
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMeasuring local pH at interfaces from molecular tumbling: A concept for designing EPR-active pH-sensitive labels and probes†
Maxim A.
Voinov
 ,
Nicholas
Nunn
,
Roshan
Rana
,
Atli
Davidsson
,
Alex I.
Smirnov
and
Tatyana I.
Smirnova
*
,
Nicholas
Nunn
,
Roshan
Rana
,
Atli
Davidsson
,
Alex I.
Smirnov
and
Tatyana I.
Smirnova
*
Department of Chemistry, North Carolina State University, 2620 Yarbrough Drive, Raleigh, NC 27695, USA. E-mail: tismirno@ncsu.edy
First published on 18th April 2024
Abstract
Molecular probes and indicators are broadly employed for pH measurements in bulk media and at interfaces. The underlying physical principle of pH measurements of most of these probes is based on a change in the electronic structure that, for example, results in a shift of the emission peak of the fluorescence probes, changes in NMR chemical shifts due to the affected electronic shielding, or magnetic parameters of pH-sensitive nitroxides as measured by EPR. Here we explore another concept for measuring local protonation state of molecular tags based on changes in rotational dynamics of electron spin-bearing moieties that are readily detected by conventional continuous wave X-band EPR. Such changes are especially pronounced at biological interfaces, such as lipid bilayer membranes, due to the probe interactions with adjacent charges and polarizable dipoles. The concept was demonstrated by synthesizing a series of pH-sensitive nitroxides and spin-labelled phospholipids. EPR spectra of these newly synthesized nitroxides exhibit relatively small – about 0.5 G – changes in isotropic nitrogen hyperfine coupling constant upon reversible protonation. However, spin-labelled phospholipids incorporated into lipid bilayers demonstrated almost 6-fold change in rotational correlation time upon protonation, readily allowing for pKa determination from large changes in EPR spectra. The demonstrated concept of EPR-based pH measurements leads to a broader range of potential nitroxide structures that can serve as molecular pH sensors at the desired pH range and, thus, facilitates further development of spin-labelling EPR methods to study electrostatic phenomena at chemical and biological interfaces.
Introduction
Effect of reversible protonation on EPR spectra of free radicals bearing protonatable groups was first reported in the middle of 1960s.1–3 It was shown that the protonated and non-protonated radical species can be readily differentiated by their EPR spectra due to changes in magnetic parameters, such as isotropic nitrogen hyperfine coupling constant Aiso and isotropic g-factor giso, upon protonation. This observation suggested that EPR spectroscopy of free radicals could be employed for studying proton transfer and related phenomena in chemical and biological systems. However, practical applications of the observed effect required designing and synthesizing free radicals that, in addition to measurable changes in magnetic parameters upon protonation, would also be stable under experimental conditions. Nitroxide-type free radicals are good candidates for such applications. Indeed, some of these nitroxides have been shown to undergo reversible protonation at the oxygen atom of the N–O˙ moiety in concentrated acids or non-aqueous solutions without the loss of the paramagnetism.4–6 However, in strongly acidic aqueous solutions most of the nitroxides undergo disproportionation to form EPR-silent oxoammonium salt and hydroxylamine7,8 making them unsuitable for experiments in aqueous media. Years later it was discovered that in some nitroxides, and particularly those bearing strongly basic groups, a positive charge resulting from protonation of these groups makes the consecutive protonation of a weakly basic N–O˙ moiety, and, hence, disproportionation, much less favourable. This finding paved the way to design and application of such nitroxides as EPR active pH probes in physicochemical and biophysical studies. For example, 4-amino derivatives of the 2,2,6,6-tetramethylpiperidine nitroxide Tempo were employed for measuring pH gradients across membranes based on the differences in membrane-binding properties of protonated and non-protonated nitroxide species.9,10 However, small effects of protonation on magnetic parameters of the EPR spectra in such nitroxides (ΔAiso = 0.13 G,11 the difference between Aiso of the non-protonated, R˙, and protonated, R˙H+, forms of the nitroxide) severely limited their practical application as EPR pH probes. Nitroxides of the 2-imidazoline series 1 (Fig. 1) show pH-induced spectral changes in physiological pH range and a relatively large – up to ΔAN1 ≈2.40 G and ΔAN3 ≈1.40 G – difference in the nitrogen hyperfine coupling constants of the protonated and non-protonated forms.12 Unfortunately, the EPR spectra of these nitroxides are rather complex due to hyperfine couplings to three non-equivalent nitrogen atoms. This complicates analysis of protonation phenomena. | ||
| Fig. 1 pH-Sensitive 2-imidazoline, 3-imidazoline and 1,3-imidazolidine nitroxides. | ||
Broad applications of protonatable nitroxides in biophysical studies became possible only in the early eighties after two groups of researchers independently discovered the effect of pH on EPR spectra of imidazolidine nitroxides 2 and 4 and 3-imidazoline 3 (Fig. 1).13,14 For these nitroxides the difference between AisoR˙ and AisoR˙H+ was found to be ΔAN ≈1.0 G. Such a large value of ΔAN simplified analysis of the nitroxide protonation phenomena from fast motion EPR spectra consisting of three sharp, equally spaced, hyperfine lines arising from hyperfine coupling to 14N nucleus. Since then, a significant progress was made in both synthesis of pH-sensitive nitroxide spin labels and probes and also development of associated EPR methods for studies of proton transfer and local electrostatic phenomena in a broad range of biological and chemical systems (for reviews see ref. 11 and 15–17).
Today, pH-sensitive EPR probes are finding numerous applications for monitoring local pH in biomedical studies. pH regulation is critical for a myriad of physiological functions like cellular metabolism, blood pH regulation, and a function of the digestive system where a disruption of pH balance is linked to various pathological conditions.18 Despite a significant decay of the EPR signal intensity in biological environment, nitroxides in conjunction with low-frequency EPR spectroscopy and imaging techniques have been shown to enable reproducible and accurate noninvasive pH determination in vitro and in vivo.19–21 The use of pH-sensitive nitroxides in low-field EPR spectroscopy and imaging is based exclusively on the difference in hyperfine constants between protonated and non-protonated forms and has been shown to provide high-precision measurements of pH19,22–24 with accuracy as high as 0.1 pH units.23,25
Nitroxide probes with low pKa values have been used for monitoring acidic pH in the stomach of rodents in experiments in vivo and to follow such changes upon administration of different antacid medications.20,26–28 The use of EPR in tissue with pH homeostasis close to neutral, such as tumor tissues, was enabled by designing probes with higher pKa values, extended half-life in vivo, and by modification of membrane permeability of probes to ensure an extracellular localization.29–31 Such probes have been applied to studies of tumor tissue acidosis in live mice by enabling quantitative visualization of regional changes in extracellular pH associated with altered tumor metabolism.29,32 More recently, such probes were shown to be able to detect an increased extracellular acidification in tumor tissue following treatment of an animal with a metabolism targeting inhibitor.33 Recently, development of pH-sensitive EPR probes moved beyond nitroxide compounds with syntheses of new dual-function trityl radicals capable to report on both pH and O2 concentration in tumor tissues.34–37
pH-Sensitive nitroxides are also finding applications in other fields, such as materials and nanoscience. For example, measurements of pKa of pH-sensitive nitroxides were utilized to evaluate acidic and electrostatic properties of porous materials like mesoporous molecular sieves,38,39 Xerogels,40 nanoporous anodic aluminum oxide (AAO),41 and silica nanoparticles-water interfaces.42
Without risking to move too far off course of the study, we also would like to mention an increased interest in use of pH-sensitive EPR-active compounds as smart control agents in chemical synthesis.43 For example, pH-switchable alkoxyamines have been used to control the polymerization of styrene and butyl acrylate44 and the protonation state of the nitroxide has been shown to influence rates of nitroxide-mediated polymerization (NMP).45–47
As illustrated above, continued synthetic efforts over the last two decades yielded a selection of pH-sensitive probes and labels covering a broad pH range. However, analysis of the literature data shows that most if not all pH-sensitive nitroxides synthesized to this date are based on modifications of the same two core structures – 3-imidazoline 3 and 1,3-imidazolidine 4 (Fig. 1). Undoubtedly, there is an explanation for that: the protonatable amidino and tertiary amino functionalities in the structures of 3-imidazoline 3 and 1,3-imidazolidine 4 heterocycles, respectively, are located two σ-bonds away from the EPR-active N–O˙. Such design is close to the optimal one based on several considerations. Firstly, the locations of the protonatable functionalities close to the spin-bearing N–O˙ moiety cause rather significant ΔAN shifts of ∼0.8–1.4 G upon the change in the protonation state of the basic groups. The change in Aiso of about 1 G simplifies accurate quantification of the individual components of EPR spectra corresponding to the protonated or non-protonated forms of the nitroxide, especially when the spin probe/label tumbling rate falls into the fast motion regime. Moving the protonatable functionality further away from the N–O˙ group significantly reduces the ΔAN value (for example, 4-amino-2,2,6,6-tetramethylpiperidine 1-oxyl was found to have ΔAN of only ≈0.13 G (ref. 11)) and, as a result, lowers the accuracy of the pKa determination. We note here that the accuracy of the ΔAN measurement can be significantly improved by using least-squares simulation of EPR spectra (for a description of fitting software see,48–51 for practical applications, see ref. 52 and 53). Secondly, an electron-withdrawing effect of the N–O˙ group at this distance from the protonatable moiety remains to be moderate so the pKa values of probes would fall within the practically useful pKa range (i.e., 3÷8 units of pH). Note that the strong electron withdrawing effect of the N–O˙ group causes a reduction of the intrinsic pKa's of basic functionalities by several units of pKa.14,54 If the protonatable group is located to the N–O˙ moiety closer than in 3 or 4, the pKa of the probe will be too low for practical applications. Taken together, these two factors significantly limit available options in designing new EPR pH probes with desirable properties, such as pKa range, partitioning coefficients between polar and apolar phases, probe binding affinities, etc., that can be tailored to specific applications.
