
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A manganese-based catalyst system for general oxidation of unactivated olefins, alkanes, and alcohols†
Dennis
Verspeek
 a,
Sebastian
Ahrens
a,
Xiandong
Wen
bc,
Yong
Yang
bc,
Yong-Wang
Li
bc,
Kathrin
Junge
*a and
Matthias
Beller
*a
a,
Sebastian
Ahrens
a,
Xiandong
Wen
bc,
Yong
Yang
bc,
Yong-Wang
Li
bc,
Kathrin
Junge
*a and
Matthias
Beller
*a
aLeibniz-Institute für Katalyse e.V., Albert-Einstein-Straße 29a, 18059 Rostock, Germany. E-mail: kathrin.junge@catalysis.de; matthias.beller@catalysis.de
bState Key Laboratory of Coal Conversion, Institute of Coal Chemistry, Chinese Academy of Sciences, Taiyuan, 030001, China
cNational Energy Center for Coal to Liquids, Synfuels China Co., Ltd, Huairou District, Beijing, 101400, China
First published on 27th February 2024
Abstract
Non-noble metal-based catalyst systems consisting of inexpensive manganese salts, picolinic acid and various heterocycles enable epoxidation of the challenging (terminal) unactivated olefins, selective C–H oxidation of unactivated alkanes, and O–H oxidation of secondary alcohols with aqueous hydrogen peroxide. In the presence of the in situ generated optimal manganese catalyst, epoxides are generated with up to 81% yield from alkenes and ketone products with up to 51% yield from unactivated alkanes. This convenient protocol allows the formation of the desired products under ambient conditions (room temperature, 1 bar) by employing only a slight excess of hydrogen peroxide with 2,3-butadione as a sub-stoichiometric additive.
Introduction
Finding and designing more efficient and environmentally friendly catalytic reactions continues to be an important task for synthetic chemists in industry and academia. To a greater extent, achieving this task is becoming more and more difficult as already existing synthetic routes, especially in the area of bulk chemical syntheses, have been optimized for decades. Nowadays, not only the best product yield but also other factors such as minimizing the amount of generated waste, avoiding excess use of reagents and additives, utilizing Earth-abundant catalysts, and circumventing any risk stemming from the use of toxic, corrosive or hazardous materials determine the quality of a given synthesis.1 The latter is especially true for oxidation reactions, as most (highly concentrated) oxidants pose serious safety risks.2 In this respect, the use of molecular oxygen, hydrogen peroxide or tert-butyl hydroperoxide is clearly preferred compared to, for example, hypervalent iodine species, hypochlorite or toxic metal oxides, e.g., OsO4.3 More specifically, the ultimate clean oxidant for liquid phase oxidation at ambient pressure is aqueous hydrogen peroxide, which unfortunately can be easily decomposed, especially by non-noble metal salts, thus limiting its general applicability. To improve the selectivity and prevent decomposition reactions of peroxides, N-heterocyclic compounds have been used as (co-)ligands,4–8 additives,9 or bases10 in metal-catalysed oxidation reactions. In fact, several multidentate ligands, e.g., pincer-type or tetradentate ligands, showed higher selectivities in oxidation reactions with oxidants like hydrogen peroxide or tert-butyl hydroperoxide (TBHP).4,5,11–19 In addition, the application of structurally simpler pyridine derivatives is a useful tool if a base is needed to deprotonate a peroxide species to enhance its nucleophilicity.10,14 Other examples include functionalized N-heterocycles, such as picolinic acid derivatives, that have found application in iron- or manganese-catalysed oxidation reactions, e.g., (ep)oxidation of olefins,5,20–22 alcohol oxidation,23 or C–H oxidation of (unactivated) alkanes.4,23,24Furthermore, picolinic acid derivatives have been used for (noble)metal-catalysed reactions in the fields of water oxidation,25 photochemistry,26,27 and others.28–32
As part of our ongoing efforts regarding the valorization of terminal aliphatic olefins, we recently reported a novel protocol for manganese-catalysed epoxidation of olefins.14 Here, the addition of quinoline was crucial to obtain high selectivity towards the desired epoxide products. Although N-heterocycles of similar structures are known to promote analogous metal-catalysed oxidation reactions,5,19,33 the exact role of quinoline has not been revealed. However, we postulated a mechanism where quinoline acts as a base to deprotonate TBHP. Following our previous findings4,5,14 regarding the employment of N-heterocycles as additives and inspired by the works of Browne and co-workers,8,20,21,23,34–38 Stack,22 and others10,24,39 employing picolinic acid as a ligand (see Scheme 1), we had the idea to combine both features in one catalyst system for the valorization of terminal aliphatic olefins as well as other oxidation reactions. Despite many developments in (non)noble metal-catalysed epoxidation reactions in recent years,5,9,15,16,40–53 this approach, i.e., combining a picolinate-based manganese system with N-heterocycles, has not been implemented. Furthermore, product degradation, oxidant decomposition and/or free-diffusing radicals still make terminal aliphatic olefins difficult to be epoxidized in high yields under benign and acid-free conditions.54,55 To address these issues, we propose manganese–picolinate complexes39,56,57 in combination with different N-heterocycles as active and selective catalysts for diverse oxidation reactions.
 | ||
| Scheme 1 Selected examples and applications (blue) of iron and manganese catalysts with picolinic acid and/or N-heterocycles as (co)-ligands and/or additives. | ||
Results and discussion
Based on our previously reported system,14 we envisioned the use of Mn(OTf)2 as a metal precursor, picolinic acid as a simple and cheap ligand with quinoline as an additive as the starting point of our investigation. Firstly, the epoxidation of 1-octene 1a as the model, yet challenging54,55 substrate using a combination of hydrogen peroxide with 2,3-butadione as the peroxide activator20 was performed in aqueous acetonitrile at room temperature. The epoxidation of terminal aliphatic olefins, e.g., propylene, with aqueous hydrogen peroxide is of high industrial relevance and currently used on a >600![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tons/a scale. Thus, a systematic variation of reaction parameters, i.e., catalysts, additives, oxidants, and their respective ratios was performed. In the first numerical variation, a 37% yield of 1,2-epoxyoctane 2a at 79% conversion was obtained by employing 0.25 mol% Mn(OTf)2, 5 mol% picolinic acid, 5 mol% quinoline, 0.5 equivalents of 2,3-butadione, and 5 equivalents of H2O2 (30% aq.) (see Table S1† for more details).
000 tons/a scale. Thus, a systematic variation of reaction parameters, i.e., catalysts, additives, oxidants, and their respective ratios was performed. In the first numerical variation, a 37% yield of 1,2-epoxyoctane 2a at 79% conversion was obtained by employing 0.25 mol% Mn(OTf)2, 5 mol% picolinic acid, 5 mol% quinoline, 0.5 equivalents of 2,3-butadione, and 5 equivalents of H2O2 (30% aq.) (see Table S1† for more details).
