
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
π-Expanded azaullazines: synthesis of quinolino-azaullazines by Povarov reaction and cycloisomerisation†
Jonas
Polkaehn
a,
Richard
Thom
a,
Peter
Ehlers
 a,
Alexander
Villinger
a and
Peter
Langer
*ab
a,
Alexander
Villinger
a and
Peter
Langer
*ab
aInstitute of Chemistry, University Rostock, Albert-Einstein-Str. 3a, 18059 Rostock, Germany. E-mail: peter.langer@uni-rostock.de; Fax: +49 381 498 6412; Tel: +49 381 498 6410
bLeibniz Institute for Catalysis (LIKAT) at the University Rostock, Albert-Einstein-Str. 29a, 18059 Rostock, Germany
First published on 14th February 2024
Abstract
Doping and extension of polycyclic aromatic hydrocarbons (PAHs) by simple and efficient synthetic methods is of increased demand for the development of novel and improved organic electronics. Diarylindolizino[6,5,4,3-ija]quinolino[2,3-c][1,6]naphthyridines (quinolino-azaullazines) were prepared by combination of Pd catalyzed cross-coupling with Povarov and cycloisomerisation reactions. The products contain an electron-rich ullazine and an electron-poor quinoline moiety and show intramolecular charge transfer properties that can be tuned by the substitution pattern. The optical properties were studied experimentally and further elaborated by (TD)DFT calculations.
Introduction
Indolizino[6,5,4,3-ija]quinolines (ullazines), first synthesized and studied by Balli et al. in 1983,1 are of considerable current interest in the field of materials science, due to their application as organic light emitting diodes (OLEDs), organic field-effect transistors (OFETs) or dye-sensitized solar cells (DSSC).2,3 Due to the electronic influence of the pyrrole moiety, ullazines represent electron-rich molecules that exhibit strong intramolecular charge transfer (ICT).2 Several synthetic methods have been developed in recent decades to further modify the properties of ullazines. Various approaches are based on the modification of the ullazine core structure by doping with heteroatoms.4,5 Other methods are focused on the variation of the substitution pattern and substituents.6 Furthermore, the expansion of the π-system represents a promising approach to modify the optical and electronic properties of ullazines (Fig. 1).7,8–15 In 2012, Ren and co-workers published the synthesis of dibenzoullazines by Friedel–Crafts arylation of aryl triazenes.8 Similarly substituted dibenzoullazines were obtained by Pd-catalyzed twofold annulation reactions of arynes in 2021.9 Müllen et al. and Nozaki et al. reported in 2015 the synthesis of π-extended ullazines by cycloaddition of polycyclic aromatic azomethine ylides.10,11 The most common strategy for the synthesis of π-expanded ullazines is based on employment of azomethine ylides. Other examples have been reported by Feng et al.,12 Nozaki et al.13 and Stępień et al.14 Moreover, Stępień et al. were able to synthesize such compounds by cyclodehydrohalogenation of α,α-disubstituted N-arylpyrroles.14 Using photochemical cyclodehydrochlorination, Morin and coworkers synthesized π-extended ullazines annulated to two pyridine or two thiophene moieties in 3,4- and 8,9-position.15 | ||
| Fig. 1 Examples of π-expanded ullazines. | ||
A well-known methodology for the preparation of polycyclic aromatic heterocycles is the Povarov reaction, a hetero-Diels–Alder reaction that was developed in 1967.16 In recent years, there has been renewed interest in this methodology.17 In 2020, Jana et al. reported the application of the Povarov reaction for the synthesis of dibenzo[a,c]acridines from 2′-alkynylbiaryl-2-carbaldehydes using catalytic amounts of FeCl3 (Fig. 2).18 Recently, we reported the synthesis of benzo[j]naphtho[2,1,8-def][2,7]phenanthrolines, π-expanded aza-pyrenes, by combination of a Povarov with a cycloisomerization reaction.19 Herein, we wish to report the synthesis of what are, to the best of our knowledge, hitherto unknown diarylindolizino[6,5,4,3-ija]quinolino[2,3-c][1,6]naphthyridines by combination of Povarov and cycloisomerization reactions. The incorporation of an electron-rich, five-membered pyrrole ring will alter the optoelectronic properties of this polycyclic aromatic scaffold and will be studied in detail by experimental and theoretical methods. In addition, the non-symmetric π-expansion of azaullazines will be studied in comparison to previously reported symmetric dibenzoullazines.
 | ||
| Fig. 2 Recent examples of Povarov reactions. | ||
Synthesis
Vilsmeier–Haack reaction of 3,5-dibromo-4-(1H-pyrrol-1-yl)pyridine (1)5 afforded formylated products 2a and 2b in 33% and 62% isolated yields, respectively (Scheme 1). During the optimization of, different reaction conditions were tested (ESI, Table 1†). It turned out that the use of 2 eq. of POCl3 in DMF at 100 °C and 3 h reaction time provided the best results. The regiosiomeric mixture could be readily separated by column chromatography. Therefore, although the desired regioisomer 2a was formed as the minor product, the method was useful in our hands to prepare sufficient material to continue the synthesis. | ||
| Scheme 1 Synthesis of 2a and 2b; (i): POCl3 (2.0 eq.), DMF, 100 °C, 3 h. | ||
With 2a in hand, an optimization of the following double Sonogashira reaction was carried out for the synthesis of 3a (ESI, Table 2†). The best yields were obtained by using PdCl2(PPh3)2 (0.05 eq.) as the catalyst and cataCXium A (0.1 eq.) as the ligand. These conditions were then applied for the synthesis of Sonogashira products 3a–e were obtained in moderate to very good yields (Scheme 2).
 | ||
| Scheme 2 Synthesis of 3a–e; (i): alkyne (3 eq.), PdCl2(PPh3)2 (0.05 eq.), cataCXium A (0.1 eq.), CuI (0.05 eq.), HNiPr2, 1,4-dioxane, 90 °C, 24 h. | ||
Subsequently, the Povarov reaction of 3a with aniline was studied to give product 4 (Table 1). We initially applied the conditions reported by Jana et al. (FeCl3, toluene, 100 °C) and obtained 4 in a very good yield of 86%.18 In contrast, employment of In(OTf)3 gave a diminished yield (41%) and Brønsted acids were completely ineffective for this reaction.
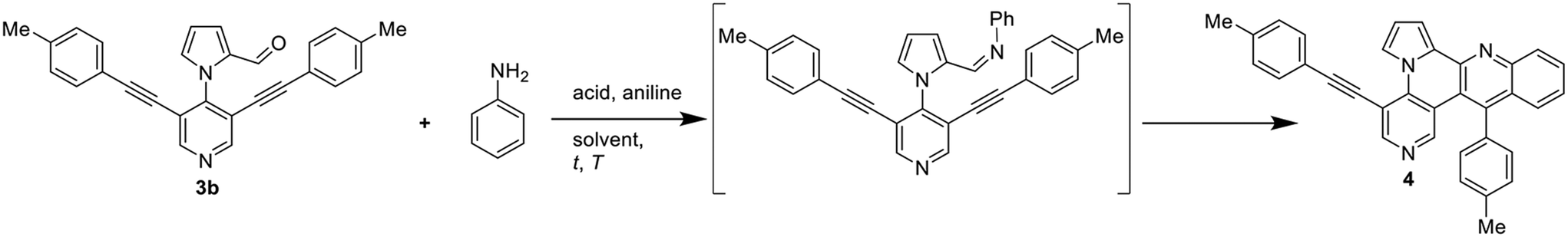
Subsequently, we studied the cycloisomerization of 4 to give quinolino-azaullazine 5a and tested p-toluenesulfonic acid (p-TsOH) and methanesulfonic acid (MsOH) which were previously used for related reactions (Table 2).5,20 Employment of p-TsOH gave a better yield of 62% as compared to MsOH under the same concentration of acid (30 eq.). Reducing the amount of acid from 30 to 20 equivalents resulted in a slightly improved yield of 67%, further reduction to 10 equivalents resulted in diminished yield (43%). During the purification of 4, we observed an alkyne-carbonyl-metathesis (ACM) reaction as competitive side reaction of the Povorav reaction.21 Purification from this side-product was very difficult and, hence, we attempted to directly convert 3b to 5a in a one-pot procedure without isolation of Povarov product 4. To our delight, this idea proved to be successful and allowed to isolate the desired quinolino-azaullazine 5a in 47% overall yield.

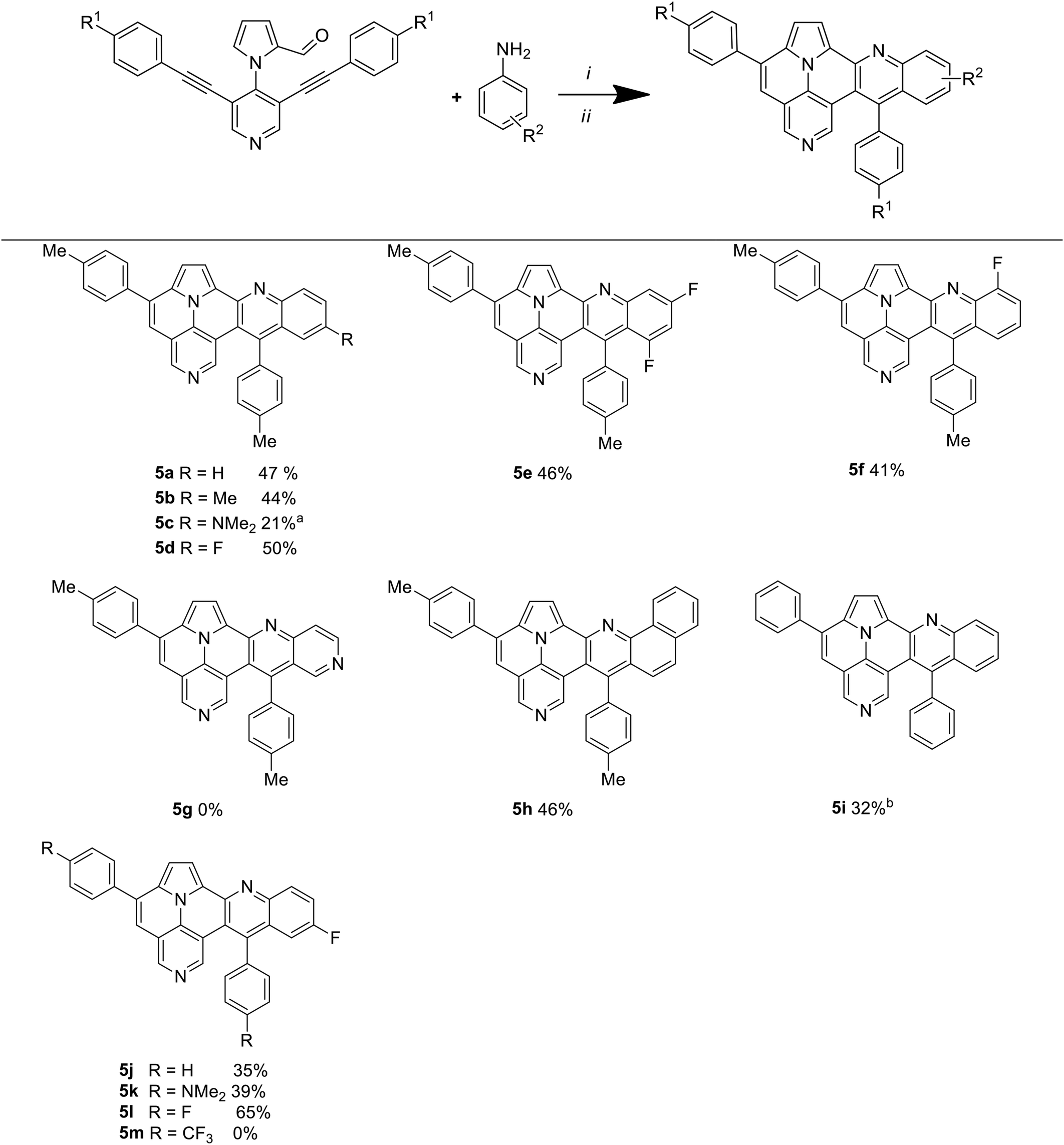
Next, we were interested in the preparative scope of our methodology. Hence, the substitution pattern of the arylacetylene and of the aniline were varied (Table 3). The reaction of 3a–e with various anilines afforded quinolino-azaullazines 5a–f,h–l in mostly moderate to good yields. All yields refer to a one-pot process in which four new bonds were formed in a single step without the need of purification of intermediates (formation of the imine, Povarov reaction and cycloisomerization). The reaction of aniline with 3e, containing the strongly electron donating NMe2 substituent, gave product 5c in a reduced yield of 21%. However, the reaction of 3e with reaction with 4-fluoroaniline afforded the corresponding product 5k in a better yield of 39%. The best yield (product 5l, 65%) was observed for the reaction of 3c, containing an electron-withdrawing fluoride substituent, with 4-fluoroaniline. Heterocyclic 4-aminopyridine did not undergo a Povarov reaction (5g). The use of 1-naphthylamine provided product 5h in good yield. Comparison of the yields of products derived from different substituents attached to the alkyne (5a, 5j and 5k) did not reveal a significant influence of the substitution pattern, except from the fact that the reaction of 4-fluoroaniline with 3d, containing a trifluoromethyl group, proved to be entirely unsuccessful. In summary, the aniline has a higher influence on the yield that the substituent attached to the alkyne.