Previously, we introduced phospholipids covalently modified with pH-sensitive nitroxides at the head group as EPR spin probes for studying interfacial electrostatic phenomena in detergent micelles and lipid bilayers.55–57 In such systems these lipid-based probes position the pH-sensitive nitroxide group at the aqueous side of the interfacial region while the lipid tail is inserted into the lipid phase. For lipid bilayers above and below the main phase transition temperature, the probes demonstrated pH-dependent X-band (9.5 GHz) EPR spectra that fall into intermediate or slow-motion regime in both protonated and non-protonated forms. When recorded at intermediate pH values, the spectra of protonated and non-protonated forms of the nitroxides were not fully resolved as in the fast motion limit; however, the changes in the EPR line shapes were significant to allow for accurate determination of a fraction of protonated nitroxide from least-squares simulations assuming a slow-exchange two-component model.55–57 It should be noted here that the changes in EPR line shapes in such systems are primarily caused by a reduction of the tumbling rate of the nitroxide upon protonation rather than effects of the nitroxide protonation state on its magnetic parameters. Indeed, the maximum reported change in the isotropic nitrogen hyperfine coupling constant Aiso upon protonation of nitroxides of ΔAiso = 1.35 G (ref. 11) is significantly smaller than the typical X-band peak-to-peak line widths – about 5 G or more – of the individual spectral components of spin-labelled phospholipids embedded into a lipid bilayer. Such large linewidths make the observations of the nitroxide protonation through changes in magnetic parameters practically impossible. However, significant changes in EPR line shapes arising from a more restricted rotation of the probe are still observed. These changes in rotational dynamics are attributed to electrostatic interactions of a charged (protonated) form of the nitroxide with a charged bilayer interface and/or a change in the effective hydrodynamic radius/hydrogen bonding network of the probe resulting from protonation. The latter effect was previously observed in electrically neutral Triton X-100 micelles.55–57 Thus, one can hypothesize that the nitroxides with the ionizable functionality located sufficiently far from the N–O˙ group to exert a minimal or no effect at all on Aiso could still serve as EPR pH probes in sterically restricting environments, such as lipid bilayers, detergent micelles, or lipid–protein interface, as long as the tumbling of the charged and the neutral forms differs significantly. Proving this hypothesis would provide synthetic chemists with more freedom in the challenging task of designing pH-sensitive nitroxide probes and, as a result, significantly expand the arsenal of pH-sensitive probes for biophysical and physicochemical EPR studies.
We note here that back in 1974 Barratt and Laggner observed pH effects on EPR line shapes of spin-labelled fatty acids incorporated into lecithin multilamellar vesicles (MLVs).58 The authors explained the effect by an increase in anisotropy of molecular tumbling arising from about 6 Å dislocation of the probe along the membrane normal upon an ionization of the probe carboxylic group remote to the nitroxide.58 Such repartitioning probes were then used to investigate surface electrostatic properties of lipid bilayers and lipid–protein interactions by analysing the fractions and rotational dynamics of multicomponent EPR spectra of spin-labelled steric acids arising upon ionization of the carboxylic group.59–61 A method of determination of the membrane potential from partitioning of small nitroxides between aqueous and lipid phases resulting in different EPR spectra due to changes in molecular tumbling was also developed.62
The general approach to pH determination from EPR spectra described here does not require any probe repartitioning as it based on a universal observation that diffusion of a charged molecular probe is slowed down by the counterions that have to move together with the probe.63 This effect is magnified at interfaces where other charges or dipoles are present and where nitroxide X-band EPR spectra of membrane probes fall into an intermediate motion regime making the spectra very sensitive to small changes in the rotational correlation time. Thus, in this proof-of-concept study we focused on the synthesis and an initial EPR characterization of pH-sensitive nitroxide spin labels and spin-labelled phospholipids designed within this new concept – to report on their ionization state through changes in the rotational dynamics observed by EPR. Spin labels were characterized by X-band EPR in aqueous solutions, whereas spin-labelled phospholipids were titrated in model multilamellar vesicles composed of either 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (POPG) or 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC). Also, we demonstrate that EPR-active pH-sensitive phospholipids can be synthesized in one step directly from the parent phosphatidylethanolamine (PE) lipid via a reductive amination reaction of the proper nitroxide containing either ketone or aldehyde group.
Results and discussion
Synthesis
Thiol-methanethiosulfonate click reaction was chosen as the leading method for covalent modification of the lipid polar head group with pH-sensitive spin label(s). This choice was justified by many years of successful application of this click chemistry in site-directed spin labelling (SDSL) EPR studies,64 superior selectivity of this reaction with respect to SH-group, commercial availability of a headgroup thiol-modified phospholipid 1,2-dipalmitoyl-sn-glycero-3-phosphothioethanol, and also by our previous experience in using this reaction in the protein spin labelling and the synthesis of spin-labelled phospholipids.55–57,65 Dimethylamino-substituted pyrrolidine nitroxide 10 was used as a core molecular structure. A synthetic route to the target spin label 12 is shown in Scheme 1.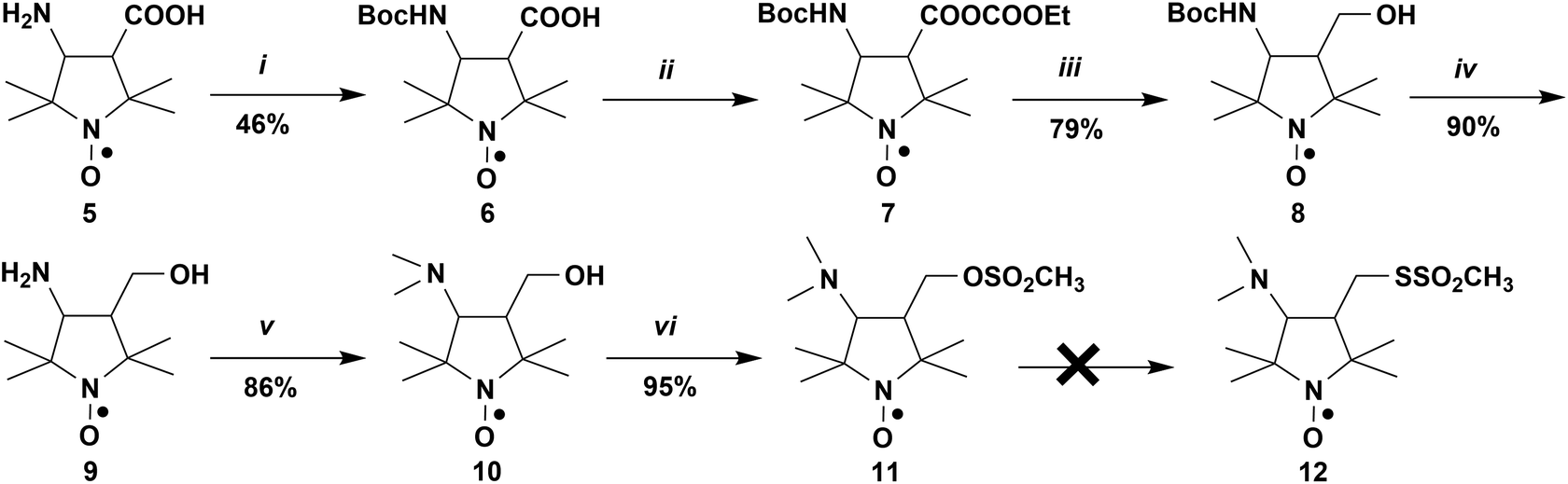 | ||
Scheme 1 (i) Di-tert-butyl dicarbonate, Et3N, CH3CN; (ii) ClCOOEt, Et3N, ether, −10 °C; (iii) NaBH4, EtOH, −15 °C; (iv) H2O/CH3OH, 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v, reflux; (v) HCOH, HCOOH, 60 °C; (vi) Et3N, CH3SO2Cl, CH2Cl2, −10 °C. :1 v/v, reflux; (v) HCOH, HCOOH, 60 °C; (vi) Et3N, CH3SO2Cl, CH2Cl2, −10 °C. | ||
The initial compound amino acid 5 (POAC) was synthesized according to literature procedures.66,67 Boc-protection of the primary amino group, followed by preparation of the mixed anhydride 7 and its reduction with NaBH4, afforded the N-Boc-protected amino alcohol 8. Deprotection of the latter in a boiling water–methanol (6:1 v/v) mixture68 gave the amino alcohol 9 in good yield. Alkylation of the primary amino group of 9 was carried out using the Eschweiler–Clarke reaction to give the tertiary amine 10; hydroxyl group of the latter was activated through formation of the corresponding mesylate 11. Nucleophilic substitution of the OSO2CH3 group in 11 with bromide, iodide, or azide afforded corresponding derivatives 13, 14, and 15 (Scheme 2). Strain-promoted azide–alkyne cycloaddition of 15 with 3-amino-1-(11,12-didehydrodibenzo[b,f]azocin-5(6H)-yl)propan-1-one (DBCO-amine) readily afforded triazole adduct 16, most likely as a mixture of syn- and anti-isomers69,70 (Scheme 2). However, attempts to obtain the methanethiosulfonyl derivative 12 from 11 have failed. Reaction of the latter with sodium methanethiosulfonate did not afford thiosulfonate nitroxide 12. At room temperature 11 did not react with NaSSO2CH3 both in DMSO and EtOH; at the elevated temperature (70 °C), an inseparable mixture of products has formed. We speculate that thiosulfonate anion might be too “soft”, compared to Br−, I−, and N3−, to attack the “hard” electrophilic carbon adjacent to the mesylate group. Besides, the geminal methyl groups in position 2 of the pyrrolidine heterocycle could create an unfavourable steric environment for the attack by a relatively bulky methanethiosulfonate. Attempts to obtain 12 through the reaction of NaSSO2CH3 with halogen derivatives 13 and 14 were also unsuccessful.