To improve the selectivity and activity, we then embarked on in-depth metal precursor screening. In general, weakly coordinating anions are especially effective in manganese-catalysed oxidation or epoxidation reactions.58 Thus, manganese(II) perchlorate, triflate, and triflimide all produced virtually identical results of 77–79% conversion and 37% epoxide yield (Table 1, entries 1–3). Switching to hexafluoropenta-2,4-dione as an anion, a slightly higher conversion and a yield of 40% was obtained (Table 1, entry 5). We then employed stronger coordinating anions in this protocol. To our delight, both Mn(II) acetate and acetylacetonate produced better yields than the initially employed precursors, giving almost full conversion of the starting material and yields of 40–45% of the desired epoxide 2a (Table 1, entries 6–8).
|
|
||||
|---|---|---|---|---|
| Entry | Precursor | Conv. (1a) [%] | Yield (2a) [%] | Sel. (2a) [%] |
| Conversion and yield determined by GC analysis with hexadecane as IST. Reaction conditions: 0.5 mmol substrate (0.250 M), 0.25 mol% precursor, 5 mol% picolinic acid, 5 mol% quinoline, 0.5 eq. of 2,3-butadione, MeCN (2 mL), 25 °C, 2 h slow addition of H2O2 (30% aq., 5 eq., diluted in MeCN) via a syringe pump.a 2.5 eq. of H2O2 (30% aq.) was used. | ||||
| 1 | Mn(OTf)2 | 79 | 37 | 47 |
| 2 | Mn(ClO4)2 | 77 | 37 | 48 |
| 3 | Mn(NTf2)2 | 78 | 37 | 47 |
| 4 | Fe(ClO4)3 | 34 | 0 | 0 |
| 5 | Mn(F6-acac)2 | 83 | 40 | 48 |
| 6 | Mn(OAc)2 | 99 | 45 | 45 |
| 7 | Mn(acac)2 | 99 | 42 | 42 |
| 8 | Mn(acac)3 | 99 | 40 | 40 |
| 9 | MnSO4 | 99 | 50 | 50 |
| 10 | Mn(NO3)2 | 99/87a | 48/43a | 48/49a |
| 11 | MnBr2 | 99 | 42 | 42 |
| 12 | MnCl 2 | 99/97 | 51/49 | 51/51 |
| 13 | MnF3 | >99 | 40 | 40 |

Similar results were obtained with Mn(II) bromide and Mn(III) fluoride (Table 1, entries 11 and 13). Surprisingly, inexpensive Mn(II) chloride and MnSO4 and Mn(NO3)2 gave best results with around 50% yield of 1,2-epoxyoctane 2a (Table 1, entries 9, 10 and 12). To better distinguish between the best working precursors, we tested MnCl2 and Mn(NO3)2 again with only 2.5 equivalents of oxidant and found that MnCl2 yielded almost identical results as before, while Mn(NO3)2 gave a slightly reduced conversion and a lower yield. Therefore, for all further experiments, MnCl2 was used as the metal precursor.
At this point, it should be also noted that a related iron system showed significantly lower conversion and no desired product yield in the present protocol (Table 1, entry 4). Notably, reducing the amount of the oxidant even further to 1.0 equivalent with MnCl2 as the precursor, we still achieved 66% conversion and 26% yield of 2a (see the ESI, Table S2,† entry 3), indicating the high selectivity of this system against hydrogen peroxide decomposition.
To study the influence of picolinic acid ligands and ligand concentration, we considered these latter results (66% conversion and 26% yield) to be more suitable for observing both positive and negative effects. Starting with an initial [PicOH]:[Mn] ratio of 20:1, we consecutively reduced the amount of picolinic acid by a factor of ten up to 0.5 mol%, i.e., a ratio of 2:1. Interestingly, 4 equivalents of picolinic acid with respect to the metal gave the best result and slightly increased yield (35%) of 1,2-epoxyoctane 2a (see Fig. S2†). Noteworthily, in the absence of picolinic acid, around 30% conversion was observed but no epoxide formation was detected.
Presumably, the starting material undergoes complete oxidative decomposition as no major side products were observed by GC analysis. A control experiment utilizing picolinic acid-N-oxide also did not result in any product formation. Hence, the formation of this species as the active ligand can be excluded under catalytic conditions.
Next, we investigated the influence of the substitution pattern on picolinic acid (see Table 2). Both electron-donating substituents (3-Me, 4-Me, and 5-Me) and electron-withdrawing substituents, i.e., 3-Cl and 3-CF3 provided product 2a in similar yields of ∼27%. 5-Fluoropicolinic acid proved less suitable, yielding 22% of epoxide. Finally, blocking the 6-position, either by employing quinoline-2-carboxylic acid or 6-fluoropicolinic acid, led to no product formation whatsoever, as in the absence of any ligand. Therefore, we assume that the active complex does not form if the 6-position of the ligand is blocked, which is in accordance with the works of Stack.22 The same result was observed for 4-oxazolecarboxylic acid, indicating that no active complex is formed.
| Conversion and yield determined by GC analysis with hexadecane as IST. Reaction conditions: 0.5 mmol substrate (0.250 M), 0.25 mol% manganese(II)chloride, 1 mol% picolinic acid derivative, 5 mol% quinoline, 0.5 eq. of 2,3-butadione, MeCN (2 mL), 25 °C, 2 h slow addition of H2O2 (30% aq., 1 eq., diluted in MeCN) via a syringe pump. |
|---|

|
To investigate the influence of the N-heterocycle, the model reaction was performed in the presence of several quinolines, pyridines and other heterocycles (Table 3). Applying 2-methylquinoline gave a slightly improved yield of epoxide 2a (42%) compared to quinoline. In contrast, the introduction of a methyl group at the 8-position of quinoline severely hindered the reaction and only yielded 21% of epoxide (for a more detailed discussion about this difference see the ESI†). Other quinoline derivatives yielded the epoxide in similar yields of 33–37%. Pyridines proved to be similarly or slightly less efficient than quinolines with bulky 2-phenylpyridine providing the epoxide only in low yield (18%). While imidazoles yielded the desired products in yields below 30%, 2-methyloxazoline proved suitable similar to quinoline. Here, 2-phenyloxazoline was also less efficient. Lastly, various benzimidazoles provided the desired products in almost identical yields of slightly above 30% with little effects of methyl substituents being observed. Considering the negative effect of very bulky substituents in the vicinity of the nitrogen-atom, a coordination of the heterocycle to the metal centre during the catalytic reaction seems reasonable. Additionally, we also employed two simple bases NaOAc and NaOH for comparison. While the former is suitable, though less effective compared to 2-methylquinoline, the latter provided a poor yield of epoxide. Taken together, these results suggest that the employed heterocycle fulfils multiple roles in this reaction, i.e., not only being a basic additive but also acting as a potentially stabilizing co-ligand for the metal catalyst.
| Conversion and yield determined by GC analysis with hexadecane as IST. Reaction conditions: 0.5 mmol substrate (0.250 M), 0.25 mol% manganese(II)chloride, 1 mol% picolinic acid, 5 mol% N-heterocycle or base, 0.5 eq. of 2,3-butadione, MeCN (2 mL), 25 °C, 2 h slow addition of H2O2 (30% aq., 1 eq., diluted in MeCN) via a syringe pump. |
|---|

|
Having identified suitable heterocycles, we then varied the amount of the employed 2-methylquinoline. As expected, employing (sub)stoichiometric amounts of 2-methylquinoline in relation to picolinic acid, much lower conversions and yields of epoxide are obtained as the postulated manganese–picolinate complex cannot be formed if the picolinic acid is not (fully) deprotonated. With 2.5 mol% or more, i.e., 2.5 equivalents of 2-methylquinoline in relation to picolinic acid, comparable results are achieved.
However, employing more than 5 mol% does not further improve the best yield of 42% obtained so far (see Fig. S3†), which is why we settled for a MnCl2:PicOH:2-methylquinoline ratio of 1:4:20.
