
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stereoselective synthesis of D-glycero-D-manno-heptose-1β,7-bisphosphate (HBP) from D-mannurono-2,6-lactone†
Yuta
Shinotsuka
a,
Riko
Nakajima
a,
Kohei
Ogawa
a,
Kaede
Takise
a,
Yutaka
Takeuchi
b,
Hiroshi
Tanaka
 b and
Kaname
Sasaki
*a
b and
Kaname
Sasaki
*a
aDepartment of Chemistry, Toho University, 2-2-1 Miyama, Funabashi 274-8510, Japan. E-mail: Kaname.sasaki@sci.toho-u.ac.jp
bDepartment of Chemical Science and Engineering, Tokyo Institute of Technology, 2-12-1-H101, Ookayama, Muguro-ku, Tokyo 152-8552, Japan
First published on 20th February 2024
Abstract
The synthesis of D-glycero-D-manno-heptose-1β,7-bisphosphate (HBP) from D-mannose is described. This synthetic approach is notable for the elongation of the seventh carbon, employing mannurono-2,6-lactone, the substrate-controlled establishment of the C-6 configuration, and the nucleophilic introduction of phosphate at the C-1 position through the utilization of 4,6-O-benzylidene-α-triflate.
The outer lipopolysaccharide (LPS) of Gram-negative bacteria harbors highly conserved heptoses,1 with D-glycero-D-manno-heptose-1β,7-bisphosphate (HBP) serving as a pivotal biosynthetic intermediate (Fig. 1). The Gram-negative bacterial enzyme D-glycero-D-manno-heptose-1,7-bisphosphate phosphatase (GmhB) engages with HBP, effecting the removal of the 7-position phosphate,2 while HldE catalyzes its conversion to ADP-D-glycero-β-D-heptose (ADP-DD-Hep). In the presence of HldD, an alternative enzymatic interconversion occurs, yielding ADP-L-glycero-β-heptose (ADP-LD-Hep), which serves as a glycosyl donor for LPS.3
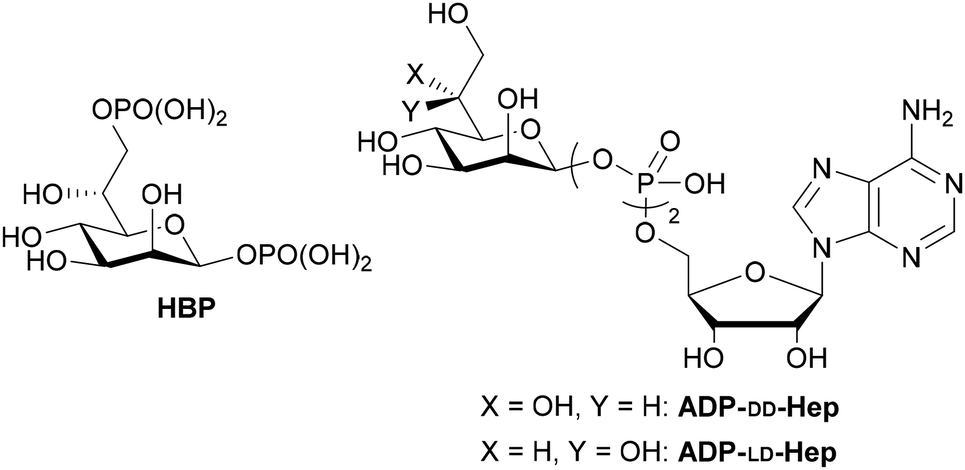 | ||
| Fig. 1 HBP and ADP-DD/LD-Hep. | ||
Gray-Owen et al. identified HBP as pathogen-associated molecular patterns (PAMPs).4 The molecule translocates into the host cytoplasm, eliciting TIFA-dependent innate immune responses. Shao et al. later ascertained the conversion of HBP to ADP-heptose 7-phosphate in the host cytoplasm, inducing TIFA-dependent inflammatory reactions. Notably, ADP-LD/DD-Hep are implicated in NF-κB activation and cytokine expression independently of HBP.5 ALPK1 has been identified as a pattern recognition receptor (PRR) for ADP-LD/DD-Hep. These recently discovered innate immune activators hold promise for the development of novel immunostimulants.
