
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances in Si-rhodamine-based fluorescent probes for live-cell imaging
Hisashi
Ohno
 a,
Eita
Sasaki
ab,
Sota
Yamada
a and
Kenjiro
Hanaoka
*ab
a,
Eita
Sasaki
ab,
Sota
Yamada
a and
Kenjiro
Hanaoka
*ab
aGraduate School of Pharmaceutical Sciences, Keio University, Tokyo 105-8512, Japan. E-mail: khanaoka@keio.jp
bFaculty of Pharmacy, Keio University, Tokyo 105-8512, Japan
First published on 6th March 2024
Abstract
Fluorescence imaging is a powerful technique for visualizing biological events in living samples with high temporal and spatial resolution. Fluorescent probes emitting far-red to near infrared (NIR) fluorescence are particularly advantageous for in vivo imaging due to their high tissue permeability and low autofluorescence, as well as their suitability for multicolor imaging. Among the far-red to NIR fluorophores, Si-rhodamine is one of the most practical fluorophores for the development of tailor-made NIR fluorescent probes because of the relative ease of synthesis of various derivatives, the unique intramolecular spirocyclization behavior, and the relatively high water solubility and high photostability of the probes. This review summarizes these features of Si-rhodamines and presents recent advances in the synthesis and applications of far-red to NIR fluorescent probes based on Si-rhodamines, focusing on live-cell imaging applications such as fluorogenic probes, super-resolution imaging and dye–protein hybrid-based indicators.
 Hisashi Ohno | Hisashi Ohno received his B.S. in 2016 from the Department of Chemistry, Faculty of Science, Hokkaido University, and his Ph.D. in 2022 from the Graduate School of Pharmaceutical Sciences, University of Tokyo. Since 2022, he has been working with Prof. Hanaoka at the Graduate School of Pharmaceutical Sciences, Keio University. His research interests include the development of new small-molecule fluorescent probes for drug discovery. |
 Eita Sasaki | Eita Sasaki received his B.S. and M.S. in Pharmaceutical Sciences from the University of Tokyo in 2003 and 2005, respectively. He earned his Ph.D. in Chemistry at the University of Texas at Austin in 2011. Following a postdoctoral work with Prof. Donald Hilvert at the ETH Zurich, he became an Assistant Professor at the Graduate School of Agricultural and Life Sciences, University of Tokyo, in 2017. Since 2021, he has been a Senior Assistant Professor at the Faculty of Pharmacy of Keio University. His research interests include the development of fluorescent probes and the engineering of self-assembling proteins. |
 Sota Yamada | Sota Yamada received his B.S. in 2015 from the Faculty of Pharmaceutical Sciences at Nagoya City University. He studied at the Graduate School of Pharmaceutical Sciences of Kyoto University and became a Project Assistant Professor at the Graduate School of Pharmaceutical Sciences, Keio University, in 2019. Since 2021, he has been working with Prof. Hanaoka at the Graduate School of Pharmaceutical Sciences, Keio University. His research interests include the development of nanoprobes for drug delivery systems. |
 Kenjiro Hanaoka | Kenjiro Hanaoka received his B.S. in 2000 from the Faculty of Pharmaceutical Sciences, University of Tokyo, and his Ph.D. in 2005 from the Graduate School of Pharmaceutical Sciences, University of Tokyo. During 2005–2007, he worked with Prof. Thomas Kodadek at the University of Texas, Southwestern Medical Center. He became an Assistant Professor at the Graduate School of Pharmaceutical Sciences, University of Tokyo, in 2007, and was promoted to Associate Professor in 2011. He became Full Professor at Keio University in 2021. His research interests cover the development of chemical biology tools that can visualize or regulate biological phenomena. |
1. Introduction
Fluorescence imaging is a powerful technique for the visualization of biological phenomena in living cells and bodies with high temporal and spatial resolution. For fluorescence imaging, probes that show changes in photophysical properties such as fluorescence intensity and excitation/emission wavelength in response to the target biomolecules are required.1 Since the first development of fluorescent probes for Ca2+ by Tsien et al.,2 many other probes have been developed and applied for biological experiments.3 Such small-molecule fluorescent probes are generally easier to use than fluorescent proteins because they do not require genetic manipulation, and so they have been widely used in biological studies.In the development of fluorescent probes, rhodamine dyes (Fig. 1a) have often been utilized as fluorescent cores for various fluorescent probes, because these dyes have many advantageous properties for fluorescence imaging, including high water solubility, high fluorescence quantum yields and high molar extinction coefficients.4 Moreover, many fluorescence-switching mechanisms have been developed for them.5 While typical rhodamine dyes containing the xanthene structure with an oxygen atom show green to red fluorescence (approximately λem = 500–600 nm), Fu et al. reported a silicon-substituted xanthene fluorophore, 2,7-bis(dimethylamino)-9,9-dimethyl-9-sila-9H-anthracenium (TMDHS), as a new near-infrared fluorophore (approximately λem = 650 nm) in 2008 (Fig. 1b).6 Our group then developed a series of Si-rhodamines (SiRs) as far-red to near-infrared (NIR) fluorescent cores for fluorescent probes working in the wavelength range of approximately λem = 600–740 nm (Fig. 1c).7 Since then, we and others have developed an extensive range of far-red to NIR fluorescent probes based on Si-rhodamines.8,9 Fluorescent probes working in this long wavelength region are particularly useful for multicolor imaging.9 In addition, NIR fluorescence (λem = 650–900 nm) is especially suitable for fluorescence imaging of organs or animals, as tissue transparency is high and autofluorescence is low in this wavelength region.10 These dyes also possess other advantageous properties for live-cell fluorescence imaging: (i) the relative ease of synthesis of various derivatives, (ii) their unique intramolecular spirocyclization behavior, and (iii) their relatively high water solubility and high photostability.