 | ||
| Scheme 2 (i) NaN3, DMSO, 70 °C; (ii) LiBr (or LiI), DMSO, 70 °C; (iii) DBCO-amine. | ||
To make the steric requirements for the nucleophilic substitution less demanding, a handle between the pyrrolidine heterocycle and the leaving group was introduced. This was accomplished by reacting the hydroxymethyl derivative 10 with either 2-bromoethyl isocyanate or 2-(2-bromoethoxy)tetrahydro-2H-pyran (Scheme 3). Tetrahydropyranyl protecting group in 17 was removed with aqueous acetic acid;71 the resulting hydroxyethyl derivative 18 was converted to the mesylate 19. Reactions of 19 and the bromo derivative 21 with NaSSO2CH3 afforded the methanethiosulfonate spin labels 20 and 22, respectively.
 | ||
| Scheme 3 (i) Br(CH2)2NCO, EtOAc; (ii) NaSSO2CH3, DMSO, rt; (iii) Br(CH2)2OTHP, KOH, DMSO, rt; (iv) CH3COOH–H2O mixture (4:1 v/v), 50–55 °C, 3 h; (v) CH3SO2Cl, Et3N, CH2Cl2, −10 °C; (vi) NaSSO2CH3, DMSO, 70 °C. | ||
Spin-labelled phospholipids 20-PTE and 22-PTE (Fig. 2) were synthesized through the thiol-methanethiosulfonate click reaction of 20 and 22, respectively, with a thiol-modified phospholipid 1,2-dipalmitoyl-sn-glycero-3-phosphothioethanol (PTE).
 | ||
| Fig. 2 Chemical structures of spin-labelled phospholipids. | ||
pH-Sensitive spin-labelled phospholipid 23-DOPE was synthesized using a different approach – through a reductive amination of the ketone nitroxide 2372 with 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) lipid (Scheme 4). This spin-labelling methodology presents a rather advantageous way to designing pH-sensitive EPR-active phospholipids from PE lipids because the amino functionality of the phosphoethanolamine residue becomes the protonatable group of the pH-sensitive spin-labelled phospholipid. We speculate that this methodology could also be applied for spin-labelling of protein lysine residues.
 | ||
| Scheme 4 Synthesis of the spin-labelled phospholipid via a reductive amination reaction. | ||
EPR characterization of novel nitroxides
Nitroxides and spin-labelled phospholipids were EPR titrated (X-band, 9.5 GHz) in aqueous solutions or lipid suspensions, respectively, to determine their pKa's. Thiosulfonate group is known to be hydrolytically labile at basic pH73 and pKa of the spin label is expected to change after a covalent attachment to a molecule of interest. For these reasons, spin labels 22 and 20 were titrated in the forms of adducts with 2-mercaptoethanol 24 and 25, respectively (Fig. 3). | ||
| Fig. 3 Chemical structures of 2-mercaptoethanol adducts. | ||
The side chain structures in these adducts (Fig. 3) are similar to those of spin labels attached to the thiol group of the PTE lipid (Fig. 2), and, as we have previously reported,55–57 the hydroxyethyl moiety of these compounds is mimicking the inductive effect of the PTE head group attachment rather well. Similarly, nitroxide 26, synthesized via the reductive amination of the ketone nitroxide 23 with 2-aminoethanol (Scheme 5), was used as a water-soluble model of the spin-labelled phospholipid 23-DOPE for EPR characterization in aqueous solutions. Azide 15 was EPR-characterized both as such, and also in a form of the triazole adduct with DBCO-amine 16 (Scheme 2). The DBCO derivatives are widely used in various copper-free alkyne–azide conjugation and labelling protocols,74–76 although this type of conjugation has a limited utility in biophysical EPR studies because of the bulkiness of the linker.
 | ||
| Scheme 5 Reductive amination of the ketone nitroxide 23. | ||
For characterization of spin-labelled phospholipids in lipid bilayers, multilamellar vesicles (MLV's) composed of either POPG or POPC were doped with 20-PTE, 22-PTE, or 23-DOPE at 1 mol% using experimental protocols described elsewhere.56,57
Aqueous solutions of all the nitroxides synthesized in this work exhibit typical isotropic three-line X-band (9.5 GHz) EPR spectra (ESI, S69–S76†). Unlike the X-band EPR spectra of many other pH-sensitive nitroxides, the spectra of the newly synthesized compounds did not show characteristic splitting of the high-field nitrogen hyperfine component at intermediate pH values. This splitting results from a difference in nitrogen hyperfine coupling constants of protonated and non-protonated forms of a nitroxide (typically, ΔAiso ≈ 0.80–1.30 G)52,53 and, is indicative of a slow, on the EPR time scale, chemical exchange. According to the previously established slow-exchange condition that relates the pKa of the nitroxide probe and the type of the chemical exchange:77
 | (1) |
For nitroxides 10–26 . Then for these nitroxides pKa values would fall into a range 3.9 < pKa < 10.1, and such EPR species are expected to be in a slow chemical exchange regime. However, none of the fast motion EPR spectra of the novel nitroxides revealed a splitting in the high field nitrogen hyperfine coupling components but only some extra broadening resulting in a decrease in the peak-to-peak amplitude (ESI, S69–S76†). Some of the reasons for the absence of splitting include a lack of spectral resolution as the two mI = −1 lines of ΔBp–p ≈ 1.2 G in widths are separated by only ΔB ≈ ΔAiso ≈ 0.50 G and/or a higher than estimated exchange rate. For example, as it was shown for pH-sensitive nitroxides of the 3-imidazoline and imidazolidine series, the R˙ ⇆ R˙H+ exchange rate could be increased by the presence of a buffer, causing the two high-field lines to coalesce. However, replacement of 50 mM phosphate buffer with 50 mM NaCl solution did not show any effect on the shape of the high-field EPR line (see ESI, S70†). Thus, we speculate that protonation of the functionality in the side chain (e.g., in nitroxides 10–26) is less sterically inhibited compared to that of the one being a part of the heterocycle, such as in 3-imidazoline and imidazolidine nitroxides. Our speculation is supported by the existing correlation between bimolecular constant of the proton exchange reaction and the steric accessibility of the protonation site.78
. Then for these nitroxides pKa values would fall into a range 3.9 < pKa < 10.1, and such EPR species are expected to be in a slow chemical exchange regime. However, none of the fast motion EPR spectra of the novel nitroxides revealed a splitting in the high field nitrogen hyperfine coupling components but only some extra broadening resulting in a decrease in the peak-to-peak amplitude (ESI, S69–S76†). Some of the reasons for the absence of splitting include a lack of spectral resolution as the two mI = −1 lines of ΔBp–p ≈ 1.2 G in widths are separated by only ΔB ≈ ΔAiso ≈ 0.50 G and/or a higher than estimated exchange rate. For example, as it was shown for pH-sensitive nitroxides of the 3-imidazoline and imidazolidine series, the R˙ ⇆ R˙H+ exchange rate could be increased by the presence of a buffer, causing the two high-field lines to coalesce. However, replacement of 50 mM phosphate buffer with 50 mM NaCl solution did not show any effect on the shape of the high-field EPR line (see ESI, S70†). Thus, we speculate that protonation of the functionality in the side chain (e.g., in nitroxides 10–26) is less sterically inhibited compared to that of the one being a part of the heterocycle, such as in 3-imidazoline and imidazolidine nitroxides. Our speculation is supported by the existing correlation between bimolecular constant of the proton exchange reaction and the steric accessibility of the protonation site.78
For accurate measurements of Aiso the experimental spectra were modelled as single-component fast-motion nitroxide spectra. The line shape of each of the nitrogen hyperfine components was approximated by a Voigt function48 with Gaussian and Lorentzian contributions to the linewidth and the effective isotropic hyperfine constant being adjustable parameters of the fit. Isotropic nitrogen hyperfine coupling parameters of the synthesized nitroxides and titration data are summarized in Table 1.