Noteworthy contributions include Sauvageau et al.'s synthesis of probes and derivatives to elucidate the inflammatory pathway induced by HBP,6,7 and Vincent et al.'s synthesis of biotinylated analogs for identifying pathogen recognition receptor (PRRs) of bacterial metabolites,8 and hydrolysis-resistant HBPs by substituting the anomeric oxygen with fluoromethylene.9 We were also captivated by its intriguing bioactivity, prompting us to embark on the synthesis of the D-glycero-D-manno-heptose skeleton and HBP through an innovative strategy.
From a synthetic chemistry standpoint, the rare heptoses have emerged as compelling synthetic targets. The envisaged synthetic challenges encompassed (i) the achievement of stereoselective construction of the β-phosphoric acid at the anomeric position, (ii) the incorporation of the seventh carbon, and (iii) the establishment of the D-glycero configuration at the 6-position.
Concerning HBP, the phosphoric acid at the anomeric position has been reported through the two distinct strategies (Scheme 1). The first total synthesis was reported by Fujimoto et al.,10 followed by subsequent syntheses reported by Zamyatina et al.11 and Vincent et al.12 These syntheses utilized the Mitsunobu reaction for the generation of anomeric β-phosphate from hemiacetal (Scheme 1-I). However, challenges were encountered in achieving optimal stereoselectivity, likely attributed to the presence of an anomeric mixture in the reaction substrates and the concurrent SN1 reaction.
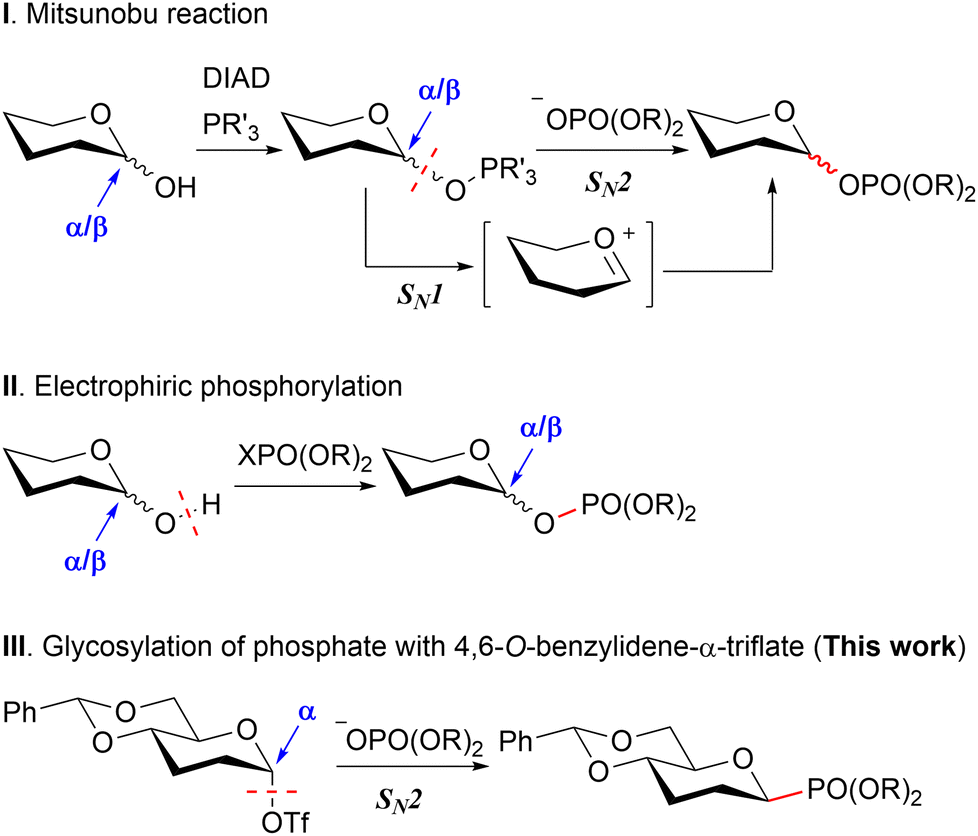 | ||
| Scheme 1 Two strategies have been employed for constructing the anomeric phosphate to date: the Mitsunobu reaction, which advances from a sugar electrophile with an anomeric mixture (I), and electrophilic phosphorylation originating from a sugar nucleophile with an anomeric mixture (II). The approach elucidated in this study involves glycosylation with a sugar electrophile employing the α-anomer presumably through an SN2 mechanism (III). Functional groups irrelevant to the discussion are omitted. | ||
Zamyatina et al.11 and Sauvageau et al.13 independently achieved electrophilic phosphorylation of the C-1 hemiacetal, displaying a more promising level of stereoselectivity. However, their stereoselectivities are believed to be governed by the configuration of hemiacetals, a characteristic challenging to control through external factors, and may be sensitive, for better or worse, to the substrate's structure, including the combination of protecting groups (Scheme 1-II).
