E. coli C41 (DE3) cells. Both host strains gave soluble recombinant protein, C41 (DE3) was used to obtain P450Sky2 for crystallization. A single transformant colony was used to inoculate Luria–Bertani media containing 50 μg mL−1 kanamycin and grown overnight at 37 °C and 220 rpm. About 10 mL of overnight culture was added to 1 L Terrific Broth media supplemented with 50 μg mL−1 kanamycin, 125 μg mL−1 thiamine and trace metals (50 μM FeCl3, 20 μM CaCl2, 10 μM MnCl2, 10 μM ZnCl2, 2 μM CoCl2, 2 μM CuCl2 and 2 μM NiSO4). Cultures were grown at 37 °C and 200 rpm. When the culture reached A600 ∼1.5, the temperature was reduced to 25 °C and induced with the addition of isopropyl 1-thio-D-galactopyranoside (1 mM final concentration). To increase synthesis of heme, the prosthetic group for P450Sky2, 30 μg mL−1 δ-aminolevulinic acid was added to each flask. The cells were grown for 48 hours and harvested by centrifugation.
Cell pellets were resuspended in buffer A (50 mM potassium phosphate buffer pH 7.5, 100 mM NaCl and 10 mM imidazole) and lysed using microfluidizer. This lysate was then centrifuged at 15000 rpm for 1 hour at 4 °C. The supernatant was loaded on a previously equilibrated Ni2+-nitrilotriacetic acid agarose column and was washed with buffer A containing 35 mM imidazole. The protein was eluted with buffer A containing 200 mM imidazole. The eluted protein was concentrated, and buffer exchanged into buffer B (50 mM potassium phosphate buffer pH 7.5 and 100 mM NaCl). The protein was further purified using S200 Sephacryl column in buffer B.
A Cary 300 UV-visible spectrophotometer was used to record UV-visible spectra at room temperature. P450Sky2 concentration was determined from reduced CO difference spectrum using a molar extinction coefficient (ε450) of 91 mM−1 cm−1.26
P450Sky2 crystallization
To determine crystallization conditions, high-throughput crystal screening was done using MCGS2 screening kit from Anatrace at room temperature. A SPT labtech Mosquito robot was used to dispense 300 nL drops of 30 mg mL−1 of P450Sky2 and screening solution and mixed in a 1:1 ratio. These were then equilibrated against 100 μL of the screening solution at room temperature. Crystals were observed in wells containing 0.2 M ammonium acetate, 0.1 M Tris·HCl buffer pH 8.5 and 25% (w/v) PEG 3350. Crystals were transferred to cryoprotectant buffer consisting of 0.2 M ammonium acetate, 0.1 M Tris·HCl buffer pH 8.5, 25% (w/v) PEG 3350 and 25% (v/v) glycerol, mounted on nylon loops and flash frozen using liquid nitrogen.
Diffraction data were collected at the Stanford synchrotron radiation beamline 12-2. XDS,27 or MOSFLM28 was used to index and integrate the raw data, and Aimless29 was used for scaling. The structure was determined by molecular replacement with Phaser30 and cytochrome P450 PksS (Protein Data Bank entry 4YZR) as the search model. Phenix31 was used to carry out further refinements.
Results
Prevalence of P450Sky2 gene in streptomycete genomes
BlastP searches revealed that homologues of P450Sky2 are encoded by uncharacterized skyllamycin clusters from other streptomycete genomes (Table S2 and Fig. S4†). These P450s show 88 to 99% sequence identity with P450Sky2. The clusters are incompletely sequenced but their prevalence suggests that many streptomycetes are capable of synthesizing this new skyllamycin analogue. This conservation of the gene for P450Sky2 suggests that the enzyme has an important biological function. P450Sky2 belongs to the CYP107 family. Many members of this group function in secondary metabolism in streptomycetes.32Analysis of oxy-skyllamycin by mass spectrometry
The S. nodosus skyllamycin gene cluster is normally silent under laboratory conditions but moderate yields of the lipopeptide can be obtained by overexpressing a gene for a LuxR-type transcriptional activator in an amphI mutant deficient in amphotericin biosynthesis.24 Oxy-skyllamycin was purified by two chromatographic HPLC steps as described in Experimental procedures (Fig. 2). Skyllamycin A and oxy-skyllamycin samples were analysed by LC-MS and MS–MS. Skyllamycin A has a mass of 1482.6 Da, whereas oxy-skyllamycin had a mass of 1498.6 Da. | ||
| Fig. 2 HPLC trace of oxy-skyllamycin after purification by two semi-preparative HPLC operations (see Experimental). This analytical run was carried out using a gradient of 60 to 90% methanol in 0.1% formic acid and oxy-skyllamycin was detected at 310 nm. | ||
The Süssmuth group found that NH3 treatment of skyllamycin A cleaves the depsipeptide ester bond and the α-hydroxyglycine residue, to give the acyl octapeptide fragment shown in Fig. 3.13 We repeated the NH3 treatment with skyllamycin A and oxy-skyllamycin. This gave the expected acyl-octapeptide fragment of skyllamycin A, m/z = 1185.5285 ([M + H]+, calculated 1185.5251) (Fig. S5A†). Identical cleavage of oxy-skyllamycin with NH3 gave a fragment that was greater in size by 15.9942 mass units, m/z = 1201.5227 ([M + H]+, calculated 1201.5201) (Fig. S6A†). MS–MS analysis was performed on the two peptides with masses ([M + H]+) of 1185.5 and 1201.5. A summary of the fragment ions identified is shown in Table 1 (full data are shown in Fig. S5 and S6†).