| Compound | A isoR˙, G | A isoR˙H+, G | ΔAiso, G | pKa |
|---|---|---|---|---|
| 10 | 15.98 ± 0.02 | 15.47 ± 0.02 | 0.51 ± 0.03 | 6.25 ± 0.01 |
| 15 | 15.78 ± 0.03 | 15.27 ± 0.02 | 0.51 ± 0.04 | 5.80 ± 0.02 |
| 16 | 15.68 ± 0.03 | 15.17 ± 0.03 | 0.51 ± 0.04 | 4.44 ± 0.02 |
| 18 | 15.97 ± 0.02 | 15.45 ± 0.02 | 0.51 ± 0.03 | 6.39 ± 0.01 |
| 24 | 15.89 ± 0.04 | 15.37 ± 0.05 | 0.52 ± 0.06 | 5.51 ± 0.03 |
| 25 | 15.95 ± 0.01 | 15.44 ± 0.02 | 0.51 ± 0.02 | 6.22 ± 0.06 |
| 26 | 16.06 ± 0.02 | 15.57 ± 0.01 | 0.49 ± 0.02 | 6.73 ± 0.01 |
All pH-sensitive nitroxides demonstrate a small but readily detectable ΔAiso ≈0.50 G difference between the isotropic nitrogen hyperfine coupling constants of the protonated and non-protonated forms and pKa values in the range of ∼4–7 units of pH depending on the structure of the side chain. We note that adduct 16 exhibits the lowest aqueous pKa = 4.44 ± 0.02, which could be either a result of an electron-withdrawing effect of the triazole moiety, or a destabilizing electrostatic effect of the protonated 3-aminopropylamide side chain (pKa = 8.81 ± 0.10 (ref. 79)), and/or an electron-withdrawing effect of the protonated triazole moiety (triazoles have been reported to be weakly basic80–82), or a combination of both effects. To support EPR determination of pKa we conducted a potentiometric titration of the compound 10. Analysis of the potentiometric measurements (see ESI, Fig. S1†) yielded pKa = 6.27 ± 0.06, which is in good agreement with the EPR data (pKa = 6.25 ± 0.01).
To demonstrate utility of spin-labelled phospholipids 20-PTE, 22-PTE, and 23-DOPE as EPR probes for biological membranes, EPR titration of these lipids in model multilamellar vesicles (MLVs) composed either of anionic phospholipid POPG or zwitterionic phospholipid POPC in fluid (17 °C) bilayer phases was performed. EPR spectra of all these spin-labelled lipids fall into an intermediate-to-slow motion regime (Fig. 4, left). It could be seen that, similar to our previous data for titration of spin-labelled lipids in model lipid bilayers,55–57 a decrease in pH resulted in a gradual appearance of a more immobilized spectral component. We attribute the latter component to a protonated form of the nitroxide that is expected to have a slower tumbling (vs. uncharged nitroxide) due to electrostatic interactions with the negatively charged bilayer interface.
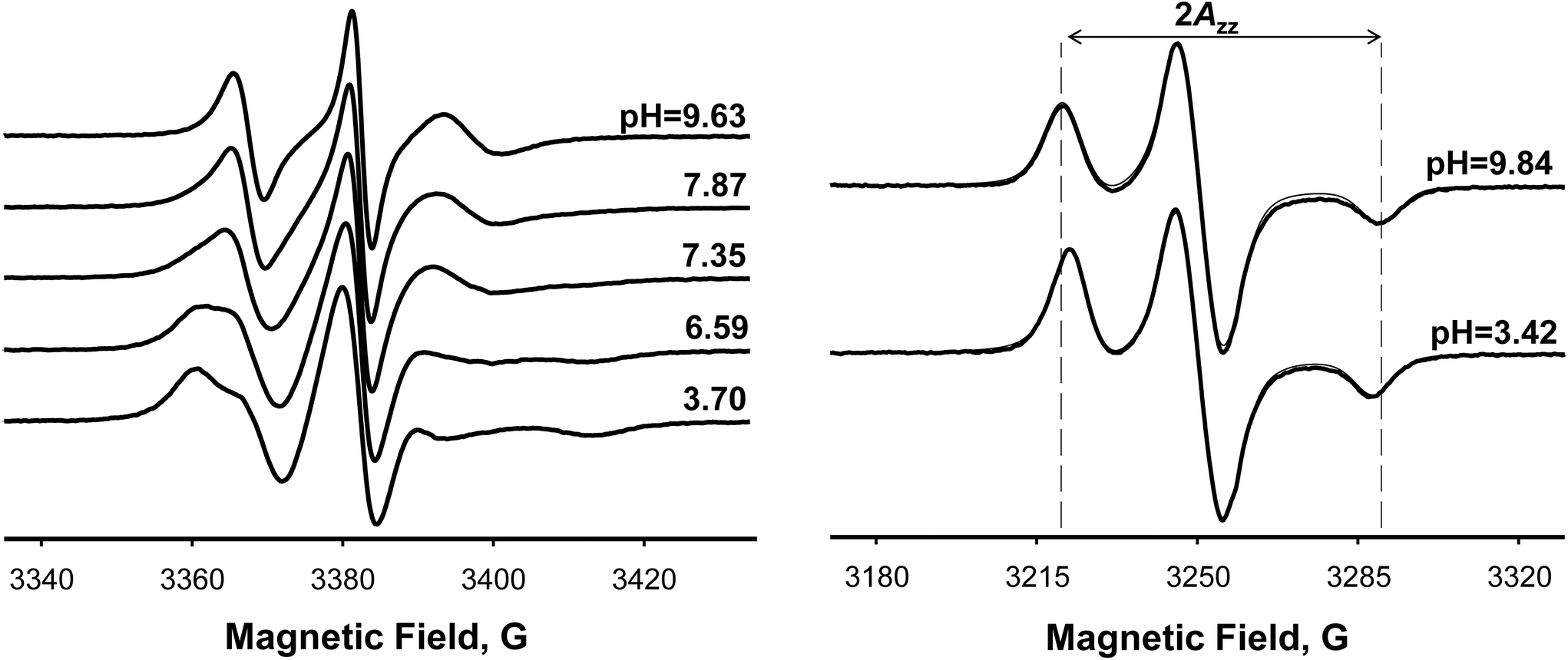 | ||
| Fig. 4 Left: Representative X-band (9.5 GHz) EPR spectra from a titration of multilamellar POPG vesicles doped with 1 mol% of the spin-labelled phospholipid 20-PTE. Spectra were measured at 17 °C. Right: Experimental rigid-limit (77 K) X-band EPR spectra from POPG MLVs doped with 1 mol% of the spin-labeled phospholipid 20-PTE in non-protonated (top) and protonated (bottom) forms shown as thick black lines are superimposed with simulated spectra (thin black lines), which are almost indistinguishable from the experimental spectra. Approximate magnetic field positions corresponding to 2Azz nitrogen hyperfine splitting of the non-protonated form are shown as dashed lines and pH values are indicated next to the spectra. | ||
A clear demonstration that the protonation affects magnetic parameters of spin-labelled phospholipids such as 20-PTE was obtained by measuring rigid limit EPR spectra at 77 K. Fig. 4 (right) compares rigid limit X-band EPR spectra of 20-PTE in POPG MLVs at pH = 9.84 (top) when this nitroxide is expected to be in the non-protonated form with a spectrum at pH = 3.42 (bottom) when this nitroxide is fully protonated. The positions of the outer nitrogen hyperfine lines corresponding to 2Azz of the non-protonated nitroxide are shown as dashed lines to demonstrate that this magnetic parameter is decreased upon protonation (compare top and bottom spectra in Fig. 4, right). This effect was quantified by least-squares simulation of the spectra using Pepper function of EasySpin.83,84 The Pepper function is designed for simulations and fitting of CW EPR spectra in the absence of rotational averaging such as those observed for single crystals or the crystals grounded into a powder. Thus, this function was selected for simulating spectra obtained at 77 K.85 The simulated spectra shown as thin lines in Fig. 4 (right) are superimposed with the experiment (thick black lines) and are nearly identical. We note that x- and y-axis principal components of both g-matrix and A-tensor of the nitroxides are typically not resolved in the rigid limit X-band EPR spectra unless the radicals are perdeuterated.86 Thus, the main changes caused by local electric fields and hydrogen bonding are expected in Azz component,87,88 which for 20-PTE in POPG MLVs was found to change from Azz ≈ 34.34 G to ≈32.84 G upon protonation. Therefore, ΔAiso = ⅓(ΔAxx + ΔAyy + ΔAzz) ≈ ⅓ΔAzz = 0.50G, which is in an excellent agreement with ΔAiso = 0.50 ± 0.01 G, determined from the fast motion spectra (Table 1).