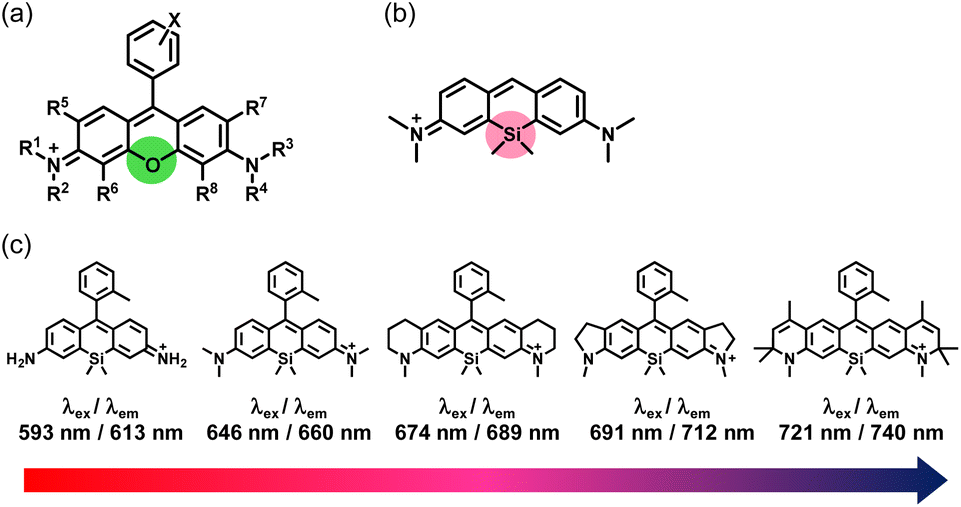 | ||
| Fig. 1 Structures of (a) typical rhodamines, (b) TMDHS and (c) a series of our developed Si-rhodamines. | ||
Here, we summarize the properties of Si-rhodamine-based fluorescent probes, and review recent advances in their synthesis and applications, focusing especially on the advantages of these dyes for live-cell fluorescence imaging.
2. Synthesis of SiRs
2.1 Symmetric SiRs
Many far-red to NIR fluorescent probes based on symmetric SiRs have been developed, and most of them have been synthesized by route 1 or route 2 (Fig. 2), using the nucleophilic addition of metalated aryl species to Si-xanthone as a key reaction. Route 1 is the originally reported synthetic scheme for SiRs, and in this route, N,N-dimethyl-3-bromoaniline 1 is first converted to the aniline homodimer 2 by reaction with formaldehyde.11 Then, a Si atom is introduced by reaction with dichlorodimethylsilane via di-lithiation of 2 through Li halogen exchange. The key intermediate, Si xanthone, is then obtained by oxidation of the xanthene ring. | ||
| Fig. 2 Reported synthetic routes for a representative symmetric SiR, 2-COOH SiR650.15 | ||
In order to improve the relatively low yield (30–40%) of the oxidation reaction at the 9-position of the xanthene ring, an alternative synthetic pathway affording a higher yield of Si xanthone (80–90% in each step) was developed (Fig. 2, route 2).12 In this route, a Si atom was introduced at the first step, i.e., the dimethyl-3-bromoaniline 1 was lithiated and reacted with dichlorodimethylsilane, affording bis(5-dimethylamino)dimethylsilane 4. Compound 4 was then dibrominated, dilithiated, and reacted with dimethylcarbamoyl chloride, affording the Si xanthone in high yield.
The nucleophilic addition of metalated aryl species to Si-xanthone often requires harsh aryllithium reagents rather than milder arylmetal reagents such as Grignard reagents because the ketone electrophile of Si-xanthone is relatively electron rich due to the two terminal dimethylamino groups, and the arylmetal nucleophile is often electron poor owing to the ester group at the ortho-position, which is necessary to install a carboxyl group in the dye structures. Thus, this reaction is incompatible with many functional groups unless complex protection strategies are considered. To overcome this problem, direct conversion of bis(5-dimethylamino-2-bromophenyl)dimethylsilane 5 to 2-COOH SiR650 has been developed (route 3).13 The dibromide undergoes metal/bromide and metal/metal exchange to give a bis(aryl Grignard) intermediate, which can then react with anhydrides or ester electrophiles. This synthetic strategy enabled efficient syntheses of various SiRs, such as 2-COOH SiR650. Subsequently, the double nucleophilic addition of aryllanthanum reagents was reported to be more efficient for this direct conversion.14
The synthetic strategies shown in Fig. 2 have enabled the synthesis of a wide variety of SiRs by selecting the appropriate combination of 2-bromoaniline derivatives, dichlorosilane derivatives, and aryllithiums (Fig. 3). For example, NH2, NMe2, azetidine, indoline and quinoline structures can be introduced at the terminal positions of the xanthene ring.7,13,16 Various functional groups such as –Me and –COOH can be introduced into the upper benzene ring, or the benzene ring itself can be substituted with heterocycles (thiophene) and alkynes.17,18 Furthermore, various substituents on the Si atom have been reported, which provide a new way to functionalize the dyes for dye attachment, providing fluorogenic labelling agents for nuclear DNA, SNAP-tag and HaloTag.19
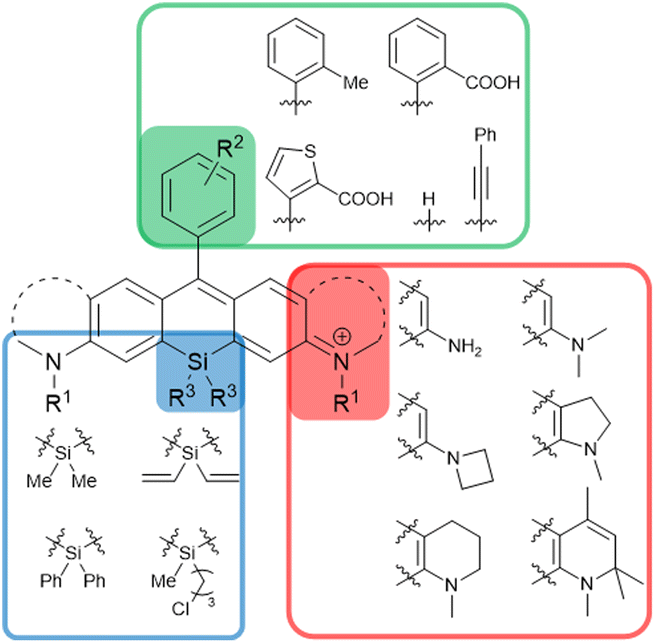 | ||
| Fig. 3 SiR derivatives synthesisable via the routes shown in Fig. 2. | ||
2.2 Asymmetric SiRs
Asymmetric SiRs are expected to be very useful as scaffolds of activatable fluorescent probes, because they enable precise modulation of the absorption and fluorescence wavelengths. In addition, different functionalities can be introduced on the two sides of the xanthene ring of asymmetric rhodamine scaffolds. Consequently, our group developed a novel synthetic scheme for the preparation of a wide variety of asymmetric SiRs by using the heterodimerization reaction between p-hydroxymethyl anilines and anilines as the key reaction (Fig. 4).20 The synthesized aniline heterodimers can be converted to asymmetric Si-xanthones by dilithiation, reaction with dichlorodimethylsilane, and then oxidation at the xanthene 9-position. | ||
| Fig. 4 (a) The key reaction for the synthesis of various asymmetric SiRs. (b) Representative structures of asymmetric SiRs synthesized using the approach shown in (a). | ||
These asymmetric SiRs indeed proved to be efficient scaffolds for fluorescent probes. For example, we have developed a novel far-red to NIR fluorescent probe for hypoxia, azoSiR640, by the diazotization of SiR640 (Fig. 5a).20 Non-fluorescent azoSiR640 is converted to strongly fluorescent SiR640 under hypoxic conditions through cleavage of the azo group by biological reductases, and was used to visualize hepatic and kidney ischemia in mice in vivo. We also reported an asymmetric Si rhodamine-based fluorescent probe for dipeptidyl peptidase-4 (DPP-4) activity, EP-SiR640 (Fig. 5b).21 This probe is based on a new molecular design for NIR fluorescent probes and targets the exopeptidase activity by utilizing the 110 nm blue shift of the fluorescence upon amidation of the N atom of the xanthene moiety. EP-SiR640, which is a conjugate of SiR640 and a dipeptide, glutamate-proline (EP), shows a large fluorescence increment (669-fold) upon reaction with DPP-4. Moreover, our group reported an off/on-type NIR fluorescent probe for HaloTag labelling, Halo SiR-5 (Fig. 5c).22 Before bonding to the HaloTag protein, the fluorescence of Halo SiR-5 is quenched via the twisted intramolecular charge transfer (TICT) mechanism due to the free rotation of the terminal amino group on the xanthene ring. However, the probe becomes highly fluorescent when bound to the HaloTag protein because the rotation of the terminal amino group is inhibited by the steric repulsion of the amino acid residues on the protein surface. We further demonstrated that Halo SiR-5 can visualize HaloTag-expressing neurons in 3D fluorescence imaging of the whole brain, using tissue-clearing technology.