Our strategy for accomplishing β-phosphoric acid involved nucleophilic substitution, i.e. glycosylation of the phosphate, employing 4,6-O-benzylidene-α-triflate esters (Scheme 1-III). We posited that a stereo-inversive glycosylation of the phosphate, entailing the α-triflate ester and capitalizing on the disarming influence of 4,6-O-benzylidene,14 could adeptly orchestrate the assembly of the β-phosphate moiety. In essence, the disarming effect judiciously stabilizes the α-triflate ester, precluding the generation of cationic intermediates. This effect suppresses the SN1 reaction, known for its confounding impact on stereoselectivity, thereby fostering the development of β-stereoselectivity.
As for introducing the seventh carbon and constructing the asymmetric carbon at C-6, there were fundamentally three strategies (Fig. 2): one involving the construction of the asymmetric carbon while forming a C-6–O-6 bond (path a),15–18 while forming a C-6–C-7 bond (path b),19,20 and while forming a C–H bond (path c). Since the last strategy reliably yields the Felkin–Anh adduct, it has been employed to rectify the configuration when the C-6–C-7 forming strategy resulted in a mixture.10,21,22 The limited adoption of this strategy may be attributed to the intricate access to precursors for the reduction reaction, 6-oxoheptosides. In essence, a straightforward method for acquiring 6-oxoheptoside was deemed essential. Our approach involved nucleophilic carbon elongation at the C-6 position of mannurono-2,6-lactone, a compound we have successfully synthesized in large quantities in four steps from readily available D-mannose,23,24 incorporating differentially protected oxygen functional groups (Scheme 2). The carbonyl carbon of the lactone, can be reduced with NaBH4, despite its conventional inactivity towards esters, indicating the remarkable electrophilicity of the highly oxidized 6-position, likely attributable to the strain it bears.23,24 The unique electrophilicity was expected to play a pivotal role in preventing excessive C1 nucleophile addition to the product, the ketone.