As established in the literature,20 diketones such as 2,3-butadione can form hydroxy–hydroperoxy adducts with hydrogen peroxide. These adducts are able to oxidize the metal catalyst, e.g., manganese(II/III) species that will then transfer the oxygen atom(s) to the substrate, generating the desired product. Besides 2,3-butadione, we also tested two other ketone additives in this reaction. Here, methyl pyruvate yielded the desired epoxide in 33% yield, whereas pyruvonitrile was less efficient, giving only 13% yield of 1,2-epoxyoctane under the employed reaction conditions. Performing the reaction without the ketone additive led to no product formation, whatsoever. Also, reducing or increasing the amount of 2,3-butadione to 0.25 or 1.0 equivalent, respectively, did not improve the reaction efficiency (see Table S4† for more information).
After having determined the optimal ratios and stoichiometry of all employed additives, the catalyst amount was varied at a 1:4:20 ratio of MnCl2:PicOH:2-methylquinoline. Increasing the amount of catalyst to 1 mol% led to slightly lower conversion of the starting material and accordingly lower yields (Table 4, entry 1). This behaviour can be explained by increased H2O2 disproportionation as described in other oxidation reactions.38 In contrast, lowering the amount of catalyst to only 0.05 mol% Mn, still achieved 38% epoxide yield. Simply changing the reaction solvent to a more polar mixture (MeCN:H2O = 75:25, vol%:vol%) again provided 42% yield of 2a, possibly due to better solubility of the manganese precursor and picolinic acid (Table 4, entry 6). However, using larger amounts of water led to solubility problems of the starting material and poor conversions. Using EtOH or an EtOH:H2O (75:25) mixture as reaction solvent led to poor results, giving only 7% and 12% yields, respectively (possibly due to EtOH oxidation competing with the substrate and/or blocking of the catalyst, see below) (Table 4, entries 7 and 8).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | H2O2 [eq.] | MnCl2 [mol%] | Solvent | Time [h] | Conv. (1a) [%] | Yield (2a) [%] | Sel. (2a) [%] |
| Conversion and yield determined by GC analysis with hexadecane as the IST. Reaction conditions: 0.5 mmol substrate (0.250 M), X mol% MnCl2:PicOH:2-methylquinoline (1:4:20), 0.5 eq. of 2,3-butadione, solvent (2 mL), 25 °C, 2 h slow addition of H2O2 (30% aq., X eq., diluted in MeCN) via a syringe pump. |
|||||||
| 1 | 1.0 | 1.0 | MeCN | 2 | 71 | 31 | 44 |
| 2 | 1.0 | 0.25 | MeCN | 2 | 85 | 42 | 49 |
| 3 | 1.0 | 0.125 | MeCN | 2 | 79 | 37 | 47 |
| 4 | 1.0 | 0.05 | MeCN | 2 | 79 | 38 | 48 |
| 5 | 1.0 | 0.01 | MeCN:H2O (95:5) |
2 | 75 | 31 | 41 |
| 6 | 1.0 | 0.05 | MeCN:H2O (75:25) |
2 | 82 | 42 | 51 |
| 7 | 1.0 | 0.05 | EtOH | 2 | 35 | 7 | 20 |
| 8 | 1.0 | 0.05 | EtOH:H2O (75:25) |
2 | 44 | 12 | 27 |
| 9 | 1.25 | 0.05 | MeCN:H2O (75:25) |
2 | 87 | 44 | 51 |
| 10 | 1.5 | 0.05 | MeCN:H2O (75:25) |
2 | 92 | 50 | 54 |
| 11 | 2.0 | 0.05 |
MeCN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :H
2
O (75:25) :H
2
O (75:25)
|
2 | 97 | 61 | 63 |
| 12 | 2.25 | 0.05 | MeCN:H2O (75:25) |
2 | 99 | 59 | 60 |
| 13 | 2.5 | 0.05 | MeCN:H2O (75:25) |
2 | >99 | 57 | 57 |
| 14 | 1.5 | 0.05 | MeCN:H2O (75:25) |
4 | 89 | 47 | 53 |
| 15 | 1.5 | 0.01 | MeCN:H2O (95:5) |
4 | 83 | 40 | 48 |

Finally, the amount of the employed hydrogen peroxide (30% aq.) was studied (Table 4, entries 9–13). Using 1.0 equivalent of H2O2 in the presence of 0.05 mol% Mn already gave 42% yield of the desired product 2a. Interestingly, by employing 2.0 equivalents of H2O2, we obtained a significantly higher yield of 61% and selectivity towards epoxide 2a of 63% (Table 4, entry 11). A further increase in H2O2 led to full conversion of 1a, however, the yields of 2a could not be improved (Table 4, entries 12 and 13).
Scope
Investigating the scope of this manganese-catalysed oxidation protocol, we first employed different terminal and linear alkenes as substrates. 1-Hexene (1b), 1-heptene (1c) and 1-octene (1a) were all converted to their corresponding epoxides 2a–c in good yields of 61–65%. These results are superior to previously reported protocols for aliphatic olefins (either in terms of yield or in terms of sustainability), where either much higher (noble-metal) catalyst loadings, more expensive ligands, or higher amounts of less benign oxidants, or (corrosive) additives were necessary.5,9,11,14,17,20,22,53,59–62 Of note, around 80% yields for these substrates can be achieved with non-noble metals (as demonstrated by Stack and co-workers);22 however, here, the less benign oxidant peracetic acid was employed which resulted in undesirable by-product formation. Further extension of the chain length, however, led to a decrease in conversion and correspondingly lower yields. With 1-decene 1d and 1-dodecene 1e, moderate yields of 56% and 37% were achieved, respectively, probably due to lower solubility of the starting materials in the polar MeCN:H2O (75:25) solvent mixture. Indeed, employing a less polar solvent mixture (MeCN:H2O = 95:5) for these substrates led to slightly varying yields of 51% of 1,2-epoxydecane 2d and 45% of 1,2-epoxydodecane 2e.
Applying di- and tri-substituted olefins showed an interesting trend: with 2-methyl-1-heptene 1f, an improved yield of 71% of the desired epoxide 2f was obtained, while with 2-methyl-2-heptene 1g, only 49% of epoxide 2g was obtained. Disubstituted olefins are more nucleophilic and therefore more reactive, accounting for better performance. Though the electronic properties of trisubstituted olefins are even more nucleophilic, here, steric influence starts to interfere with the reaction, demonstrating the selectivity of this catalytic system for sterically less demanding olefins.
Testing cyclic olefins, the reaction proceeded with much higher selectivity. With cyclohexene 1h and cyclooctene 1i, the desired epoxides 2h and 2i were obtained in ∼80% yield. In both cases, no allylic oxidation products were observed, indicating that this reaction does not proceed via a radical/Fenton-type reactivity pathway.
Investigating dienes as substrates, we first employed 1,7-octadiene 1j under the standard reaction conditions. Here, 88% conversion and 21% of diepoxide 2j-2 were observed with about 30% of the mono-epoxide 2j-1 product. Obviously, with dienes, the total concentration of olefinic functionalities is twice as high as that with simple olefins. Therefore, we doubled the amount of hydrogen peroxide to 4 equivalents. Interestingly, this did not change the result. However, reducing the amount of the employed substrate 1j to 0.25 mmol, i.e., operating with the same concentration of olefinic functionalities as that under the optimized conditions, a significant increase of the yield and selectivity was observed. In this case, full conversion of the starting material 1j was observed, and no mono-epoxide 2j-1 remained after 2 h reaction time, obtaining 49% of the desired di-epoxide 2j-2. It should be noted that such di-epoxidation reactions have been scarcely investigated but offer interesting possibilities for oligomerisation and polymerisation.