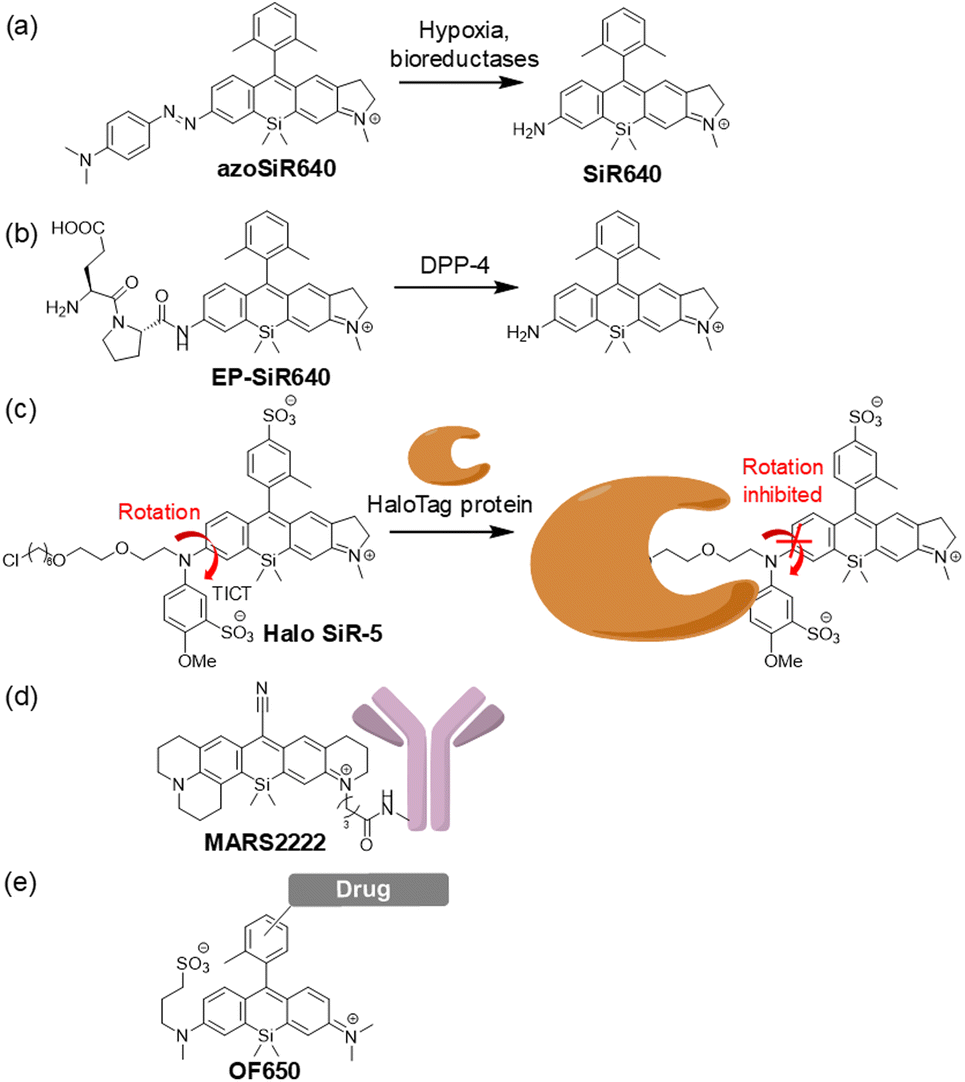 | ||
| Fig. 5 NIR optical probes based on the asymmetric SiR scaffold. NIR fluorescent probes for (a) hypoxia, (b) the enzymatic activity of DPP-4 and (c) fluorogenic labelling of the HaloTag protein, (d) the labelling probe for Raman imaging and (e) the unbiased always-on NIR fluorescent probe–drug conjugate. | ||
Other groups have also developed various fluorescent/Raman probes based on the asymmetric SiR scaffold. Miao et al. reported the NIR dye MARS2222-NHS, containing an asymmetric Si-pyronine scaffold (Fig. 5d), for electronic pre-resonant stimulated Raman scattering microscopy.23 MARS2222-NHS has a cyano group at position 9 of the xanthene ring to generate the Raman signal, so in contrast to typical SiRs as shown in Fig. 3 and 4, a linker cannot be introduced into the upper benzene moiety. Instead, a linker was introduced into one of the terminal amino groups of the asymmetric Si-pyronine scaffold in MARS2222-NHS, using the synthetic scheme shown in Fig. 4a. Miao et al. performed immunostaining of α-tubulin and fibrillarin with MARS2222-labelled antibodies and demonstrated multiplexed Raman imaging. Moreover, Wang et al. reported an unbiased always-on NIR fluorescent probe, OF650 (Fig. 5e).24 OF650 has a sulfo group connected via a linker to the terminal amino group of the xanthene ring. This probe has an overall charge of zero and shows high water solubility. They demonstrated that the combination of OF650 with OF550, which has an O atom at the 10 position of the xanthene ring, can be used for ratiometric fluorescence imaging to enable quantitative imaging of drug distribution in tissues.