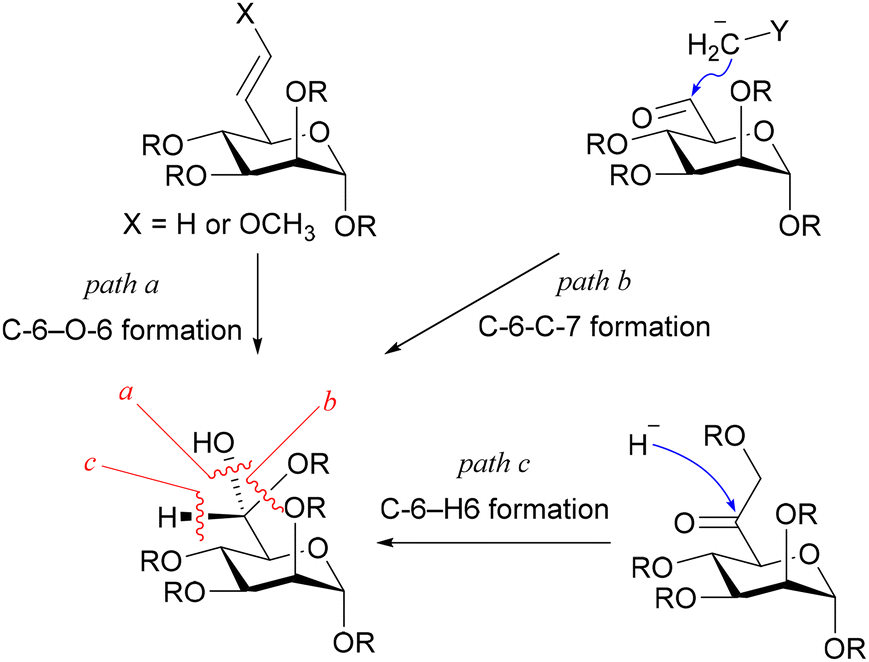 | ||
| Fig. 2 Three strategies for synthesizing D-glycero-D-manno-heptose. The stereogenic center at C-6 has been constructed while forming the C-6–O-6 bond (path a), the C-6–C-7 bond, where stereoselectivity was not observed (path b), or the C-6–H-6 bond, whose precursors seem burdensome to access (path c). | ||
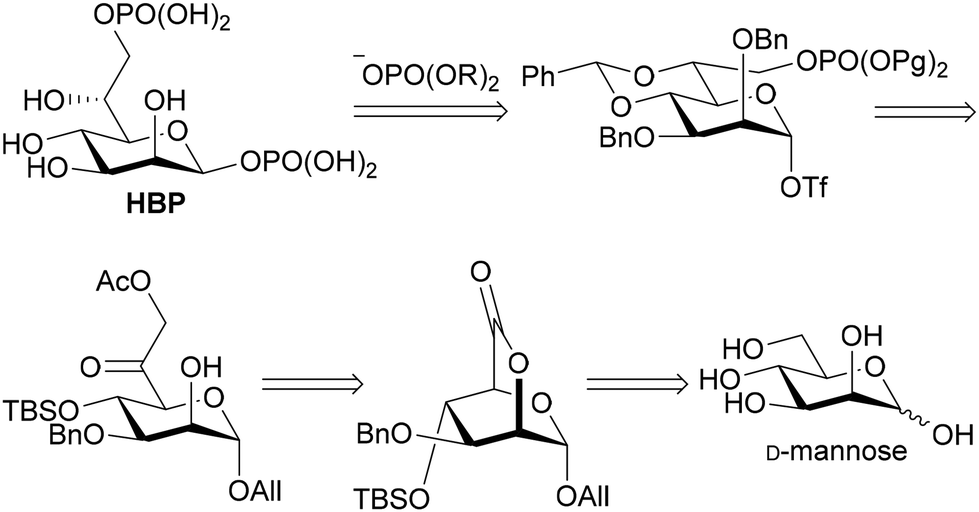 | ||
| Scheme 2 Retrosynthetic analysis of HBP. | ||
Indeed, as shown in Scheme 3, 2,6-lactone 1 could be synthesized from commercially available D-mannose in four steps,23 and 2 was acquired with a yield of 74% through the nucleophilic introduction of the C-7 carbon at −95 °C, utilizing lithium methyl iodide prepared in situ from CH2I2 and CH3Li. As anticipated, no superfluous addition reactions to the generated ketone were observed. Additional experiments are required to validate the contribution of the intermediate hemiacetal to the inhibition of the excess addition. Nevertheless, it is certain, at least to some extent, that the unusual electrophilicity of the 2,6-lactone structure enables this reaction. Introduction of the oxygen functional group at C-7 was accomplished through the utilization of KOAc, resulting in the formation of compound 3 with a yield of 86%. Subsequently, 3 was solubilized in THF, followed by the addition of NaBH4 at 0 °C. The ensuing reaction, conducted for 1 hour at room temperature, yielded the C-6 reduced product 4 as the primary product, concomitant with the formation of 4′ resulting from the migration of the acetyl group. To ascertain the diastereoselectivity, the ratio was determined by subsequent acetylation to be approximately 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (for detail, see ESI†). The substrate's stereodirectionality was remarkably robust, to the extent that even when employing chiral borohydrides like TarB-NO2 or CBS, the stereoselectivity remained unchanged, and the formation of the L-glycero-isomer hardly occurred. The observed stereoselectivity aligns with the Felkin–Anh model (Fig. 3). The addition reaction of borohydride appears to proceed while circumventing the bulky substituent at the C-4 position.