Due to its industrial relevance in the fragrance industry, we also investigated the selective mono- and di-epoxidation of cyclooctadiene 1k (COD). When employing only 1.5 eq. of H2O2 (30% aq.) to prevent over-oxidation to di-epoxide 2k-2, 88% conversion was achieved, and the desired mono-epoxide 2k-1 was isolated in 62% yield. Halving the substrate concentration and using 5 eq. of peroxide, we were able to selectively obtain di-epoxide 2k-2 as single major product in 55% isolated yield. To further demonstrate the applicability of this system, we also performed a multi-gram scale (5 g of substrate) reaction of the mono-epoxidation of COD. Here, we isolated 3.1 g of the desired product 2k-1 (55% yield).
As mentioned vide supra, cyclic olefins are more reactive than terminal olefins, thus, we employed 4-vinyl-cyclohexene 1l as the starting material to investigate the selectivity. Under standard conditions, 97% conversion of diene 1l was achieved with 53% yield of the ring epoxidation product 2l-1 (dr 1:1.3) and 16% yield of di-epoxide 2l-2 (dr 2.5:1). No sole side-chain epoxidation product was observed. Reducing the amount of oxidant to 1.5 equivalents increased the reaction efficiency by obtaining the same yield of the desired ring epoxide 2l-1 but less overoxidation to di-epoxide 2l-2 was observed. Again, when employing only 0.25 mmol of diene and 5 equivalents of oxidant, full conversion and 47% yield of di-epoxide 2l-2 were obtained as the single major product (Table 5).
| Conversion and yield determined by GC analysis with hexadecane as IST. Reaction conditions: 0.5 mmol substrate (0.250 M), 0.05 mol% MnCl2, 0.2 mol% picolinic acid, 1 mol% 2-methylquinoline, 0.5 eq. of 2,3-butadione, MeCN:H2O (75:25, 2 mL), 25 °C, 2 h slow addition of H2O2 (30% aq., 2 eq., diluted in MeCN) via a syringe pump. aMeCN:H2O (95:5) as solvent. b0.25 mmol of substrate employed. cYield determined by NMR analysis with dibromomethane as the IST. dSame results were obtained employing 4 eq. of H2O2. e1.5 eq. of H2O2 were employed. fIsolated yield. g5 eq. of H2O2 were employed. h5 g scale reaction. i1.5 eq. of 2,3-butadione were employed, n.d.: not determined. |
|---|

|
Though this protocol was initially optimised for aliphatic alkenes, we also employed aromatic alkenes as substrates under the same conditions. In the case of styrene 1m, we observed a reduced conversion of 40% and a 34% yield of styrenoxide 2m. Though the yield is comparably low, a high selectivity of 85% was achieved here. This prompted us to further investigate styrene as the model substrate for aromatic olefins. As only low conversion was observed, we reduced the concentration of styrene to 0.125 M. This change led to much better results, approximately doubling the conversion and yield to 84% and 69%, respectively. Further increasing the amount of 2,3-butadione did not lead to full conversion. Investigating the effect of electron-withdrawing and electron-donating substituents at the 4-position of styrene did not reveal significant changes in the outcome. With both 4-F- and 4-MeO-substituents (see substrates 1n and 1o), the same conversions of 75% were achieved, while similar yields of 60% and 64% were obtained, respectively, demonstrating the robustness of this system towards electronic effects of substituted aromatic substrates.
Switching from styrenes to allylbenzene 1p, we obtained 77% conversion and 43% of the desired epoxide 2p under standard conditions. Employing 0.25 mmol of substrate led to almost full conversion (97%); however, a lower selectivity compared to styrene was obtained, giving the desired product 2p in 52% yield. Trace amounts of benzylic oxidation products were observed here.
To further expand the applications of this protocol, we then turned our attention to the epoxidation of naturally occurring alkenes, e.g., terpenes. Here, we first employed (−)-limonene 1q as a substrate, using only 1.5 equivalents of oxidant under otherwise standard reaction conditions. In this case, we obtained 41% of the ring epoxidation compound 2q-1 as the major product and 11% of the di-epoxide product 2q-2. Fine tuning the reaction conditions to obtain the di-epoxide as major product was easily accomplished first by halving the substrate concentration which resulted in a roughly one to one mixture of both products and consecutively raising the amount of H2O2 (30% aq.) to 5 equivalents, which then yielded the desired di-epoxide product 2q-2 in 45% yield as the sole major product. Next, we subjected α-pinene 1r to our epoxidation reaction conditions. In this case, 83% conversion but only 27% yield of the desired product 2r were obtained, while minor amounts of other unselective oxidation/decomposition products were detected upon GC-MS analysis, e.g., campholenic aldehyde. Since aldehydes are easily oxidized to the corresponding carboxylic acids, this would account for the lower selectivity with this substrate, as the formation of large amounts of acids negatively impede the performance of this catalyst system. In the case of myrcene 1s, high conversion of all three C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds (∼76% after 1 h and ∼83% after 2 h), but unselective product formation was detected.
C double bonds (∼76% after 1 h and ∼83% after 2 h), but unselective product formation was detected.
In addition to terpenes, we also investigated the “mushroom alcohol” 1-octene-3-ol 1t and the analogous ketone 1u as substrates. Interestingly, with the former substrate, NMR analysis indicated the formation of three major products. First, the epoxidation of the CC double bond to the corresponding hydroxy-epoxide diastereomers 2t-1 (d.r. 1:1) is observed with 24% yield. In addition, the O–H group is also further oxidized to the ketone epoxide 2t-2 in 12% yield. As this class of compound easily undergoes epoxide ring-opening, the corresponding diol 2t-3 is formed with 5% yield. A similar reaction outcome was observed with 1-octene-3-one 1u as the substrate. Finally, the fatty acid ester ethyl oleate 1v was employed as substrate and the desired epoxide product 2v was isolated in 66% yield, again demonstrating the high selectivity of this system towards aliphatic unactivated CC double bonds (see Table 6).
| Conversion and yield determined by GC analysis with hexadecane as IST. Reaction conditions: 0.5 mmol substrate (0.250 M), 0.05 mol% MnCl2, 0.2 mol% picolinic acid, 1 mol% 2-methylquinoline, 0.5 eq. of 2,3-butadione, MeCN:H2O (75:25, 2 mL), 25 °C, 2 h slow addition of H2O2 (30% aq., 2 eq., diluted in MeCN) via a syringe pump. aMeCN:H2O (95:5) as solvent. b0.25 mmol of substrate employed. cYield determined by NMR analysis with dibromomethane as the IST. d1.5 eq. of H2O2 were employed. eIsolated yield. f5 eq. of H2O2 were employed. |
|---|

|
Besides epoxidation, selective aliphatic C–H oxidation with non-noble metal catalysts is even more challenging. Obviously, such transformations allow the implementation of functional groups, i.e., hydroxy or carbonyl groups, into unfunctionalized compounds, thus profoundly changing the physical (and biological) properties of the starting materials.63 Therefore, we also investigated C–H oxidation reactions with the present catalytic protocol as trace amounts of C–H oxidation products were observed when employing allylbenzene as the substrate.
Also, similar systems for oxidation of C–H bonds in alkanes have been reported in the literature.23 As the model substrate for C–H functionalization reactions, we chose cyclohexane 3a due to its industrial relevance and equivalence of all present C–H bonds. In fact, “KA oil”, a mixture of cyclohexanone and cyclohexanol, is used as a precursor for adipic acid whose production exceeds three million tons per anum and is still growing annually.64 After a short optimization (see the ESI and Table S5† for more information), we were delighted to obtain 43% yield of cyclohexanone 5a from cyclohexane with complete selectivity for ketone 5a over alcohol 4a.