Asymmetric SiRs are excellent scaffolds for NIR fluorescent probes because it is easy to introduce multiple functional groups, linkers, enzyme recognition structures, etc. at the terminal amino group of the xanthene ring. So, these scaffolds can be utilized to develop complex functional fluorescent probes.
3. Intramolecular spirocyclization behavior
3.1 Intramolecular spirocyclization equilibrium
One of the notable characteristics of SiRs for fluorescence imaging is that their LUMO energy level is much lower than those of conventional O-rhodamines (Fig. 6a).6 The lower LUMO energy level leads to a smaller HOMO–LUMO gap, which is the reason for the red shift of absorbance and fluorescence wavelengths, and also facilitates intramolecular spirocyclization due to the increased nucleophilicity at the 9-position of the xanthene. So, SiR derivatives with nucleophilic functional groups, such as COOH and hydroxymethyl (HM) groups, at the 2-position of the upper benzene moiety exist in an equilibrium between open and closed forms in aqueous solutions under physiological conditions (Fig. 6b). For example, comparing 2-COOH SiR650 with 2-Me SiR650, the former has a lower molar absorption coefficient because it is in a spirocyclization equilibrium between the open (fluorescent) and closed (nonfluorescent) forms, while the latter can take only the open form (Fig. 6c).11,24 The spirocyclization equilibrium between the open and closed forms is one of the unique features of SiRs, and various probes have been developed in which changes of this equilibrium are utilized to modulate the fluorescence. Here, to illustrate the utility of these spirocyclization-based SiR probes, we describe their application in super-resolution imaging and fusion protein-based chemical sensors. | ||
| Fig. 6 (a) HOMO and LUMO energy levels of pyronine Y and TMDHS. (b) The intramolecular spirocyclization equilibrium of SiRs. (c) Molar extinction coefficients (ε) of 2-Me SiR650 and 2-COOH SiR650. | ||
3.2 Application to super-resolution imaging
By utilizing the equilibrium intramolecular spirocyclization of SiRs, a new type of spontaneously blinking fluorophore, HMSiR, has been constructed (Fig. 7a).25 Most molecules of HMSiR exist in a non-fluorescent state at physiological pH, but spontaneously and reversibly switch (blink) to the fluorescent open form with a suitable lifetime for super-resolution imaging using the single-molecule localization microscopy (SLM) technique. So, unlike other super-resolution imaging methods using conventional organic fluorophores such as STED,26 PALM,27 STORM28 and SIM,29 HMSiR does not require any additive or prior laser irradiation to induce the dark state. To confirm the utility of HMSiR, time-lapse SLM of microtubules in live cells was performed. | ||
| Fig. 7 Structures of HMSiR analogues employed for super-resolution imaging using SLM. | ||
Later, several other groups reported HMSiR analogues with improved fluorescence quantum yield and photostability. For example, Chi et al. developed an HMSiR analogue, HM-DS655, possessing azetidine moieties (Fig. 7b).30 The in silico design of this dye was based on the fact that ΔGC–O (the change of Gibbs free energy from the closed form to the open form) shows a strong correlation with pKcycl. The authors predicted the pKcycl values from DFT calculations of candidate compounds, and selected the structures with pKcycl ≤ 6, which are appropriate for spontaneous blinking (Table 1). They performed 3D-STORM imaging of the nucleus in living HeLa cells using HM-DS655.
Tyson et al. developed a bright and near-IR-emitting fluorescent dye, Yale676sb, providing another example of an HMSiR analogue (Fig. 7c).31 Yale676sb was designed to show a longer wavelength of fluorescence, while maintaining the same pKcycl value as HMSiR, by introducing both ring structures and electron-withdrawing fluoroethyl groups on the amino groups of the xanthene ring. Two-colour super-resolution imaging at one excitation wavelength was achieved by co-staining ER and mitochondria with Yale 676sb and HMSiR, respectively, in U2-OS cells.
Morozumi et al. reported a spontaneously blinking Si-pyronine, SiP650, based on intermolecular nucleophilic addition/dissociation (Fig. 7d).32 The 9 position of Si-pyronine is susceptible to nucleophilic attack by intracellular reduced glutathione, and the addition/dissociation reaction between the Si-pyronine and intracellular reduced glutathione could be utilized for SLM. They demonstrated that SiP650-Halo, which consists of the SiP650 structure with a Halo ligand at the terminal amino group, could be applied to live-cell SLM of β-tubulin.
Finally, Werther et al. developed a fluorogenic and self-blinking HMSiR analogue, f-HM-SiR (Fig. 7e).33f-HM-SiR is strongly quenched in the initial state and shows a fluorescence enhancement through the inverse electron demand Diels Alder reaction (DAinv) between 1,2,4,5-tetrazine and ring-strained alkyne. Since f-HM-SiR can selectively stain organelles labelled with various dienophiles and its fluorescence is turned on only when the click reaction conjugates are formed, the artifact fluorescence signal caused by nonspecific accumulation of probe molecules can be greatly reduced. In fact, f-HM-SiR could selectively stain BCN (bicyclo[6.1.0]non-4-yne)- or TCO ((E)-cyclooct-2-en-1-ol)-labelled organelles, enabling long-term (>200 s) live-cell SLM with high spatiotemporal resolution.