 | ||
| Fig. 3 Structure of linear acyl-octapeptide fragment remaining after cleavage of skyllamycin A with NH3.13 The red lines indicate further cleavages observed during MS–MS.13,14 | ||
| Peptide | b ions | y ions | Origin | Identity |
|---|---|---|---|---|
| Oxy-skyllamycin | 963.4 | Cleavage after O-Me-β-OH-Tyr | b7 – 2H2O | |
| Skyllamycin A | 947.4 | Cleavage after O-Me-β-OH-Tyr | b7 – 2H2O | |
| Oxy-skyllamycin | 915.4 | Cleavage after Thr | y7 | |
| Skyllamycin A | 915.4 | Cleavage after Thr | y7 | |
| Oxy-skyllamycin | 788.3 | Cleavage after Pro | b6 – H2O | |
| Skyllamycin A | 772.3 | Cleavage after Pro | b6 – H2O | |
| Oxy-skyllamycin | 709.3 | Cleavage after β-OH-Phe | b5 – H2O | |
| Skyllamycin A | 693.3 | Cleavage after β-OH-Phe | b5 – H2O | |
| Oxy-skyllamycin | 476.2 | Cleavage after β-OH-Phe | y3 – H2O | |
| Skyllamycin A | 476.2 | Cleavage after β-OH-Phe | y3 – H2O | |
| Oxy-skyllamycin | 288.1 | Cleavage after Thr | b1 | |
| Skyllamycin A | 272.1 | Cleavage after Thr | b1 | |
| Oxy-skyllamycin | 187.1 | O-containing propenylcinnamate | ||
| Skyllamycin A | 171.1 | Propenylcinnamate fragment |
The results were compared with published MS–MS data on naturally occurring skyllamycins A to E and analogs obtained by genetic manipulation of Strep. acta13–15 (Fig. S6†). For skyllamycin A and the S. nodosus oxy-skyllamycin, the spectra contained identical y7 and y3 fragment ions that retain the C-terminal tryptophan amide. The N-terminal b1, b5, b6, and b7 fragment ions could be identified in MS–MS spectra for the skyllamycin A fragment. In the S. nodosus oxy-skyllamycin spectra, each of these signals was replaced by an ion with a mass that was greater by 16 mass units. The propenylcinnamate fragment of skyllamycin A was identified as an ion with a mass of 171.1 whereas the corresponding fragment of oxy-skyllamycin has a mass of 187.1. These results clearly demonstrate that the extra oxygen atom in the new S. nodosus skyllamycin is located in the cinnamoyl unit.