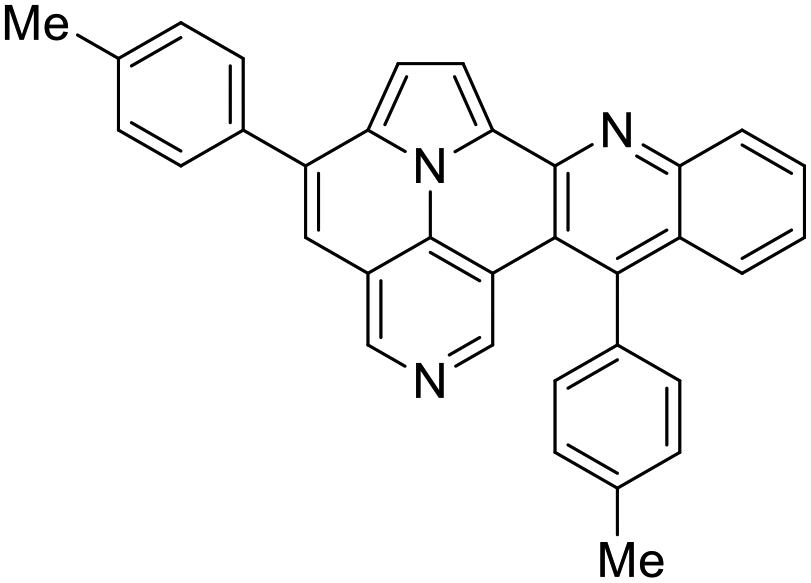
The structure of 5d was independently confirmed by X-ray crystal structure analysis (Fig. 3). Crystallization was carried out in a mixture of heptane and dichloromethane (DCM). For a better visualization, the co-crystallized DCM has been removed. Both p-tolyl residues are twisted out of plane of the core structure by dihedral angles of 45° and 78°. Moreover, the crystal lattice shows a slipped antiparallel π–π-stacking with a spacing of 3.44 Å and 3.41 Å between the core structures, respectively.
 | ||
| Fig. 3 X-ray structures of 5d. | ||
Physical properties
The photophysical properties of selected derivatives were determined by steady-state absorption and photoluminescence spectra (PL) (Fig. 4). The UV/VIS-spectra indicate strong absorption bands at approx. 250–300 nm and weaker bands at approx. 450–500 nm for all compounds. Comparing the different aniline-derived moieties, a slight influence of the substituents is evident. Thus, 5a and 5d show very similar absorption values and shapes of the absorption bands while π-extended compound 5h exhibits somewhat higher extinction coefficients. In contrast, 5c has a visibly lower absorption intensity accompanied with loss of fine structure. Additionally, a red shift of the maxima is observed, which can be attributed to the strong donor properties of the N,N-dimethylamio unit. Compounds 5d, 5j and 5k provide almost identical absorption spectra, due to the twisted orientation of the phenyl rings. Incorporation of a strong donor substituent, as present in 5k, leads to an altered absorption spectrum as well as loss of fine structure. The redshift, on the other hand, is not as pronounced as for 5c. | ||
| Fig. 4 UV/Vis- (top/bottom, left) and PL-spectra (top/bottom, right, λex = 450 nm) of shown compounds in DCM (c = 10−5 M) at 20 °C. | ||
The PL-spectra, 5d, 5j and 5k also show identical band structures and an emission maximum at 525 nm. Only donor-substituted derivative 5k exhibits a red shift by 14 nm as compared to the other derivatives. Similar to the absorption properties, variation of the substituents directly attached to the heterocyclic core structure alters the PL-spectra to a greater extent. Compared to unsubstituted derivative 5a (λem = 515 nm), N,N-dimethylamino-substituted compound 5c shows the strongest red shift (λem = 539 nm), followed by fluorine-substituted 5d (λem = 525 nm), while the strongest blue shift occurs for 5h (λem = 505 nm).
Most compounds show quantum yields between 19% and 28%. The highest value was determined for 5h (Φ = 0.35). In contrast, the two N,N-dimethylamino-compounds 5c (Φ = 0.19) and 5k (Φ = 0.20) show the lowest quantum yields. The spectroscopic data are presented in Table 4.
| 5a | 5c | 5d | 5h | 5j | 5k | 5l | |
|---|---|---|---|---|---|---|---|
| a Determined from the intersection of the normalized absorption and emission spectra. b Fluorescence standard: quinine hemisulfate monohydrate in 0.05 M H2SO4 (Φ = 0.52).22 | |||||||
| λ 1,abs [nm] | 471 | 503 | 477 | 470 | 477 | 482 | 480 |
| ε λ 1 [104 L mol−1 cm−1] | 1.1 | 0.7 | 0.8 | 1.6 | 0.8 | 0.7 | 0.9 |
| λ 2,abs [nm] | 448 | 480 | 454 | 447 | 477 | 455 | 455 |
| ε λ 2 [104 L mol−1 cm−1] | 1.0 | 0.7 | 0.8 | 1.3 | 0.8 | 0.7 | 0.9 |
| λ 1,em [nm] | 515 | 539 | 525 | 505 | 524 | 539 | 524 |
E
optg
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) [eV]
[eV] |
2.56 | 2.42 | 2.53 | 2.59 | 2.53 | 2.50 | 2.53 |
| Φ | 0.26 | 0.19 | 0.27 | 0.35 | 0.25 | 0.20 | 0.28 |
Since the absorption and emission spectra of 5c and 5k indicated the strongest influence of the substituents on the core structure, these compounds as well as reference compound 5a were additionally investigated by time-dependent density functional theory (TD-DFT) calculations using Gaussian 09 to get insight into transition characters.23 The results show that the S1 ← S0 excitation for 5a and 5c is described by a HOMO → LUMO transition with high oscillator strength. The same applies to 5k, but with additional minor contribution of HOMO−1 → LUMO and lower oscillator strength for this transition. However, the S2 ← S0 transition, which for 5k again consisted of a HOMO → LUMO and HOMO−1 → LUMO transition, has a significantly higher oscillator strength as compared to 5a and 5c. Hence, the orbital contribution of these transitions is reversed. In agreement with the experimental results, TD-DFT calculations reveal the greatest impact for derivative 5k, containing a strong π-donor attached to the heterocyclic core structure.
Comparison of non-symmetric quinolino-azaullazine 5a with symmetric dibenzoullazines of Gao and Morin and of isoelectronic quinolino-azapyrene reveal further insights into the optical properties of 5a (Table 5).9,15,19 Absorption and emission maxima of 5a are both strongly, bathochromically shifted compared to all three structurally related compounds. The quantum yield of 5a is reduced to approximately half of the value of quinolino-azapyrene, while extinction coefficients are rather similar for both compounds. A similar trend is observed for 2-aza-pyrene and 2-aza-ullazine without fused quinoline moieties.5,20 In a smaller extend, quantum yield of 5a is also reduced compared to symmetric dibenzoullazine reported by Gao and co-workers.9 However, in impact on the quantum yield of 5a by the presence of additional tolyl groups and hence increased non-radiative decay compared to dibenzoullazine can currently not ruled out.



To get an understanding of the redox properties, compound 5a was studied by cyclic voltammetry (CV) (Fig. S1 and S2†). Compound 5a possesses an irreversible oxidation potential at 0.72 V with an onset potential of 0.61 V, as well as a quasi-reversible oxidation potential at 1.09 V. An irreversible reduction potential was detected at −2.09 V with an onset potential of 1.90 V and a second reduction potential at 3.11 V. The HOMO energy was calculated from the onset potential and is with −5.41 eV somewhat higher than that of previously reported quinolino-azapyrene 6 which is a result of the donor-capacity of the ullazine substructure of 5a.19 The determined LUMO energy is −2.90 eV.
We performed density functional theory (DFT) calculations to obtain further insights into the electronic structures and transition characters. The calculations were performed for compounds 5a, 5c, 5i and 5k. Quinolino-azapyrene 6 and quinolino-ullazine 7 were additionally studied to compare the impact of different core structures (Fig. 5). The calculated HOMO–LUMO gap of the quinolino-azapyrene system is considerably larger, which is mainly due to increased HOMO energies, whereas the LUMO-energies are largely unchanged, highlighting the donating ability of the involved ullazine and azaullazine moiety. Similar HOMO–LUMO-gap energies were determined both for ullazine and azaullazine based structures 5a, 5i and 7. However, an improved stabilization of both HOMO and LUMO orbitals is observed in case of azaullazines 5a and 5i with respect to ullazine 7, which is obviously a result of the presence of the additional nitrogen atom. The NMe2-groups attached to the heterocyclic core structure (5c) as well as the NMe2-groups attached to the phenyl ring (5k) lead to reduced band gaps and elevated HOMO and LUMO energies. A more distinct effect is observed for 5c as compared to 5k. However, HOMO and LUMO energies are more destabilized by incorporation of one NMe2-group directly to the core structure (5c) than it does for 5k with NMe2-groups attached to each of the two phenyl rings. Furthermore, an ICT character for 5k can be assumed from its frontier orbitals, derived from its certain push–pull substitution pattern and its optical properties.
 | ||
| Fig. 5 Frontier orbitals of selected compounds and energy levels calculated at the B3LYP/6-31G(d,p) level of theory within IEFPCM in DCM. | ||
The potential occurrence of ICT properties was further validated by solvatochromic studies. Hence, the optical properties in solvents of different polarity were studied for NMe2-containing compounds 5c and 5k, as well as for 5a (Fig. 6). All absorption spectra show similar behavior in nonpolar cyclohexane. A definite fine structure of the absorption bands, as typical for various acenes24 and acridines,25 can be observed for all investigated derivatives. This fine structure is lost when the polarity of the solvents increases. This indicates the presence of an ICT effect in all three derivatives 5a,c,k and suggests that the ICT character is already present in the heterocyclic core structure, regardless of the substituent. This assumption is supported by the emission spectra, in which all compounds show two maxima and one shoulder at higher wavelength in cyclohexane. In more polar solvents, the emissions appear broadened with a single maximum. Furthermore, a bathochromic shift of emission maxima is visible for these solvents. The shift is ∼40 nm for 5a and for 5c between cyclohexane and ethanol, indicating that the NMe2-group of 5c has only negligible effect on the ICT character of the core structure. In contrast, a stronger shift of 59 nm can be observed for 5k. For all compounds, a decrease of the emission in more polar solvents is observed and accompanied by a reduction of their quantum yields (ESI, Table 5†). However, 5k again behaves quite differently from 5a and 5c and shows only very weak fluorescence in acetonitrile and ethanol with quantum yields ≤ 1%, what might be explained by the occurrence of a twisted intramolecular charge transfer (TICT). This effect appears in polar acetonitrile and ethanol, suggesting that the polarity of the solvent may sufficient to stabilize a TICT. This twisted structure may explain the quenching of the fluorescence through the radiation-free TICT-relaxation.26
 | ||
| Fig. 6 Top: UV/Vis-spectra of 5a (left), 5c (middle), 5k (right); Bottom: PL-spectra 5a (left), 5c (middle), 5k (right), t, λex = 340 nm; spectra measured with toluene, dichloromethane, acetonitrile and ethanol (c = 10−5 M at 20 °C). | ||
Further investigations of the ICT-character were performed by calculations of the dipole moments of the ground (S0) and excited state (S1) of 5a, 5c and 5k. These calculations confirm the results of the studies related to solvatochromism. Compound 5k (μS0 = 4.4193; μS1 = 26.2043) indicates the strongest increase in the dipole moment, followed by 5a (μS0 = 1.0280; μS1 = 10.6564) and 5c (μS0 = 4.1596; μS1 = 6.0825).