3.3 Application to fluorogenic probes
In 2013, Johnsson's group reported that SiR analogues with two carboxyl groups at the upper benzene ring can exhibit a fluorogenic character when bound to proteins.34 The open form-to-closed form ratio of these SiRs was affected by the dielectric constant of the surrounding environment. When the dielectric constant is low, i.e., in the hydrophobic environment, the closed form of the dye is dominant, and when the dielectric constant is high, i.e., in the hydrophilic environment, the open form of the dye is dominant. Based on this property, they developed SiR-SNAP (Fig. 8a) and SiR-Halo, which showed a dramatically increased proportion of the open form upon binding to SNAP-tag and HaloTag, respectively. SiR-SNAP showed no significant background signal in a wide range of fluorescence imaging applications such as confocal fluorescence microscopy and super-resolution imaging (GSDIM, STORM and STED).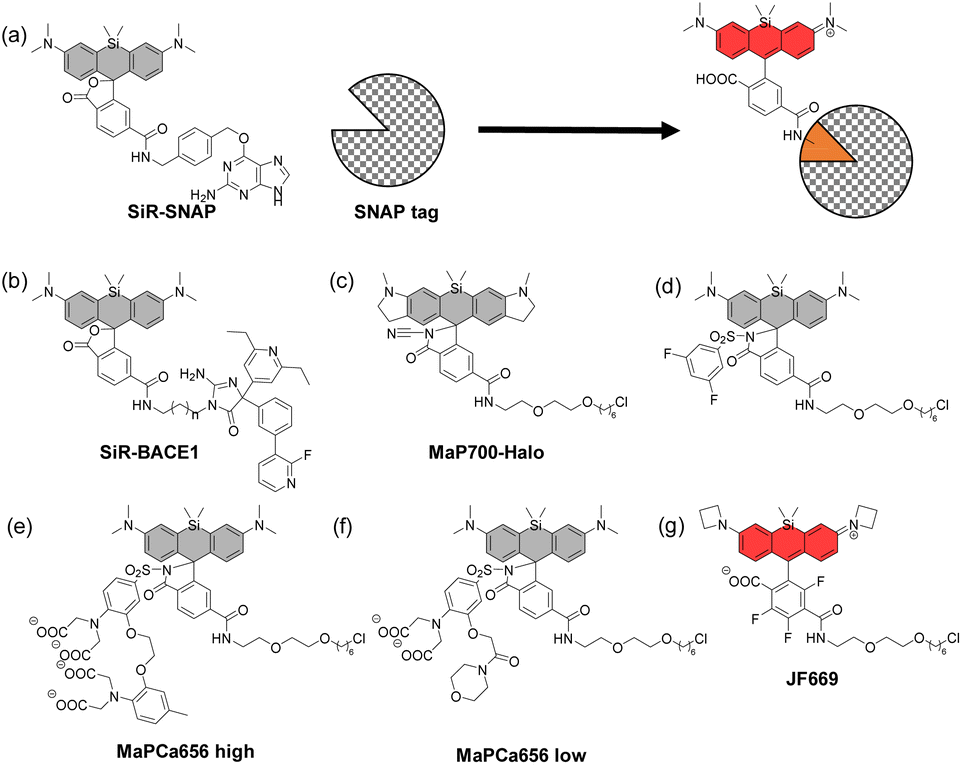 | ||
| Fig. 8 Fluorogenic protein-binding SiR derivatives. | ||
Based on the same mechanism, various fluorogenic probes which emit fluorescence upon binding to the target proteins have been reported. For example, SiR-BACE1, a NIR fluorescent probe that can detect β-site APP-cleaving enzyme 1 (BACE1), a major player in the pathogenesis of Alzheimer's disease, has been developed (Fig. 8b).35 SiR-BACE1 consists of SiR650 and the BACE1 inhibitor S-39, and it takes an open form emitting NIR fluorescence only when it binds to BACE1. SiR-BACE1 successfully visualized endogenous BACE1 in fixed neuronal tissue of mice.
A cell-permeable fluorogenic SiR analogue, MaP700-Halo, has also been reported (Fig. 8c).36 In this molecular design, an electron-deficient amide such as acylcyanamide, acyl sulfonamide (Fig. 8d)37 or acyl sulfamide is introduced into the 2-position of the upper benzene moiety, instead of a carboxyl group. MaP700-Halo having an acylcyanamide moiety hardly emitted fluorescence in water, and the fluorescence intensity increased up to 650-fold upon binding to the HaloTag. This probe further showed a low non-specific background signal compared to the probe having a carboxyl group at the 2-position of the upper benzene ring.
More recently, SiR-based-fluorogenic probes with additional functions have been reported. For example, MaPCa 656 high and MaPCa656 low (Fig. 8e and f) consist of the SiR dye with a Halo ligand and a Ca2+ chelator (BAPTA and MOBHA, respectively) linked via an acyl sulfonamide.38 They take open forms only when bound to the HaloTag protein and function as calcium indicators. Binding to HaloTag enables the control of their intracellular localization. The probes were applied to rat hippocampal neurons, where the high-affinity indicator MaPCa 656 high could detect a single action potential under no-wash conditions. On the other hand, the low-affinity indicator MaPCa656 low was successfully localized to the ER, where it could detect a calcium efflux isochronal to an increase in cytosolic calcium. They also coupled MaPCa 656 high with H-Luc, which is a chimera between HaloTag and the furimazine-dependent luciferase NanoLuc, and developed the first far-red bioluminescent calcium indicator. The use of H-Luc-MaPCa 656 high in cells indicates the feasibility of applying it to bioassays in HTS approaches.
Finally, the structural optimization of SiR-based-fluorogenic probes has also been performed in terms of KL–Z, the equilibrium constant of the intramolecular spirocyclization in a dioxane/water mixture. For example, a fluorogenic SiR analogue, JF669, with a fine-tuned KL–Z has been reported (Fig. 8g).39 In some SiR analogues, the open form is almost non-existent due to the high electrophilicity of the xanthene ring, so that the fluorescence intensity is low. On the other hand, the percentage of the open form was increased in JF669 by tri-fluorination on the upper benzene moiety. The SNAP ligand-conjugated JF669 could visualize the nucleus-localized SNAP-tag-histone variant H2A in living U2OS cells.
4. High photostability
Cyanine dyes such as Cy5 and Cy7 have been widely used as classical NIR fluorophores, but their photostability is poor, resulting in a decrease in fluorescence intensity during fluorescence imaging. Thus, fluorescent dyes with high photostability are needed to withstand long-term (and strong) photoirradiation during procedures such as super-resolution imaging and time-lapse imaging. SiRs, like other rhodamine derivatives, have high cell-membrane permeability and good photostability,7 making them promising fluorochromes for such applications. In addition, SiRs are promising dye scaffolds for photocaged compounds because of their resistance to non-specific decomposition. In the following sections, we present recent examples of protein–dye hybrid-type fluorescent probes using SiRs, which could overcome the relatively low photostability of fluorescent proteins, and photocaged SiRs suitable for super-resolution fluorescence imaging.4.1 Protein–dye hybrid-type fluorescent probes
Many protein-based fluorescent indicators based on fluorescent proteins have been developed.40,41 These protein-based fluorescent indicators have the advantage that they can be specifically expressed in various subcellular compartments and can utilize the evolved molecular recognition motifs found in nature. However, most fluorescent proteins can only be excited at wavelengths shorter than 550 nm, and red-shifted fluorescent proteins are still difficult to optimize.42 Since the use of tag proteins such as HaloTag and SNAP-tag is a well-established method to label target proteins with SiRs (Fig. 9a), several protein-based NIR fluorescent indicators utilizing the SiR-HaloTag or SiR-SNAP-tag conjugate have been reported. Here, we summarize recent developments in fluorescent indicators based on SiR-labelled fusion proteins. | ||
| Fig. 9 SiR-labelled protein-based fluorescent indicators. | ||
Hiblot et al. reported H-Luc/S-Luc, which is a fusion protein of NanoLuc and HaloTag/SNAP-tag (Fig. 9b).43 The glow-type luciferase NanoLuc has been widely used in bioimaging because of its extraordinary brightness, but its short wavelength (emission maximum of 460 nm) is not ideal for bioimaging. So, H-Luc and S-Luc, which exhibit high BRET (bioluminescence resonance energy transfer) efficiency, were developed by fusing circular permutated versions of NanoLuc with HaloTag7 or SNAP-tag. H-Luc and S-Luc can be labelled with fluorescent dyes such as SiR650 and they exhibit high BRET efficiency, extending the fluorescence wavelength up to ∼650 nm. By using this system, NIR fluorescence could be observed in HeLa cells expressing H-Luc labelled with SiR650 in the nucleus.