Consequently, we subjected various alkanes to this slightly modified catalytic protocol. Using cyclododecane 3b, we obtained cyclododecanone 5b (a precursor to laurolactam) in 31% yield as the sole major product (the limiting factor here seems to be the solubility). Employing cyclooctane 3c, we observed a high conversion of 93% and a good yield of 51% of the desired ketone product 5c. Again, only traces of alcohol 4c were detected. Next, we tested alkanes bearing aromatic rings as substrates. Here, tetrahydronaphthalene 3d performed similarly well with 72% conversion and 43% of the corresponding ketone 5d, while small amounts of alcohol 4d were detected in this case. Switching to non-cyclic alkanes, such as ethylbenzene 3e bearing an activated benzylic position, a different reactivity is expected. Indeed, 38% conversion was observed, resulting in a mixture of 13% phenylethanol 4e and 24% acetophenone 5e. Applying a more polar solvent mixture, slightly improved this result, giving 46% conversion and 29% of acetophenone 5e. In the case of n-octane 3f, 50% conversion resulting in a 1:1:1 mixture of the three possible ketone products 5f-1–3 with 33% combined yield with no alcohol formation observed (see Table 7).
| Conversion and yield determined by GC analysis with hexadecane as IST. Reaction conditions: 0.25 mmol substrate (0.125 M), 0.1 mol% MnCl2, 0.4 mol% picolinic acid, 2 mol% 2-methylquinoline, 1 eq. of 2,3-butadione, MeCN:H2O (95:5, 2 mL), 25 °C, 2 h slow addition of H2O2 (30% aq., 4 eq., diluted in MeCN) via a syringe pump. aMeCN:H2O (75:25) as solvent. |
|---|

|
Finally, we investigated the oxidation of alcohols with the present protocol since we also observed O–H oxidation employing 1-octene-3-ol as the epoxidation substrate (see Table 8) and mainly ketones resulted from C–H oxidation. First, we compared primary and secondary alcohols to verify the standing thesis that primary alcohols are indeed not tolerated under present reaction conditions due to the formation of carboxylic acids. With 2-octanol 4f as the substrate, we were delighted to achieve 86% conversion and 79% yield of 2-octanone 5f under standard epoxidation reaction conditions. In contrast, oxidation of 1-octanol 4g did not take place selectively under the standard reaction conditions and low conversion (30%) and ca. 20% of octanoic acid 6g were detected. Since this catalytic system relies on the deprotonation of picolinic acid by the 2-methylquinoline additive to form the active complex, the formation of significant amounts of acid obviously impedes the catalytic activity. Consequently, various secondary alcohols were subjected to our catalytic protocol. When employing cyclohexanol 4a and cyclooctanol 4c as substates, identical yields of 68% of the desired ketones 5a and 5c were obtained. Using the less polar cyclododecanol 4b as substrate, a reduced yield of 47% was obtained. However, this was improved upon by switching to a less polar solvent mixture (MeCN:H2O = 95:5), resulting in 59% yield of cyclododecanone 5b. Furthermore, phenylethanol 4e proved to be an excellent substrate with almost full conversion and selectivity, yielding acetophenone 5e in 92% yield. Lastly, with tetrahydronaphthalene-1-ol 4d, 96% conversion and 61% yield of 1-tetralone 5d were achieved. Here, small amounts of over-oxidation products, e.g., the diketone, were observed upon GC analysis, accounting for the lower mass balance. In general, however, higher mass balances are achieved with O–H oxidation reactions compared to C–H oxidation or epoxidation reactions with this catalytic system.
| Conversion and yield determined by GC analysis with hexadecane as IST. Reaction conditions: 0.5 mmol substrate (0.250 M), 0.05 mol% MnCl2, 0.2 mol% picolinic acid, 1 mol% 2-methylquinoline, 0.5 eq. of 2,3-butadione, MeCN:H2O (75:25, 2 mL), 25 °C, 2 h slow addition of H2O2 (30% aq., 2 eq., diluted in MeCN) via a syringe pump. aMeCN:H2O (95:5) as solvent. b0.25 mmol of substrate employed. |
|---|

|
Mechanistic investigations and proposal
Upon investigating the scope of our catalytic protocol, several interesting information on the activity of this novel system was obtained. To gain more knowledge about the detailed action of this manganese catalyst, we investigated the involvement of radical species by conducting control experiments employing the radical scavengers TEMPO and BHT. Both compounds impede the reactivity but do not block the catalyst. For example, the addition of 5 mol% TEMPO reduces the catalytic activity by 20% (see Scheme S1 and the ESI† for more information). To prove whether the observed over-oxidation products are a result of radical side-reactions, we reacted 1,2-epoxyoctane 2a under the standard reaction conditions and found that 80% of 2a could be recovered after 2 hours. Interestingly, performing the same experiment in the presence of TEMPO, we found that 2a could be completely recovered. Therefore, we assume that the partial degradation of the epoxide products is a result of unwanted radical reactions.Next, we recorded a kinetic profile of the epoxidation of 1-octene 1a to identify possible intermediates or follow-up products that might be formed in small amounts during the reaction. In accordance with related studies,35 it is apparent that both the substrate consumption and the product formation follow an approximately linear course. Nevertheless, in the beginning, substrate conversion is slightly faster than the product formation, indicating that the active epoxidation catalytic species might not be formed immediately upon H2O2 addition. Therefore, the selectivity towards the desired product 2a at the beginning of the reaction is about 40% until it rises to ∼60% after 40 minutes and remains constant for the rest of the reaction (see Fig. 1). Additionally, no major side-products or decrease in the yield of the product were observed. Therefore, we assume that substrate over-oxidation or degradation takes place at the very beginning of the reaction, as the active catalytic species is not yet formed. This is also in agreement with previous works.5,14 Furthermore, we recorded the kinetic profile of the C–H oxidation of cyclohexane 3a to cyclohexanone 5a to compare both oxidative transformations. Here, at the beginning of the reaction, a lower selectivity is observed that reaches ∼60% after 60 minutes and stays in the range of 60–70% for the remaining reaction time. Again, the lower selectivity towards the desired product at the beginning of the reaction indicates a lag period during which the active catalytic species is not yet formed. In contrast to 1-octene epoxidation, no quantitative conversion of cyclohexane 3a is achieved under the present reaction conditions. Finally, there is no accumulation of cyclohexanol 4a as an intermediate as only trace amounts of the alcohol are observed during the whole reaction time (see Fig. 2). Taking these results and previous works34 into consideration, we propose similar reaction pathways and reactive intermediates for both types of oxidation reactions.