Analysis by NMR spectroscopy
The purified oxy-skyllamycin was characterized further by nuclear magnetic resonance (NMR) spectroscopy. As an aid to the interpretation of the current data it is noted that skyllamycin A has been extensively characterized by NMR.10 NMR analysis has also been carried out for skyllamycin C,15 an analogue of skyllamycin A in which the C2–C3 trans double bond in the cinnamate chain is reduced and a methyl group is absent from the peptide backbone (Fig. S6†). The NMR spectra for oxy-skyllamycin were collected in deuterated methanol (facilitating analysis by removing signals for protons attached to heteroatoms) and were compared with reported 1H and 13C spectral data for skyllamycin A.10–12,15Overall, the proton NMR spectrum of the new S. nodosus oxy-skyllamycin was remarkably similar to that of skyllamycin A (Table S3 and Fig. S7–S10†). Compared to data obtained by Giltrap using the same solvent,11,12 most signals for corresponding fragments were within a 0.1 ppm difference (Table S3†). Outliers included the aromatic β-OH-O-Me-Tyr signals, which for oxy-skyllamycin were coincident, the D-Trp aromatic signals, and an α-CH that resonated at slightly different frequencies (ca. ±0.1 ppm). However, the obvious difference was the absence of signals at 6.39, 5.77 and 1.51 ppm, which result from the propenyl hydrogen atoms at C-10, C-11, and C-12 of the cinnamoyl chain of skyllamycin A. The absent signals were replaced by resonances at 3.95, 2.76–2.79, and 0.49 ppm for oxy-skyllamycin. These latter spectral differences are consistent with a chemical change in skyllamycin A's propenyl unit to a methyl substituted epoxide. Unfortunately, the signals at 3.95, 2.76–2.79 and 0.49 ppm were broad and coupling constants could not be extracted. However, the relationship between the hydrogen atoms from this three-carbon unit was confirmed using 1H–1H-gCOSY and TOCSY experiments (Fig. S11–S20†). Then carbon resonances for protonated carbon atoms in oxy-skyllamycin were identified from the 1H–13C-HSQC spectrum. Taken together with the MS–MS data, these results clearly show that oxy-skyllamycin contains an epoxide in the cinnamoyl chain of the lipodepsipeptide natural product (Fig. 4). Since the coupling constants for the β-methyl epoxide unit could not be extracted the relative stereochemistry (i.e., cis or trans) of the disubstituted epoxide cannot be determined. However, the pronounced upfield shift (0.49 ppm) of the methyl group is notable and is likely consistent with a phenyl-ring shielding effect for a cis-epoxide stereochemistry.
 | ||
| Fig. 4 Structure of oxy-skyllamycin from S. nodosus. The molecule is identical to skyllamycin A except for the C10–C11 epoxide in the substituted cinnamoyl chain. | ||
Identification of the gene responsible for epoxide formation
The S. nodosus oxy-skyllamycin cluster includes genes for two cytochrome P450 enzymes. One is a homologue of the P450 involved in formation of the β-hydroxy-Leu, β-hydroxy-Phe and β-hydroxy-O-methyl Tyr residues that appear in the peptide macrocycle of skyllamycin A.16,17 The function of the second P450, P450Sky2, was unknown. Gene disruption was carried out to investigate whether P450Sky2 catalyzes formation of the epoxide in oxy-skyllamycin.The chromosomal P450Sky2 gene was replaced with an inactive version containing a 591 base-pair internal deletion, as detailed in the Experimental section. Gene replacement was achieved in S. nodosus NM, which is proficient in amphotericin biosynthesis but synthesizes modest amounts of oxy-skyllamycin after activation of the BGC. Two independent P450Sky2 deletion mutants were obtained (Fig. 5).
 | ||
| Fig. 5 Inactivation of P450Sky2 gene. A. A 3211 bp region containing the P450Sky2 gene was amplified by PCR and cloned into a pUC118 plasmid. An internal Bgl II fragment of 591 bp was deleted from the P450 gene to create an in-frame deletion. KC-UCD1 mediated gene replacement was carried out as described.40 B. Analysis of gene replacement mutants by PCR. The P450Sky2 region was amplified by PCR with primers SChk1 and SChk2. The original strain gave a PCR product of 1109 bp (lane 1) whereas gene replacement mutants gave a product of 518 bp (lanes 2 and 3). | ||
Both S. nodosus NM and S. nodosus NM ΔP450Sky2 were transformed with pIAGO-LuxR. Methanol extracts of production cultures were analysed by HPLC and LC-MS. The results revealed that inactivation of P450Sky2 completely abolished synthesis of oxy-skyllamycin and resulted in production of low but detectable amounts of skyllamycin A. Extracted ion chromatograms for oxy-skyllamycin and skyllamycin A are shown in Fig. 6. These results show that P450Sky2 inactivation results in production of skyllamycin A (Fig. 6A) and loss of oxy-skyllamycin production (Fig. 6C). In contrast, the parent strain S. nodosus NM produces oxy-skyllamycin (Fig. 6D) but not skyllamycin A (Fig. 6B).