:1 (for detail, see ESI†). The substrate's stereodirectionality was remarkably robust, to the extent that even when employing chiral borohydrides like TarB-NO2 or CBS, the stereoselectivity remained unchanged, and the formation of the L-glycero-isomer hardly occurred. The observed stereoselectivity aligns with the Felkin–Anh model (Fig. 3). The addition reaction of borohydride appears to proceed while circumventing the bulky substituent at the C-4 position.
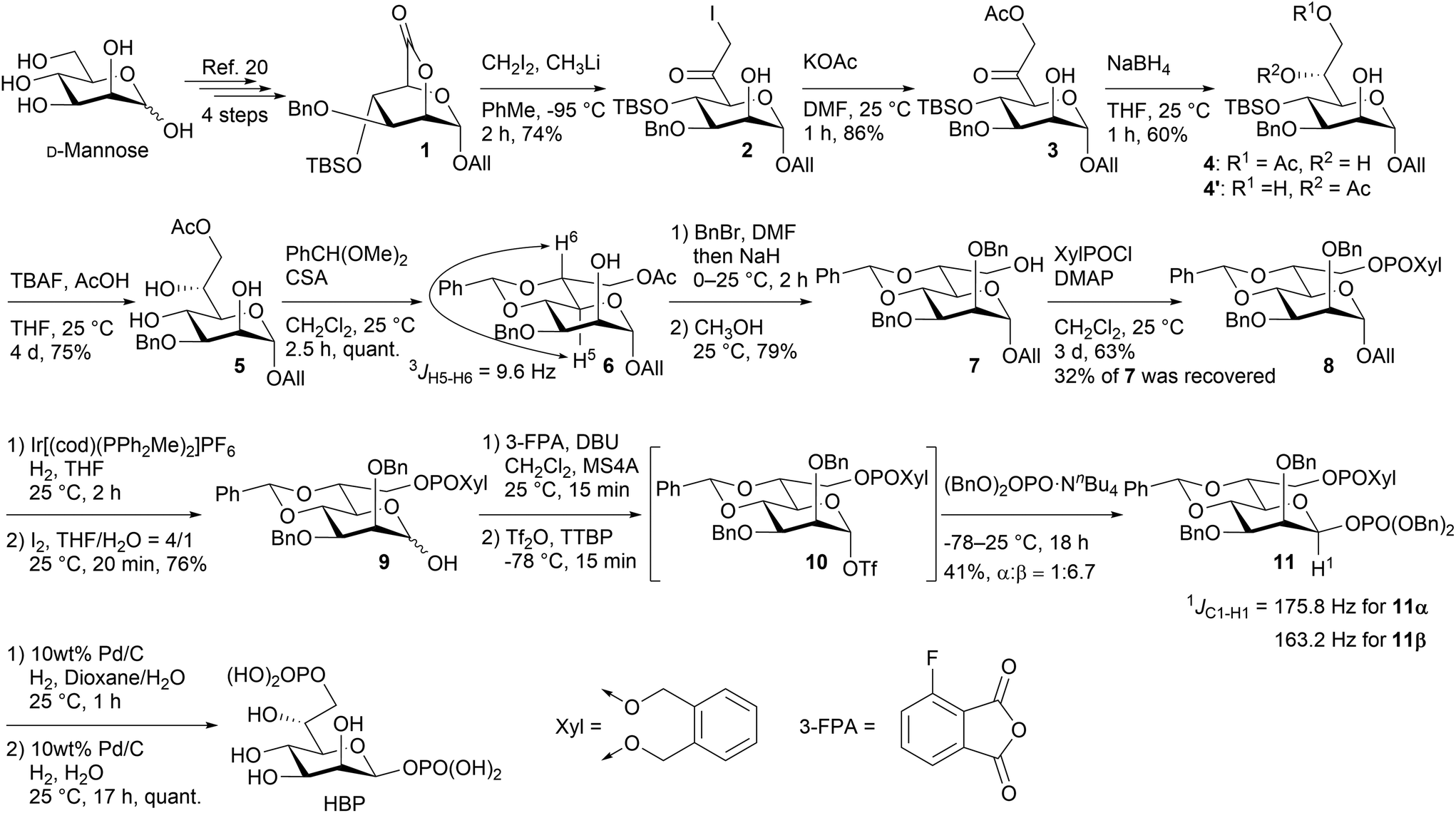 | ||
| Scheme 3 Synthesis of HBP. | ||
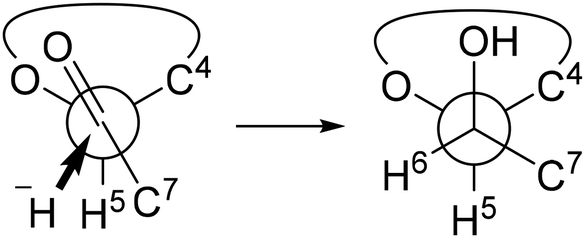 | ||
| Fig. 3 A plausible explanation for the observed stereoselectivity in the reduction of 3. | ||
The stereochemistry at the C-6 position was ascertained to be the D-glycero configuration, elucidated through 4,6-O-benzylidenation resulting in the formation of 6, with 3JH5–H6 exhibiting an antiperiplanar coupling at 9.6 Hz. The 4,6-O-benzylidene group, incorporated for the purpose of configuration confirmation, was subsequently employed in the construction of the anomeric β-phosphate. This tethering group has exhibited notable efficacy in the synthesis of 1,2-cis-configured equatorial glycosides, particularly β-mannosides, renowned for their intricate synthetic pathways.14 The resolution to the synthetic challenges resides in the disarming effect, wherein cationic intermediates originating from electrophiles bearing 4,6-O-benzylidene groups undergo destabilization, leading to an SN2-like nucleophilic substitution of the leaving group situated in the axial position.14 The 4,6-O-benzylidene-based synthetic strategy for equatorial glycosides has extended to heptosides,25 and the potential derivation of 6 could furnish the corresponding electrophile utilized in that context. Specifically, we postulate that the introduction of the β-phosphate moiety could be facilitated through nucleophilic substitution of the α-triflate ester, stabilized by the 4,6-O-benzylidene group.