A representative example of a recently developed protein–dye hybrid fluorescent probe is the SiR-labelled HaloTag fusion protein with a Ca2+ or voltage-sensitive motif, HaloCaMP or HASAP (or HArclight), respectively (Fig. 9c).44 Based on the fluorogenic character of SiRs having a carboxyl group at the 2-position of the upper benzene ring, the fluorescence intensity of SiR in these probes increases only when the recognition domain for Ca2+ or voltage is close to the labelled SiR. The Ca2+ indicator, HaloCaMP, showed bright fluorescence owing to fine-tuning of the electrophilicity of the xanthene ring of the SiR. This probe could detect changes in Ca2+ concentration in cultured primary rat hippocampal neurons, and the HaloCaMP variants were substantially brighter, quicker to respond and more sensitive in neurons than the fluorescent protein-based far-red genetically encoded calcium ion indicator (GECI), NIR-GECO1.42 The voltage sensors, HASAP and HArclight, were also capable of detecting the action potentials in cultured primary rat hippocampal neurons.
More recently, a chemogenetic platform of FRET pairs, ChemoX, has been reported. This system enables the development of biosensors with high dynamic ranges attributed to the reversible interaction of fluorescent proteins and fluorescently labelled HaloTag (Fig. 9d).45 The HaloTag protein was fused to the C-terminus of EGFP so that the labelled-SiR on HaloTag is in close proximity to EGFP, and the interaction interface between EGFP and HaloTag was further optimized by introducing five mutations to achieve a near-quantitative FRET efficiency. Based on this scaffold, the authors developed biosensor systems for Ca2+, ATP and NAD+ by introducing the recognition domain for each target molecule into the linker between EGFP and HaloTag. They also demonstrated that minimal modifications of these biosensors enabled their readout to be switched to fluorescence intensity, fluorescence lifetime or bioluminescence.
4.2 Photocaged compounds
The controlled switching of fluorophores between non-fluorescent and fluorescent states is widely used for super-resolution fluorescence microscopy. Most photoactivatable probes developed to date require the introduction of highly hydrophobic and bulky photolabile protecting groups such as o-nitrobenzyl and nitroso groups (an example is shown in Fig. 10a).46 This approach results in the release of highly cytotoxic aldehydes during photo-uncaging. To overcome this issue, caging-group-free photoactivatable probes have recently been developed. For example, photoactivation via the excited state of the xanthene ring itself has been reported. This allows the design of photoactivatable probes with smaller molecular size, which is important for the development of NIR probes with improved water solubility and cell membrane permeability. | ||
| Fig. 10 Structures of recently reported photoactivatable SiR analogues. | ||
For example, the photoactivatable SiR analogue, PA-JF646, has a diazoketone structure, and this probe was converted to 2Me-substituted JF646 by photoirradiation (Fig. 10b).47 The simultaneous usage of PA-JF646 and PA-JF594, which is a PA-JF646 analogue having an O atom instead of a Si atom, enabled multicolor single-particle tracking by super-resolution microscopy. Moreover, the photoactivatable Si-pyronine analogue, PA-SiR, has also been reported (Fig. 10c).48 UV irradiation of PA-SiR induced protonation, where the proton is probably derived from the water solvent, and conversion to the fluorescent Si-pyronine, which reverted to PA-SiR over time. PA-SiR enabled live-cell SLM of the outer mitochondrial membrane, and cumulative single-particle tracks of the β-2 adrenergic receptor were obtained. Furthermore, a photoactivatable SiR analogue, DH-SiR, which is a reduced leuco-form of SiR, has been reported (Fig. 10d).49 DH-SiR is converted to SiR by photo-oxidation upon photoirradiation at 400 nm. SLM imaging with DH-SiR could visualize mitochondrial dynamics. Furthermore, caging-group-free photoactivatable probes, PaXs, have been reported.50 Based on the radical character of xanthone in the excited state, PaXs bearing an olefin at the 1-position of xanthone to trap the generated radical were designed. Although the representative PaX absorbs only below 500 nm, it was converted to the fluorescent pyronine-type PaX-CF by intramolecular cyclization via radical formation of xanthone in the excited state upon photoirradiation (Fig. 10e). This PaX was suitable for a wide range of optical microscopy and super-resolution microscopy techniques.
5. Conclusions
This review summarizes recent advances in the field of far-red to NIR fluorescent probes based on SiRs. Probes working in this wavelength region are indispensable tools for fluorescence imaging in living systems due to their low autofluorescence, high tissue penetration, and suitability for multicolor imaging. Indeed, the SiRs are among the most widely used far-red to NIR fluorophores because of their exceptional brightness and photostability.Since the first report of the Si-xanthene scaffold about 15 years ago, various far-red to NIR fluorescent probes based on SiRs have been developed and utilized for a wide range of applications including super-resolution imaging. One of the important technical advances in the development of SiRs has been the improvement of synthetic methods, providing access to both symmetric and asymmetric SiRs in high yields. SiR derivatives having various substructures can be easily synthesized simply by combining the corresponding reagents. It is now possible to design tailor-made NIR fluorescent probes based on the SiR scaffold for desired imaging applications. The advanced synthesis technology for SiRs has also enabled fine-tuning of probe designs and properties, leading to the development of various photocaged probes and dye–protein hybrid-based indicators.
The unique spirocyclization property of SiRs has also been employed to expand their range of applications. SO2-rhodamine derivatives with an electrophilic –SO2 group at the 10-position of the xanthene ring were recently reported as rhodamine derivatives working in the longer-wavelength NIR region, but they exist only in the closed form under physiological conditions due to the strong electrophilicity of the xanthene ring.51 On the other hand, the more appropriate electrophilicity of the xanthene ring of SiRs allows tautomerization between the open and closed forms in water, and this property of SiRs can be used not only for off/on switching of fluorescence, but also for SLM imaging due to their dynamic self-blinking nature.