 | ||
| Fig. 6 Analysis of skyllamycins from NM and NM ΔP450Sky2 strains containing pIAGO-LuxR. Panels C and D shows the oxy-skyllamycin extracted ion chromatograms obtained after analysis of extracts from S. nodosus NM ΔP450Sky2 pIAGO-LuxR and S. nodosus NM pIAGO-LuxR, respectively. S. nodosus NM pIAGO-LuxR produces oxy-skyllamycin, S. nodosus NM ΔP450Sky2 pIAGO-LuxR does not. Panels A and B show the skyllamycin A extracted ion chromatograms obtained after analysis of extracts from S. nodosus NM ΔP450Sky2 pIAGO-LuxR and S. nodosus NM pIAGO-LuxR, respectively. S. nodosus NM ΔP450Sky2 pIAGO-LuxR produces skyllamycin A, S. nodosus NM pIAGO-LuxR does not. | ||
P450Sky2 overproduction and characterization
Recombinant P450Sky2 enzyme was produced in Escherichia coli and purified to homogeneity as described in the Experimental section. The UV-visible spectrum of oxidized P450Sky2 (Fig. 7) has a characteristic low-spin Soret absorption at 418 nm, and α- and β-bands at 566 nm and 538 nm, respectively. The reduced-CO difference spectrum displays a characteristic P450 spectral maximum with a Soret peak at 447 nm (Fig. 7). | ||
| Fig. 7 Absolute UV-visible spectra of 8.7 μM substrate-free P450Sky2 in 50 mM potassium phosphate buffer pH 7.5. As expected, a low-spin spectrum is observed for oxidized substrate-free protein. Upon bubbling with CO followed by sodium dithionite reduction, characteristic P450 band is formed with a fraction of P420. Formation of P420 in substrate-free P450s is not uncommon. | ||
Overall P450Sky2 structural analysis
The P450Sky2 crystal structure was solved and refined at 1.43 Å (Table S4†). Crystals belong to space group P1211 with a monomer in the asymmetric unit. P450Sky2 exhibits the traditional P450 triangular fold (Fig. 8A). The heme iron is axially coordinated to the protein with a conserved cysteine (Cys354). The heme is sandwiched between the I and L helices. Right on top of I helix are the F and G helices which are connected by FG loop and form a wide-open active site. Compared to other P450s this disordered loop has a few missing amino acids. | ||
| Fig. 8 Crystal structure of P450Sky2. (A) P450Sky2 exhibits the traditional P450 fold consisting of α-helical and β-sheet domains which is observed in other P450s. (B) 2Fo-Fc electron density map contoured at 1.0σ with water-bound hexacoordinated iron heme and octanoic acid in the active site. | ||
A water molecule, at a distance of 2.3 Å, is coordinated to the heme iron (Fig. 8B) which is characteristic of a substrate-free P450. Upon closer investigation of the active site, a long continuous Y-shaped electron density above this water molecule was observed. This is clearly a fatty acid molecule from E. coli cells33 with the 16-carbon palmitic acid being the most likely candidate. However, only eight carbons could be modelled so octanoic acid was used in the refinement (Fig. 8B) with the carboxylate group close to the water coordinated heme iron. The distance between heme iron and carboxylate oxygen of octanoic acid is 4.1 Å. The presence of similarly positioned fatty acid in the active site has been found in other P450 structures.34,35 This very likely has no functional significance. Rather, the open and mostly hydrophobic substrate access channel of different P450s can bind a variety of nonpolar molecules and in the case of P450Sky2, it is a fatty acid.
A detailed view of the amino acid side chains lining the active site is shown in Fig. 9.