In addition, we performed protonation studies with trifluoromethanesulfonic acid (TFA) in DCM for compound 5a. As shown in Fig. 7, protonation of 5a results in a bathochromic shift in the absorption and emission spectra. The first absorption maximum shifts from 471 nm for the free base 5a to 534 nm for protonated species and an isosbestic point at a wavelength of 482 nm can be detected. The PL-spectra show similar results: the emission maximum shifts from 515 nm to 586 nm, with the first emission maximum decreasing and the second increasing simultaneously. In addition, protonation results, beside the bathochromic shift, also in a decrease of fluorescence. However, changes and shapes of the absorption and PL spectra during the protonation experiments prohibit conclusion whether only one or both pyridinic nitrogen are protonated.
 | ||
| Fig. 7 Absorption and PL spectra of 5a in dependence of the TFA concentration. | ||
Finally, we performed nuclear-independent chemical shift (NICS) calculations for the quinolino-azaullazine core structure (5′) to gain insight into the aromatic behavior of the compound. Therefore, NICS(1.25)zz values were calculated for each ring to investigate the local aromaticity.27 Furthermore, ring currents were calculated using the BC-Wizard program developed by Gershoni-Poranne et al.28 The calculations were performed in comparison to quinolino-azapyrene 6′ and quinolino-ullazine 7′ (Fig. 8).19 All three compounds display a global diatropic ring current, as well as negative NICSzz values in all rings. In addition, all studied compounds exhibit two semiglobal ring currents, one in the quinoline moiety, the other in the benzo-isoquinoline or pyrrolo-quinoline moiety. However, the connecting ring of both moieties is not involved in any semiglobal ring current, thus the molecules consist of two aromatic subunits connected by a ring with non-aromaticity in case of 5′ and 7′, and low aromaticity for 6′, which is confirmed by the NICSzz values. In the two ullazine structures, this ring shows even lower aromaticity than in the aza-pyrene structure. In addition, the ring current maps show that there is a difference mainly in the benzoisoquinoline or pyrroloquinoline moiety, which is due to the different molecular structure. Thus, 6′ shows a stronger semiglobal current, whereas 5′ and 7′ exhibit a stronger local ring current focused on the pyrrole unit. Comparing the structures of ullazine 7′ and azaullazine 5′, only negligible differences are noticed, as both have almost the same ring currents and similar NICSzz values. Hence, the impact on aromaticity by the presence of an additional nitrogen of 7′ as compared to 5′ is very small. However, a pronounced influence is observed by incorporation of an ullazine as compared to a related azapyrene sub-structure.
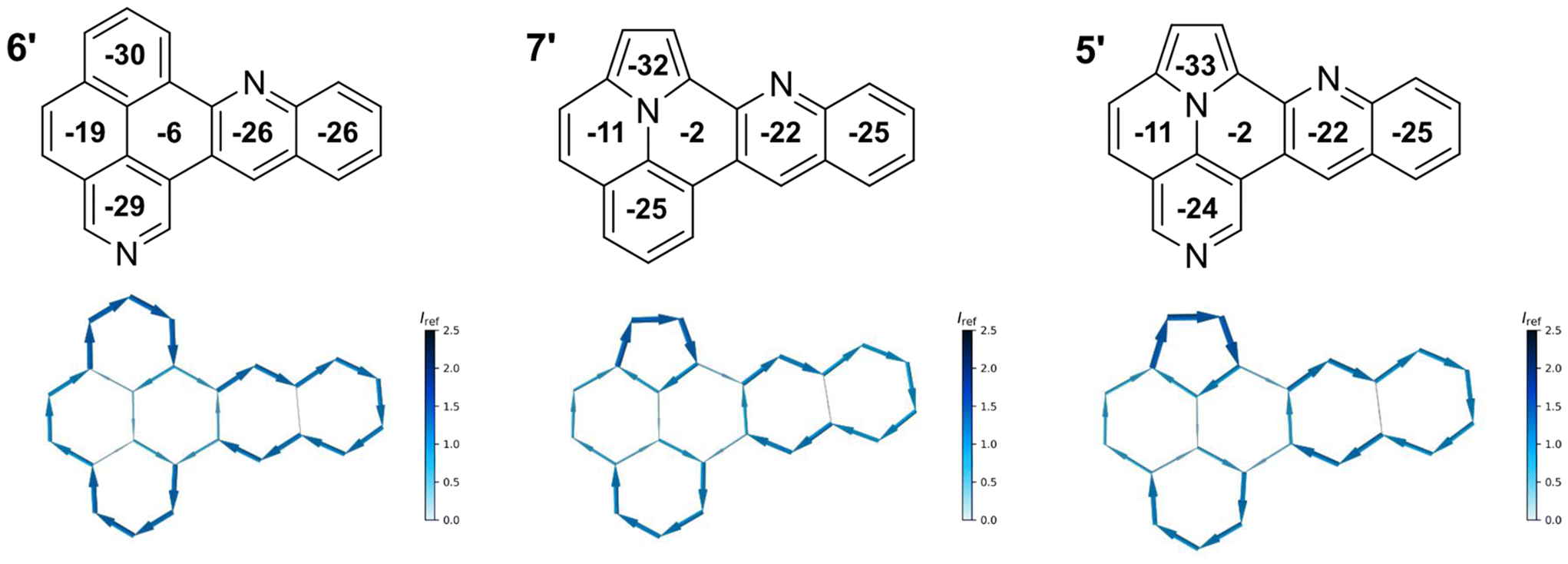 | ||
| Fig. 8 NICS calculations for benzo[j]naphtho[2,1,8-def][2,7]phenanthroline (6′); indolizino[6,5,4,3-ija]quinolino[2,3-c]quinoline (7′); indolizino[6,5,4,3-ija]quinolino[2,3-c][1,6]naphthyridine (5′). Top: Scheme of molecule with respective NICS(1.25)zz values in the center of each ring. Bottom: NICS2BC graphs (current was calculated form NICS(1.25)zz strength relative to Iref (ring current of benzene, 11.5 nA T−1)).28 | ||
Conclusion
Hitherto unknown quinolino-azaullazines were prepared by combination of Pd catalyzed cross-coupling with Povarov reactions. The synthetic strategy provides a convenient access to various functionalized molecules, which allowed for a fine-tuning of the optical properties. Especially the variation of the aniline component in the Povarov reaction has an important impact on the optoelectronic properties of the products. All investigated compounds exhibit strong absorption in visible light as well as greenish fluorescence with quantum yields between 19% and 35%. Noteworthy is the introduction of strongly donating NMe2-substituents, which cause extensive alternation of the optical properties not only attached to the core structure, but also on the phenyl substituents. Solvatochromism studies revealed that an ICT character is already present in the core structure, independently from the substitution pattern. However, the presence of a NMe2-group at the phenyl groups even improved the ICT properties, which suggests the presence of TICT. Further modification of the optical properties was achieved by protonation, which causes a red shift of both absorption and emission. Cyclic voltammetry investigation revealed a more facile oxidation as compared to quinolino-azapyrenes, due to the strong donor capacity of the azaullazine core structure. These observations were confirmed by DFT calculations. NICS calculations disclosed that the novel aza-PAHs prepared have a weak global ring current and can be best described by the presence of two independent aromatic systems consisting of a pyrroloquinoline and a quinoline unit connected by a non-aromatic ring. Further studies will focus on the synthesis of related heteroatom doped PAHs by means of the Povarov reaction.Experimental section
General information
The nuclear magnetic resonance spectra (1H/13C/19F NMR) were recorded on a Bruker AVANCE 300 III, 250 II, or 500. The analyzed chemical shifts δ are referenced to residual solvents signals of the deuterated solvents CDCl3 (δ = 7.26 ppm/77.0 ppm). Multiplicities due to spin–spin correlation are reported as follows, s = singlet, d = doublet, dd = double doublet, ddd = doublets of doublets, m = multiplet, and further described through their coupling constants J. Infrared spectra (IR) were measured as attenuated total reflection (ATR) experiments with a Nicolet 380 FT-IR spectrometer. The signals have been characterized through their wave numbers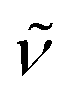 and their corresponding absorption as very strong (vs), strong (s), medium (m) or weak (w). UV/Vis spectra were recorded on a Cary 60 UV-vis spectrophotometer and emission spectra with an Agilent Cary Eclipse fluorescence spectrophotometer. Cyclic voltammograms were measured at room temperature in DCM or THF (c = 10−3 M) with 0.1 M n-Bu4NPF6 as a supporting electrolyte, glassy carbon working electrode, ANE2 (Ag/AgNO3 0.01 M in CH3CN) as reference electrode and Pt counter-electrode (0.5 mm diameter platinum wire) with ferrocene (c = 10−3 M, in CH3CN) as an external standard at a scan rate of 100 mV s−1 or 200 mV s−1. The potentiostat used was a PalmSense EmStat 3 blue or a Parstat 4000 from Ametek. The working electrode is a 3 mm diameter (length 80, 6.35 mm outer diameter) glassy carbon disk electrode in a KeI-F coating that was polished on a polishing pad in aqueous alumina slurry (0.03 μm alumina powder). The solvents were deoxygenated by purging with argon. The potential is given vs. Fc/Fc+. The direction of scan is reductive with a starting potential of 1.5 V and a switching potential of −1.5 V for DCM and for THF a starting potential of 0 V and a switching potential of −3.2 V. CVs are plotted using the IUPAC. Basic and high-resolution mass spectra (MS/HRMS) were measured on instruments which are paired with a preceding gas chromatograph (GC) or liquid chromatograph (LC). The samples have been ionized through electron impact ionization (EI) on an Agilent 6890/5973 or Agilent 7890/5977 GC-MS equipped with a HP-5 capillary column using helium carrier gas or by applying electron spray ionization (ESI) on an Agilent 1200/6210 Time-of-Flight (TOF) LC-MS. Melting points (mp) were determined by a Micro-Hot-Stage GalenTM III Cambridge Instruments and are not corrected. X-ray single-crystal structure analysis was performed on a Bruker Apex Kappa-II CCD diffractometer.
and their corresponding absorption as very strong (vs), strong (s), medium (m) or weak (w). UV/Vis spectra were recorded on a Cary 60 UV-vis spectrophotometer and emission spectra with an Agilent Cary Eclipse fluorescence spectrophotometer. Cyclic voltammograms were measured at room temperature in DCM or THF (c = 10−3 M) with 0.1 M n-Bu4NPF6 as a supporting electrolyte, glassy carbon working electrode, ANE2 (Ag/AgNO3 0.01 M in CH3CN) as reference electrode and Pt counter-electrode (0.5 mm diameter platinum wire) with ferrocene (c = 10−3 M, in CH3CN) as an external standard at a scan rate of 100 mV s−1 or 200 mV s−1. The potentiostat used was a PalmSense EmStat 3 blue or a Parstat 4000 from Ametek. The working electrode is a 3 mm diameter (length 80, 6.35 mm outer diameter) glassy carbon disk electrode in a KeI-F coating that was polished on a polishing pad in aqueous alumina slurry (0.03 μm alumina powder). The solvents were deoxygenated by purging with argon. The potential is given vs. Fc/Fc+. The direction of scan is reductive with a starting potential of 1.5 V and a switching potential of −1.5 V for DCM and for THF a starting potential of 0 V and a switching potential of −3.2 V. CVs are plotted using the IUPAC. Basic and high-resolution mass spectra (MS/HRMS) were measured on instruments which are paired with a preceding gas chromatograph (GC) or liquid chromatograph (LC). The samples have been ionized through electron impact ionization (EI) on an Agilent 6890/5973 or Agilent 7890/5977 GC-MS equipped with a HP-5 capillary column using helium carrier gas or by applying electron spray ionization (ESI) on an Agilent 1200/6210 Time-of-Flight (TOF) LC-MS. Melting points (mp) were determined by a Micro-Hot-Stage GalenTM III Cambridge Instruments and are not corrected. X-ray single-crystal structure analysis was performed on a Bruker Apex Kappa-II CCD diffractometer.