To demonstrate that the protonation of pH-sensitive nitroxides attached to the lipid polar head results in large changes in rotational tumbling, representative EPR spectra of 20-PTE incorporated at 1 mol% into POPG MLVs and measured at 17 °C were least-squares simulated using chili function of EasySpin.84,85 The Chili function is designed for simulations and fitting of CW-EPR spectra in the slow-motion regime, and, hence, was used for the 17 °C spectra. Simulations of EPR spectra by the Chili function is based on a solution of the Stochastic Liouville Equation (SLE). For simplicity, an isotropic rotational model was assumed, and magnetic parameters obtained from the rigid limit simulations of Fig. 4 were slightly adjusted to account for fast residual librations resulting in some pre-averaging of the anisotropic magnetic parameters. Fig. 5 shows that the best simulations (thin lines) closely follow the experimental spectra (thick lines). The spectra of non-protonated nitroxide yielded effective correlation time τ ≈ 1.4 ns while rotational tumbling of the fully protonated form at pH = 3.42 demonstrated significantly longer correlation time of ≈8.3 ns.
 | ||
| Fig. 5 Experimental (thick line) and least-squares simulated (thin) X-band EPR spectra of multilamellar POPG vesicles doped with 20-PTE at 1 mol%. Experimental spectra were measured at 17 °C and pH = 9.84 (non-protonated form, top) and pH = 3.42 (fully protonated, bottom). The effective rotational correlation time increased from τ ≈ 1.4 ns for the non-protonated nitroxide to τ ≈ 8.3 ns for the protonated form. | ||
Although simulations of slow-motion EPR spectra of spin-labelled lipids demonstrated a good agreement with experiment (Fig. 5), such simulations are known to be time consuming because of multiple parameters involved. Thus, in order to decrease the number of adjustable parameters and simplify the simulations, the spectra at intermediate pH values were simulated as a superposition of two components, each corresponding to the protonated and non-protonated forms of the nitroxide (i.e., a slow exchange model was assumed). These two reference spectra we measured experimentally, and the weights of the individual components were adjusted during the fitting and then used to calculate a fraction f = IR/(IR + IRH+) of the non-protonated form from the corresponding double integrals of the individual components. Details of the decomposition procedure are described elsewhere.56,57Fig. 6 shows an example of such a two-component decomposition of an EPR spectrum of 20-PTE in POPG MLVs. The residual of the fit – a difference between the experimental and simulated spectra – reveals only minor deviations, thus, demonstrating the applicability of the slow exchange model. To determine the pKa of the probe, the fraction of the non-protonated form, ƒ, was plotted as a function of pH and the experimental titration data were fitted to the Henderson–Hasselbalch equation. Fig. 7 shows such plots for titration of 22-PTE incorporated in multilamellar vesicles composed of POPC or POPG at 17 °C, and the corresponding fits to the Henderson–Hasselbalch equation. Titration data for the three spin-labelled phospholipids incorporated in POPC or POPG multilamellar vesicles are summarized in Table 2.
 | ||
| Fig. 6 Example of a least-squares decomposition of the experimental X-band EPR spectrum from multilamellar POPG vesicles doped with 1 mol% 20-PTE. Spectrum was measured at 17 °C and pH = 6.97. (A) Experimental X-band EPR spectrum; (B) and (C) – least-squares simulated spectra of the protonated and non-protonated forms of the nitroxide, respectively; (D) residual of the fit – the difference between the experimental and simulated spectra. | ||
 | ||
| Fig. 7 Experimental EPR titration data for multilamellar POPC (○) and POPG (●) vesicles doped with spin-labelled lipid 22-PTE at 1 mol%. Data were acquired at 17 °C. The fits to the Henderson–Hasselbalch equation are shown as solid lines and the best-fit pKa values are indicated next to the plots. | ||
Notably, unlike 22-PTE and 23-DOPE, the experimental titration plot for 20-PTE in multilamellar POPC vesicles contains several reproducible outlier data points at pH below the pKa of the probe (Fig. 8, left panel). Since this model system has no titratable groups other than tertiary amino group of the nitroxide (pKa of the phosphate group in POPC is too low, pKa ≈ 1.0 (ref. 89)), we ruled out a possibility of the second pKa transition. Furthermore, when EPR titration of 20-PTE was conducted in 200 nm unilamellar POPC vesicles (ULVs), the titration plot (Fig. 8, right panel) showed no deviations from the Henderson–Hasselbalch titration curve. This indicates that the observed deviation was caused by the multilamellar nature of the vesicles. This observation can be rationalized as follows. Fig. 8 (left panel) shows that at pH slightly below the probe pKa the fraction of the non-protonated form of the nitroxide ceased to follow the typical S-shaped titration curve, indicating that the fraction of the protonated form is accumulating slower than expected. In terms of the acid–base equilibrium, this means that there is a local factor that disfavours further positive charge development at the nitroxide moieties. One of such factors could be a positive charge of the choline quaternary ammonium groups from the leaflet of the adjacent lamella. The observed effect seems to be electrostatic in nature and comes into play when the amount of the positively charged nitroxide residues reaches a certain number (about 50% of the total amount in our case). We can safely assume that below this critical R˙H+ concentration the lipid system is capable of negating the electrostatic repulsion and accommodating these newly developed positive charges through conformational changes of phospholipid headgroups.90,91 However, above this critical R˙H+ concentration, conformational changes in the headgroup region cannot negate the electrostatic repulsion any more, and further addition of acid results in formation of a smaller than expected fraction of the protonated nitroxides. Note that the upper half of the titration curve reaches plateau at pH ≈ pKa + 2.0, which is typical for titration. The lower half, on the other hand, reaches the plateau at pH ≈ pKa − 3.0, which is approximately one unit of pH lower than expected. Clearly, similar deviation from a single pKa titration curve is not possible for the unilamellar vesicles, which have no adjacent lamellas. The answer to the question “why this effect was observed only for 20-PTE but not 22-PTE or 23-DOPE” lies, most likely, in the structure and the length of the tether linking the nitroxide moiety to the phospholipid. We speculate that the observed phenomenon is a combination of the length and the conformational flexibility of the tether that makes the protonatable group readily affected by the charged choline residue. One way to check this hypothesis in the future will be to study MLVs containing a fraction of lipids with the headgroups modified by PEG (polyethylene glycol) because PEG coatings of liposomes is known to shield the surface from aggregation.92
 | ||
| Fig. 8 Experimental EPR titration data for the spin-labeled lipid 20-PTE in multilamellar (left) and 200 nm unilamellar (right) POPC vesicles. Data were acquired at 17 °C. The Henderson–Hasselbalch titration curves are shown as solid lines. Note that the experimental data in the left panel represent a combination of the data from two independent EPR titration experiments carried out over entire pH range. | ||
It should be noted here that the pKa of 20-PTE in 200 nm ULVs is significantly lower than MLVs prepared from the same lipids (ΔpKa = 0.92 ± 0.07). Previously, we have observed a similar, but less sizable effect upon titration of another spin-labelled phospholipid, IKMTSL-PTE, in DMPG vesicles.57 This effect was attributed to the difference in average packing density of lipids in the ULVs vs. MLVs that are composed of many lamellae of varying local curvatures. An interaction between adjacent lamellae could be another factor. Indeed, we have shown that the surface electrostatic potential of phosphatidylcholine MLVs such as those composed of DMPC is negative (e.g., Ψ ≈ −100 mV at 17 °C (ref. 56)). Then the presence of the adjacent lamellas in MLVs is expected to increase the negative surface potentials of the individual MLV bilayers vs. surface potential in ULVs and shift the observed local pKa to higher values. However, further detailed investigation of this effect appears to be beyond the focus of the current study.
Conclusions
A concept for designing pH-sensitive nitroxides for studying electrostatic phenomena at biological interfaces based on changes in the shape of EPR spectra arising from a difference in molecular tumbling rates of the protonated and non-protonated forms, was demonstrated. Following this concept, a new series of protonatable nitroxide spin labels and probes of the pyrrolidine type was designed to exhibit a suitable range of pKa values but not necessarily have a large effect of protonation on magnetic parameters of the nitroxide (i.e., Aiso). The rotational motion of nitroxides at the biological interfaces, such as lipid bilayer surfaces, becomes significantly more restricted when these spin probes acquire an electric charge, and the fraction of such molecules can be readily derived by a straightforward decomposition of EPR spectra into two components. Since determination of the fractions of nitroxides in the charged-uncharged equilibria is based on the effects of molecular motion, such pH-sensitive probes may even show no measurable changes in magnetic parameters in order for the method described here to work.The protonatable functionality in the newly synthesized nitroxides is not a part of the nitroxide heterocycle, as in the pH-sensitive nitroxides of the 3-imidazoline and 1,3-imidazolidine series but is located in the side chain of the pyrrolidine nitroxide. These nitroxides demonstrated a rather small difference between the Aiso of the protonated and non-protonated forms, ΔAiso ≈ 0.50 G, and the pKa values varying within the range from pKa = 4.44 ± 0.02 to pKa = 6.73 ± 0.01. Click reaction between the new methanethiosulfonate spin labels and a head group thiol-modified phospholipid 1,2-dipalmitoyl-sn-glycero-3-phosphothioethanol (PTE) was employed for synthesizing the EPR-active pH-sensitive phospholipids. Yet another type of the EPR-active pH-sensitive phospholipid was synthesized via one-step direct reductive amination of the ketone nitroxide with phosphatidylethanolamine lipid (DOPE). These new spin-labelled phospholipids report on their ionization state through the changes in the rotational dynamics. Indeed, the intermediate-to-slow-motion X-band EPR spectra of these spin-labelled phospholipids, doped to POPG or POPC MLVs at 1 mol%, demonstrated a gradual appearance of a more immobilized spectral component upon decrease in pH. The observed pH effect was attributed to the fact that the tumbling of the protonated nitroxide characterized by an effective rotational correlation time τ was significantly slowed down from τ ≈ 1.4 ns for uncharged nitroxide to τ ≈ 8.3 ns for the charge one due to the electrostatic interactions with the charged bilayer interface. The pKa values of these new spin-labelled phospholipids were found to vary within a rather broad range from pKa = 4.61 ± 0.03 to pKa = 8.23 ± 0.03, depending on the structure of the protonatable head group and the composition of the lipid bilayer.
Overall, the feasibility of the concept of measuring local electrostatic phenomena at biological interfaces has been demonstrated. Spin-labelled phospholipids synthesized within the proposed approach were shown to exhibit rather pronounced pH-induced changes in EPR spectra characterized by protonation transitions with pKa values varying within a rather broad pH range. The latter makes these phospholipids a valuable spectroscopic EPR probes for studying proton transfer-related and electrostatic phenomena in detergent micelles and at biological interfaces, such, for example, as lipid bilayer-water or lipid bilayer-membrane protein interface. We believe that the results of this proof-of-concept work will untie chemist's hands and open new avenues in designing the EPR active pH probes for biophysical studies.