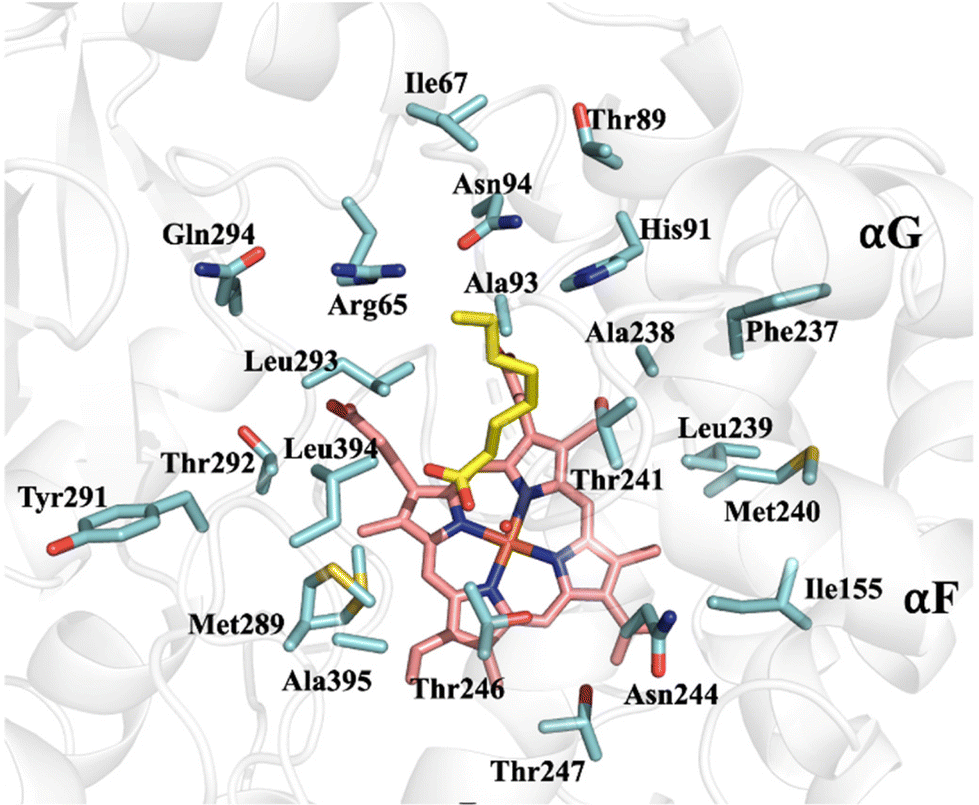 | ||
| Fig. 9 Active site residues in P450Sky2. In the Avm43, DmlF and EpcF epoxidases, 15 of these 22 amino acids are conserved, 5 are conservatively substituted, and Tyr291 is replaced with Asn (see Fig. S21–S23†). In the P450Sky2 homologues encoded in uncharacterized skyllamycin clusters (Table S2†), 20 of these amino acids are conserved, four of the five homologues had the substitution Ala395 → Val and one had Thr292 → Ala. | ||
Molecular modeling of the skyllamycin A – P450Sky2 complex
Considerable effort over the coming years will be required to investigate fully the enzymatic activity of P450Sky2 in vitro. It is unclear at this time whether the natural substrate is skyllamycin A, or an earlier intermediate such as propenyl-cinnamoyl-ACP or a propenyl-cinnamoyl-peptidyl-PCP (Fig. S1 and S2†). Attempts were made to investigate whether P450Sky2 binds or epoxidizes skyllamycin A. These experiments could not be completed because of insufficient quantities of substrate material, and neither binding nor in vitro enzymatic activity have been convincingly demonstrated so far. Substrate binding might be impaired by the fatty acid bound in the active site. Reconstituting enzyme activity may require identification of the correct redox partner systems.While the preliminary binding studies were inconclusive, it was possible to model skyllamycin A into the wide-open active site cavity of P450Sky2 (Fig. 10A) using Pymol (pymol.org). 4,5-De-epoxypimaricin bound PimD36 (PDB ID: 2XBK) was used as the starting model because PimD, like P450Sky2, epoxidizes its substrate. The C10–C11 alkene of skyllamycin A (Fig. 1) was aligned with the C4–C5 alkene of 4,5-de-epoxypimaricin in the active site. The distance between the heme iron and modelled C10–C11 of skyllamycin A is 4 Å and 5 Å, respectively (Fig. 10B). As shown in Fig. 10A, the whole molecule of skyllamycin A (spheres) fits well in the active site of P450Sky2 suggesting that skyllamycin A itself can enter the protein and form oxy-skyllamycin upon catalysis. Since, the substrate was modelled in the open structure of P450Sky2, we cannot say at this time that this is true representation of a protein–substrate complex since P450s can undergo substantial structural changes when a substrate binds.
 | ||
| Fig. 10 Skyllamycin A modeled into the P450Sky2 structure. (A) P450Sky2 exhibits a wide active site cavity in which skyllamycin A (spheres) can be readily docked. (B) Distance between heme-Fe and modeled C10–C11 of skyllamycin A is 4 Å and 5 Å, respectively. | ||
Discussion
Oxy-skyllamycin is the sixth naturally occurring skyllamycin to be identified and could be named skyllamycin F. The way is now open to investigate the effect of the epoxide on the various biological activities. For example, further genetic engineering of S. nodosus should yield epoxide-containing analogues of the biofilm-disrupting agent skyllamycin B.At least four other CCNPs have been structurally characterized in which the cinnamoyl chain contains a C10–C11 epoxide (Fig. S21†). These are NC1,37 atrovimycin1 nyuzenamide C38 and epoxinnamide.39 In the case of atrovimycin, the epoxide is hydrolysed to give a vicinal dihydroxylated cinnamic acyl chain.1Streptomyces HS-NF-1222A produces epoxy cinnamoyl-threonine, a compound that has weak acaricidal activity40 (Fig. S22†). Homologues of P450Sky2 are present in the BGCs for atrovimycin (Avm43),1 nyuzenamide C (DmlF),38 and epoxinnamide (EpcF)39 (Fig. S23†).