Analytical data
:1). 1H NMR (300 MHz, CDCl3) δ = 9.49 (d, 4J = 1.0 Hz, 1H), 8.69 (s, 2H), 7.09 (dd, 3J = 3.9 Hz, 4J = 1.6 Hz, 1H), 6.81 (ddd, 3J = 2.7 Hz, 4J = 1.6 Hz, 4J = 1.0 Hz, 1H), 6.49 (dd, 3J = 4.0 Hz, 3J = 2.7 Hz, 1H). 13C NMR (75 MHz, CDCl3) δ = 178.2 (CHO), 151.3 (CH), 145.9, 131.7 (C), 129.1, 123.6 (CH), 121.2 (C), 112.3 (CH). IR (ATR, cm−1):  , 1484 (m), 1399 (m), 1356 (s), 1199 (m), 1037 (m), 999 (m), 886 (m), 779 (s), 750 (s), 715 (m), 610 (m), 577 (s). MS (EI, 70 eV): m/z (%) = 330 ([M]+, 1), 252 (11), 251 (98), 250 (15), 249 (100), 170 (21), 142 (8), 130 (5), 115 (9), 114 (7). HRMS (EI): calculated for C10H679Br2N2O ([M]+) 327.88414, found 327.88451; calculated for C10H679Br81BrN2O ([M]+) 329.88209, found 329.88255; calculated for C10H681Br2N2O ([M]+) 331.88005, found 331.88075.
:1 heptane:ethyl acetate) as a colourless solid (0.673 g, 62%). Mp 100–110 °C. Rf 0.14 (heptane/ethyl acetate 3:1). 1H NMR (300 MHz, CDCl3) δ = 9.89 (dd, 4J = 0.4 Hz, 1H), 8.81 (s, 2H), 7.34 (dd, 4J = 2.1 Hz, 4J = 1.6 Hz, 1H), 6.87 (ddd, 3J = 3.1 Hz, 4J = 1.6 Hz, 4J = 0.4 Hz, 1H), 6.74 (ddd, 3J = 3.0 Hz, 4J = 2.1 Hz, 4J = 0.7 Hz, 1H). 13C NMR (75 MHz, CDCl3) δ = 185.2 (CHO), 152.0 (CH), 145.1 (C), 128.9 (CH), 128.3 (C), 123.6 (CH), 120.5 (C), 109.3 (CH). IR (ATR, cm−1):
, 1484 (m), 1399 (m), 1356 (s), 1199 (m), 1037 (m), 999 (m), 886 (m), 779 (s), 750 (s), 715 (m), 610 (m), 577 (s). MS (EI, 70 eV): m/z (%) = 330 ([M]+, 1), 252 (11), 251 (98), 250 (15), 249 (100), 170 (21), 142 (8), 130 (5), 115 (9), 114 (7). HRMS (EI): calculated for C10H679Br2N2O ([M]+) 327.88414, found 327.88451; calculated for C10H679Br81BrN2O ([M]+) 329.88209, found 329.88255; calculated for C10H681Br2N2O ([M]+) 331.88005, found 331.88075.
:1 heptane:ethyl acetate) as a colourless solid (0.673 g, 62%). Mp 100–110 °C. Rf 0.14 (heptane/ethyl acetate 3:1). 1H NMR (300 MHz, CDCl3) δ = 9.89 (dd, 4J = 0.4 Hz, 1H), 8.81 (s, 2H), 7.34 (dd, 4J = 2.1 Hz, 4J = 1.6 Hz, 1H), 6.87 (ddd, 3J = 3.1 Hz, 4J = 1.6 Hz, 4J = 0.4 Hz, 1H), 6.74 (ddd, 3J = 3.0 Hz, 4J = 2.1 Hz, 4J = 0.7 Hz, 1H). 13C NMR (75 MHz, CDCl3) δ = 185.2 (CHO), 152.0 (CH), 145.1 (C), 128.9 (CH), 128.3 (C), 123.6 (CH), 120.5 (C), 109.3 (CH). IR (ATR, cm−1): 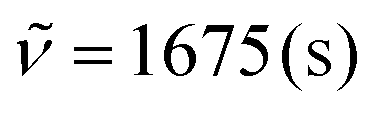 , 1508 (s), 1467 (s), 1294 (m), 1257 (m), 1210 (m), 1152 (m), 1084 (m), 1041 (s), 816 (m), 732 (s), 618 (m), 591 (m). MS (EI, 70 eV): m/z (%) = 332 (34), 331 (54), 330 ([M]+, 69), 329 (100), 328 (36), 327 (50), 142 (7), 141 (10), 130 (6), 128 (6), 114 (7). HRMS (ESI-TOF): calculated for C10H7Br2N2O ([M + H]+) 328.8925, found 328.8931.
:1). 1H NMR (300 MHz, CDCl3) δ = 9.60 (d, 4J = 0.8 Hz, 1H), 8.80 (s, 2H), 7.34–7.28 (m, 10H), 7.24 (dd, 3J = 3.9 Hz, 4J = 1.6 Hz, 1H), 7.19 (ddd, 3J = 2.6 Hz, 4J = 1.6 Hz, 4J = 0.9 Hz, 1H), 6.58 (dd, 3J = 3.9 Hz, 3J = 2.7 Hz, 1H). 13C NMR (75 MHz, CDCl3) δ = 178.2 (CHO), 151.8 (CH), 148.2, 132.9 (C), 131.6, 130.5, 129.2, 128.3, 122.4 (CH), 121.7, 118.8 (C), 111.3 (CH), 97.2, 81.6 (C
, 1508 (s), 1467 (s), 1294 (m), 1257 (m), 1210 (m), 1152 (m), 1084 (m), 1041 (s), 816 (m), 732 (s), 618 (m), 591 (m). MS (EI, 70 eV): m/z (%) = 332 (34), 331 (54), 330 ([M]+, 69), 329 (100), 328 (36), 327 (50), 142 (7), 141 (10), 130 (6), 128 (6), 114 (7). HRMS (ESI-TOF): calculated for C10H7Br2N2O ([M + H]+) 328.8925, found 328.8931.
:1). 1H NMR (300 MHz, CDCl3) δ = 9.60 (d, 4J = 0.8 Hz, 1H), 8.80 (s, 2H), 7.34–7.28 (m, 10H), 7.24 (dd, 3J = 3.9 Hz, 4J = 1.6 Hz, 1H), 7.19 (ddd, 3J = 2.6 Hz, 4J = 1.6 Hz, 4J = 0.9 Hz, 1H), 6.58 (dd, 3J = 3.9 Hz, 3J = 2.7 Hz, 1H). 13C NMR (75 MHz, CDCl3) δ = 178.2 (CHO), 151.8 (CH), 148.2, 132.9 (C), 131.6, 130.5, 129.2, 128.3, 122.4 (CH), 121.7, 118.8 (C), 111.3 (CH), 97.2, 81.6 (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C). IR (ATR, cm−1):
C). IR (ATR, cm−1):  , 1480 (m), 1403 (m), 1354 (m), 1036 (m), 890 (m), 781 (s), 748 (s), 686 (s), 573 (m), 548 (m). MS (EI, 70 eV): m/z (%) = 372 ([M]+, 19), 345 (25), 344 (100), 343 (47), 342 (40), 341 (11), 340 (14), 316 (8), 315 (9), 266 (10), 171 (5), 158 (4). HRMS (ESI-TOF): calculated for C26H17N2O ([M + H]+) 373.1341, found 373.1349.
:1), 1H NMR (500 MHz, CDCl3) δ = 9.59 (d, 4J = 0.8 Hz, 1H), 8.77 (s, 2H), 7.23 (dd, 3J = 4.0 Hz, 4J = 1.6 Hz, 1H), 7.20 (d, 3J = 8.2 Hz, 4H), 7.18–7.17 (m, 1H), 7.11 (d, 3J = 7.9 Hz, 4H), 6.56 (dd, 3J = 4.0 Hz, 3J = 2.7 Hz, 1H), 2.34 (s, 6H). 13C NMR (126 MHz, CDCl3) δ = 178.2 (CHO), 151.5 (CH), 147.9, 139.5 (C), 132.9, 131.5, 130.4, 129.1 (CH), 119.0, 118.7 (C), 111.2 (CH), 97.6, 81.1 (CC), 21.5 (CH3). IR (ATR, cm−1):
, 1480 (m), 1403 (m), 1354 (m), 1036 (m), 890 (m), 781 (s), 748 (s), 686 (s), 573 (m), 548 (m). MS (EI, 70 eV): m/z (%) = 372 ([M]+, 19), 345 (25), 344 (100), 343 (47), 342 (40), 341 (11), 340 (14), 316 (8), 315 (9), 266 (10), 171 (5), 158 (4). HRMS (ESI-TOF): calculated for C26H17N2O ([M + H]+) 373.1341, found 373.1349.
:1), 1H NMR (500 MHz, CDCl3) δ = 9.59 (d, 4J = 0.8 Hz, 1H), 8.77 (s, 2H), 7.23 (dd, 3J = 4.0 Hz, 4J = 1.6 Hz, 1H), 7.20 (d, 3J = 8.2 Hz, 4H), 7.18–7.17 (m, 1H), 7.11 (d, 3J = 7.9 Hz, 4H), 6.56 (dd, 3J = 4.0 Hz, 3J = 2.7 Hz, 1H), 2.34 (s, 6H). 13C NMR (126 MHz, CDCl3) δ = 178.2 (CHO), 151.5 (CH), 147.9, 139.5 (C), 132.9, 131.5, 130.4, 129.1 (CH), 119.0, 118.7 (C), 111.2 (CH), 97.6, 81.1 (CC), 21.5 (CH3). IR (ATR, cm−1):  , 1508 (m), 1484 (m), 1407 (s), 1356 (m), 1030 (m), 886 (m), 814 (s), 785 (s), 754 (s), 743 (s), 577 (s), 528 (s). MS (EI, 70 eV): m/z (%) = 400 ([M]+, 14), 373 (29), 372 (100), 371 (23), 370 (11), 369 (9), 357 (10), 356 (17), 178 (9), 177 (9). HRMS (ESI-TOF): calculated for C28H21N2O ([M + H]+) 401.1654, found 401.1643.