As summarized in this review, the unique properties of SiRs, including longer wavelength fluorescence, high photostability, and intramolecular spirocyclization equilibrium, have led to their extensive application in fluorescence imaging of biological samples. In recent years, it has become possible to combine the optical properties of SiRs with the dynamics of proteins, as exemplified by protein–dye hybrid-type fluorescent probes, and this has further increased the diversity of molecular design. However, the SiRs used in protein–dye hybrid fluorescent probes reported so far are limited to symmetric derivatives with relatively simple structures, leaving much room for structural optimization of SiRs. In the future, it is expected that the molecular structure of protein–dye hybrid-type fluorescent probes will be fine-tuned by precisely designing both dyes and proteins to further improve their functional characteristics, such as higher sensitivity and brightness. Given the establishment of efficient synthetic methods and a detailed understanding of the fluorogenic properties of SiRs, we expect that the SiRs will continue to find new uses in the field of analytical science, especially in the context of complex biological systems.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by JSPS KAKENHI Grant Numbers JP23H02613, JP21H05262, JP23K17389 and JP23K20040 to K. H., a grant from the Japan Agency for Medical Research and Development (AMED) (JP23ak0101182h0003, JP23wm0325046s0103 and JP23gm1510012s0201) to K. H., JST CREST to K. H., Nakatani Foundation for Advancement of Measuring Technologies in Biomedical Engineering, The Uehara Memorial Foundation, to K. H., Program for the Advancement of Next Generation Research Projects (Keio University) and Academic Development Fund (Keio University Academic Development Funds) to K. H.References
- T. Ueno and T. Nagano, Nat. Methods, 2011, 8, 642–645 CrossRef CAS PubMed.
- G. Grynkiewicz, M. Poenie and R. Y. Tsien, J. Biol. Chem., 1985, 260, 3440–3450 CrossRef CAS PubMed.
- B. N. G. Giepmans, S. R. Adams, M. H. Ellisman and R. Y. Tsien, Science, 2006, 312, 217–224 CrossRef CAS PubMed.
- M. Beija, C. A. M. Afonso and J. M. G. Martinho, Chem. Soc. Rev., 2009, 38, 2410–2433 RSC.
- H. Kobayashi, M. Ogawa, R. Alford, P. L. Choyke and Y. Urano, Chem. Rev., 2010, 110, 2620–2640 CrossRef CAS PubMed.
- M. Fu, Y. Xiao, X. Qian, D. Zhao and Y. Xu, Chem. Commun., 2008, 1780–1782 RSC.
- Y. Koide, Y. Urano, K. Hanaoka, W. Piao, M. Kusakabe, N. Saito, T. Terai, T. Okabe and T. Nagano, J. Am. Chem. Soc., 2012, 134, 5029–5031 CrossRef CAS PubMed.
- Y. Kushida, T. Nagano and K. Hanaoka, Analyst, 2015, 140, 685–695 RSC.
- T. Ikeno, T. Nagano and K. Hanaoka, Chem. – Asian J., 2017, 12, 1435–1446 CrossRef CAS PubMed.
- R. Weissleder and V. Ntziachristos, Nat. Med., 2003, 9, 123–128 CrossRef CAS PubMed.
- Y. Koide, Y. Urano, K. Hanaoka, T. Terai and T. Nagano, ACS Chem. Biol., 2011, 6, 600–608 CrossRef CAS PubMed.
- A. N. Butkevich, G. Lukinavičius, E. D'Este and S. W. Hell, J. Am. Chem. Soc., 2017, 139, 12378–12381 CrossRef CAS PubMed.
- J. B. Grimm, T. A. Brown, A. N. Tkachuk and L. D. Lavis, ACS Cent. Sci., 2017, 3, 975–985 CrossRef CAS PubMed.
- A. N. Butkevich, Org. Lett., 2021, 23, 2604–2609 CrossRef CAS PubMed.
- K. Numasawa, K. Hanaoka, N. Saito, Y. Yamaguchi, T. Ikeno, H. Echizen, M. Yasunaga, T. Komatsu, T. Ueno, M. Miura, T. Nagano and Y. Urano, Angew. Chem., Int. Ed., 2020, 59, 6015–6020 CrossRef CAS PubMed.
- Y. Kushida, K. Hanaoka, T. Komatsu, T. Terai, T. Ueno, K. Yoshida, M. Uchiyama and T. Nagano, Bioorg. Med. Chem. Lett., 2012, 22, 3908–3911 CrossRef CAS PubMed.
- A. N. Butkevich, G. Y. Mitronova, S. C. Sidenstein, J. L. Klocke, D. Kamin, D. N. H. Meineke, E. D'Este, P.-T. Kraemer, J. G. Danzl, V. N. Belov and S. W. Hell, Angew. Chem., Int. Ed., 2016, 55, 3290–3294 CrossRef CAS PubMed.
- T. Pastierik, P. Sebej, J. Medalová, P. Stacko and P. Klán, J. Org. Chem., 2014, 79, 3374–3382 CrossRef CAS PubMed.
- D. N. Rao, X. Ji and S. C. Miller, Chem. Sci., 2022, 13, 6081–6088 RSC.
- K. Hanaoka, Y. Kagami, W. Piao, T. Myochin, K. Numasawa, Y. Kuriki, T. Ikeno, T. Ueno, T. Komatsu, T. Terai, T. Nagano and Y. Urano, Chem. Commun., 2018, 54, 6939–6942 RSC.
- Y. Hoshino, K. Hanaoka, K. Sakamoto, M. Yasunaga, T. Kojima, D. Kotani, A. Nomoto, E. Sasaki, T. Komatsu, T. Ueno, H. Takamaru, Y. Saito, Y. Seto and Y. Urano, RSC Chem. Biol., 2022, 3, 859–867 RSC.
- K. Hanaoka, S. Iwaki, K. Yagi, T. Myochin, T. Ikeno, H. Ohno, E. Sasaki, T. Komatsu, T. Ueno, M. Uchigashima, T. Mikuni, K. Tainaka, S. Tahara, S. Takeuchi, T. Tahara, M. Uchiyama, T. Nagano and Y. Urano, J. Am. Chem. Soc., 2022, 144, 19778–19790 CrossRef CAS PubMed.
- Y. Miao, N. Qian, L. Shi, F. Hu and W. Min, Nat. Commun., 2021, 12, 4518 CrossRef CAS PubMed.
- L. G. Wang, A. R. Montaño, J. R. Combs, N. P. McMahon, A. Solanki, M. M. Gomes, K. Tao, W. H. Bisson, D. A. Szafran, K. S. Samkoe, K. M. Tichauer and S. L. Gibbs, Nat. Chem., 2023, 15, 729–739 CrossRef CAS PubMed.
- S. Uno, M. Kamiya, T. Yoshihara, K. Sugawara, K. Okabe, M. C. Tarhan, H. Fujita, T. Funatsu, Y. Okada, S. Tobita and Y. Urano, Nat. Chem., 2014, 6, 681–689 CrossRef CAS PubMed.