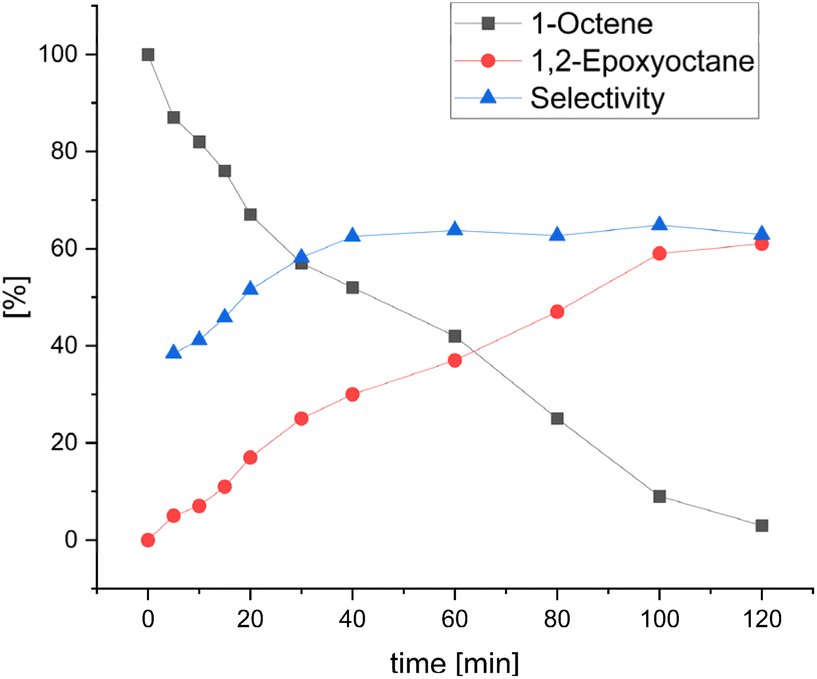 | ||
| Fig. 1 Kinetic profile of manganese-catalysed epoxidation of 1-octene. Conversion and yield determined by GC analysis with hexadecane as IST. Reaction conditions: 0.5 mmol substrate (0.250 M), 0.05 mol% MnCl2, 0.2 mol% picolinic acid, 1 mol% 2-methylquinoline, 0.5 eq. of 2,3-butadione, MeCN:H2O (75:25, 2 mL), 25 °C, 2 h slow addition of H2O2 (30% aq., 2 eq., diluted in MeCN) via a syringe pump. For each point in time, a separate reaction was set up and analysed after the indicated slow addition time. | ||
 | ||
| Fig. 2 Kinetic profile of manganese-catalysed C–H oxidation of cyclohexane. Conversion and yield determined by GC analysis with hexadecane as IST. Reaction conditions: 0.25 mmol substrate (0.125 M), 0.1 mol% MnCl2, 0.4 mol% picolinic acid, 2 mol% 2-methylquinoline, 1 eq. of 2,3-butadione, MeCN:H2O (95:5, 2 mL), 25 °C, 2 h slow addition of H2O2 (30% aq., 4 eq., diluted in MeCN) via a syringe pump. For each point in time, a separate reaction was set up and analysed after the indicated slow addition time. | ||
While investigating the scope, we observed that aromatic alkenes required higher catalyst loadings than aliphatic alkenes to achieve comparable yields. Furthermore, allylbenzene was preferentially oxidized to the epoxide, although (benzylic) C–H oxidation is also possible. Also, 1-octene-3-ol was primarily oxidized to the corresponding epoxide though in lower yield due to several follow-up oxidations. These results show that epoxidation seems to be preferred over C–H and O–H oxidation, while O–H oxidation is preferred compared to C–H oxidation. To prove these assumptions, competition experiments of 1-octene with selected other substrate classes were performed. First, equal amounts of 1-octene 1a and styrene 1m (0.25 mmol each) were subjected to our standard reaction conditions. Interestingly, styrene performed similarly well in this competition experiment (60% yield of 2m), while 1-octene was converted in poor yield (8%) to 1,2-epoxyoctane 2a.
Although this system was optimized for the epoxidation of aliphatic alkenes, their aromatic, activated counterparts are more reactive when both substrates are employed in a single reaction. In the second set up, we compared 1-octene 1a as an epoxidation substrate to 2-octanol 4f as an alcohol oxidation substrate. Here, 1,2-epoxyoctane 2a was formed in a similar yield as before from the former substrate (56%), while 2-octanone 5f was obtained in a somewhat reduced yield of 56%, confirming the previously observed trend that epoxidation takes precedent over O–H oxidation when both functional groups are present. Finally, subjecting equal amounts of 1-octene 1a and cyclohexane 3a to our standard reaction conditions, 1,2-epoxyoctane 2a was again obtained in a similar yield as before (54%) while cyclohexanone 5a was only obtained in 10% yield (18% when the less polar solvent mixture is used, see Scheme 2). Taken together, these results show that the presented manganese catalyst system preferentially oxidizes alkenes in the presence of alcohols and in the presence of C–H oxidation substrates. Furthermore, under optimized reaction conditions, 1-octene requires lower catalyst loading and fewer peroxide equivalents than styrene; however, with higher loadings and more peroxide, styrene outcompetes 1-octene as a substrate when both compounds are present in the same reaction set-up.
 | ||
| Scheme 2 Competitions experiments of 1-octene with selected other substrate classes. Conversion and yield determined by GC analysis with hexadecane as IST. aMeCN:H2O (95:5) as solvent. | ||
Based on all these observations, we propose the following catalytic cycle for this newly developed oxidation catalyst: in the first step A, the generation of the postulated [(PicO)2MnX2]2− - complex occurs, enabled by deprotonation of PicOH by 2-methylquinoline resulting in the negatively charged species I, with two protonated quinolyl species [2-MQ-H]+ as counterions. Here, the nature of the two ligands X− occupying the two additional coordination sites of the manganese centre remains unclear. Two chloride ligands derived from the precursor or OH-groups from hydrolysis of MnCl2 to Mn(OH)2 and solvent coordination (H2O, MeCN) seem to be possible.
In the second step B, one of the ligands X− is exchanged by the coordination of the co-ligand, 2-methylquinoline, leading to the formation of species II (though species I and II are possibly in equilibrium). Here, the formal charge of X− would be compensated by the present protonated quinolyl species [2-MQ-H]+. In accordance with the literature, 2,3-butadione and hydrogen peroxide are in equilibrium (C) with 3-hydroxy-3-hydroperoxybutanone.34 In the following step D, this formed adduct substitutes the remaining X− ligand, resulting in H2O or HCl elimination, which in turn is deprotonated by another 2-methylquinoline, forming an additional [2-MQ-H]+ and manganese species III. Considering the results from the co-ligand screening, where 8-methylquinoline exhibited a much worse performance than 2-methylquinoline, the formation of species III could be severely hindered by the steric effect of the 8-methyl group in the case of 8-MQ as the co-ligand. Additionally, the presence of TEMPO could either compete with picolinic acid as the ligand, or impede step D, by coordinating to the manganese centre and preventing 3-hydroxy-3-hydroperoxybutanone from coordinating, thus accounting for the negative effect TEMPO had on the reaction outcome.
Species III, in which manganese is still in the oxidation state (II) undergoes heterolysis of the O–O bond from the coordinated 3-hydroxy-3-hydroperoxybutanone, resulting in the formation of species IV with a manganese(IV) centre (step E). This step is facilitated by the present acidic counter-cation [2-MQ-H]+ which further activates the O–O bond by either forming a hydrogen bond or even promoting protonolysis65 of species III resulting in the immediate regeneration of the 2-MQ.