The manipulation of the protective groups at the O-2 and O-7 positions was executed with precision to preclude undesired migration, resulting in the formation of 7; benzyl bromide was introduced to 6 before the addition of NaH to forestall the migration of the acetyl group, followed by the addition of methanol in the same vessel to effectuate deacetylation. Subsequently, the phosphorylation of the O-7 position was performed, leading to 8. While the synthesis of the phosphoryl chloride proved straightforward,26 its reactivity exhibited a diminutive nature, and a portion of the material 7 was recovered. Subsequently, 8 underwent deprotection of the allyl group situated at the O-1 position to afford the hemiacetal 9. While Gin et al. previously documented glycosylation involving anomeric hydroxy groups as electrophiles using Ph2SO and Tf2O,27 our study successfully applied the protocol established by Kim et al., utilizing phthalic anhydride and Tf2O for our nucleophilic β-phosphorylation.28 The synthesis of β-phosphate 11 was accomplished through the putative intermediate α-triflate 10, yielding a moderate overall yield and exhibiting high stereoselectivity (α:β = 1:6.7). The configuration was ascertained through 1JC1–H1, a dependable and widely employed method for mannosides,29 revealing a coupling constant of 163.2 Hz for the predominant β-anomer and 175.8 Hz for the minor α-anomer. Notably, among the byproducts, a disaccharide linked by phthalate was observed, a concern previously emphasized in Kim et al.'s original work. Ultimately, global deprotection employing Pd–C furnished the desired HBP. The liberation of phosphoric acid occurred rapidly, leading to an increase in the substrate's polarity. Consequently, transitioning the reaction solvent from hydrous dioxane to water expedited the comparatively sluggish deprotection of the benzylidene group.
Conclusions
We have successfully synthesized HBP utilizing the readily available hexose, D-mannose, as the starting material. The heptose skeleton was derived through controlled one-carbon elongation, leveraging 2,6-lactone as a pivotal intermediate and capitalizing on the heightened electrophilicity of the C-6 position. The D-glycero-D-manno skeleton was meticulously assembled through substrate-controlled stereoselective C-6 reduction. Additionally, the β-phosphate moiety was incorporated via nucleophilic substitution of 4,6-O-benzylidene-α-triflate. While this synthetic approach might not inherently surpass preceding routes in overall yield or procedural steps, it presents the potential advantage of simplified purification due to its notable stereoselectivities.Author contributions
K. S. conceived the project with contributions from all authors. Y. S., R. N., K. O., K. T., Y. T., and H. T. conducted the experiments and gathered the data. All authors analyzed the data, contributed to manuscript preparation, and have provided approval for the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI grant number JP22K05338. We express our gratitude to Prof. Yoichi Habata and Prof. Shunsuke Kuwahara of Department of Chemistry, Toho University, for generously allowing us to use their pressurized reaction vessel for catalytic hydrogen reduction.References
- R. F. Maldonado, I. Sá-Correia and M. A. Valvano, FEMS Microbiol. Rev., 2016, 40, 480–493 CrossRef CAS PubMed.
- L. Wang, H. Huang, H. H. Nguyen, K. N. Allen, P. S. Mariano and D. Dunaway-Mariano, Biochemistry, 2010, 49, 1072–1081 CrossRef CAS PubMed.
- M. A. Valvano, P. Messner and P. Kosma, Microbiology, 2002, 148, 1979–1989 CrossRef CAS PubMed.
- R. G. Gaudet, A. Sintsova, C. M. Buckwalter, N. Leung, A. Cochrane, J. Li, A. D. Cox, J. Moffat and S. D. Gray-Owen, Science, 2015, 348, 1251–1255 CrossRef CAS PubMed.
- P. Zhou, Y. She, N. Dong, P. Li, H. He, A. Borio, Q. Wu, S. Lu, X. Ding, Y. Cao, Y. Xu, W. Gao, M. Dong, J. Ding, D.-C. Wang, A. Zamyatina and F. Shao, Nature, 2018, 561, 122–126 CrossRef CAS PubMed.
- I. A. Adekoya, C. X. Guo, S. D. Gray-Owen, A. D. Cox and J. Sauvageau, J. Immunol., 2018, 201, 2385–2391 CrossRef PubMed.