:1). 1H NMR (300 MHz, CDCl3) δ = 9.54 (d, 4J = 0.8 Hz, 1H), 8.73 (s, 2H), 7.27–7.17 (m, 5H), 7.12 (ddd, 3J = 2.6 Hz, 4J = 1.6 Hz, 4J = 0.9 Hz, 1H), 6.98–6.91 (m, 4H), 6.53 (dd, 3J = 3.9 Hz, 3J = 2.7 Hz, 1H). 13C NMR (75 MHz, CDCl3) δ = 178.2 (CHO), 163.0 (d, 1J = 251.4 Hz, C), 151.7 (CH), 148.1 (C), 133.6 (d, 3J = 8.6 Hz, CH), 132.9 (C), 130.4, 122.4 (CH), 118.7 (C), 117.8 (d, 4J = 3.5 Hz, C), 115.8 (d, 2J = 22.2 Hz, C), 111.3 (C), 96.2 (CC), 81.4 (d, 5J = 1.5 Hz, CC). 19F NMR (282 MHz, CDCl3): δ = −108.8. IR (ATR, cm−1):
, 1508 (m), 1484 (m), 1407 (s), 1356 (m), 1030 (m), 886 (m), 814 (s), 785 (s), 754 (s), 743 (s), 577 (s), 528 (s). MS (EI, 70 eV): m/z (%) = 400 ([M]+, 14), 373 (29), 372 (100), 371 (23), 370 (11), 369 (9), 357 (10), 356 (17), 178 (9), 177 (9). HRMS (ESI-TOF): calculated for C28H21N2O ([M + H]+) 401.1654, found 401.1643.
:1). 1H NMR (300 MHz, CDCl3) δ = 9.54 (d, 4J = 0.8 Hz, 1H), 8.73 (s, 2H), 7.27–7.17 (m, 5H), 7.12 (ddd, 3J = 2.6 Hz, 4J = 1.6 Hz, 4J = 0.9 Hz, 1H), 6.98–6.91 (m, 4H), 6.53 (dd, 3J = 3.9 Hz, 3J = 2.7 Hz, 1H). 13C NMR (75 MHz, CDCl3) δ = 178.2 (CHO), 163.0 (d, 1J = 251.4 Hz, C), 151.7 (CH), 148.1 (C), 133.6 (d, 3J = 8.6 Hz, CH), 132.9 (C), 130.4, 122.4 (CH), 118.7 (C), 117.8 (d, 4J = 3.5 Hz, C), 115.8 (d, 2J = 22.2 Hz, C), 111.3 (C), 96.2 (CC), 81.4 (d, 5J = 1.5 Hz, CC). 19F NMR (282 MHz, CDCl3): δ = −108.8. IR (ATR, cm−1):  , 1504 (s), 1407 (s), 1226 (s), 1154 (m), 1092 (m), 832 (s), 783 (m), 742 (s), 575 (m), 528 (s), 492 (s). MS (EI, 70 eV): m/z (%) = 408 ([M]+, 17), 381 (26), 380 (100), 379 (43), 378 (36), 377 (7), 376 (9), 352 (6), 351 (7), 313 (4), 284 (9), 189 (4). HRMS (ESI-TOF): calculated for C26H15F2N2O ([M + H]+) 409.1152, found 409.1149.
:1). 1H NMR (300 MHz, CDCl3) δ = 9.52 (d, 4J = 0.9 Hz, 1H), 8.75 (s, 2H), 7.48 (d, 3J = 8.1 Hz, 4H), 7.30 (d, 3J = 7.9 Hz, 4H), 7.17 (dd, 3J = 3.9 Hz, 4J = 1.6 Hz, 1H), 7.10 (ddd, 3J = 2.6 Hz, 4J = 1.6 Hz, 4J = 0.9 Hz, 1H), 6.52 (dd, 3J = 3.9 Hz, 3J = 2.7 Hz, 1H). 13C NMR (75 MHz, CDCl3) δ = 178.2 (CHO), 152.4 (CH), 148.9, 132.9 (C), 131.9 (CH), 130.9 (q, 2J = 32.8 Hz, C), 130.5 (CH), 125.4 (C), 125.3 (q, 3J = 3.8 Hz, CH), 123.6 (q, 1J = 272.2 Hz, C), 123.0 (CH), 118.3 (C), 111.5 (CH), 95.6, 83.6 (CC). 19F NMR (282 MHz, CDCl3): δ = −63.0. IR (ATR, cm−1):
, 1504 (s), 1407 (s), 1226 (s), 1154 (m), 1092 (m), 832 (s), 783 (m), 742 (s), 575 (m), 528 (s), 492 (s). MS (EI, 70 eV): m/z (%) = 408 ([M]+, 17), 381 (26), 380 (100), 379 (43), 378 (36), 377 (7), 376 (9), 352 (6), 351 (7), 313 (4), 284 (9), 189 (4). HRMS (ESI-TOF): calculated for C26H15F2N2O ([M + H]+) 409.1152, found 409.1149.
:1). 1H NMR (300 MHz, CDCl3) δ = 9.52 (d, 4J = 0.9 Hz, 1H), 8.75 (s, 2H), 7.48 (d, 3J = 8.1 Hz, 4H), 7.30 (d, 3J = 7.9 Hz, 4H), 7.17 (dd, 3J = 3.9 Hz, 4J = 1.6 Hz, 1H), 7.10 (ddd, 3J = 2.6 Hz, 4J = 1.6 Hz, 4J = 0.9 Hz, 1H), 6.52 (dd, 3J = 3.9 Hz, 3J = 2.7 Hz, 1H). 13C NMR (75 MHz, CDCl3) δ = 178.2 (CHO), 152.4 (CH), 148.9, 132.9 (C), 131.9 (CH), 130.9 (q, 2J = 32.8 Hz, C), 130.5 (CH), 125.4 (C), 125.3 (q, 3J = 3.8 Hz, CH), 123.6 (q, 1J = 272.2 Hz, C), 123.0 (CH), 118.3 (C), 111.5 (CH), 95.6, 83.6 (CC). 19F NMR (282 MHz, CDCl3): δ = −63.0. IR (ATR, cm−1):  , 1613 (m), 1484 (m), 1405 (m), 1319 (s), 1164 (s), 1102 (s), 1063 (s), 1016 (s), 839 (s), 781 (m), 740 (s), 575 (m). MS (EI, 70 eV): m/z (%) = 508 ([M]+, 23), 481 (27), 480 (100), 479 (30), 478 (13), 461 (7), 411 (9), 410 (19), 363 (5), 334 (6), 195 (7). HRMS (ESI-TOF): calculated for C28H15F6N2O ([M + H]+) 509.1089, found 509.1096.
:1). 1H NMR (300 MHz, CDCl3) δ = 9.57 (d, 4J = 0.7 Hz, 1H), 8.68 (s, 2H), 7.23 (dd, 3J = 3.9 Hz, 4J = 1.6 Hz, 1H), 7.20–7.14 (m, 5H), 6.59–6.53 (m, 5H), 2.95 (s, 12H). 13C NMR (75 MHz, CDCl3) δ = 178.2 (CHO), 150.4 (C), 150.3 (CH), 146.4, 132.8 (C), 132.8, 132.5, 130.4 (CH), 119.5 (C), 111.5, 110.8 (CH), 108.2 (C), 99.1, 80.2 (CC), 39.9 (CH3). IR (ATR, cm−1):
, 1613 (m), 1484 (m), 1405 (m), 1319 (s), 1164 (s), 1102 (s), 1063 (s), 1016 (s), 839 (s), 781 (m), 740 (s), 575 (m). MS (EI, 70 eV): m/z (%) = 508 ([M]+, 23), 481 (27), 480 (100), 479 (30), 478 (13), 461 (7), 411 (9), 410 (19), 363 (5), 334 (6), 195 (7). HRMS (ESI-TOF): calculated for C28H15F6N2O ([M + H]+) 509.1089, found 509.1096.
:1). 1H NMR (300 MHz, CDCl3) δ = 9.57 (d, 4J = 0.7 Hz, 1H), 8.68 (s, 2H), 7.23 (dd, 3J = 3.9 Hz, 4J = 1.6 Hz, 1H), 7.20–7.14 (m, 5H), 6.59–6.53 (m, 5H), 2.95 (s, 12H). 13C NMR (75 MHz, CDCl3) δ = 178.2 (CHO), 150.4 (C), 150.3 (CH), 146.4, 132.8 (C), 132.8, 132.5, 130.4 (CH), 119.5 (C), 111.5, 110.8 (CH), 108.2 (C), 99.1, 80.2 (CC), 39.9 (CH3). IR (ATR, cm−1):  , 1603 (s), 1564 (s), 1519 (s), 1480 (s), 1360 (s), 1232 (m), 1168 (s), 806 (s), 783 (s), 740 (s), 577 (m), 523 (m). MS (EI, 70 eV): m/z (%) = 458 ([M]+, 100), 457 (23), 431 (15), 430 (57), 429 (22), 414 (22), 229 (14), 215 (12), 214 (25), 206 (12), 192 (11). HRMS (ESI-TOF): calculated for C30H27N4O ([M + H]+) 459.2185, found 459.2189.
:1). 1H NMR (300 MHz, CDCl3) δ = 9.37 (dd, 3J = 3.1 Hz, 4J = 1.5 Hz, 1H), 8.63 (s, 1H), 8.46 (s, 1H), 8.14 (d, 3J = 8.4 Hz, 1H), 7.71 (ddd, 3J = 8.4 Hz, 3J = 6.7 Hz, 4J = 1.4 Hz, 1H), 7.66 (dd, 3J = 3.8 Hz, 4J = 1.5 Hz, 1H), 7.57 (dd, 3J = 8.6 Hz, 4J = 1.5 Hz, 1H), 7.54–7.50 (m, 2H), 7.43 (d, 3J = 7.7 Hz, 2H), 7.37 (ddd, 3J = 8.4 Hz, 3J = 6.7 Hz, 4J = 1.3 Hz, 1H), 7.32–7.19 (m, 4H), 6.80 (dd, 3J = 3.8 Hz, 3J = 3.1 Hz, 1H), 2.53 (s, 3H), 2.41 (s, 3H). 13C NMR (75 MHz, CDCl3) δ = 153.7, 148.2, 145.6, 144.8, 139.6, 138.8, 135.2 (C), 131.3 (C), 131.3, 130.7, 130.2, 129.4, 129.2, 128.8 (CH), 127.1 (C), 126.9, 125.7, 120.3 (CH), 119.3, 115.9 (C), 113.5, 109.5 (CH), 97.9, 85.6 (CC), 21.6, 21.5 (CH3). IR (ATR, cm−1):
, 1603 (s), 1564 (s), 1519 (s), 1480 (s), 1360 (s), 1232 (m), 1168 (s), 806 (s), 783 (s), 740 (s), 577 (m), 523 (m). MS (EI, 70 eV): m/z (%) = 458 ([M]+, 100), 457 (23), 431 (15), 430 (57), 429 (22), 414 (22), 229 (14), 215 (12), 214 (25), 206 (12), 192 (11). HRMS (ESI-TOF): calculated for C30H27N4O ([M + H]+) 459.2185, found 459.2189.
:1). 1H NMR (300 MHz, CDCl3) δ = 9.37 (dd, 3J = 3.1 Hz, 4J = 1.5 Hz, 1H), 8.63 (s, 1H), 8.46 (s, 1H), 8.14 (d, 3J = 8.4 Hz, 1H), 7.71 (ddd, 3J = 8.4 Hz, 3J = 6.7 Hz, 4J = 1.4 Hz, 1H), 7.66 (dd, 3J = 3.8 Hz, 4J = 1.5 Hz, 1H), 7.57 (dd, 3J = 8.6 Hz, 4J = 1.5 Hz, 1H), 7.54–7.50 (m, 2H), 7.43 (d, 3J = 7.7 Hz, 2H), 7.37 (ddd, 3J = 8.4 Hz, 3J = 6.7 Hz, 4J = 1.3 Hz, 1H), 7.32–7.19 (m, 4H), 6.80 (dd, 3J = 3.8 Hz, 3J = 3.1 Hz, 1H), 2.53 (s, 3H), 2.41 (s, 3H). 13C NMR (75 MHz, CDCl3) δ = 153.7, 148.2, 145.6, 144.8, 139.6, 138.8, 135.2 (C), 131.3 (C), 131.3, 130.7, 130.2, 129.4, 129.2, 128.8 (CH), 127.1 (C), 126.9, 125.7, 120.3 (CH), 119.3, 115.9 (C), 113.5, 109.5 (CH), 97.9, 85.6 (CC), 21.6, 21.5 (CH3). IR (ATR, cm−1): 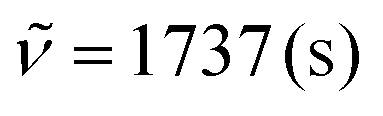 , 1597 (s), 1504 (s), 1451 (s), 1356 (s), 1205 (s), 1028 (s), 960 (m), 812 (s), 762 (s), 690 (s), 604 (m). MS (EI, 70 eV): m/z (%) = 473 ([M]+, 100), 472 (87), 471 (21), 470 (9), 459 (7), 458 (24), 457 (22), 456 (11), 229 (10), 228 (12). HRMS (ESI-TOF): calculated for C34H24N3 ([M + H]+) 474.1970, found 474.1978.