Experimental section
Materials and methods
All chemicals and solvents were purchased from VWR International (Radnor, PA), Fisher Scientific (Waltham, MA), or Sigma-Aldrich (St Louis, MO), unless otherwise indicated, and used without additional purification. All solvents were reagent grade and used as received. 1,2-Dipalmitoyl-sn-glycero-3-phosphothioethanol (PTE), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (POPG), and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) were purchased from Avanti Polar Lipids (Alabaster, AL) as chloroform solutions (>99% pure) and used without further purification. 1-Oxyl-2,2,5,5-tetramethyl-pyrrolidin-3-one 23 was a generous gift from Dr Igor A. Kirilyuk (Novosibirsk Institute of Organic Chemistry, Russia). Preparative thin layer chromatography was performed on a PTLC plate (Kieselgel 60 F254; Merck, Whitehouse, NJ). MS analysis was carried out using a high-resolution mass spectrometer Thermo Fisher Scientific Exactive™ Plus MS, a benchtop full-scan Orbitrap mass spectrometer, equipped with Heated Electrospray Ionization (HESI). Samples were introduced into the mass spectrometer via TriVersa NanoMate® (Advion Interchim Scientific, Ithaca, NY). The mass spectrometer was operated in a positive ion mode. Infrared spectra were obtained with a Jasco FT/IR-4100 spectrometer (Jasco Inc., Easton, MD). ULVs were characterized by dynamic light scattering (DLS, Zetasizer, Malvern, Westborough, MA).EPR measurements
X-band (9.5 GHz) continuous wave (CW) EPR spectra were recorded with a Varian (Palo Alto, CA) Century Series E-109 spectrometer interfaced to a PC. The sample temperature was maintained with stability better than ±0.02 °C and a gradient below 0.07 °C cm−1 over the sample region by a digital variable temperature accessory described previously.93 Aqueous solutions or lipid suspensions were drawn into a poly(tetrafluoroethylene) capillary (i.d. = 0.81 mm, o.d. = 1.12 mm, NewAge Industries, Inc., Southampton, PA), and the capillary was folded and inserted into a i.d. = 3 mm, o.d. = 4 mm clear fused quartz EPR tube open from both ends (VWR International). Typical spectrometer settings were as follows: the modulation amplitude was set to a quarter or a half of the narrowest line peak-to-peak line width; time constant, 64 ms; incident microwave power, 2 mW; sweep time, 30 s; and scan width, 100 G. Typically, between 10 and 50 individual scans were acquired and averaged out.EPR titration experiments
EPR titration experiments in aqueous solution were carried out in 50 mM phosphate buffer solutions adjusted to a required pH. In all the experiments the pH values were measured with a SimpHony pH meter (VWR International) equipped with InLab microcombination pH electrode (Mettler-Toledo GmbH, Greifensee, Switzerland) three-point calibrated using two sets of standard VWR (VWR International) buffer solutions: one at pH = 1.68, 4.0, and 7.0 and another at pH = 4.0, 7.0, and 10.0. Before the measurements, both the samples and the standard buffer solution were equilibrated at a required temperature using a circulating bath Model 9710 (PolyScience, Niles, IL) equipped with a digital temperature controller. For EPR titration and pH equilibration of MLVs, approximately 50–100 μl of lipid dispersion was placed into a 1.5 ml Eppendorf tube and the pH of the solution was adjusted by titration with a 0.3 or 0.05 M HCl solution or a 0.1 M NaOH solution. To ensure pH equilibration inside the MLVs after adjusting the pH, the dispersion was subjected to ten consecutive freeze–thaw cycles between liquid nitrogen and a water bath maintained at the temperature of the EPR experiment. The sample was vortexed occasionally, and the pH was measured at the temperature of the EPR experiment. For titration and pH equilibration of ULVs, the pH of the lipid dispersion was adjusted by titration with a 0.3 or 0.05 M HCl solution or a 0.1 M NaOH solution, and the sample was allowed to equilibrate for 10–15 min at the temperature of experiment with occasional vortexing.Potentiometric titration
Potentiometric titration in aqueous solution was carried out at room temperature (21 °C) as follows. 3 mM solution of the nitroxide 10 in DI water was acidified with HCl down to pH = 3.66 (completely protonated form of the nitroxide) and titrated with 10 mM NaOH solution. Experimental data and the fit are shown in Fig. S1 of the ESI.† The pH of the solution was measured with a SimpHony pH meter equipped with InLab microcombination pH electrode three-point calibrated using standard VWR buffer solutions at pH = 1.68, 4.0, and 7.0.3-Cyano-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrole 1-oxyl was synthesized according to the literature procedure.66
:50 (v/v), and then with hexane–EtOAc, 40:60 (v/v), as eluents, to give 3-amino-4-cyano-2,2,5,5-tetramethylpyrrolidine 1-oxyl (5.06 g, 62.6%). Also, 0.14 g (1.9%) of the starting nitrile was recovered.
Step 2. Sodium borohydride (1.25 g, 33 mmol) was added to EtOH (75 ml) cooled to −15 °C, and an ether solution of the mixed anhydride 7 from Step 1 was added dropwise upon stirring and cooling. The reaction mixture was allowed to stay for 2 h (slowly warmed up to the room temperature). The reaction mixture was evaporated to dryness, water (20 ml) was added to the residue, and the mixture was extracted with chloroform. Organic extract was dried over Na2SO4. The product was purified on silica gel, with CHCl3 containing 4.5 v/v% of CH3OH as eluent to give 0.75 g of hydroxymethyl derivative 8 as a light yellow solid (79% yield after two steps). mp 196–198 °C (dec) (hexane–EtOAc). FT-IR (KBr, λmax, cm−1): 3339 (br), 3375 (br), 2985, 2970, 2934, 2877, 2813, 1686, 1518, 1362, 1301, 1288, 1167, 1052, 1004, 860, 755. HRMS-ESI: m/z calculated for C14H27N2O4 [M]+ 287.19653, found 287.19621.
:2 v/v, Rf = 0.69) to give 13 as a yellow crystalline solid, yield 73%, mp 90–92 °C (hexane–EtOAc). FT-IR (KBr, λmax, cm−1): 3026, 2988, 2973, 2932, 2922, 2871, 2837, 2794, 2786, 1481, 1470, 1449, 1359, 1281, 1253, 1223, 1175, 1077, 1061, 1044, 991, 962, 879, 648. HRMS-ESI: m/z calcd for C11H23BrN2O [M + H]+ 278.09883 (79Br) and 280.09733 (81Br), found 278.09836 and 280.09573.
:2 v/v) afforded 14 as a light yellow powder, yield 33%, mp 99–102 °C (dec) (hexane). FT-IR (KBr, λmax, cm−1): 3026, 2970, 2932, 2917, 2871, 2837, 2799, 1481, 1372, 1359, 1212, 1186, 1173, 1071, 1055, 1039, 957, 877. HRMS-ESI: m/z calcd for C11H23IN2O [M + H]+ 326.08496, found 326.08464.
:2 v/v) as an eluent (Rf = 0.49) gave 15 as a yellow solid, yield 93%. mp 48–50 °C (hexane–EtOAc). FT-IR (KBr, λmax, cm−1): 2979, 2969, 2933, 2833, 2794, 2781, 2098, 1481, 1451, 1370, 1358, 1283, 1243, 1225, 1174, 1074, 1038, 897, 817. HRMS-ESI: m/z calcd for C11H23N5O [M + H]+ 241.18971, found 241.18917.
:3:0.5 v/v) as an eluent) showed no presence of the starting materials. After a chromatographic purification, compound 16 was obtained as a crystalline solid, Rf = 0.31 (silica gel deactivated with Et3N, CHCl3–CH3OH–Et3N (100:3:0.5 v/v) as an eluent), yield 0.0057 g (98%). FT-IR (neat, λmax, cm−1): 3419, 2975, 2925, 1649, 1457, 1380, 1199, 1002, 967, 938, 781. HRMS-ESI: m/z calcd for C29H39N7O2 [M + H]+ 517.31597, found 517.31558; m/z calcd for C29H40N7O2 [M + 2H]2+ 259.16163, found 259.16149.
:1 v/v) was heated at 50–55 °C for 3 h (monitored by TLC, silica gel, CHCl3 + 1.5% v/v CH3OH). After the reaction was completed, the reaction mixture was added dropwise upon vigorous stirring to a saturated NaHCO3 solution (10 ml), after that solid NaOH was added to bring the pH up to pH = 10–11. The basic solution was extracted with EtOAc, organic extract was washed with brine, and dried over Na2SO4. After concentration under a reduced pressure the residue was purified on silica gel with CHCl3 + 2% v/v CH3OH as an eluent to give 18 as a yellow oil, yield 100 mg (94%). FT-IR (neat, λmax, cm−1): 3414, 2973, 2929, 2873, 2786, 1732, 1646, 1455, 1380, 1359, 1281, 1244, 1212, 1180, 1124, 1074, 1055, 1039. HRMS-ESI: m/z calcd for C13H28N2O3 [M + H]+ 260.20944, found 260.20984.