- D. Williams, M. P. Jamshidi, F. St Michael, K. Chisholm, A. Cox and J. Sauvageau, J. Org. Chem., 2021, 86, 2184–2199 CrossRef CAS PubMed.
- L. Liang, J. Cao, T.-Y. W. Wei, M.-D. Tsai and S. P. Vincent, Org. Biomol. Chem., 2021, 19, 4943–4948 RSC.
- L. Liang, T.-Y. W. Wei, P.-Y. Wu, W. Herrebout, M.-D. Tsai and S. P. Vincent, ChemBioChem, 2020, 21, 2982–2990 CrossRef CAS PubMed.
- S. Inuki, T. Aiba, S. Kawakami, T. Akiyama, J.-I. Inoue and Y. Fujimoto, Org. Lett., 2017, 19, 3079–3082 CrossRef CAS PubMed.
- A. Borio, A. Hofinger, P. Kosma and A. Zamyatina, Tetrahedron Lett., 2017, 58, 2826–2829 CrossRef CAS.
- L. Liang and S. P. Vincent, Tetrahedron Lett., 2017, 58, 3631–3633 CrossRef CAS.
- J. Sauvageau, M. Bhasin, C. X. Guo, I. A. Adekoya, S. D. Gray-Owen, S. Oscarson, L. Guazzelli and A. Cox, Carbohydr. Res., 2017, 450, 38–43 CrossRef CAS PubMed.
- D. Crich, Acc. Chem. Res., 2010, 43, 1144–1153 CrossRef CAS PubMed.
- J. S. Brimacombe and A. K. M. S. Kabir, Carbohydr. Res., 1986, 150, 35–51 CrossRef CAS.
- J. S. Brimacombe and A. K. M. Kabir, Carbohydr. Res., 1986, 152, 329–334 CrossRef CAS.
- N. C. R. van Straten, N. M. A. J. Kriek, C. M. Timmers, S. C. M. Wigchert, G. A. van der Marel and J. H. van Boom, J. Carbohydr. Chem., 1997, 16, 947 CrossRef CAS.
- S. K. Mulani, K.-C. Cheng and K.-K. T. Mong, Org. Lett., 2015, 17, 5536–5539 CrossRef CAS PubMed.
- S. Picard and D. Crich, Tetrahedron, 2013, 69, 5501–5510 CrossRef CAS.
- M. Kim, B. Grzeszczyk and A. Zamojski, Tetrahedron, 2000, 56, 9319–9337 CrossRef CAS.
- S. Jarosz, S. Skóra, A. Stefanowicz, M. Mach and J. Frelek, J. Carbohydr. Chem., 1999, 18, 961–974 CrossRef CAS.
- J. A. Read, R. A. Ahmed and M. E. Tanner, Org. Lett., 2005, 7, 2457–2460 CrossRef CAS PubMed.
- Y. Hashimoto, S. Tanikawa, R. Saito and K. Sasaki, J. Am. Chem. Soc., 2016, 138, 14840–14843 CrossRef CAS PubMed.
- K. Sasaki and Y. Hashimoto, Synlett, 2017, 28, 1121–1126 CrossRef CAS.
- D. Crich and A. Banerjee, J. Am. Chem. Soc., 2006, 128, 8078–8086 CrossRef CAS PubMed.
- J. I. Murray, R. Woscholski and A. C. Spivey, Chem. Commun., 2014, 50, 13608–13611 RSC.
- B. A. Garcia and D. Y. Gin, J. Am. Chem. Soc., 2000, 122, 4269–4279 CrossRef CAS.
- J. Y. Baek, B.-Y. Lee, R. Pal, W.-Y. Lee and K. S. Kim, Tetrahedron Lett., 2010, 51, 6250–6254 CrossRef CAS.
- K. Bock and C. Pedersen, J. Chem. Soc., Perkin Trans. 2, 1974, 293–297, 10.1039/P29740000293.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ob00139g |
| This journal is © The Royal Society of Chemistry 2024 |