This work provides the first experimental evidence that P450Sky2 functions as an epoxide-forming P450. The crystal structure is the first for a P450 that acts on the substituted cinnamoyl chain of a lipopeptide. Molecular modeling indicated that skyllamycin A can associate with the P450 in a structural pose that orientates its propenyl substituent group towards the heme co-factor. However, further work will be required to identify the timing of the epoxidation event in the biosynthetic pathway and redox partners for P450Sky2.
The P450Sky2 structure may be of interest in areas outside natural product biosynthesis. Epoxide-forming P450s are exploited for conversion of substrates such as β-methylstyrene to chiral epoxides that are useful for synthesis of valuable pharmaceuticals and fine chemicals.41–43 Epoxidases also function in mammalian metabolism of plant-derived phenylpropenes, which are extensively used as flavour and fragrance compounds. Examples include anethole and β- and α-asarones. These compounds have many potentially valuable biological activities but conversion of their propenyl side chains to epoxides is associated with carcinogenicity.44–46
Conclusions
In summary, we have purified and determined the structure of oxy-skyllamycin, a new skyllamycin analogue from the amphotericin B producer, S. nodosus. Oxy-skyllamycin differs from skyllamycin A in that it contains an epoxide in the substituted cinnamoyl lipid side chain. Targeted gene disruption revealed that the P450Sky2 enzyme acts on the propenyl-cinnamoyl moiety to form a C10–C11 epoxide. This confirms the proposed functions of homologous cytochrome P450s encoded in BGCs for other epoxidized cinnamoyl-containing peptides. The high-resolution structure of P450Sky2 will assist efforts to investigate how epoxide stereochemistry is determined in these natural products.Data availability
Structure factors and coordinates have been deposited with the Protein Data Bank under accession code 8FZ8 and will be released upon publication. NMR data are lodged with Biological Magnetic Resonance Data Bank, ID = BMRbig86.Author contributions
P. C. and D. C. L. conceptualization; Y. S., J. A. A., V. C. M., H. M., M. H., J. M., P. E., Y. O., P. C., D. C. L., T. L. P. investigation; S. L. K. funding acquisition; P. C., S. L. K., T. L. P., supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Y. S. was supported by a Ph. D. studentship from the -China Scholarship Council scheme. M. H. received a Ph. D. studentship from the Irish Research Council for Science Engineering and Technology (IRCSET). Mass spectrometry facilities were funded as part of the Comprehensive Molecular Analysis Platform (CMAP) initiative under the Science Foundation Ireland (SFI) Research Infrastructure Programme, reference 18/RI/5702, and supported by UCD School of Chemistry. P. E. gratefully acknowledges support from SFI (12/RC/2275_P2 and 16/RC/3889). Research at Swansea University was supported by the European Regional Development Fund/Welsh European Funding Office via the BEACON project (S. L. K.). The crystallographic work was supported by National Institute of Health Grants GM131920 (T. L. P.). The authors thank SSRL beamline staff for their assistance during remote X-ray diffraction data collection.References
- Q. Liu, Z. Liu, C. Sun, M. Shao, J. Ma, X. Wei, T. Zhang, W. Li and J. Ju, Org. Lett., 2019, 21, 2634–2638 CrossRef CAS PubMed.
- S. Omura, D. Van der Pyl, J. Inokoshi, Y. Takahashi and H. Takeshima, J. Antibiot., 1993, 46, 222–228 CrossRef CAS PubMed.
- C. Sun, Z. Yang, C. Zhang, Z. Liu, J. He, Q. Liu, T. Zhang, J. Ju and J. Ma, Org. Lett., 2019, 21, 1453–1457 CrossRef CAS PubMed.
- S. Um, S. H. Park, J. Kim, H. J. Park, K. Ko, H.-S. Bang, S.-K. Lee, J. Shin and D.-C. Oh, Org. Lett., 2015, 17, 1272–1275 CrossRef CAS PubMed.
- Z. Yu, S. Vodanovic-Jankovic, M. Kron and B. Shen, Org. Lett., 2012, 14, 4946–4949 CrossRef CAS PubMed.