:1). 1H NMR (300 MHz, CDCl3) δ = 8.74 (s, 1H), 8.13 (ddd, 3J = 8.5 Hz, 4J = 1.3 Hz, 5J = 0.6 Hz, 1H), 8.03 (s, 1H), 7.95 (d, 3J = 4.2 Hz, 1H), 7.73–7.64 (m, 3H), 7.52–7.46 (m, 3H), 7.39–7.27 (m, 5H), 7.14 (s, 1H), 7.09 (d, 3J = 4.2 Hz, 1H), 2.57 (s, 3H), 2.47 (s, 3H). 13C NMR (75 MHz, CDCl3) δ = 148.1, 146.4 (C), 145.0, 144.8 (CH), 144.8, 138.9, 138.7, 135.3, 134.8, 134.3, 134.0 (C), 130.9, 130.3, 129.5 (CH), 129.2 (C), 128.6, 128.0 (CH), 127.3 (C), 127.0 (CH), 126.2 (C), 125.7 (CH), 119.8, 117.5, 117.2 (C), 115.3, 111.2, 107.1 (CH), 21.6, 21.3 (CH3). IR (ATR, cm−1):
, 1597 (s), 1504 (s), 1451 (s), 1356 (s), 1205 (s), 1028 (s), 960 (m), 812 (s), 762 (s), 690 (s), 604 (m). MS (EI, 70 eV): m/z (%) = 473 ([M]+, 100), 472 (87), 471 (21), 470 (9), 459 (7), 458 (24), 457 (22), 456 (11), 229 (10), 228 (12). HRMS (ESI-TOF): calculated for C34H24N3 ([M + H]+) 474.1970, found 474.1978.
:1). 1H NMR (300 MHz, CDCl3) δ = 8.74 (s, 1H), 8.13 (ddd, 3J = 8.5 Hz, 4J = 1.3 Hz, 5J = 0.6 Hz, 1H), 8.03 (s, 1H), 7.95 (d, 3J = 4.2 Hz, 1H), 7.73–7.64 (m, 3H), 7.52–7.46 (m, 3H), 7.39–7.27 (m, 5H), 7.14 (s, 1H), 7.09 (d, 3J = 4.2 Hz, 1H), 2.57 (s, 3H), 2.47 (s, 3H). 13C NMR (75 MHz, CDCl3) δ = 148.1, 146.4 (C), 145.0, 144.8 (CH), 144.8, 138.9, 138.7, 135.3, 134.8, 134.3, 134.0 (C), 130.9, 130.3, 129.5 (CH), 129.2 (C), 128.6, 128.0 (CH), 127.3 (C), 127.0 (CH), 126.2 (C), 125.7 (CH), 119.8, 117.5, 117.2 (C), 115.3, 111.2, 107.1 (CH), 21.6, 21.3 (CH3). IR (ATR, cm−1):  , 1539 (m), 1502 (m), 1445 (m), 1358 (m), 1179 (m), 1109 (m), 1041 (m), 892 (m), 816 (s), 758 (s), 734 (s), 610 (m), 490 (s). MS (EI, 70 eV): m/z (%) = 473 ([M]+, 100), 472 (69), 471 (72), 470 (17), 457 (17), 456 (18), 236 (46), 235 (16), 228 (37), 227 (24), 226 (12), 220 (12). HRMS (ESI-TOF): calculated for C34H24N3 ([M + H]+) 474.1970, found 474.1977.
:1). 1H NMR (300 MHz, CDCl3) δ = 8.74 (s, 1H), 8.05 (d, 3J = 8.6 Hz, 1H), 7.99 (s, 1H), 7.95 (d, 3J = 4.2 Hz, 1H), 7.71–7.66 (m, 2H), 7.54 (dd, 3J = 8.6 Hz, 4J = 2.0 Hz, 1H), 7.52–7.47 (m, 2H), 7.37–7.32 (m, 2H), 7.32–7.27 (m, 2H), 7.24–7.22 (m, 1H), 7.15 (s, 1H), 7.10 (d, 3J = 4.1 Hz, 1H), 2.58 (s, 3H), 2.47 (s, 3H), 2.41 (s, 3H). 13C NMR (75 MHz, CDCl3) δ = 146.8, 145.7 (C), 145.0, 144.7 (CH), 144.1, 138.8, 138.7, 135.7, 135.5, 134.9, 134.3, 134.0 (C), 132.7, 130.9, 129.6 (CH), 129.0 (C), 128.7, 128.4, 128.0 (CH), 127.3, 126.3 (C), 125.6 (CH), 119.8, 117.7, 117.2 (C), 115.2, 110.8, 107.0 (CH), 21.9, 21.6, 21.4 (CH3). IR (ATR, cm−1):
, 1539 (m), 1502 (m), 1445 (m), 1358 (m), 1179 (m), 1109 (m), 1041 (m), 892 (m), 816 (s), 758 (s), 734 (s), 610 (m), 490 (s). MS (EI, 70 eV): m/z (%) = 473 ([M]+, 100), 472 (69), 471 (72), 470 (17), 457 (17), 456 (18), 236 (46), 235 (16), 228 (37), 227 (24), 226 (12), 220 (12). HRMS (ESI-TOF): calculated for C34H24N3 ([M + H]+) 474.1970, found 474.1977.
:1). 1H NMR (300 MHz, CDCl3) δ = 8.74 (s, 1H), 8.05 (d, 3J = 8.6 Hz, 1H), 7.99 (s, 1H), 7.95 (d, 3J = 4.2 Hz, 1H), 7.71–7.66 (m, 2H), 7.54 (dd, 3J = 8.6 Hz, 4J = 2.0 Hz, 1H), 7.52–7.47 (m, 2H), 7.37–7.32 (m, 2H), 7.32–7.27 (m, 2H), 7.24–7.22 (m, 1H), 7.15 (s, 1H), 7.10 (d, 3J = 4.1 Hz, 1H), 2.58 (s, 3H), 2.47 (s, 3H), 2.41 (s, 3H). 13C NMR (75 MHz, CDCl3) δ = 146.8, 145.7 (C), 145.0, 144.7 (CH), 144.1, 138.8, 138.7, 135.7, 135.5, 134.9, 134.3, 134.0 (C), 132.7, 130.9, 129.6 (CH), 129.0 (C), 128.7, 128.4, 128.0 (CH), 127.3, 126.3 (C), 125.6 (CH), 119.8, 117.7, 117.2 (C), 115.2, 110.8, 107.0 (CH), 21.9, 21.6, 21.4 (CH3). IR (ATR, cm−1):  , 1354 (m), 1183 (m), 1109 (m), 1036 (m), 884 (m), 814 (s), 802 (s), 781 (s), 711 (m), 602 (m), 488 (s). MS (EI, 70 eV): m/z (%) = 487 ([M]+, 100), 486 (55), 485 (11), 472 (18), 471 (24), 470 (11), 457 (6), 244 (9), 236 (23). HRMS (EI): calculated for C35H25N3 ([M]+) 487.20430, found 487.20439.
:1). 1H NMR (500 MHz, CDCl3) δ = 8.73 (s, 1H), 8.05 (d, 3J = 9.3 Hz, 1H), 8.03 (s, 1H), 7.90 (d, 3J = 4.1 Hz, 1H), 7.70 (d, 3J = 8.0 Hz, 2H), 7.47 (d, 3J = 7.7 Hz, 2H), 7.40 (dd, 3J = 9.3 Hz, 4J = 2.8 Hz, 1H), 7.34 (d, 3J = 7.7 Hz, 2H), 7.31 (d, 3J = 8.0 Hz, 2H), 7.13 (s, 1H), 7.11 (d, 3J = 4.1 Hz, 1H), 6.43 (d, 4J = 2.8 Hz, 1H), 2.91 (s, 6H), 2.56 (s, 3H), 2.47 (s, 3H). 13C NMR (126 MHz, CDCl3) δ = 147.9 (C), 144.9, 144.4 (CH), 143.6, 142.8, 141.4, 138.6, 138.4, 136.1, 135.0, 134.2, 134.1 (C), 130.9, 129.5, 129.4 (CH), 128.9 (C), 128.6 (CH), 128.5 (C), 128.0 (CH), 126.8 (C), 120.7 (CH), 119.9, 117.9, 117.3 (C), 114.6, 109.6, 106.8, 104.1 (CH), 40.5, 21.6, 21.3 (CH3). IR (ATR, cm−1):
, 1354 (m), 1183 (m), 1109 (m), 1036 (m), 884 (m), 814 (s), 802 (s), 781 (s), 711 (m), 602 (m), 488 (s). MS (EI, 70 eV): m/z (%) = 487 ([M]+, 100), 486 (55), 485 (11), 472 (18), 471 (24), 470 (11), 457 (6), 244 (9), 236 (23). HRMS (EI): calculated for C35H25N3 ([M]+) 487.20430, found 487.20439.
:1). 1H NMR (500 MHz, CDCl3) δ = 8.73 (s, 1H), 8.05 (d, 3J = 9.3 Hz, 1H), 8.03 (s, 1H), 7.90 (d, 3J = 4.1 Hz, 1H), 7.70 (d, 3J = 8.0 Hz, 2H), 7.47 (d, 3J = 7.7 Hz, 2H), 7.40 (dd, 3J = 9.3 Hz, 4J = 2.8 Hz, 1H), 7.34 (d, 3J = 7.7 Hz, 2H), 7.31 (d, 3J = 8.0 Hz, 2H), 7.13 (s, 1H), 7.11 (d, 3J = 4.1 Hz, 1H), 6.43 (d, 4J = 2.8 Hz, 1H), 2.91 (s, 6H), 2.56 (s, 3H), 2.47 (s, 3H). 13C NMR (126 MHz, CDCl3) δ = 147.9 (C), 144.9, 144.4 (CH), 143.6, 142.8, 141.4, 138.6, 138.4, 136.1, 135.0, 134.2, 134.1 (C), 130.9, 129.5, 129.4 (CH), 128.9 (C), 128.6 (CH), 128.5 (C), 128.0 (CH), 126.8 (C), 120.7 (CH), 119.9, 117.9, 117.3 (C), 114.6, 109.6, 106.8, 104.1 (CH), 40.5, 21.6, 21.3 (CH3). IR (ATR, cm−1):  , 1504 (m), 1440 (m), 1341 (m), 1055 (m), 892 (m), 814 (s), 800 (s), 781 (s), 587 (m), 495 (m). MS (EI, 70 eV): m/z (%) = 516 ([M]+, 100), 501 (6), 500 (7), 486 (11), 472 (10), 471 (14), 400 (11), 258 (22), 257 (8). HRMS (EI): calculated for C36H28N4 ([M]+) 516.23085, found 516.23130.