Alternatively, 2-MQ would be regenerated in a consecutive step by deprotonation of [2-MQ-H]+ with concomitant regeneration of 2,3-butadione and formation of H2O as the oxidant by-products. High-valent manganese oxo-species IV, which is stabilised by the present donor-ligand 2-methylquinoline,10,66 is presumed to be the active oxidation catalyst, thus oxidizing the present alkene to the corresponding epoxide (step F). Upon regeneration of the manganese(II) species II, the free coordination site is stabilized again by ligand X− (see Scheme 3).
 | ||
| Scheme 3 Mechanistic proposal for the manganese-catalysed (ep)oxidation reaction. | ||
Conclusions
In summary, we demonstrated the general potential of an easily accessible manganese-based catalyst system for the selective oxidation of olefins, alkanes, and alcohols, which are of importance for bulk chemicals as well as naturally occurring feedstocks. To the best of our knowledge, this non-noble metal catalyst system offers the highest efficiency of any acid-free in situ system especially for the epoxidation of unactivated terminal aliphatic olefins with yields of up to 65%. Additionally, unactivated (cyclic) alkanes can be converted selectively with yields of up to 51% to their corresponding ketones, streamlining industrially relevant processes, e.g., adipic acid production. Furthermore, the role of the employed N-heterocycles was investigated in detail. On the one hand, 2-methylquinoline acts as a base deprotonating picolinic acid and generating the active catalyst system. On the other hand, it can be regarded as a co-ligand which has a beneficial effect on the reaction outcome.Experimental
Important safety note
Hydrogen peroxide may cause explosion upon contact with metal catalysts. Therefore, we are working with the safer 30% aqueous hydrogen peroxide solution instead of the higher concentrated 50% solution.General procedure for the epoxidation of olefins
An 8 mL glass vial equipped with a Teflon coated stirring bar was charged with stock solutions of MnCl2 (0.25 μmol, 31.5 μg, 0.05 mol% in 250 μL H2O), picolinic acid (1 μmol, 0.123 mg, 0.2 mol% in 250 μL H2O) and freshly distilled 2-methylquinoline (5.0 μmol, 0.716 mg, 1.0 mol% in 250 μL MeCN). The resulting mixture was stirred for 5 minutes. Next, a solution of 2,3-butadione (0.25 mmol, 43 mg, 0.5 eq. in 250 μL MeCN) was added. The resulting mixture was further diluted with MeCN to a total volume of 2 mL (MeCN:H2O = 75:25) and stirred for additional 5 min. Then, 1-octene (0.5 mmol, 56.1 mg, 0.250 M) was added. Next, a solution of hydrogen peroxide (H2O2) (1.0 mmol, 2.0 eq., 104 μL, 30% aq.) in MeCN (906 μL) was added via a syringe pump to the reaction mixture over a course of 2 h.
For GC analysis, the reaction mixture was then diluted with EtOAc, filtered, and analysed using hexadecane (30 μL) as an internal standard to determine the conversion and yield by 5-point calibration of the respective compounds.
The same procedure was applied for alcohol oxidation. For C–H oxidation of alkanes, a slightly modified protocol was applied (see the ESI† for more information).
Author contributions
D. V. conceived and conceptualised the project, co-wrote and co-edited the manuscript, performed the experiments, and analysed the data; S. A. co-performed the experiments; X. W., Y. Y. and Y.-W. L. supported the project with funding acquisition; K. J. and M. B. provided the infrastructure, performed discussions of the project results, and co-wrote and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the analytical department from LIKAT for their help and services. Also, we acknowledge the funding from Synfuels China.References
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, 1998, vol. 29, pp. 14821–14842 Search PubMed.
- S. Caron, R. W. Dugger, S. G. Ruggeri, J. A. Ragan and D. H. B. Ripin, Chem. Rev., 2006, 106, 2943–2989 CrossRef CAS PubMed.
- F. Cavani and J. H. Teles, ChemSusChem, 2009, 2, 508–534 CrossRef CAS PubMed.
- S. Mao, D. Verspeek, X. Wen, Y. Yang, Y.-W. Li, K. Junge and M. Beller, ChemCatChem, 2023, e202300735 CrossRef CAS.
- S. Mao, S. Budweg, A. Spannenberg, X. Wen, Y. Yang, Y.-W. Li, K. Junge and M. Beller, ChemCatChem, 2021, e202101668, DOI:10.1002/cctc.202101668.
- D. Ros, T. Gianferrara, C. Crotti and E. Farnetti, Front. Chem., 2020, 8, 810 CrossRef CAS.
- S. Jana, P. De, C. Dey, S. G. Dey, A. Dey and S. S. Gupta, Chem. Sci., 2023, 14, 10515–10523 RSC.
- D. Pijper, P. Saisaha, J. W. de Boer, R. Hoen, C. Smit, A. Meetsma, R. Hage, R. P. van Summeren, P. L. Alsters, B. L. Feringa and W. R. Browne, Dalton Trans., 2010, 39, 10375–10381 RSC.
- C. Coperet, H. Adolfsson and K. B. Sharpless, Chem. Commun., 1997, 1565–1566 RSC.
- A. Neshat, M. Kakavand, F. Osanlou, P. Mastrorilli, E. Schingaro, E. Mesto and S. Todisco, Eur. J. Inorg. Chem., 2020, 2020, 480–490 CrossRef CAS.
- C. Miao, B. Wang, Y. Wang, C. Xia, Y. M. Lee, W. Nam and W. Sun, J. Am. Chem. Soc., 2015, 138, 936–943 CrossRef PubMed.
- W. Wang, D. Xu, Q. Sun and W. Sun, Chem. – Asian J., 2018, 13, 2458–2464 CrossRef CAS PubMed.
- F. Zhu, G. Yang, A. J. Zoll, E. V. Rybak-Akimova and X. Zhu, Catalysts, 2020, 10, 285 CrossRef CAS.
- D. Verspeek, S. Ahrens, A. Spannenberg, X. Wen, Y. Yang, Y.-W. Li, K. Junge and M. Beller, Catal. Sci. Technol., 2022, 12, 7341–7348 RSC.
- W. Zhang, J. L. Loebach, S. R. Wilson and E. N. Jacobsen, J. Am. Chem. Soc., 1990, 112, 2801–2803 CrossRef CAS.
- E. N. Jacobsen, W. Zhang, A. R. Muci, J. R. Ecker and L. Deng, J. Am. Chem. Soc., 1991, 113, 7063–7064 CrossRef CAS.
- M. C. White, A. G. Doyle and E. N. Jacobsen, J. Am. Chem. Soc., 2001, 123, 7194–7195 CrossRef CAS PubMed.
- M. S. Chen and M. C. White, Science, 2007, 318, 783–787 CrossRef CAS PubMed.
- Q.-W. Zhang, J. A. A. W. Elemans, P. B. White and R. J. M. Nolte, Chem. Commun., 2018, 54, 5586–5589 RSC.
- J. J. Dong, P. Saisaha, T. G. Meinds, P. L. Alsters, E. G. Ijpeij, R. P. van Summeren, B. Mao, M. Fañanás-Mastral, J. W. de Boer, R. Hage, B. L. Feringa and W. R. Browne, ACS Catal., 2012, 2, 1087–1096 CrossRef CAS.
- P. Saisaha, D. Pijper, R. P. van Summeren, R. Hoen, C. Smit, J. W. de Boer, R. Hage, P. L. Alsters, B. L. Feringa and W. R. Browne, Org. Biomol. Chem., 2010, 8, 4444–4450 RSC.
- R. A. Moretti, J. Du Bois and T. D. P. Stack, Org. Lett., 2016, 18, 2528–2531 CrossRef CAS PubMed.
- J. J. Dong, D. Unjaroen, F. Mecozzi, E. C. Harvey, P. Saisaha, D. Pijper, J. W. de Boer, P. Alsters, B. L. Feringa and W. R. Browne, ChemSusChem, 2013, 6, 1774–1778 CrossRef CAS PubMed.
- S. Kiani, A. Tapper, R. J. Staples and P. Stavropoulos, J. Am. Chem. Soc., 2000, 122, 7503–7517 CrossRef CAS.
- H. A. Younus, I. Yildiz, N. Ahmad, H. S. Mohamed, G. Khabiri, S. Zhang, F. Verpoort, P. Liu and Y. Zhang, Appl. Organomet. Chem., 2022, 36, e6538 CrossRef CAS.