- M. Hashimoto, K. Hayashi, M. Murai, T. Fujii, M. Nishikawa, S. Kiyoto, M. Okuhara, M. Kohsaka and H. Imanaka, J. Antibiot., 1992, 45, 1064–1070 CrossRef CAS PubMed.
- M. Bae, H. Kim, K. Moon, S. J. Nam, J. Shin, K. B. Oh and D. C. Oh, Org. Lett., 2015, 17, 712–715 CrossRef CAS PubMed.
- M. R. Ul Karim, Y. In, T. Zhou, E. Harunari, N. Oku and Y. Igarashi, Org. Lett., 2021, 23, 2109–2113 CrossRef PubMed.
- J. Zhu, S. Zhang, D. L. Zechel, T. Paululat and A. Bechthold, ACS Chem. Biol., 2019, 14, 1793–1801 CrossRef CAS PubMed.
- S. Toki, T. Agatsuma, K. Ochiai, Y. Saitoh, K. Ando, S. Nakanishi, N. A. Lokker, N. A. Giese and Y. Matsuda, J. Antibiot., 2001, 54, 405–414 CrossRef CAS PubMed.
- A. M. Giltrap, F. P. J. Haeckl, K. L. Kurita, R. G. Linington and R. J. J. Payne, Chem. – Eur. J., 2017, 23, 15046–15049 CrossRef CAS PubMed.
- A. M. Giltrap, F. P. J. Haeckl, K. L. Kurita, R. G. Linington and R. J. J. Payne, J. Org. Chem., 2018, 83, 7250–7270 CrossRef CAS PubMed.
- S. Pohle, C. Appelt, M. Roux, H.-P. Fiedler and R. D. Süssmuth, J. Am. Chem. Soc., 2011, 133, 6194–6205 CrossRef CAS PubMed.
- J. Bracegirdle, P. Hou, V. V. Nowak, D. F. Ackerley, R. A. Keyzers and J. G. Owen, J. Nat. Prod., 2021, 84, 2536–2543 CrossRef CAS PubMed.
- G. Navarro, A. T. Cheng, K. C. Peach, W. M. Bray, V. S. Bernan, F. H. Yildiz and R. G. Linington, Antimicrob. Agents Chemother., 2014, 58, 1092–1099 CrossRef PubMed.
- S. Uhlmann, R. D. Süssmuth and M. J. Cryle, ACS Chem. Biol., 2013, 8, 2586–2596 CrossRef CAS PubMed.
- K. Haslinger, C. Brieke, S. Uhlmann, L. Sieverling, R. D. Süssmuth and M. J. Cryle, Angew. Chem., Int. Ed., 2014, 53, 8518–8522 CrossRef CAS PubMed.
- V. Schubert, F. Di Meo, P. L. Saaidi, S. Bartoschek, H. P. Fiedler, P. Trouillas and R. D. Süssmuth, Chem. – Eur. J., 2014, 20, 4948–4955 CrossRef CAS PubMed.
- J. Yu, J. Song, C. Chi, T. Liu, T. Geng, Z. Cai, W. Dong, C. Shi, X. Ma, Z. Zhang, X. Ma, B. Xing, H. Jin, L. Zhang, S. Dong, D. Yang and M. Ma, ACS Catal., 2021, 11, 11733–11741 CrossRef CAS.
- J. Shi, Y. Shi, J. C. Li, W. Wei, Y. Chen, P. Cheng, C. L. Liu, H. Zhang, R. Wu, B. Zhang, R. H. Jiao, S. Yu, Y. Liang, R. X. Tan and H. M. Ge, J. Am. Chem. Soc., 2022, 144, 7939–7948 CrossRef CAS PubMed.
- Z. Deng, J. Liu, T. Li, H. Li, Z. Liu, Y. Dong and W. Li, Angew. Chem., Int. Ed., 2021, 60, 153–158 CrossRef CAS PubMed.
- J. Shi, C. L. Liu, B. Zhang, W. J. Guo, J. Zhu, C.-Y. Chang, E. R. Zhao, R. H. Jiao, R. X. Tan and H. M. Ge, Chem. Sci., 2019, 10, 4839–4846 RSC.
- Z. Yang, C. Sun, Z. Liu, Q. Liu, T. Zhang, J. Ju and J. Ma, J. Nat. Prod., 2020, 83, 1666–1673 CrossRef CAS PubMed.
- P. Sweeney, C. D. Murphy and P. Caffrey, Appl. Microbiol. Biotechnol., 2016, 100, 1285–1295 CrossRef CAS PubMed.
- S. B. Zotchev and P. Caffrey, Methods Enzymol., 2009, 459, 243–258 CAS.