:1). 1H NMR (300 MHz, CDCl3) δ = 8.75 (s, 1H), 8.14–8.07 (m, 1H), 8.03 (s, 1H), 7.91 (d, 3J = 4.2 Hz, 1H), 7.71–7.64 (m, 2H), 7.54–7.41 (m, 3H), 7.38–7.32 (m, 2H), 7.31–7.27 (m, 2H), 7.14 (s, 1H), 7.11–7.03 (m, 2H), 2.57 (s, 3H), 2.47 (s, 3H). 13C NMR (75 MHz, CDCl3) δ = 160.0 (d, 1J = 246.9 Hz, C), 145.7 (d, 3J = 6.1 Hz, C), 145.1 (d, 3J = 6.7 Hz, CH), 144.3 (d, 4J = 2.6 Hz, C), 139.2, 138.8, 134.9, 134.7, 134.3, 134.0 (C), 131.1, 131.1, 131.0, 129.6 (CH), 129.2 (C), 128.5 (CH), 128.1 (C), 128.0 (CH), 126.0 (C), 120.7 (d, 2J = 26.3 Hz, CH), 119.8, 117.7, 117.2 (C), 115.3, 111.1 (CH), 110.1 (d, 2J = 23.8 Hz, CH), 107.1 (CH), 21.6, 21.3 (CH3). 19F NMR (282 MHz, CDCl3): δ = −113.0. IR (ATR, cm−1):
, 1504 (m), 1440 (m), 1341 (m), 1055 (m), 892 (m), 814 (s), 800 (s), 781 (s), 587 (m), 495 (m). MS (EI, 70 eV): m/z (%) = 516 ([M]+, 100), 501 (6), 500 (7), 486 (11), 472 (10), 471 (14), 400 (11), 258 (22), 257 (8). HRMS (EI): calculated for C36H28N4 ([M]+) 516.23085, found 516.23130.
:1). 1H NMR (300 MHz, CDCl3) δ = 8.75 (s, 1H), 8.14–8.07 (m, 1H), 8.03 (s, 1H), 7.91 (d, 3J = 4.2 Hz, 1H), 7.71–7.64 (m, 2H), 7.54–7.41 (m, 3H), 7.38–7.32 (m, 2H), 7.31–7.27 (m, 2H), 7.14 (s, 1H), 7.11–7.03 (m, 2H), 2.57 (s, 3H), 2.47 (s, 3H). 13C NMR (75 MHz, CDCl3) δ = 160.0 (d, 1J = 246.9 Hz, C), 145.7 (d, 3J = 6.1 Hz, C), 145.1 (d, 3J = 6.7 Hz, CH), 144.3 (d, 4J = 2.6 Hz, C), 139.2, 138.8, 134.9, 134.7, 134.3, 134.0 (C), 131.1, 131.1, 131.0, 129.6 (CH), 129.2 (C), 128.5 (CH), 128.1 (C), 128.0 (CH), 126.0 (C), 120.7 (d, 2J = 26.3 Hz, CH), 119.8, 117.7, 117.2 (C), 115.3, 111.1 (CH), 110.1 (d, 2J = 23.8 Hz, CH), 107.1 (CH), 21.6, 21.3 (CH3). 19F NMR (282 MHz, CDCl3): δ = −113.0. IR (ATR, cm−1):  , 1541 (m), 1457 (s), 1339 (m), 1230 (m), 1166 (s), 1041 (m), 822 (s), 800 (s), 734 (s), 600 (s), 490 (s). MS (EI, 70 eV): m/z (%) = 491 ([M]+, 100), 490 (71), 489 (18), 487 (13), 476 (18), 475 (19), 245 (10), 238 (18), 237 (10). HRMS (EI): calculated for C34H22N3F ([M]+) 491.17923, found 491.18050.
:1). 1H NMR (300 MHz, CDCl3) δ = 8.69 (s, 1H), 7.86 (d, 3J = 4.2 Hz, 1H), 7.73 (s, 1H), 7.66–7.60 (m, 2H), 7.54–7.48 (m, 1H),7.47–7.41 (m, 2H), 7.36–7.30 (m, 4H), 7.12 (s, 1H), 7.02 (d, 3J = 4.2 Hz, 1H), 6.83–6.73 (m, 1H), 2.56 (s, 3H), 2.47 (s, 3H). 13C NMR (126 MHz, CDCl3) δ = 162.6 (dd, 1J = 252.0 Hz, 3J = 14.2 Hz, C), 159.8 (dd, 1J = 264.4 Hz, 3J = 14.4 Hz, C), 149.4 (dd, 3J = 14.7 Hz, 4J = 1.8 Hz, C), 146.0 (C), 145.3, 145.1 (CH), 144.7, 138.8 (C), 136.8 (d, 4J = 4.4 Hz), 134.4, 134.0, 133.9 (C), 130.5 (CH), 129.7 (C), 129.6, 127.9 (CH), 127.3 (d, 4J = 4.1 Hz, CH), 125.3, 119.6 (C), 117.9, 117.2 (C), 115.8 (CH), 114.7 (dd, 3J = 6.1 Hz, 3J = 2.0 Hz, C), 112.2 (CH), 108.6 (dd, 2J = 20.1 Hz, 4J = 4.9 Hz, CH), 107.3 (CH), 102.9 (pt, 2J = 28.4 Hz, CH), 21.6, 21.3 (CH3). 19F NMR (282 MHz, CDCl3): δ = −106.5 (d, 4JF,F = 9.0 Hz), −99.9 (d, 4JF,F = 9.0 Hz). IR (ATR, cm−1):
, 1541 (m), 1457 (s), 1339 (m), 1230 (m), 1166 (s), 1041 (m), 822 (s), 800 (s), 734 (s), 600 (s), 490 (s). MS (EI, 70 eV): m/z (%) = 491 ([M]+, 100), 490 (71), 489 (18), 487 (13), 476 (18), 475 (19), 245 (10), 238 (18), 237 (10). HRMS (EI): calculated for C34H22N3F ([M]+) 491.17923, found 491.18050.
:1). 1H NMR (300 MHz, CDCl3) δ = 8.69 (s, 1H), 7.86 (d, 3J = 4.2 Hz, 1H), 7.73 (s, 1H), 7.66–7.60 (m, 2H), 7.54–7.48 (m, 1H),7.47–7.41 (m, 2H), 7.36–7.30 (m, 4H), 7.12 (s, 1H), 7.02 (d, 3J = 4.2 Hz, 1H), 6.83–6.73 (m, 1H), 2.56 (s, 3H), 2.47 (s, 3H). 13C NMR (126 MHz, CDCl3) δ = 162.6 (dd, 1J = 252.0 Hz, 3J = 14.2 Hz, C), 159.8 (dd, 1J = 264.4 Hz, 3J = 14.4 Hz, C), 149.4 (dd, 3J = 14.7 Hz, 4J = 1.8 Hz, C), 146.0 (C), 145.3, 145.1 (CH), 144.7, 138.8 (C), 136.8 (d, 4J = 4.4 Hz), 134.4, 134.0, 133.9 (C), 130.5 (CH), 129.7 (C), 129.6, 127.9 (CH), 127.3 (d, 4J = 4.1 Hz, CH), 125.3, 119.6 (C), 117.9, 117.2 (C), 115.8 (CH), 114.7 (dd, 3J = 6.1 Hz, 3J = 2.0 Hz, C), 112.2 (CH), 108.6 (dd, 2J = 20.1 Hz, 4J = 4.9 Hz, CH), 107.3 (CH), 102.9 (pt, 2J = 28.4 Hz, CH), 21.6, 21.3 (CH3). 19F NMR (282 MHz, CDCl3): δ = −106.5 (d, 4JF,F = 9.0 Hz), −99.9 (d, 4JF,F = 9.0 Hz). IR (ATR, cm−1):  , 1543 (s), 1399 (m), 1339 (s), 1205 (s), 1135 (s), 1041 (m), 997 (m), 845 (s), 818 (s), 787 (s), 732 (s), 558 (s), 492 (m). MS (EI, 70 eV): m/z (%) = 509 ([M]+, 100), 508 (84), 507 (12), 494 (21), 493 (29), 420 (6), 329 (7), 247 (13). HRMS (EI): calculated for C34H21N3F2 ([M]+) 509.16981, found 509.17052.
:1); 1H NMR (500 MHz, CDCl3) δ = 8.70 (s, 1H), 7.97 (d, 3J = 4.2 Hz, 2H), 7.69–7.64 (m, 2H), 7.50 (d, 3J = 7.7 Hz, 2H), 7.37–7.30 (m, 5H), 7.25–7.20 (m, 2H), 7.11 (s, 1H), 7.05 (d, 3J = 4.1 Hz, 1H), 2.57 (s, 3H), 2.47 (s, 3H). 13C NMR (126 MHz, CDCl3) δ = 157.3 (d, 1J = 256.6 Hz, C), 146.3 (d, 4J = 2.6 Hz, C), 145.1, 145.0 (CH), 144.8, 139.1, 138.8 (C), 138.5 (d, 2J = 11.9 Hz, C), 135.1, 134.6, 134.2, 133.9 (C), 131.0, 129.6 (CH), 129.3 (C), 128.9 (d, 5J = 1.7 Hz, C), 128.5, 128.0 (CH), 125.9 (C), 124.7 (d, 3J = 7.8 Hz, CH), 122.7 (d, 4J = 4.5 Hz, CH), 119.7, 117.9, 117.1 (C), 115.4 (CH), 113.9 (d, 2J = 18.6 Hz, CH), 112.1, 107.3 (CH), 21.6, 21.3 (CH3). 19F NMR (282 MHz, CDCl3): δ = −126.0. IR (ATR, cm−1):
, 1543 (s), 1399 (m), 1339 (s), 1205 (s), 1135 (s), 1041 (m), 997 (m), 845 (s), 818 (s), 787 (s), 732 (s), 558 (s), 492 (m). MS (EI, 70 eV): m/z (%) = 509 ([M]+, 100), 508 (84), 507 (12), 494 (21), 493 (29), 420 (6), 329 (7), 247 (13). HRMS (EI): calculated for C34H21N3F2 ([M]+) 509.16981, found 509.17052.
:1); 1H NMR (500 MHz, CDCl3) δ = 8.70 (s, 1H), 7.97 (d, 3J = 4.2 Hz, 2H), 7.69–7.64 (m, 2H), 7.50 (d, 3J = 7.7 Hz, 2H), 7.37–7.30 (m, 5H), 7.25–7.20 (m, 2H), 7.11 (s, 1H), 7.05 (d, 3J = 4.1 Hz, 1H), 2.57 (s, 3H), 2.47 (s, 3H). 13C NMR (126 MHz, CDCl3) δ = 157.3 (d, 1J = 256.6 Hz, C), 146.3 (d, 4J = 2.6 Hz, C), 145.1, 145.0 (CH), 144.8, 139.1, 138.8 (C), 138.5 (d, 2J = 11.9 Hz, C), 135.1, 134.6, 134.2, 133.9 (C), 131.0, 129.6 (CH), 129.3 (C), 128.9 (d, 5J = 1.7 Hz, C), 128.5, 128.0 (CH), 125.9 (C), 124.7 (d, 3J = 7.8 Hz, CH), 122.7 (d, 4J = 4.5 Hz, CH), 119.7, 117.9, 117.1 (C), 115.4 (CH), 113.9 (d, 2J = 18.6 Hz, CH), 112.1, 107.3 (CH), 21.6, 21.3 (CH3). 19F NMR (282 MHz, CDCl3): δ = −126.0. IR (ATR, cm−1): 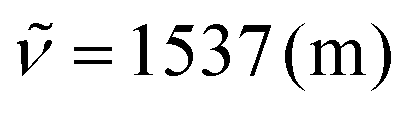 , 1445 (m), 1339 (m), 1241 (m), 1119 (m), 1041 (m), 892 (m), 816 (s), 760 (s), 579 (m), 486 (m). MS (EI, 70 eV): m/z (%) = 491 ([M]+, 100), 474 (9), 473 (45), 229 (1). HRMS (EI): calculated for C34H22N3F ([M]+) 491.17923, found 491.17838.