- T. Shimamura, N. Yoshimura, H. Otsuka, M. Yoshida and A. Kobayashi, J. Photochem. Photobiol., A, 2023, 436, 114412 CrossRef CAS.
- D. Kim, M. Ahn, K.-R. Wee and D. W. Cho, Phys. Chem. Chem. Phys., 2022, 24, 13074–13082 RSC.
- M. V. Dimitrijević, L. E. Mihajlović-Lalić, S. Grgurić-Šipka, T. M. Mihajlov-Krstev, D. L. Miladinović and J. M. Poljarević, J. Coord. Chem., 2023, 76, 783–797 CrossRef.
- C. Gao, C. Liu, A. Said, H. Niu, D. Wang, G. Wang, C.-H. Tung and Y. Wang, Dalton Trans., 2022, 51, 3706–3712 RSC.
- F. Huo and Y. Lu, Chem. Eng. J., 2022, 440, 135804 CrossRef CAS.
- F. Lucio-Martínez, Z. Garda, B. Váradi, F. K. Kálmán, D. Esteban-Gómez, É. Tóth, G. Tircsó and C. Platas-Iglesias, Inorg. Chem., 2022, 61, 5157–5171 CrossRef PubMed.
- Z. Yang, Y. Cui, B. Pan and J. J. Pignatello, Environ. Sci. Technol., 2023, 57(47), 18918–18928 CrossRef CAS.
- B. Meunier, Chem. Rev., 1992, 92, 1411–1456 CrossRef CAS.
- J. B. Kasper, P. Saisaha, M. de Roo, M. J. Groen, L. Vicens, M. Borrell, J. W. de Boer, R. Hage, M. Costas and W. R. Browne, ChemCatChem, 2023, 15, e202201072 CrossRef CAS PubMed.
- P. Saisaha, J. J. Dong, T. G. Meinds, J. W. de Boer, R. Hage, F. Mecozzi, J. B. Kasper and W. R. Browne, ACS Catal., 2016, 6, 3486–3495 CrossRef CAS.
- P. Saisaha, J. W. de Boer and W. R. Browne, Chem. Soc. Rev., 2013, 42, 2059–2074 RSC.
- F. Mecozzi, J. J. Dong, P. Saisaha and W. R. Browne, Eur. J. Org. Chem., 2017, 6919–6925 CrossRef CAS PubMed.
- J. B. Kasper, L. Vicens, C. M. de Roo, R. Hage, M. Costas and W. R. Browne, ACS Catal., 2023, 13, 6403–6415 CrossRef CAS.
- D. Huang, W. Wang, X. Zhang, C. Chen, F. Chen, Q. Liu, D. Liao, L. Li and L. Sun, Eur. J. Org. Chem., 2004, 1454–1464 CrossRef CAS.
- H. Zhang, Q. Yao, L. Lin, C. Xu, X. Liu and X. Feng, Adv. Synth. Catal., 2017, 359, 3454–3459 CrossRef CAS.
- T. Katsuki and K. B. Sharpless, J. Am. Chem. Soc., 1980, 102, 5974–5976 CrossRef CAS.
- K. Matsumoto, Y. Sawada, B. Saito, K. Sakai and T. Katsuki, Angew. Chem., 2005, 117, 5015–5019 CrossRef.
- A. Berkessel, T. Guenther, Q. Wang and J. M. Neudörfl, Angew. Chem., Int. Ed., 2013, 52, 8467–8471 CrossRef CAS PubMed.
- R. Irie, K. Noda, Y. Ito and T. Katsuki, Tetrahedron Lett., 1991, 32, 1055–1058 CrossRef CAS.
- K. Schröder, B. Join, A. J. Amali, K. Junge, X. Ribas, M. Costas and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 1425–1429 CrossRef.
- O. Cussó, M. Cianfanelli, X. Ribas, R. J. M. Klein Gebbink and M. Costas, J. Am. Chem. Soc., 2016, 138, 2732–2738 CrossRef PubMed.
- H. Egami, T. Oguma and T. Katsuki, J. Am. Chem. Soc., 2010, 132, 5886–5895 CrossRef CAS PubMed.
- Y. Shen, P. Jiang, P. T. Wai, Q. Gu and W. Zhang, Catalysts, 2019, 9, 31 CrossRef.
- W.-C. Cheng, W.-H. Fung and C.-M. Che, J. Mol. Catal. A: Chem., 1996, 113, 311–319 CrossRef CAS.
- M. K. Tse, C. Döbler, S. Bhor, M. Klawonn, W. Mägerlein, H. Hugl and M. Beller, Angew. Chem., Int. Ed., 2004, 43, 5255–5260 CrossRef CAS.
- M. K. Tse, S. Bhor, M. Klawonn, G. Anilkumar, H. Jiao, A. Spannenberg, C. Döbler, W. Mägerlein, H. Hugl and M. Beller, Chem. – Eur. J., 2006, 12, 1875–1888 CrossRef CAS.
- A. K. Yudin and K. B. Sharpless, J. Am. Chem. Soc., 1997, 119, 11536–11537 CrossRef CAS.
- A. Murphy, A. Pace and T. D. P. Stack, Org. Lett., 2004, 6, 3119–3122 CrossRef CAS PubMed.
- J. R. Coombs and J. P. Morken, Angew. Chem., Int. Ed., 2016, 55, 2636–2649 CrossRef CAS.
- A. Murphy, G. Dubois and T. D. P. Stack, J. Am. Chem. Soc., 2003, 125, 5250–5251 CrossRef CAS.
- B. Kumar, B. Singh, A. Banday, S. Tewari, V. Kumar, S. Murugavel, P. A. Joy and A. Ramanan, CrystEngComm, 2021, 23, 6703–6723 RSC.
- D.-F. Zhou, Q.-Y. Chen, H.-J. Fu and Q. Yan, Spectrochim. Acta, Part A, 2011, 81, 604–608 CrossRef CAS PubMed.
- R. M. Philip, S. Radhika, C. M. A. Abdulla and G. Anilkumar, Adv. Synth. Catal., 2021, 363, 1272–1289 CrossRef CAS.
- H. Mimoun, M. Mignard, P. Brechot and L. Saussine, J. Am. Chem. Soc., 1986, 108, 3711–3718 CrossRef CAS.
- M. V. Benjamin, S. Lane, Victoria J. DeRose and Kevin Burgess, J. Am. Chem. Soc., 2002, 124, 11946–11954 CrossRef PubMed.
- K. P. Ho, W. L. Wong, K. M. Lam, C. P. Lai, T. H. Chan and K. Y. Wong, Chemistry, 2008, 14, 7988–7996 CrossRef CAS PubMed.
- I. Garcia-Bosch, X. Ribas and M. Costas, Adv. Synth. Catal., 2009, 351, 348–352 CrossRef CAS.
- M. C. White and J. Zhao, J. Am. Chem. Soc., 2018, 140, 13988–14009 CrossRef CAS PubMed.
- J. Rios, J. Lebeau, T. Yang, S. Li and M. D. Lynch, Green Chem., 2021, 23, 3172–3190 RSC.
- J. Zhang, Y.-M. Lee, M. S. Seo, S. Fukuzumi and W. Nam, Inorg. Chem., 2022, 61, 6594–6603 CrossRef CAS PubMed.
- K. Srinivasan and J. Kochi, Inorg. Chem., 1985, 24, 4671–4679 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data of isolated compounds. See DOI: https://doi.org/10.1039/d4ob00155a |
| This journal is © The Royal Society of Chemistry 2024 |