- T. Omura and R. Sato, J. Biol. Chem., 1964, 239, 2370–2378 CrossRef CAS PubMed.
- W. Kabsch, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 125–132 CrossRef CAS PubMed.
- H. Powell, O. Johnson and A. Leslie, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 69, 1195–1203 CrossRef PubMed.
- M. D. Winn, C. C. Ballard, K. D. Cowtan, E. J. Dodson, P. Emsley, P. R. Evans, R. M. Keegan, E. B. Krissinel, A. G. Leslie and A. McCoy, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 235–242 CrossRef CAS PubMed.
- A. J. McCoy, R. W. Grosse-Kunstleve, P. D. Adams, M. D. Winn, L. C. Storoni and R. J. Read, J. Appl. Crystallogr., 2007, 40, 658–674 CrossRef CAS PubMed.
- P. D. Adams, P. V. Afonine, G. Bunkóczi, V. B. Chen, N. Echols, J. J. Headd, L.-W. Hung, S. Jain, G. J. Kapral and R. W. G. Kunstleve, Methods, 2011, 55, 94–106 CrossRef CAS PubMed.
- F. C. Mnguni, T. Padayachee, W. Chen, D. Gront, J.-H. Yu, D. R. Nelson and K. Syed, Int. J. Mol. Sci., 2020, 21, 4814 CrossRef CAS PubMed.
- E. E. Allen and D. H. Bartlett, J. Bacteriol., 2000, 182, 1264–1271 CrossRef CAS PubMed.
- T. Fujishiro, O. Shoji, S. Nagano, H. Sugimoto, Y. Shiro and Y. Watanabe, J. Biol. Chem., 2011, 286, 29941–29950 CrossRef CAS PubMed.
- S. Han, T. V. Pham, J. H. Kim, Y. R. Lim, H. G. Park, D. Jeong, C. H. Yun, Y. J. Chun, L. W. Kang and D. Kim, Biochem. Biophys. Res. Commun., 2017, 482, 902–908 CrossRef CAS PubMed.
- P. M. Kells, H. Ouellet, J. Santos-Aberturas, J. F. Aparicio and L. M. Podust, Chem. Biol., 2010, 17, 841–851 CrossRef CAS PubMed.
- M. Liu, N. Liu, F. Shang and Y. Huang, Eur. J. Org. Chem., 2016, 3943–3948 CrossRef CAS.
- J. S. An, M.-S. Kim, J. Han, S. C. Jang, J. H. Im, J. Cui, Y. Lee, S.-J. Nam, J. Shin, S. K. Lee, Y. J. Yoon and D.-C. Oh, J. Nat. Prod., 2022, 85, 804–814 CrossRef CAS PubMed.
- S. Kang, J. Han, S. C. Jang, J. S. An, I. Kang, Y. Kwon, S.-J. Nam, S. H. Shim, J.-C. Cho, S. K. Lee and D.-C. Oh, Mar. Drugs, 2022, 20, 455 CrossRef CAS PubMed.
- H. Qi, Z. Ma, Z. L. Xue, Z. Yu, Q. Y. Xu, H. Zhang, X. P. Yu and J. D. Wang, Nat. Prod. Res., 2020, 34, 2080–2085 CrossRef CAS PubMed.
- A. Li, J. Liu, S. Q. Phama and Z. Li, Chem. Commun., 2013, 49, 11572–11574 RSC.
- P. Zhao, J. Chen, N. Ma, J. Chen, X. Qin, C. Liu, F. Yao, L. Yao, L. Jin and Z. Cong, Chem. Sci., 2021, 12, 6307–6314 RSC.
- C. Zhang, P.-X. Liu, L.-Y. Huang, S.-P. Wei, L. Wang, S.-Y. Yang, X.-Q. Yu, L. Pu and Q. Wang, Chem. – Eur. J., 2016, 22, 10969–10975 CrossRef CAS PubMed.
- A. T. Cartus, S. Stegmuoller, N. Simson, A. Wahl, S. Neef, H. Kelm and D. Schrenk, Chem. Res. Toxicol., 2015, 28, 1760–1773 Search PubMed.
- S. G. Kim, A. Liem, B. C. Stewart and J. A. Miller, Carcinogenesis, 1999, 20, 1303–1307 CrossRef CAS PubMed.
- X.-L. Feng, Y. Yu, D.-P. Qin, H. Gao and X.-S. Yao, RSC Adv., 2015, 5, 5173–5182 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ob00178h |
| This journal is © The Royal Society of Chemistry 2024 |