- S. W. Hell and J. Wichmann, Opt. Lett., 1994, 19, 780–782 CrossRef CAS PubMed.
- E. Betzig, G. H. Patterson, R. Sougrat, O. W. Lindwasser, S. Olenych, J. S. Bonifacino, M. W. Davidson, J. Lippincott-Schwartz and H. F. Hess, Science, 2006, 313, 1642–1645 CrossRef CAS PubMed.
- M. J. Rust, M. Bates and X. Zhuang, Nat. Methods, 2006, 3, 793–795 CrossRef CAS PubMed.
- R. Fedosseev, Y. Belyaev, J. Frohn and A. Stemmer, Opt. Lasers Eng., 2005, 43, 403–414 CrossRef.
- W. Chi, Q. Qiao, C. Wang, J. Zheng, W. Zhou, N. Xu, X. Wu, X. Jiang, D. Tan, Z. Xu and X. Liu, Angew. Chem., Int. Ed., 2020, 59, 20215–20223 CrossRef CAS PubMed.
- J. Tyson, K. Hu, S. Zheng, P. Kidd, N. Dadina, L. Chu, D. Toomre, J. Bewersdorf and A. Schepartz, ACS Cent. Sci., 2021, 7, 1419–1426 CrossRef CAS PubMed.
- A. Morozumi, M. Kamiya, S. Uno, K. Umezawa, R. Kojima, T. Yoshihara, S. Tobita and Y. Urano, J. Am. Chem. Soc., 2020, 142, 9625–9633 CAS.
- P. Werther, K. Yserentant, F. Braun, N. Kaltwasser, C. Popp, M. Baalmann, D.-P. Herten and R. Wombacher, Angew. Chem., Int. Ed., 2020, 59, 804–810 CrossRef CAS PubMed.
- G. Lukinavičius, K. Umezawa, N. Olivier, A. Honigmann, G. Yang, T. Plass, V. Mueller, L. Reymond, I. R. Corrêa Jr., Z.-G. Luo, C. Schultz, E. A. Lemke, P. Heppenstall, C. Eggeling, S. Manley and K. Johnsson, Nat. Chem., 2013, 5, 132–139 CrossRef PubMed.
- S. Karch, J. Broichhagen, J. Schneider, D. Böning, S. Hartmann, B. Schmid, P. Tripal, R. Palmisano, C. Alzheimer, K. Johnsson and T. Huth, J. Med. Chem., 2018, 61, 6121–6139 CrossRef CAS PubMed.
- L. Wang, M. Tran, E. D'Este, J. Roberti, B. Koch, L. Xue and K. Johnsson, Nat. Chem., 2020, 12, 165–172 CrossRef CAS PubMed.
- N. Lardon, L. Wang, A. Tschanz, P. Hoess, M. Tran, E. D'Este, J. Ries and K. Johnsson, J. Am. Chem. Soc., 2021, 143, 14592–14600 CrossRef CAS PubMed.
- N. Mertes, M. Busch, M.-C. Huppertz, C. N. Hacker, J. Wilhelm, C.-M. Gürth, S. Kühn, J. Hiblot, B. Koch and K. Johnsson, J. Am. Chem. Soc., 2022, 144, 6928–6935 CrossRef CAS PubMed.
- J. B. Grimm, A. N. Tkachuk, L. Xie, H. Choi, B. Mohar, N. Falco, K. Schaefer, R. Patel, Q. Zheng, Z. Liu, J. Lippincott-Schwartz, T. A. Brown and L. D. Lavis, Nat. Methods, 2020, 17, 815–821 CrossRef CAS PubMed.
- C. Grienberger and A. Konnerth, Neuron, 2012, 73, 862–885 CrossRef CAS PubMed.
- M. Z. Lin and M. J. Schnitzer, Nat. Neurosci., 2016, 19, 1142–1153 CrossRef PubMed.
- Y. Qian, K. D. Piatkevich, B. Mc Larney, A. S. Abdelfattah, S. Mehta, M. H. Murdock, S. Gottschalk, R. S. Molina, W. Zhang, Y. Chen, J. Wu, M. Drobizhev, T. E. Hughes, J. Zhang, E. R. Schreiter, S. Shoham, D. Razansky, E. S. Boyden and R. E. Campbell, Nat. Methods, 2019, 16, 171–174 CrossRef CAS PubMed.
- J. Hiblot, Q. Yu, M. D. B. Sabbadini, L. Reymond, L. Xue, A. Schena, O. Sallin, N. Hill, R. Griss and K. Johnsson, Angew. Chem., Int. Ed., 2017, 56, 14556–14560 CrossRef CAS PubMed.
- C. Deo, A. S. Abdelfattah, H. K. Bhargava, A. J. Berro, N. Falco, H. Farrants, B. Moeyaert, M. Chupanova, L. D. Lavis and E. R. Schreiter, Nat. Chem. Biol., 2021, 17, 718–723 CrossRef CAS PubMed.
- L. Hellweg, A. Edenhofer, L. Barck, M.-C. Huppertz, M. S. Frei, M. Tarnawski, A. Bergner, B. Koch, K. Johnsson and J. Hiblot, Nat. Chem. Biol., 2023, 19, 1147–1157 CrossRef CAS.
- J. B. Grimm, T. Klein, B. G. Kopek, G. Shtengel, H. F. Hess, M. Sauer and L. D. Lavis, Angew. Chem., Int. Ed., 2016, 55, 1723–1727 CrossRef CAS PubMed.
- J. B. Grimm, B. P. English, H. Choi, A. K. Muthusamy, B. P. Mehl, P. Dong, T. A. Brown, J. Lippincott-Schwartz, Z. Liu, T. Lionnet and L. D. Lavis, Nat. Methods, 2016, 13, 985–988 CrossRef CAS PubMed.
- M. S. Frei, P. Hoess, M. Lampe, B. Nijmeijer, M. Kueblbeck, J. Ellenberg, H. Wadepohl, J. Ries, S. Pitsch, L. Reymond and K. Johnsson, Nat. Commun., 2019, 10, 4580 CrossRef PubMed.
- X. Zhang, M. Zhang, Y. Yan, M. Wang, J. Li, Y. Yu, Y. Xiao, X. Luo, X. Qian and Y. Yang, Chem. Commun., 2021, 57, 7553–7556 RSC.
- R. Lincoln, M. L. Bossi, M. Remmel, E. D'Este, A. N. Butkevich and S. W. Hell, Nat. Chem., 2022, 14, 1013–1020 CrossRef CAS PubMed.
- J. Liu, Y.-Q. Sun, H. Zhang, H. Shi, Y. Shi and W. Guo, ACS Appl. Mater. Interfaces, 2016, 8, 22953–22962 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |