
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A thiol–ene mediated approach for peptide bioconjugation using ‘green’ solvents under continuous flow†
Inés
Rabadán González
 a,
Joshua T.
McLean
a,
Nikita
Ostrovitsa
a,
Sheila
Fitzgerald
b,
Andrea
Mezzetta
c,
Lorenzo
Guazzelli
c,
Donal F.
O'Shea
b and
Eoin M.
Scanlan
*a
a,
Joshua T.
McLean
a,
Nikita
Ostrovitsa
a,
Sheila
Fitzgerald
b,
Andrea
Mezzetta
c,
Lorenzo
Guazzelli
c,
Donal F.
O'Shea
b and
Eoin M.
Scanlan
*a
aTrinity Biomedical Sciences Institute, Trinity College Dublin, 152-160 Pearse Street, Dublin 2, Ireland. E-mail: eoin.scanlan@tcd.ie
bDepartment of Chemistry, RCSI, 123 St Stephen's Green, Dublin 2, Ireland
cUniversity of Pisa, Via Bonanno 33, 56126, Pisa, Italy
First published on 21st February 2024
Abstract
Flow chemistry has emerged as an integral process within the chemical sector permitting energy efficient synthetic scale-up while improving safety and minimising solvent usage. Herein, we report the first applications of the photoactivated, radical-mediated thiol–ene reaction for peptide bioconjugation under continuous flow. Bioconjugation reactions employing deep eutectic solvents, bio-based solvents and fully aqueous systems are reported here for a range of biologically relevant peptide substrates. The use of a water soluble photoinitiator, Irgacure 2959, permitted synthesis of glycosylated peptides in fully aqueous conditions, obviating the need for addition of organic solvents and enhancing the green credentials of these rapid, photoactivated, bioconjugation reactions.
Introduction
The field of radical chemistry has undergone rapid changes since its inception 120 years ago. The early reputation of radicals as unpredictable reactive intermediates has been largely dispelled through development of reagents and additives capable of directing radicals via predictable mechanistic pathways.1 However, such reagents are often Sn, Sm or Hg based and, despite significant issues with toxicity and disposal, persist within contemporary organic syntheses. This has severely limited the potential of radical chemistries to adhere to ‘green chemistry’ principles, particularly in an industrial context.2 The thiol–ene reaction (Fig. 1), radical mediated hydrothiolation across an unsaturated bond, has the potential to be considered an archetypal green reaction. Facile generation of thiyl radicals in combination with rapid addition kinetics, permits high conversions without requirement for toxic metallic additives.3 Mild reaction conditions are tolerant to a wide range of functional groups, obviating the need for atom-inefficient protecting group strategies, thereby significantly reducing waste during complex biomolecule syntheses.4 As a consequence, the thiol–ene reaction has been widely applied across a diverse range of fields from drug discovery and chemical biology5 to materials and polymer science.6 In particular, the tolerance of the thiol–ene reaction to aqueous conditions reduces the requirement for hazardous organic solvents and aqueous thiol–ene is now widely utilised for bioconjugation applications, although harmful organic solvents are still routinely required to solubilize the radical initiators. Initial studies utilised azobisisobutyronitrile (AIBN) as a radical initiator and required heating of the reaction mixture over prolonged (>6 h) reaction times to achieve efficient product formation. These harsh conditions precluded application of radical mediated thiol–ene to sensitive unprotected biomolecules, including peptides and proteins.7–9 Dondoni and co-workers10 reported the application of 2,2-dimethoxy-2-phenylacetophenone (DPAP) for the modification of bovine serum albumin (BSA) under aqueous conditions using ultraviolet (UV) initiation, highlighting the potential of this approach for protein and peptide bioconjugation. This has been demonstrated through the modification of peptides,11–18 proteins,19 carbohydrates20–22 and the synthesis of complex multivalent structures.23–28 Glycosylation of virus-like Qβ particles by Davis and co-workers19 and the development of cysteine lipidation by Brimble and co-workers17,18 highlight the potential of aqueous thiol–ene chemistries for the synthesis of biologically pertinent peptide derivatives.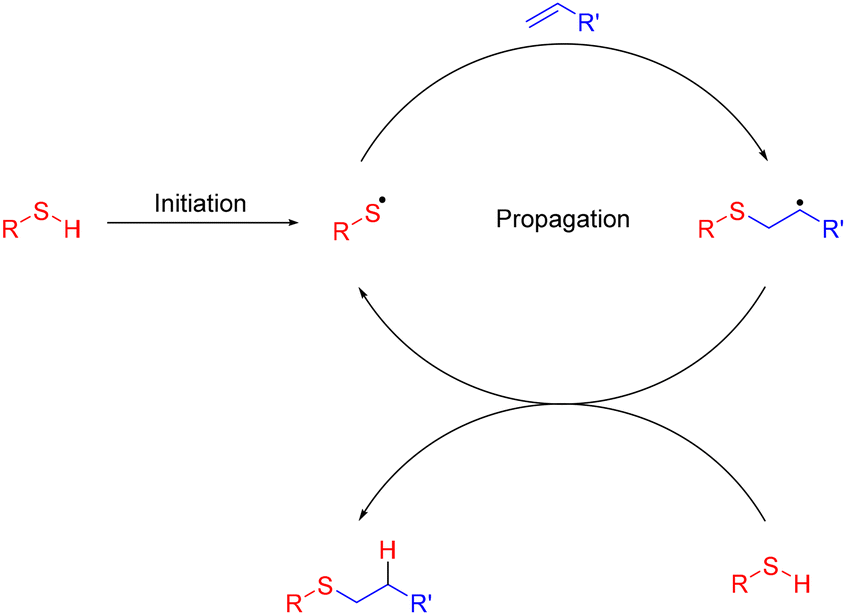 | ||
| Fig. 1 Radical chain mechanism of the radical mediated thiol–ene reaction.3 | ||
A limitation of this approach from the perspective of sustainability, is the requirement for harmful organic solvents to ensure sufficient solubility of the initiator DPAP, which possesses only sparing aqueous solubility. For substrates possessing some organic solubility, the addition of alcohol or ethereal solvents such as methanol (MeOH), butan-1-ol (BuOH) or tetrahydrofuran (THF) is sufficient to permit efficient thiol–ene addition. However, large, unprotected substrates such as peptides and proteins require the addition of aprotic polar solvents dimethylformamide (DMF), dimethylsulfoxide (DMSO) or N-methyl-2-pyrrolidine (NMP) to ensure initiator solubility. NMP and DMF are defined as substances of very high concern by the European Chemicals Agency (ECHA) and have serious and irreversible effects on human health and the environment.29 Obviating the need for these organic solvents in radical mediated thiol–ene reactions would therefore provide a significant increase in the sustainability and safety of peptide and carbohydrate bioconjugate syntheses. This may be achieved either by using reaction components that possess excellent aqueous solubility or through use of greener solvents including bio-based and deep eutectic solvents (DESs). Selection of suitable solvents is a significant factor in devising greener syntheses and limiting environmental impact.30 Water is the prototypical green solvent as it is non-toxic, readily available and cheap. Bio-based solvents derived from sustainable feedstocks and less harmful solvents provide further opportunity for greener and safer syntheses while retaining the desirable solubility characteristics of conventional organic solvents.31 DESs provide scope for solvent recycling thereby substantially limiting the creation of solvent waste in organic syntheses and are compatible with thiol–ene chemistries.32 Flow chemistry has also served to increase the sustainability and safety of chemical processes in both academic and industrial settings.33 Continuous flow setups limit the interaction of chemists with hazardous material and serve to reduce solvent usage.34 Facile scale-up in continuous flow also removes the time intensive optimisation work necessary to migrate from laboratory batch set-up to those suitable for manufacturing processes.35 As a highly efficient two-component reaction requiring only catalytic quantities of initiating species, the radical mediated thiol–ene reaction provides an ideal candidate for adaption to a continuous flow set-up. Consequently, thiol–ene chemistry under continuous flow has been an active area of research demonstrated through the synthesis of alkylic thioesters,36 lipidated cysteine derivatives,37 substituted cinchona alkaloids,38 thiomorpholines39 and polyamidoamines.40 To date however, radical mediated thiol–ene modification of biologically pertinent unprotected peptides has not been demonstrated either under continuous flow or in fully aqueous or green solvent conditions.
Results and discussion
Optimisation of radical mediated thiol–ene reactions of GSH under continuous flow conditions
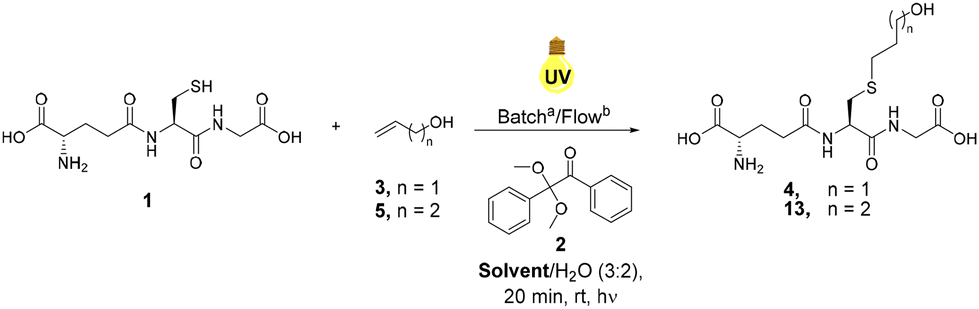
Glutathione (GSH), 1 was selected as a model thiolated peptide substrate due to the commercial availability of GSH and the therapeutic potential of GSH conjugates (GSX).14 GSH is a tripeptide featuring a glutamic acid (Glu)-cysteine (Cys) gamma amide linkage followed by a glycine (Gly) residue (Glu-Cys-Gly).41 Notably, GSX conjugates demonstrate potential as cytotoxic agents and therefore offer attractive synthetic targets.14 Conway and co-workers utilised radical mediated thiol–ene under batch conditions to synthesise novel GSX conjugates to investigate GSH binding in Kef a bacterial K+ efflux system, and our initial investigations utilised these optimised conditions.14 THF/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was used in order to ensure sufficient solubility of both GSH 1 and photoinitiator DPAP 2. Allyl alcohol 3 was selected as the alkene component due to its ready availability and good solubility in the selected solvent system. Optimisation reactions under continuous flow employed a system consisting of two HPLC pumps and a microchip reactor (1000 μL) covered by a 36 W UV lamp (365 nm). Optimisation of the radical mediated thiol–ene reaction of GSH 1 and allyl alcohol 3 is shown in Table 1. The reaction of 1 eq. of GSH 1 and 1 eq. of allyl alcohol 3 in the presence of 0.05 eq. of DPAP 2 under UV irradiation furnished thioether adduct 4 in an 83% yield over 20 min (Table 1, entry I). 1H NMR spectroscopic analysis of the crude mixture revealed residual GSH 1 within the sample. An increase to 2.5 eq. of allyl alcohol 3 (Table 1, entry II) resulted in full consumption of 1 and permitted isolation of thioether 4 in an improved 91% yield. The use of stoichiometric DPAP 2 resulted in a lower yield of 4 (Table 1, entry III), likely due to the high initiator concentration in THF diminishing mixing and subsequent reaction efficiency. Decreasing the reaction time from 20 min to 10 min resulted in a significant reduction in yield from 91% to 34% (Table 1, entry IV). However, near quantitative yields were obtained over 10 min with an increase in temperature from rt to 40 °C (Table 1, entry V).
:1) was used in order to ensure sufficient solubility of both GSH 1 and photoinitiator DPAP 2. Allyl alcohol 3 was selected as the alkene component due to its ready availability and good solubility in the selected solvent system. Optimisation reactions under continuous flow employed a system consisting of two HPLC pumps and a microchip reactor (1000 μL) covered by a 36 W UV lamp (365 nm). Optimisation of the radical mediated thiol–ene reaction of GSH 1 and allyl alcohol 3 is shown in Table 1. The reaction of 1 eq. of GSH 1 and 1 eq. of allyl alcohol 3 in the presence of 0.05 eq. of DPAP 2 under UV irradiation furnished thioether adduct 4 in an 83% yield over 20 min (Table 1, entry I). 1H NMR spectroscopic analysis of the crude mixture revealed residual GSH 1 within the sample. An increase to 2.5 eq. of allyl alcohol 3 (Table 1, entry II) resulted in full consumption of 1 and permitted isolation of thioether 4 in an improved 91% yield. The use of stoichiometric DPAP 2 resulted in a lower yield of 4 (Table 1, entry III), likely due to the high initiator concentration in THF diminishing mixing and subsequent reaction efficiency. Decreasing the reaction time from 20 min to 10 min resulted in a significant reduction in yield from 91% to 34% (Table 1, entry IV). However, near quantitative yields were obtained over 10 min with an increase in temperature from rt to 40 °C (Table 1, entry V).
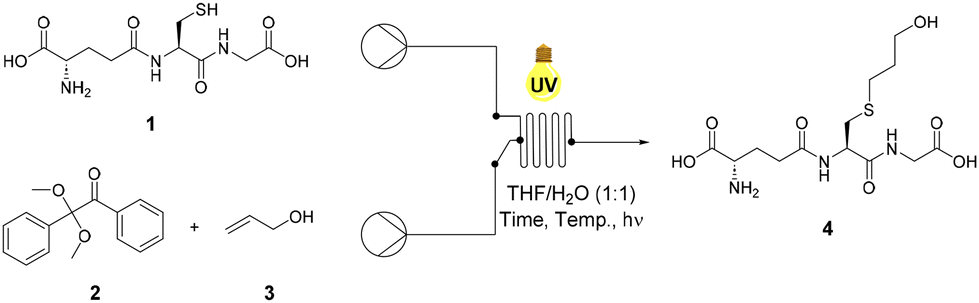
Increased temperature required the addition of a heating source under the reactor, significantly complicating reaction scale up and negatively impacting the environmental sustainability of this approach. Optimal conditions (Table 1, entry II) at rt gave GSX 4 in >90% yield and formed the basis of further investigations into substrate scope.
Substrate scope of radical mediated thiol–ene reactions of GSH
With optimal reaction conditions established under continuous flow using allyl alcohol 3, the reactivity of a series of alkene components 5–12 with GSH 1 was investigated to explore the substrate scope of the reaction (Scheme 1). Alkenes 5–12 were selected to demonstrate the applicability of thiol–ene chemistry to the synthesis of biochemically relevant conjugates from unprotected amino acid or carbohydrate starting materials. Reactions were performed in both batch conditions and under continuous flow in order to compare relative reaction efficiencies (Scheme 1). When compared to the yields obtained in batch, thiol–ene reactions performed under continuous flow resulted in increased yields of the corresponding GSXs in the majority of cases (4, 13, 15, 16, 17, 19 and 20, Scheme 1). The general increase in yields observed for GSX conjugates under continuous flow was likely due to increased mixing efficiency and a decrease in the attenuation of UV irradiation, comparable to a decrease in irradiation path length. In the case of hydroxyl GSX conjugates 4 and 13 and unprotected monosaccharide GSX conjugate 14, yields under continuous flow were higher, but the difference was less significant as yields in batch were >80%.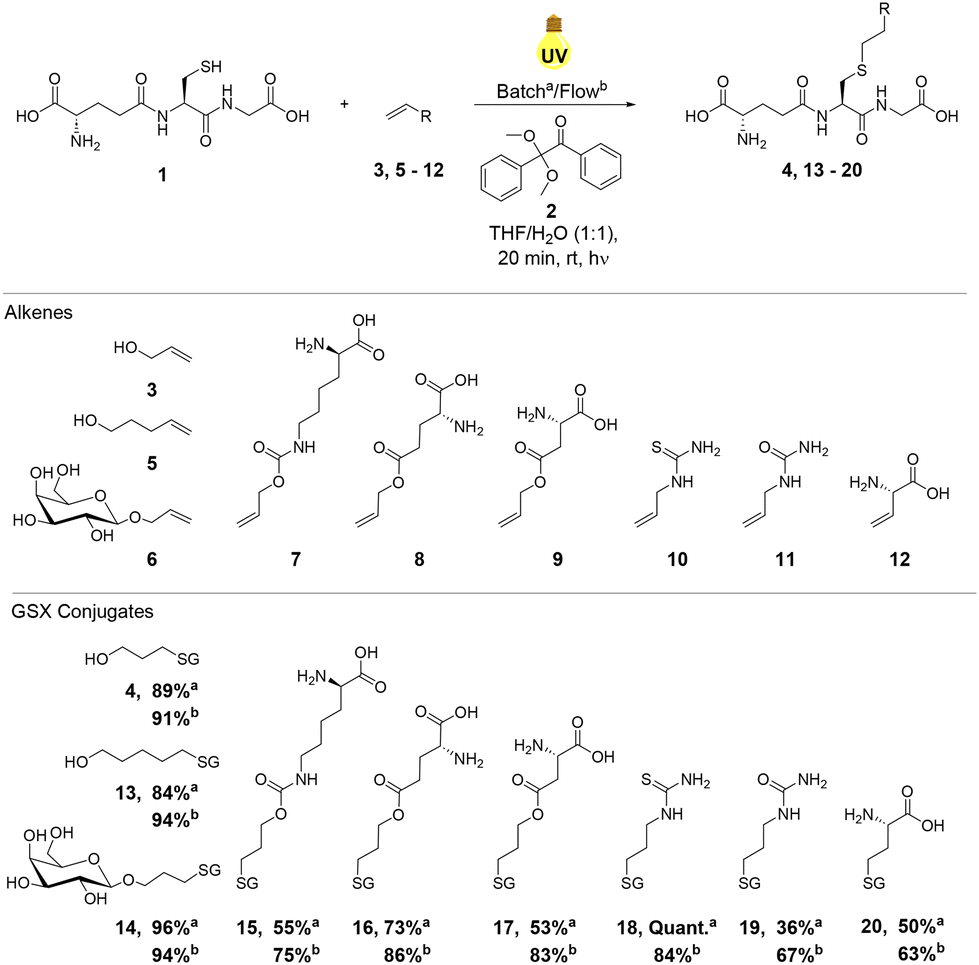 | ||
| Scheme 1 Substrate scope of GSH thiol–ene reaction under UV irradiation at rt for 20 min in (a) batch conditions and (b) under continuous flow. Yields were calculated by 1H NMR spectroscopy using dimethyl sulfone as an internal standard (1 eq.). | ||
Increases in yield were particularly enhanced (∼30% increase from batch to continuous flow) in the reactions of electron-poor alkenes 9 and 11, derived from aspartic acid and allylurea, respectively. Electron-poor alkenes suffer from decreased thiol–ene reactivity due to destabilisation of the corresponding C-centered radical intermediate (Fig. 1).42 The increase in reaction efficiency resulting from the continuous flow set up employed is therefore clearly observed in those reactions displaying slower reaction kinetics and poorer substrate reactivity. Yields of GSX conjugates 15, 16 and 20 (Scheme 1) increased under continuous flow, but to a lesser degree due to the greater reactivity and faster reaction kinetics of the respective alkene precursors 7, 8 and 12 (Scheme 1). In the case of compound 18, quantitative conversion to the corresponding thioether was observed for the batch reaction, dropping to 87% under continuous flow conditions. The stark improvement in conversion for thiourea derivative 18 relative to the urea 19, in particular for the batch reaction may be due to improved solubility of the thiourea substrate.
Radical mediated thiol–ene reactions in DESs, bio-based and less harmful solvents
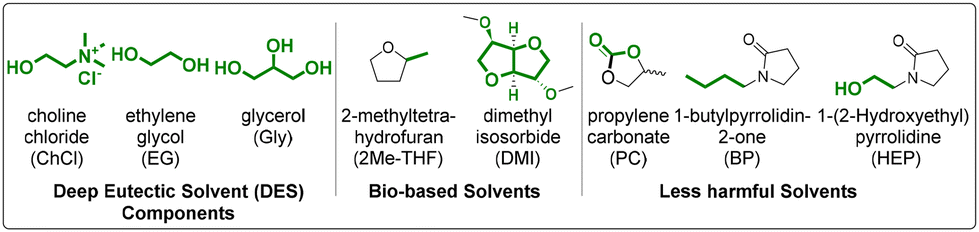
Having demonstrated the efficiency of the thiol–ene mediated GSH bioconjugation under continuous flow, we next set out to further enhance the ‘green’ credentials of the process through use of green solvents.The use of DESs in the radical mediated thiol–ene reaction has previously been reported by our labs and obviates the need for organic solvents in the reaction.32 DESs have been previously employed under continuous flow in the manufacturing of gold and silver nanocrystals, thereby indicating the feasibility of this approach.43 However, due to the higher viscosity of DESs in comparison to H2O/THF mixtures, continuous flow in a chip reactor was not possible due to considerable instrument back pressure. Based on previous work from Seeberger44 and Baker,43 a simple, high volume flow system was developed in our laboratory utilising a 2-channel syringe pump and a coil reactor (9.65 m length and 0.8 mm of internal diameter). This setup was housed in a UV oven with an extraneous sample collector for analysis (ESI†). In addition to DESs, a number of bio-based solvents were investigated (Table 2), the use of which had already been demonstrated under continuous flow,45 but not in the context of radical mediated thiol–ene chemistry. An initial study was performed using DES/H2O in a ratio 3:2 to assist substrate solubility, reduce viscosity and yet retain the DESs hydrogen bonding characteristics.32 Radical mediated thiol–ene reactions were undertaken with GSH 1 and unsaturated alcohols 3 and 5 in presence of DPAP 2 and UV irradiation under the previously optimised continuous flow conditions (Table 2). Conversion of GSH 1 to the GSXs 4 and 5 was established by RP-HPLC analysis utilising a calibration curve (ESI†). In batch reaction, conversions were between 98% and quantitative (Table 2, columns 3 and 5) in all solvent systems investigated highlighting the compatibility of DESs with radical mediated thiol–ene methodologies. Conversions under continuous flow (Table 2, columns 4 and 6) were slightly lower than those obtained in batch, although they remained >80% in all cases. This is likely due to the higher viscosity of DES, bio-based and less harmful solvents creating distortions in the flow system, preventing effective reagent mixing. The lowest reaction conversions correspond to choline chloride based DESs (Table 2, entry I and II) and in less harmful solvent HEP (Table 2, entry VI) and bio-based solvent DMI (Table 2, entry VII), solvent systems which possessed the highest viscosity. Having demonstrated the first examples of thiol–ene mediated peptide conjugation under continuous flow using both DES and biobased solvents, we next set out to achieve peptide bioconjugation in continuous flow in fully aqueous conditions.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Conv. 4 (batch)a | Conv. 4 (flow)b | Conv. 13 (batch)a | Conv. 13 (flow)b |
| a Batch reaction. b Continuous flow as stated in the table descriptor. | |||||
| I | ChCl:EG (1:2) |
>99% | 83% | 98% | 91% |
| II | ChCl:Gly (1:2) |
Quant. | 84% | >99% | Quant. |
| III | 2Me-THF | >99% | 95% | >99% | 86% |
| IV | PC | Quant. | 96% | >99% | >99% |
| V | BP | Quant. | 93% | >99% | >99% |
| VI | HEP | 98% | 96% | Quant. | Quant. |
| VII | DMI | >99% | 96% | Quant. | Quant. |
|
|||||
Substrate scope of the thiol–ene reactions of GSH in water under continuous flow
In order to fully eliminate the requirement for hazardous organic solvents in thiol–ene mediated bioconjugation under fully aqueous conditions, we investigated the use of water-soluble photoinitiator 21, Irgacure 2959, which has been utilised principally in dental applications for the crosslinking of hydrogels and polymers.46 However, 21 is not widely utilised synthetically and has not previously been used to initiate thiol–ene addition in a synthetic context. Radical mediated thiol–ene reactions of GSH 1 and alkenes 3 and 5–11 were undertaken in the presence of water-soluble initiator 21 (Scheme 2).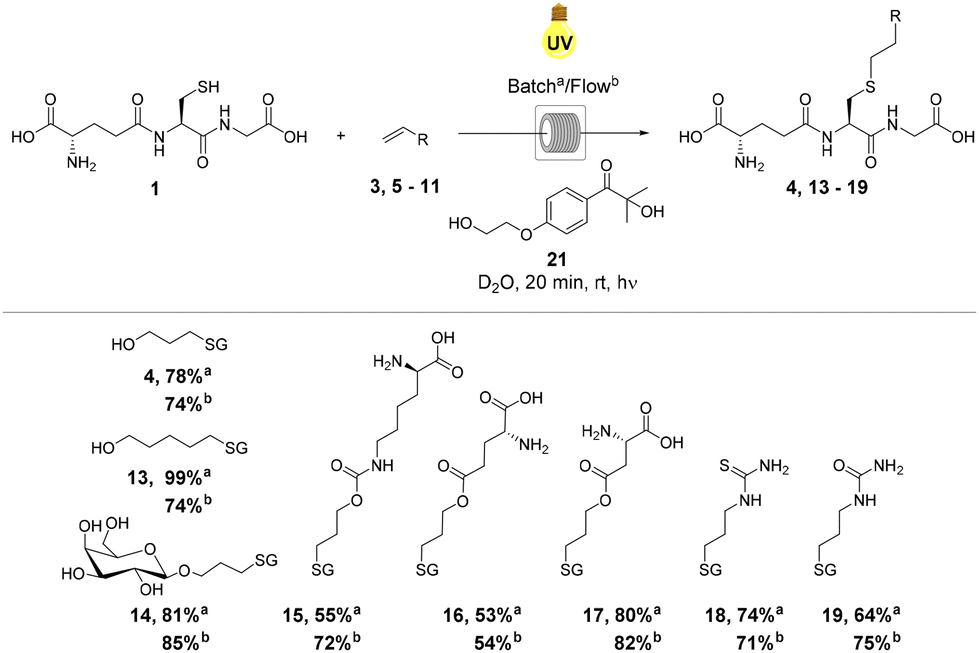 | ||
| Scheme 2 Substrate scope of radical mediated thiol–ene reactions using Irgacure 2959 21 under (a) batch conditions and (b) under continuous flow. Yields were calculated by 1H NMR spectroscopy using dimethyl sulfone as an internal standard (1 eq.). | ||
Generally, product yields obtained from reactions initiated with 21 under continuous flow were somewhat lower than the corresponding reactions initiated with DPAP 2 (Scheme 1). This is as a result of the slower rate of homolytic cleavage to form the corresponding initiating radical species of 21 in comparison to DPAP 2 (Fig. 2).47 The secondary alkyl radical species resulting from cleavage of 21 (Fig. 2, D) exhibits significantly lower stability than the stabilised benzyl radical resulting from the cleavage of DPAP 2 (Fig. 2, B), thereby limiting the rate of homolytic fission. Comparison of the crude 1H NMR spectra of the radical mediated thiol–ene reactions of GSH 1 and aspartic acid derived alkene 9 utilising DPAP 2 and initiator 21 indicated that the reaction mixture initiated by Igracure 2959 21 contained a greater proportion of signals corresponding to unreacted initiator. This suggests that the slower homolytic fission of initiator 21 is indeed responsible for the lower yields observed in comparison to reactions utilising DPAP 2. These experiments demonstrated the feasibility of radical mediated thiol–ene addition under fully aqueous conditions to furnish peptidic substrates under continuous flow, albeit with slightly lower reaction efficiency than with standard initiator DPAP 2. Nevertheless, satisfactory reaction yields were obtained in fully aqueous conditions, removing the requirement for any organic solvents, and our attention turned to application of this methodology to a biologically relevant peptide targets. Batch reactions were also performed under these conditions, furnishing conversions either comparable to or lower than those observed under continuous flow.
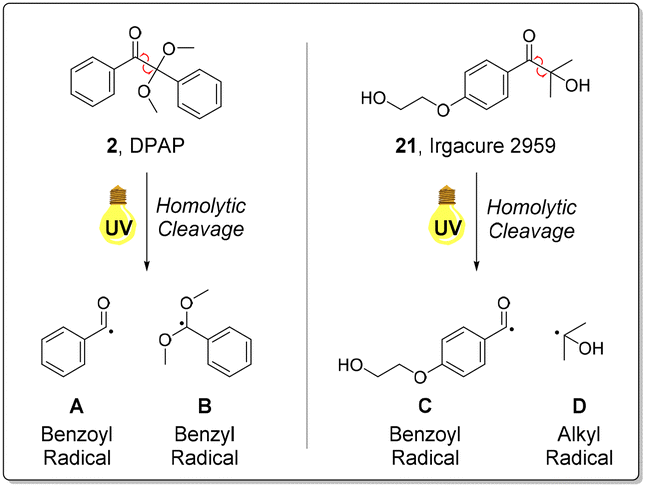 | ||
| Fig. 2 Homolytic fission of DPAP 2 and Irgacure 2959 21 to generate the corresponding benzoyl (A and C) and benzyl (B) and alkyl (D) radicals.47 | ||
Thiol–ene mediated glycosylation of biologically active peptides in water under continuous flow
Tumour targeting peptide, CREKA 22, has garnered significant interest in the literature due to its high specificity.48 CREKA 22 binds fibrin, an insoluble endogenous protein which is a key component of blood clots. The concentration of fibrin increases at sites of injury in healthy somatic cells to prevent bleeding. In tumour cells, however, it forms an integral component of the intracellular matrix.48 Conjugation of CREKA peptide 22 has primarily been utilised to facilitate the imaging of cancer cells, typically via maleimide conjugation at the Cys residue of the peptide.49 CREKA peptide 22 represents an ideal model substrate for aqueous radical mediated thiol–ene bioconjugation due to the presence of a terminal Cys residue and aqueous solubility. Allylated monosaccharide 6 was selected as the alkene component of the reaction as glycoconjugates of CREKA peptide 22 have not previously been reported in the literature and may be of considerable importance for biological applications due to the unique properties of glycoconjugates including cell uptake and lectin binding. CREKA peptide 22 was prepared via manual solid phase peptide synthesis (SPPS) and purified by RP-HPLC (ESI†). Radical mediated thiol–ene addition of galactoside 6 (20 eq.) to CREKA peptide 22 in the presence of 21 under UV irradiation in H2O + 0.1% formic acid under continuous flow was undertaken (Scheme 3). Mild acidic conditions were used to preclude oxidation of the starting material.50,51 The progress of the reaction was monitored by RP-HPLC analysis and indicated full consumption of 22 in 2 h. Subsequent freeze drying and semi-PREP HPLC analysis permitted isolation of the target glycosylated peptide 23 in 44% yield. AFP peptide 24 was selected as a second biologically relevant peptide example. Antifungal proteins (AFP) are useful for the treatment of fungal infections such as Candidiasis, Cryptococcosis and Aspergillosis.52 AFPs are known for their sustainability, high efficacy, restricted toxicity and low cost of production.52 CKYKAQ peptide 24 is an AFP sequence located in the bacterial type 3 chitin-binding domanins.53 This sequence is also located in the chitin-binding domains of sensitive fungi, such as A. giganteus.54 CKYKAQ peptide 24 was prepared via manual SPPS and purified by RP-HPLC (ESI†). The progress of the thiol–ene reaction with 6 under aqueous continuous flow conditions was monitored by RP-HPLC analysis and indicated full consumption of starting material 24 in 2 h (Scheme 4). HPLC purification permitted isolation of the target glycosylated peptide 25 in an improve 58% isolated yield, relative to 23, highlighting the potential impact of the amino acid sequence on yield under these conditions. The addition of 6 M guanidine HCl (Gdn HCl), as reported by Y. Wang and co-workers,55 to disrupt intramolecular and intermolecular hydrogen bonding and improve the solubility of reagents was investigated, however, no notable increase in the yield of products 23 or 25 was observed. The slightly reduced yields observed for the glycopeptide examples may in part be due to the higher concentrations of the peptides employed in these reactions which may promote disulfide bond formation resulting in loss of product.56 Finally, to demonstrate the applicability of our green approach for bioconjugation of larger peptide systems, we investigated the glycosylation of RGD peptide 26 which comprises 11 amino acid residues. RGD-peptides are known to be over expressed cancer cells.57 The RGD motif targets integrins overexpressed on the tumor neovasculature, enabling targeting of imaging agents, therapeutics and gene delivery systems.58 RGD peptide conjugates are therefore of considerable interest for medicinal chemistry and drug development. H-PQVT![[R with combining low line]](https://www.rsc.org/images/entities/char_0052_0332.gif)
![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif)
![[D with combining low line]](https://www.rsc.org/images/entities/char_0044_0332.gif) VFTEG-NH2 peptide 26 was synthesised by SPPS using a Liberty Blue® peptide synthetiser. In an attempt to improve ligation yields, we employed a strategy reported by Alexander and co-workers59 whereby the alkene moiety is installed on the peptide component of the thiol–ene reaction, thereby precluding any side reaction with cys residues Peptide 26 was then glycosylated with thiolated GlcNAc derivative 27 (5 eq.), which was prepared from glucosamine (ESI†). The thiol–ene mediated bioconjugation reaction of the larger RGD peptide 26 with glycosylated thiol 27 was performed, with initiator Irgacure 2959 21 (2.5 eq.), under continuous flow for 2 h in DES:H2O (3:2) (Scheme 5). The crude material was analysed and purified by RP-HPLC, furnishing RGD-peptide 28 was afforded in 62% isolated yield. The use of the DES:H2O system was identified as being optimal for the RGD peptide and compatibility of this green solvent system with initiator 21 was demonstrated. These examples represent the first example of radical mediated thiol–ene bioconjugation of biologically active peptides, in green solvents, under continuous flow and significantly reduces the environmental impact of the thiol–ene bioconjugation methodology.
VFTEG-NH2 peptide 26 was synthesised by SPPS using a Liberty Blue® peptide synthetiser. In an attempt to improve ligation yields, we employed a strategy reported by Alexander and co-workers59 whereby the alkene moiety is installed on the peptide component of the thiol–ene reaction, thereby precluding any side reaction with cys residues Peptide 26 was then glycosylated with thiolated GlcNAc derivative 27 (5 eq.), which was prepared from glucosamine (ESI†). The thiol–ene mediated bioconjugation reaction of the larger RGD peptide 26 with glycosylated thiol 27 was performed, with initiator Irgacure 2959 21 (2.5 eq.), under continuous flow for 2 h in DES:H2O (3:2) (Scheme 5). The crude material was analysed and purified by RP-HPLC, furnishing RGD-peptide 28 was afforded in 62% isolated yield. The use of the DES:H2O system was identified as being optimal for the RGD peptide and compatibility of this green solvent system with initiator 21 was demonstrated. These examples represent the first example of radical mediated thiol–ene bioconjugation of biologically active peptides, in green solvents, under continuous flow and significantly reduces the environmental impact of the thiol–ene bioconjugation methodology.
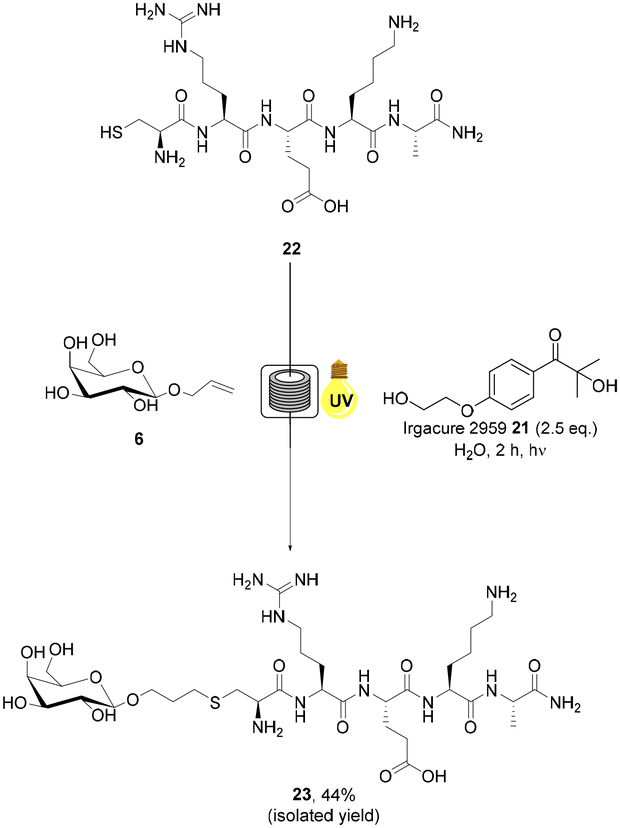 | ||
| Scheme 3 Radical mediated thiol–ene reaction of allylated monosaccharide 6 and CREKA peptide 22 under continuous flow to yield glycosylated peptide 23. | ||
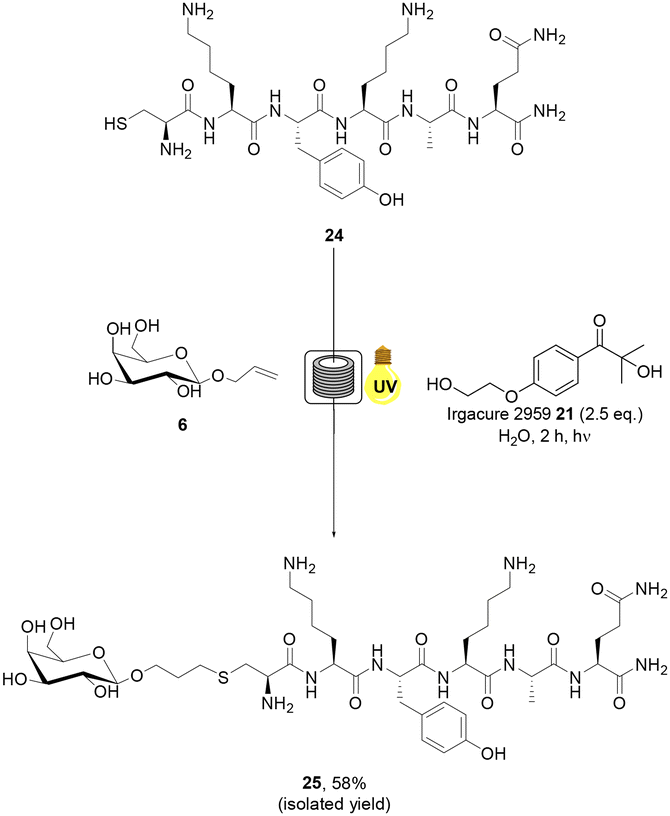 | ||
| Scheme 4 Radical mediated thiol–ene reaction of allylated monosaccharide 6 and CKYKAQ peptide 24 under continuous flow to yield glycosylated peptide 25. | ||
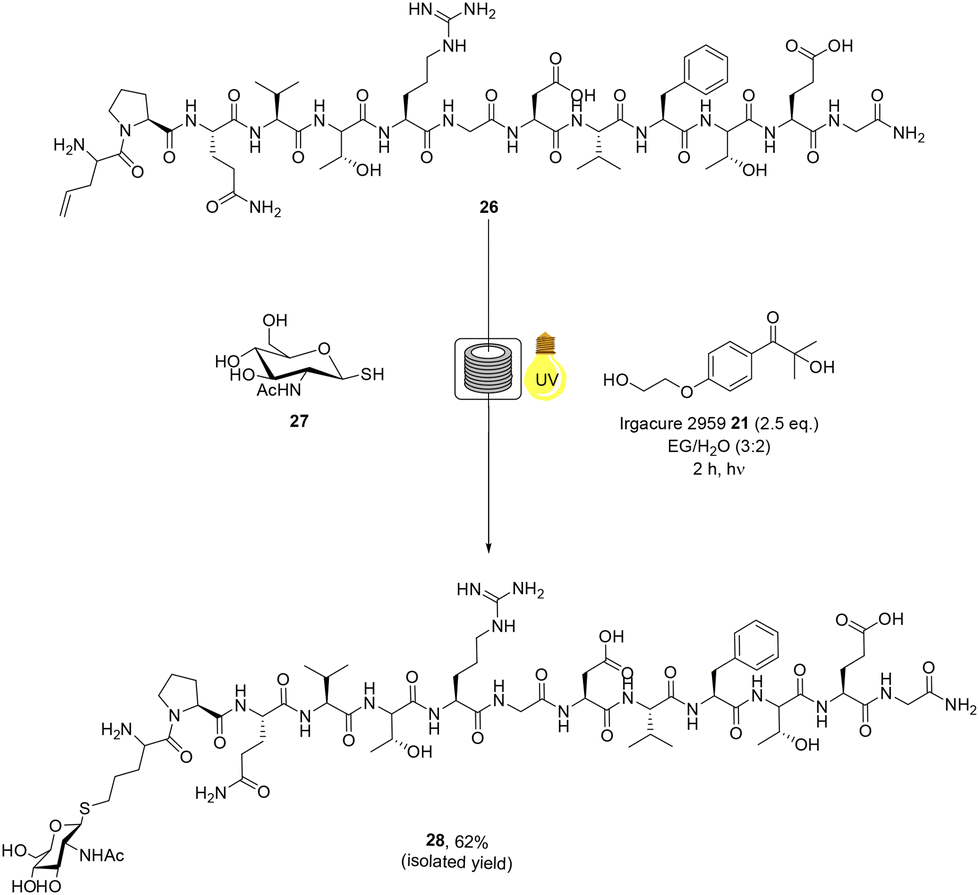 | ||
| Scheme 5 Radical mediated thiol–ene reaction of thiolated monosaccharide 27 and RGD peptide 26 under continuous flow to yield glycosylated peptide 28. | ||
Conclusions
This study has demonstrated the feasibility of radical-mediated thiol–ene chemistry on peptidic substrates under continuous flow utilising a variety of green solvents. Initial optimisation of the batch conditions of Conway and coworkers14 in THF/H2O demonstrated that a continuous flow methodology was suitable for accessing GSX conjugates. Subsequent investigation of reaction scope with a range of alkenes indicated that GSXs could be accessed in higher yields under continuous flow than the corresponding reactions in batch, thereby demonstrating the potential of continuous flow to increase the sustainability and green credentials of commonly utilised organic methodologies. A variety of novel biologically relevant GSX peptide derivatives were synthesised under continuous flow. DESs, bio-based and less harmful solvents were found to be amenable for radical mediated thiol–ene under continuous flow, thereby removing the need for toxic and environmentally damaging organic solvents. Finally, innovative use of water soluble photoinitiator Irgacure 2959 enabled the synthesis of water-soluble substrates under fully aqueous conditions, removing completely the requirement for organic solvents and significantly reducing the environmental impact of the thiol–ene bioconjugation methodology. The application of the described continuous flow strategy culminated in the glycosylation of three biologically important peptides, furnishing the respective glycoconjugates in good yield. The efficiency and generality of this green approach to radical mediated peptide bioconjugation, through the employment of a complimentary continuous flow set-up and considerate solvent selection, highlights the potential to increase the sustainability of pre-existing radical mediated processes in both academia and industry.Author contributions
I. R. G., J. T. M. and E. M. S. designed and carried out the experiments and wrote the manuscript; S. F. and D. F. O. S designed and carried out flow experiments, performed analysis and helped with writing; A. M and L. G. contributed to the experimental design, preparation of DESs and writing of the manuscript. N. O. synthesised peptides, contributed to experimental design and helped with writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the SFI Frontiers funding 19/FFP/6667.References
- K. J. Romero, M. S. Galliher, D. A. Pratt and C. R. J. Stephenson, Chem. Soc. Rev., 2018, 47, 7851–7866 RSC.
- M. S. Galliher, B. J. Roldan and C. R. J. Stephenson, Chem. Soc. Rev., 2021, 50, 10044–10057 RSC.
- F. Dénès, M. Pichowicz, G. Povie and P. Renaud, Chem. Rev., 2014, 114, 2587–2693 CrossRef PubMed.
- L. Markey, S. Giordani and E. M. Scanlan, J. Org. Chem., 2013, 78, 4270–4277 CrossRef CAS PubMed.
- N. C. Taylor, G. Hessman, H. B. Kramer and J. F. McGouran, Chem. Sci., 2020, 11, 2967–2972 RSC.
- A. B. Lowe, Polym. Chem., 2014, 5, 4820–4870 RSC.
- O. Busnel, F. Carreaux, B. Carboni, S. Pethe, S. V.-L. Goff, D. Mansuy and J.-L. Boucher, Bioorg. Med. Chem., 2005, 13, 2373–2379 CrossRef CAS.
- Y. Gao, A. Eguchi, K. Kakehi and Y. C. Lee, Org. Lett., 2004, 6, 3457–3460 CrossRef CAS.
- A. Leydet, C. Jeantet-Segonds, C. Bouchitté, C. Moullet, B. Boyer, J. P. Roque, M. Witvrouw, J. Este, R. Snoeck, G. Andrei and E. De Clercq, J. Med. Chem., 1997, 40, 350–356 CrossRef CAS.
- A. Dondoni, A. Massi, P. Nanni and A. Roda, Chem. – Eur. J., 2009, 15, 11444–11449 CrossRef CAS PubMed.
- S. R. Alexander, D. Lim, Z. Amso, M. A. Brimble and A. J. Fairbanks, Org. Biomol. Chem., 2017, 15, 2152–2156 RSC.
- M. Fiore, N. Berthet, A. Marra, E. Gillon, P. Dumy, A. Dondoni, A. Imberty and O. Renaudet, Org. Biomol. Chem., 2013, 11, 7113–7122 RSC.
- M. Fiore, M. Lo Conte, S. Pacifico, A. Marra and A. Dondoni, Tetrahedron Lett., 2011, 52, 444–447 CrossRef CAS.
- J. Healy, T. Rasmussen, S. Miller, I. R. Booth and S. J. Conway, Org. Chem. Front., 2016, 3, 439–446 RSC.
- G. Piccirillo, A. Pepe, E. Bedini and B. Bochicchio, Chem. – Eur. J., 2017, 23, 2648–2659 CrossRef CAS.
- B. Thomas, M. Fiore, G. C. Daskhan, N. Spinelli and O. Renaudet, Chem. Commun., 2015, 51, 5436–5439 RSC.
- S.-H. Yang, P. W. R. Harris, G. M. Williams and M. A. Brimble, Eur. J. Org. Chem., 2016, 2608–2616 CrossRef CAS.
- V. Yim, I. Kavianinia, M. K. Knottenbelt, S. A. Ferguson, G. M. Cook, S. Swift, A. Chakraborty, J. R. Allison, A. J. Cameron, P. W. R. Harris and M. A. Brimble, Chem. Sci., 2020, 11, 5759–5765 RSC.
- N. Floyd, B. Vijayakrishnan, J. R. Koeppe and B. G. Davis, Angew. Chem., Int. Ed., 2009, 48, 7798–7802 CrossRef CAS.
- A. Brinkø, C. Risinger, A. Lambert, O. Blixt, C. Grandjean and H. H. Jensen, Org. Lett., 2019, 21, 7544–7548 CrossRef.
- A. Marra, J. Dong, T. Ma, S. Giuntini, E. Crescenzo, L. Cerofolini, M. Martinucci, C. Luchinat, M. Fragai, C. Nativi and A. Dondoni, Chem. – Eur. J., 2018, 24, 18981–18987 CrossRef CAS PubMed.
- I. Mattsson, R. Sitdikov, A. C. M. Gunell, M. Lahtinen, T. Saloranta-Simell and R. Leino, RSC Adv., 2020, 10, 3960–3966 RSC.
- R. S. Bagul, M. M. Hosseini, T. C. Shiao and R. Roy, Can. J. Chem., 2017, 95, 975–983 CrossRef CAS.
- C. Caumes, E. Gillon, B. Legeret, C. Taillefumier, A. Imberty and S. Faure, Chem. Commun., 2015, 51, 12301–12304 RSC.
- M. Galán, E. Fuentes-Paniagua, F. J. de la Mata and R. Gómez, Organometallics, 2014, 33, 3977–3989 CrossRef.
- C. E. Gutierrez-Ulloa, M. Y. Buyanova, E. K. Apartsin, A. G. Venyaminova, F. J. de la Mata, M. Valiente and R. Gómez, Org. Biomol. Chem., 2017, 15, 7352–7364 RSC.
- M. Lo Conte, S. Staderini, A. Chambery, N. Berthet, P. Dumy, O. Renaudet, A. Marra and A. Dondoni, Org. Biomol. Chem., 2012, 10, 3269–3277 RSC.
- C. E. Peña-González, P. García-Broncano, M. F. Ottaviani, M. Cangiotti, A. Fattori, M. Hierro-Oliva, M. L. González-Martín, J. Pérez-Serrano, R. Gómez, M. Á. Muñoz-Fernández, J. Sánchez-Nieves and F. J. de la Mata, Chem. – Eur. J., 2016, 22, 2987–2999 CrossRef PubMed.
- É. Ujaczki, V. Stark-Rogel, M. Olbrich, M. Fuetsch and J. Backmann, Environ. Sci. Eur., 2022, 34, 101 CrossRef.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288–296 RSC.
- Y. Gu and F. Jérôme, Chem. Soc. Rev., 2013, 42, 9550–9570 RSC.
- M. D. Nolan, A. Mezzetta, L. Guazzelli and E. M. Scanlan, Green Chem., 2022, 24, 1456–1462 RSC.
- F. Ferlin, D. Lanari and L. Vaccaro, Green Chem., 2020, 22, 5937–5955 RSC.
- S. G. Newman and K. F. Jensen, Green Chem., 2013, 15, 1456–1472 RSC.
- D. Dallinger and C. O. Kappe, Curr. Opin. Green Sustainable Chem., 2017, 7, 6–12 CrossRef.
- P. Janssens, W. Debrouwer, K. Van Aken and K. Huvaere, ChemPhotoChem, 2018, 2, 884–889 CrossRef CAS.
- X. Wang, H. Liang, J. Jiang, Q. Wang, Y. Luo, P. Feng and C. Zhang, Green Chem., 2020, 22, 5722–5729 RSC.
- K. E. Konan, A. Abollé and F.-X. Felpin, Eur. J. Org. Chem., 2023, e202201055 CrossRef CAS.
- A. Steiner, R. C. Nelson, D. Dallinger and C. O. Kappe, Org. Process Res. Dev., 2022, 26, 2532–2539 CrossRef CAS PubMed.
- F. Wojcik, A. G. O'Brien, S. Götze, P. H. Seeberger and L. Hartmann, Chem. – Eur. J., 2013, 19, 3090–3098 CrossRef CAS PubMed.
- T. Homma and J. Fujii, Curr. Drug Metab., 2015, 16, 560–571 CrossRef CAS.
- B. H. Northrop and R. N. Coffey, J. Am. Chem. Soc., 2012, 134, 13804–13817 CrossRef CAS.
- L. Adhikari, N. E. Larm and G. A. Baker, ACS Sustainable Chem. Eng., 2020, 8, 14679–14689 CrossRef CAS.
- M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS.
- N. S. Sinclair, D. Poe, R. F. Savinell, E. J. Maginn and J. S. Wainright, J. Electrochem. Soc., 2021, 168, 020527 CrossRef CAS.
- J. R. Choi, K. W. Yong, J. Y. Choi and A. C. Cowie, BioTechniques, 2019, 66, 40–53 CrossRef CAS PubMed.
- A. Sun, X. He, X. Ji, D. Hu, M. Pan, L. Zhang and Z. Qian, Chin. Chem. Lett., 2021, 32, 2117–2126 CrossRef CAS.
- A. C. Okur, P. Erkoc and S. Kizilel, Colloids Surf., B, 2016, 147, 191–200 CrossRef CAS PubMed.
- N. Zhang, B. Ru, J. Hu, L. Xu, Q. Wan, W. Liu, W. Cai, T. Zhu, Z. Ji, R. Guo, L. Zhang, S. Li and X. Tong, J. Nanobiotechnol., 2023, 21, 77 CrossRef CAS.
- F. Li, A. Allahverdi, R. Yang, G. B. Lua, X. Zhang, Y. Cao, N. Korolev, L. Nordenskiold and C. F. Liu, Angew. Chem., Int. Ed., 2011, 50, 9611–9614 CrossRef CAS.
- F. Li, A. Allahverdi, R. Yang, G. B. J. Lua, X. Zhang, Y. Cao, N. Korolev, L. Nordenskiçld and C. Liu, Angew. Chem., 2011, 123, 9785–9788 CrossRef.
- S. Hagen, F. Marx, A. F. Ram and V. Meyer, Appl. Environ. Microbiol., 2007, 73(7), 2128–2134 CrossRef CAS.
- C. Yu-Pei, L. Yingying, C. Fangfang, W. Hongtan and Z. Shudi, Front. Microbiol., 2023, 14, 1664–302X Search PubMed.
- S. Hagen, F. Marx, A. F. Ram and V. Meyer, Appl. Environ. Microbiol., 2007, 73, 2128–2134 CrossRef CAS PubMed.
- Y. Wang, B. J. Bruno, S. Cornillie, J. M. Nogieira, D. Chen, T. E. Cheatham, C. S. Lim and D. H.-C. Chou, Chem. – Eur. J., 2017, 23, 7087 CrossRef CAS.
- A. Dondoni, A. Massi, P. Nanni and A. Roda, Chem. – Eur. J., 2009, 15, 11444–11449 CrossRef CAS PubMed.
- J. L. Sutcliffe-Goulden, M. J. O'Doherty, P. K. Marsden, I. R. Hart, J. F. Marshall and S. S. Bansal, Eur. J. Nucl. Med., 2002, 29, 754–759 CrossRef CAS PubMed.
- F. Wang, Y. Li, Y. Shen, A. Wang, S. Wang and T. Xie, Int. J. Mol. Sci., 2013, 14, 13447–13462 CrossRef.
- S. R. Alexander, D. Lim, Z. Amso, M. A. Brimble and A. J. Fairbanks, Org. Biomol. Chem., 2017, 15, 2152–2156 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ob00122b |
| This journal is © The Royal Society of Chemistry 2024 |