:30:1 v/v) was used as an eluent, 70% yield. FT-IR (neat, λmax, cm−1): 3342, 2954, 2920, 2849, 1739, 1726, 1531, 1467, 1242, 1178, 1103, 1071, 1023. HRMS-ESI: m/z calcd for C51H99N3NaO11PS2 [M + H]+ 1047.63508, found 1047.63213. 20-PTE: a mixture CHCl3–CH3OH–H2O (70:15:0.5 v/v) was used as an eluent, 44% yield. FT-IR (neat, λmax, cm−1): 2949, 2917, 2847, 2793, 1742, 1464, 1376, 1362, 1239, 1167, 1097, 1068, 1017. HRMS-ESI: m/z calcd for C50H98O10N2NaPS2 [M + H]+ 1004.62927, found 1004.62864; C50H97O10N2Na2PS2 [M + Na]+ 1026.61122, found 1026.61118.
:15:0.5 v/v) as an eluent affording 23-DOPE as slightly yellow viscous oil. Yield 21.6 mg (91%). FT-IR (neat, λmax, cm−1): 3409, 2925, 2852, 1737, 1460, 1231, 1065, 821. HRMS-ESI: m/z calcd for C49H93N2O9P [M + H]+ 884.66132, found 884.59905.
Synthesis of mercaptoethanol adducts 24 and 25 was carried out similar to the previously described procedure.55
Author contributions
T.S. was responsible for conceptualization, directing the project, funding acquisition, and editing the draft. M.A.V. was responsible for conceptualization, investigation and data curation and was responsible for the original draft. N.N., R.R., and A.D. participated in investigation, A.I.S was responsible for conceptualization, data curation and editing the draft. All authors participated in the discussion of the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is based upon work supported by the National Science Foundation under Grants No. 1508607 and 2305172. The authors are thankful to Dr Igor A. Kirilyuk (Novosibirsk Institute of Organic Chemistry, Russia) for donating 1-oxyl-2,2,5,5-tetramethyl-pyrrolidin-3-one. This work was performed in part by the Molecular Education, Technology and Research Innovation Center (METRIC) at NC State University, which is supported by the State of North Carolina.References
- A. Carrington and I. C. P. Smith, Mol. Phys., 1964, 8, 101–105 CrossRef CAS.
- H. Fischer, Mol. Phys., 1965, 9, 149–152 CrossRef CAS.
- H. Zeldes and R. Livingston, J. Chem. Phys., 1966, 45, 1946–1954 CrossRef CAS.
- H. Hogeveen, H. R. Gersmann and A. P. Praat, Recl. Trav. Chim. Pays-Bas, 1967, 86, 1063–1065 CrossRef CAS.
- B. M. Hoffman and T. B. Eames, J. Am. Chem. Soc., 1969, 91, 2169–2170 CrossRef CAS.
- V. Malatesta and K. U. Ingold, J. Am. Chem. Soc., 1973, 95, 6404–6407 CrossRef CAS.
- V. A. Golubev, E. G. Rozantsev and M. B. Neiman, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1965, 14, 1898–1904 CrossRef.
- J. H. Osiecki and E. F. Ullman, J. Am. Chem. Soc., 1968, 90, 1078–1079 CrossRef CAS.
- D. S. Cafiso and W. L. Hubbell, Biochemistry, 1978, 17, 3871–3877 CrossRef CAS PubMed.
- A. T. Quintanilha and R. J. Mehlhorn, FEBS Lett., 1978, 91, 104–108 CrossRef CAS.
- V. V. Khramtsov and L. B. Volodarsky, in Biological Magnetic Resonance: Volume 14: Spin Labeling, ed. L. J. Berliner, Springer US, Boston, MA, 2002, pp. 109–180 Search PubMed.
- E. F. Ullman, L. Call and J. H. Osiecki, J. Org. Chem., 1970, 35, 3623–3631 CrossRef CAS.
- J. F. W. Keana, M. J. Acarregui and S. L. M. Boyle, J. Am. Chem. Soc., 1982, 104, 827–830 CrossRef CAS.
- V. V. Khramtsov, L. M. Weiner, I. A. Grigoriev and L. B. Volodarsky, Chem. Phys. Lett., 1982, 91, 69–72 CrossRef CAS.
- T. I. V. Smirnova, M. A. Voinov and A. I. Smirnov, in Encyclopedia of Analytical Chemistry: Applications, Theory, and Instrumentation, ed. R. A. Meyers, John Wiley & Sons Inc., 2009 Search PubMed.
- M. A. Voinov and A. I. Smirnov, Electron Paramagn. Reson., 2011, 22, 71–106 CAS.
- M. A. Voinov and A. I. Smirnov, Methods Enzymol., 2015, 564, 191–217 CAS.
- W. Aoi and Y. Marunaka, BioMed Res. Int., 2014, 2014, 598986 Search PubMed.
- A. Sotgiu, K. Mäder, G. Placidi, S. Colacicchi, C. L. Ursini and M. Alecci, Phys. Med. Biol., 1998, 43, 1921–1930 CrossRef CAS PubMed.
- V. Khramtsov, I. Grigor'ev, M. Foster, D. Lurie and I. Nicholson, Cell. Mol. Biol., 2000, 46, 1361–1374 CAS.
- A. N. Tikhonov, R. V. Agafonov, I. A. Grigor'ev, I. A. Kirilyuk, V. V. Ptushenko and B. V. Trubitsin, Biochim. Biophys. Acta, Bioenerg., 2008, 1777, 285–294 CrossRef CAS PubMed.
- V. V. Khramtsov, Curr. Org. Chem., 2005, 9, 909–923 CrossRef CAS.
- S. Koda, J. Goodwin, V. V. Khramtsov, H. Fujii and H. Hirata, Anal. Chem., 2012, 84, 3833–3837 CrossRef CAS PubMed.
- V. V. Khramtsov, I. A. Grigor'ev, M. A. Foster and D. J. Lurie, Antioxid. Redox Signaling, 2004, 6, 667–676 CrossRef CAS PubMed.
- V. V. Khramtsov, G. L. Caia, K. Shet, E. Kesselring, S. Petryakov, J. L. Zweier and A. Samouilov, J. Magn. Reson., 2010, 202, 267–273 CrossRef CAS PubMed.
- D. I. Potapenko, M. A. Foster, D. J. Lurie, I. A. Kirilyuk, J. M. S. Hutchison, I. A. Grigor'ev, E. G. Bagryanskaya and V. V. Khramtsov, J. Magn. Reson., 2006, 182, 1–11 CrossRef CAS PubMed.
- M. A. Foster, I. A. Grigor'ev, D. J. Lurie, V. V. Khramtsov, S. McCallum, I. Panagiotelis, J. M. Hutchison, A. Koptioug and I. Nicholson, Magn. Reson. Med., 2003, 49, 558–567 CrossRef CAS PubMed.
- B. Gallez, K. Mäder and H. M. Swartz, Magn. Reson. Med., 1996, 36, 694–697 CrossRef CAS PubMed.
- A. Samouilov, O. V. Efimova, A. A. Bobko, Z. Sun, S. Petryakov, T. D. Eubank, D. G. Trofimov, I. A. Kirilyuk, I. A. Grigor'ev, W. Takahashi, J. L. Zweier and V. V. Khramtsov, Anal. Chem., 2014, 86, 1045–1052 CrossRef CAS PubMed.
- D. A. Komarov, I. Dhimitruka, I. A. Kirilyuk, D. G. Trofimiov, I. A. Grigor'ev, J. L. Zweier and V. V. Khramtsov, Magn. Reson. Med., 2012, 68, 649–655 CrossRef CAS PubMed.
- V. V. Ptushenko, L. N. Ikryannikova, I. A. Grigor'ev, I. A. Kirilyuk, B. V. Trubitsin and A. N. Tikhonov, Appl. Magn. Reson., 2006, 30, 329–343 CrossRef CAS.
- D. A. Komarov, Y. Ichikawa, K. Yamamoto, N. J. Stewart, S. Matsumoto, H. Yasui, I. A. Kirilyuk, V. V. Khramtsov, O. Inanami and H. Hirata, Anal. Chem., 2018, 90, 13938–13945 CrossRef CAS PubMed.
- R. Nakaoka, K. Kato, K. Yamamoto, H. Yasui, S. Matsumoto, I. A. Kirilyuk, V. V. Khramtsov, O. Inanami and H. Hirata, Anal. Chem., 2023, 3940–3950 CrossRef CAS PubMed.
- Y. Liu, F. A. Villamena and J. L. Zweier, Chem. Commun., 2008, 4336–4338 RSC.
- A. A. Bobko, I. Dhimitruka, J. L. Zweier and V. V. Khramtsov, J. Am. Chem. Soc., 2007, 129, 7240–7241 CrossRef CAS PubMed.
- I. Dhimitruka, A. A. Bobko, C. M. Hadad, J. L. Zweier and V. V. Khramtsov, J. Am. Chem. Soc., 2008, 130, 10780–10787 CrossRef CAS PubMed.
- A. A. Bobko, I. Dhimitruka, D. A. Komarov and V. V. Khramtsov, Anal. Chem., 2012, 84, 6054–6060 CrossRef CAS PubMed.
- E. G. Kovaleva, L. S. Molochnikov, D. O. Antonov, D. P. T. Stepanova, M. Hartmann, A. N. Tsmokalyuk, A. Marek and A. I. Smirnov, J. Phys. Chem. C, 2018, 122, 20527–20538 CrossRef CAS.
- D. O. Antonov, D. P. Tambasova, A. B. Shishmakov, I. A. Kirilyuk and E. G. Kovaleva, Gels, 2021, 7, 119 CrossRef CAS PubMed.
- E. G. Kovaleva, L. S. Molochnikov, E. L. Golovkina, M. Hartmann, I. A. Kirilyuk and I. A. Grigoriev, Microporous Mesoporous Mater., 2015, 203, 1–7 CrossRef CAS.
- E. G. Kovaleva, L. S. Molochnikov, D. Tambasova, A. Marek, M. Chestnut, V. A. Osipova, D. O. Antonov, I. A. Kirilyuk and A. I. Smirnov, J. Membr. Sci., 2020, 604, 118084 CrossRef CAS.
- V. Perelygin, M. A. Voinov, A. Marek, E. Ou, J. Krim, D. Brenner, T. I. Smirnova and A. I. Smirnov, J. Phys. Chem. C, 2019, 123, 29972–29985 CrossRef CAS.
- M. Edeleva, G. Audran, S. Marque and E. Bagryanskaya, Materials, 2019, 12, 688 CrossRef CAS PubMed.
- Y. Le Du, L. Binet, P. Hémery and L. Marx, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2871–2877 CrossRef CAS.
- M. V. Edeleva, I. A. Kirilyuk, I. F. Zhurko, D. A. Parkhomenko, Y. P. Tsentalovich and E. G. Bagryanskaya, J. Org. Chem., 2011, 76, 5558–5573 CrossRef CAS PubMed.
- D. A. Parkhomenko, M. V. Edeleva, V. G. Kiselev and E. G. Bagryanskaya, J. Phys. Chem. B, 2014, 118, 5542–5550 CrossRef CAS PubMed.
- G. Gryn'ova, L. M. Smith and M. L. Coote, Phys. Chem. Chem. Phys., 2017, 19, 22678–22683 RSC.
- A. I. Smirnov and R. L. Belford, J. Magn. Reson., Ser. A, 1995, 113, 65–73 CrossRef CAS.
- T. I. Smirnova, A. I. Smirnov, R. B. Clarkson and R. L. Bedford, J. Phys. Chem., 1995, 99, 9008–9016 CrossRef CAS.
- A. I. Smirnov, T. I. Smirnova and P. D. Morse, Biophys. J., 1995, 68, 2350–2360 CrossRef CAS PubMed.
- A. I. Smirnov and T. I. Smirnova, in EPR: Instrumental Methods, ed. L. J. Berliner and C. J. Bender, Springer US, Boston, MA, 2004, pp. 277–348 Search PubMed.
- M. A. Voinov, J. F. Polienko, T. Schanding, A. A. Bobko, V. V. Khramtsov, Y. V. Gatilov, T. V. Rybalova, A. I. Smirnov and I. A. Grigor'ev, J. Org. Chem., 2005, 70, 9702–9711 CrossRef CAS PubMed.
- K. Margita, M. A. Voinov and A. I. Smirnov, Cell Biochem. Biophys., 2017, 75, 185–193 CrossRef CAS PubMed.
- V. V. Khramtsov, L. M. Weiner, S. I. Eremenko, O. I. Belchenko, P. V. Schastnev, I. A. Grigorev and V. A. Reznikov, J. Magn. Reson., 1985, 61, 397–408 CAS.
- M. A. Voinov, I. A. Kirilyuk and A. I. Smirnov, J. Phys. Chem. B, 2009, 113, 3453–3460 CrossRef CAS PubMed.
- M. A. Voinov, I. Rivera-Rivera and A. I. Smirnov, Biophys. J., 2013, 104, 106–116 CrossRef CAS PubMed.
- M. A. Voinov, C. T. Scheid, I. A. Kirilyuk, D. G. Trofimov and A. I. Smirnov, J. Phys. Chem. B, 2017, 121, 2443–2453 CrossRef CAS PubMed.
- M. D. Barratt and P. Laggner, Biochim. Biophys. Acta, Biomembr., 1974, 363, 127–133 CrossRef CAS PubMed.
- M. Egretcharlier, A. Sanson, M. Ptak and O. Bouloussa, FEBS Lett., 1978, 89, 313–316 CrossRef CAS PubMed.
- L. I. Horvath, P. J. Brophy and D. Marsh, Biochemistry, 1988, 27, 5296–5304 CrossRef CAS PubMed.
- M. Esmann and D. Marsh, Biochemistry, 1985, 24, 3572–3578 CrossRef CAS PubMed.
- D. S. Cafiso and W. L. Hubbell, Annu. Rev. Biophys. Bioeng., 1981, 10, 217–244 CrossRef CAS PubMed.
- M. J. Stephen, J. Chem. Phys., 1971, 55, 3878–3883 CrossRef CAS.
- C. S. Klug and J. B. Feix, in Methods in Cell Biology, Academic Press, 2008, vol. 84, pp. 617–658 Search PubMed.
- M. A. Voinov, A. Ruuge, V. A. Reznikov, I. A. Grigor'ev and A. I. Smirnov, Biochemistry, 2008, 47, 5626–5637 CrossRef CAS PubMed.
- E. G. Rozantzev and L. A. Krinitzkaya, Tetrahedron, 1965, 21, 491–497 CrossRef.
- A. Rassat and P. Rey, Bull. Soc. Chim. Fr., 1967, 815–818 CAS.
- C. Zinelaabidine, O. Souad, J. Zoubir, B. Malika and A. Nour-Eddine, Int. J. Chem., 2012, 4, 73 CrossRef CAS.
- C. G. Gordon, J. L. Mackey, J. C. Jewett, E. M. Sletten, K. N. Houk and C. R. Bertozzi, J. Am. Chem. Soc., 2012, 134, 9199–9208 CrossRef CAS PubMed.
- A. V. Strizhak, K. Sharma, O. Babii, S. Afonin, A. S. Ulrich, I. V. Komarov and D. R. Spring, Org. Biomol. Chem., 2018, 16, 8559–8564 RSC.
- D. R. Williams and W. S. Kissel, J. Am. Chem. Soc., 1998, 120, 11198–11199 CrossRef CAS.
- L. A. Krinitskaya, E. G. Rozantsev and M. B. Neiman, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1965, 14, 100–102 CrossRef.
- N. S. Zefirov, N. V. Zyk, E. K. Beloglazkina and A. G. Kutateladze, Sulfur Rep., 1993, 14, 223–240 CrossRef CAS.
- H. Y. Yoon, D. Lee, D. K. Lim, H. Koo and K. Kim, Adv. Mater., 2022, 34, 28 Search PubMed.
- M. F. Debets, S. S. van Berkel, J. Dommerholt, A. J. Dirks, F. P. J. T. Rutjes and F. L. van Delft, Acc. Chem. Res., 2011, 44, 805–815 CrossRef CAS PubMed.
- E. M. Sletten and C. R. Bertozzi, Acc. Chem. Res., 2011, 44, 666–676 CrossRef CAS PubMed.
- V. V. Khramtsov, L. M. Weiner, S. I. Eremenko, O. I. Belchenko, P. V. Schastnev, I. A. Grigor'ev and V. A. Reznikov, J. Magn. Reson., 1985, 61, 397–408 CAS.
- V. V. Khramtsov and L. M. Weiner, Russ. Chem. Rev., 2007, 57, 824–838 CrossRef.
- Chemical Book, CAS DataBase List https://www.chemicalbook.com/ChemicalProductProperty_EN_CB32730210.htm.
- J. Catalan, J. L. M. Abboud and J. Elguero, Adv. Heterocycl. Chem., 1987, 41, 187–274 CrossRef CAS.
- A. Albert and P. J. Taylor, J. Chem. Soc., Perkin Trans. 2, 1989, 1903–1905 RSC.
- J. L. M. Abboud, C. Foces-Foces, R. Notario, R. E. Trifonov, A. P. Volovodenko, V. A. Ostrovskii, I. Alkorta and J. Elguero, Eur. J. Org. Chem., 2001, 2001, 3013–3024 CrossRef.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed.
- S. Stoll , easyspin, easyspin.org.
- EasySpin Academy, https://www.youtube.com/watch?v=uy_smGyxZxc&t=26s.
- A. I. Smirnov, R. Belford and R. Clarkson, in Spin Labeling: The Next Millennium, ed. L. J. Berliner, Plenum Press, New York, 2002, vol. 16, ch. 3, pp. 83–107 Search PubMed.
- A. F. Gulla and D. E. Budil, J. Phys. Chem. B, 2001, 105, 8056–8063 CrossRef CAS.
- T. I. Smirnova and A. I. Smirnov, in ESR Spectroscopy in Membrane Biophysics, ed. M. A. Hemminga and L. J. Berliner, Springer US, New York, 2007, vol. 27, ch. 6, pp. 165–251 Search PubMed.
- D. Marsh, Handbook of Lipid Bilayers, CRC Press, 2nd edn, 2013, DOI:10.1201/b11712.
- M. Roux, J. M. Neumann, R. S. Hodges, P. F. Devaux and M. Bloom, Biochemistry, 1989, 28, 2313–2321 CrossRef CAS PubMed.
- J. P. F. Doux, B. A. Hall and J. A. Killian, Biophys. J., 2012, 103, 1245–1253 CrossRef CAS PubMed.
- J. S. Suk, Q. Xu, N. Kim, J. Hanes and L. M. Ensign, Adv. Drug Delivery Rev., 2016, 99, 28–51 CrossRef CAS PubMed.
- A. M. Alaouie and A. I. Smirnov, J. Magn. Reson., 2006, 182, 229–238 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ob00167b |
| This journal is © The Royal Society of Chemistry 2024 |