:1). 1H NMR (300 MHz, CDCl3) δ = 9.47–9.43 (m, 1H), 8.65 (s, 1H), 7.99 (d, 3J = 4.1 Hz, 1H), 7.90 (s, 1H), 7.76–7.70 (m, 1H), 7.70–7.60 (m, 4H), 7.51–7.41 (m, 3H), 7.37–7.27 (m, 4H), 7.25–7.21 (m, 1H), 7.08 (d, 3J = 4.1 Hz, 1H), 7.07 (s, 1H), 2.58 (s, 3H), 2.47 (s, 3H). 13C NMR (75 MHz, CDCl3) δ = 146.4, 145.8 (C), 144.1, 143.9 (CH), 143.2, 138.7, 138.6, 135.6, 134.8, 134.1, 134.0, 133.6 (C), 130.8 (CH), 130.7 (C), 129.5, 128.7 (CH), 128.6 (C), 128.0, 127.4, 126.8 (CH), 126.6 (C), 126.5, 125.2 (CH), 124.7 (C), 123.6 (CH), 119.9, 117.5, 117.1 (C), 115.1, 110.4, 106.9 (CH), 21.6, 21.3 (CH3). IR (ATR, cm−1):
, 1445 (m), 1339 (m), 1241 (m), 1119 (m), 1041 (m), 892 (m), 816 (s), 760 (s), 579 (m), 486 (m). MS (EI, 70 eV): m/z (%) = 491 ([M]+, 100), 474 (9), 473 (45), 229 (1). HRMS (EI): calculated for C34H22N3F ([M]+) 491.17923, found 491.17838.
:1). 1H NMR (300 MHz, CDCl3) δ = 9.47–9.43 (m, 1H), 8.65 (s, 1H), 7.99 (d, 3J = 4.1 Hz, 1H), 7.90 (s, 1H), 7.76–7.70 (m, 1H), 7.70–7.60 (m, 4H), 7.51–7.41 (m, 3H), 7.37–7.27 (m, 4H), 7.25–7.21 (m, 1H), 7.08 (d, 3J = 4.1 Hz, 1H), 7.07 (s, 1H), 2.58 (s, 3H), 2.47 (s, 3H). 13C NMR (75 MHz, CDCl3) δ = 146.4, 145.8 (C), 144.1, 143.9 (CH), 143.2, 138.7, 138.6, 135.6, 134.8, 134.1, 134.0, 133.6 (C), 130.8 (CH), 130.7 (C), 129.5, 128.7 (CH), 128.6 (C), 128.0, 127.4, 126.8 (CH), 126.6 (C), 126.5, 125.2 (CH), 124.7 (C), 123.6 (CH), 119.9, 117.5, 117.1 (C), 115.1, 110.4, 106.9 (CH), 21.6, 21.3 (CH3). IR (ATR, cm−1): 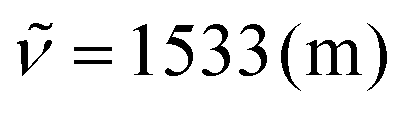 , 1459 (m), 1356 (m), 1179 (m), 1111 (m), 1039 (m), 882 (m), 820 (s), 795 (s), 762 (s), 719 (m), 488 (m). MS (EI, 70 eV): m/z (%) = 523 ([M]+, 100), 522 (44), 521 (15), 516 (10), 508 (12), 507 (14), 262 (14), 261 (13), 254 (19), 253 (11). HRMS (EI): calculated for C38H25N3 ([M]+) 523.20430, found 523.20496.
:1). 1H NMR (300 MHz, CDCl3) δ = 8.73 (s, 1H), 8.16–8.10 (m, 1H), 7.96–7.92 (m, 2H), 7.81–7.75 (m, 2H), 7.73–7.65 (m, 4H), 7.59–7.47 (m, 3H), 7.47–7.39 (m, 3H), 7.39–7.31 (m, 1H), 7.16 (s, 1H), 7.08 (d, 3J = 4.2 Hz, 1H). 13C NMR (75 MHz, CDCl3) δ = 148.1, 146.1 (C), 145.1, 145.0 (CH), 144.7, 138.3, 137.6, 134.3, 134.0 (C), 130.3, 130.2 (CH), 129.1 (C), 129.1, 128.9, 128.8, 128.7, 128.6, 128.1 (CH), 127.1 (C), 126.9 (CH), 126.2 (C), 125.8 (CH), 119.7, 117.4, 117.0 (C), 115.7, 111.3, 107.1 (CH). IR (ATR, cm−1):
, 1459 (m), 1356 (m), 1179 (m), 1111 (m), 1039 (m), 882 (m), 820 (s), 795 (s), 762 (s), 719 (m), 488 (m). MS (EI, 70 eV): m/z (%) = 523 ([M]+, 100), 522 (44), 521 (15), 516 (10), 508 (12), 507 (14), 262 (14), 261 (13), 254 (19), 253 (11). HRMS (EI): calculated for C38H25N3 ([M]+) 523.20430, found 523.20496.
:1). 1H NMR (300 MHz, CDCl3) δ = 8.73 (s, 1H), 8.16–8.10 (m, 1H), 7.96–7.92 (m, 2H), 7.81–7.75 (m, 2H), 7.73–7.65 (m, 4H), 7.59–7.47 (m, 3H), 7.47–7.39 (m, 3H), 7.39–7.31 (m, 1H), 7.16 (s, 1H), 7.08 (d, 3J = 4.2 Hz, 1H). 13C NMR (75 MHz, CDCl3) δ = 148.1, 146.1 (C), 145.1, 145.0 (CH), 144.7, 138.3, 137.6, 134.3, 134.0 (C), 130.3, 130.2 (CH), 129.1 (C), 129.1, 128.9, 128.8, 128.7, 128.6, 128.1 (CH), 127.1 (C), 126.9 (CH), 126.2 (C), 125.8 (CH), 119.7, 117.4, 117.0 (C), 115.7, 111.3, 107.1 (CH). IR (ATR, cm−1):  , 1337 (m), 1311 (m), 1183 (m), 1105 (m), 1034 (m), 888 (m), 849 (m), 808 (m), 760 (s), 701 (s), 649 (m), 474 (m). MS (EI, 70 eV): m/z (%) = 445 ([M]+, 100), 444 (80), 443 (43), 442 (20), 441 (15), 418 (8), 367 (8), 223 (10), 222 (22), 221 (14), 207 (7). HRMS (ESI-TOF): calculated for C32H20N3 ([M + H]+) 446.1657, found 446.1656.
:1). 1H NMR (300 MHz, CDCl3) δ = 8.77 (s, 1H), 8.13 (dd, 3J = 9.2 Hz, 4J = 5.5 Hz, 1H), 7.94 (d, 3J = 4.2 Hz, 1H), 7.82–7.76 (m, 2H), 7.73–7.66 (m, 3H), 7.59–7.47 (m, 4H), 7.46–7.39 (m, 3H), 7.18 (s, 1H), 7.10 (d, 3J = 4.2 Hz, 1H), 7.07–7.01 (m, 1H). 13C NMR (75 MHz, CDCl3/TFA) δ = 162.1 (d, 1J = 257.8 Hz, C), 158.2 (d, 4J = 5.8 Hz, C), 139.7 (C), 138.5 (CH), 137.6 (d, 4J = 2.0 Hz, C), 137.1, 135.2, 134.9 (C), 134.8 (CH), 134.1, 133.5 (C), 132.4, 131.1, 129.8 (CH), 128.7 (d, 3J = 8.7 Hz, C), 128.5 (d, 2J = 25.6 Hz, CH), 128.3, 126.7 (CH), 122.5 (C), 122.4 (d, 3J = 9.3 Hz, CH), 120.5, 118.6 (CH), 118.6, 118.3, 116.3 (C), 114.6 (CH), 113.1 (d, 2J = 25.1 Hz, CH). 19F NMR (282 MHz, CDCl3): δ = −112.8. IR (ATR, cm−1):
, 1337 (m), 1311 (m), 1183 (m), 1105 (m), 1034 (m), 888 (m), 849 (m), 808 (m), 760 (s), 701 (s), 649 (m), 474 (m). MS (EI, 70 eV): m/z (%) = 445 ([M]+, 100), 444 (80), 443 (43), 442 (20), 441 (15), 418 (8), 367 (8), 223 (10), 222 (22), 221 (14), 207 (7). HRMS (ESI-TOF): calculated for C32H20N3 ([M + H]+) 446.1657, found 446.1656.
:1). 1H NMR (300 MHz, CDCl3) δ = 8.77 (s, 1H), 8.13 (dd, 3J = 9.2 Hz, 4J = 5.5 Hz, 1H), 7.94 (d, 3J = 4.2 Hz, 1H), 7.82–7.76 (m, 2H), 7.73–7.66 (m, 3H), 7.59–7.47 (m, 4H), 7.46–7.39 (m, 3H), 7.18 (s, 1H), 7.10 (d, 3J = 4.2 Hz, 1H), 7.07–7.01 (m, 1H). 13C NMR (75 MHz, CDCl3/TFA) δ = 162.1 (d, 1J = 257.8 Hz, C), 158.2 (d, 4J = 5.8 Hz, C), 139.7 (C), 138.5 (CH), 137.6 (d, 4J = 2.0 Hz, C), 137.1, 135.2, 134.9 (C), 134.8 (CH), 134.1, 133.5 (C), 132.4, 131.1, 129.8 (CH), 128.7 (d, 3J = 8.7 Hz, C), 128.5 (d, 2J = 25.6 Hz, CH), 128.3, 126.7 (CH), 122.5 (C), 122.4 (d, 3J = 9.3 Hz, CH), 120.5, 118.6 (CH), 118.6, 118.3, 116.3 (C), 114.6 (CH), 113.1 (d, 2J = 25.1 Hz, CH). 19F NMR (282 MHz, CDCl3): δ = −112.8. IR (ATR, cm−1):  , 1459 (m), 1434 (m), 1356 (m), 1232 (m), 1175 (m), 1107 (m), 828 (m), 800 (m), 725 (m), 696 (s), 604 (m), 565 (m), 474 (m). MS (EI, 70 eV): m/z (%) = 463 ([M]+, 100), 462 (78), 461 (43), 460 (13), 459 (13), 436 (5), 385 (9), 232 (11), 231 (26), 230 (12), 217 (5). HRMS (ESI-TOF): calculated for C32H19FN3 ([M + H]+) 464.1563, found 464.1554.
:1). 1H NMR (500 MHz, CDCl3) δ = 8.76 (s, 1H), 8.21 (s, 1H), 8.14 (dd, 3J = 9.2 Hz, 4J = 5.5 Hz, 1H), 7.95 (d, 3J = 4.1 Hz, 1H), 7.73–7.68 (m, 2H), 7.51–7.46 (m, 1H), 7.27–7.23 (m, 1H), 7.22–7.19 (m, 2H), 7.17 (d, 3J = 4.1 Hz, 1H), 7.14 (s, 1H), 6.99–6.95 (m, 2H), 6.89–6.86 (m, 2H), 3.12 (s, 6H), 3.06 (s, 6H). 13C NMR (126 MHz, CDCl3) δ = 160.0 (d, 1J = 246.4 Hz, C), 150.8, 150.7 (C), 146.6 (d, 4J = 6.0 Hz, C), 145.3 (C), 145.1, 144.7 (CH), 144.6 (d, 4J = 2.4 Hz, C), 134.4, 133.9 (C), 130.9 (d, 3J = 8.9 Hz, CH), 129.5 (C), 129.4, 128.9 (CH), 128.8 (d, 3J = 9.2 Hz, C), 126.0, 125.4, 124.8 (C), 120.5 (d, 2J = 26.5 Hz, CH), 120.1, 118.3, 117.5 (C), 114.0, 113.5, 112.3, 110.9 (CH), 110.4 (d, 2J = 23.5 Hz, CH), 107.2 (CH), 40.4, 40.3 (CH3). 19F NMR (282 MHz, CDCl3): δ = −113.6. IR (ATR, cm−1):
, 1459 (m), 1434 (m), 1356 (m), 1232 (m), 1175 (m), 1107 (m), 828 (m), 800 (m), 725 (m), 696 (s), 604 (m), 565 (m), 474 (m). MS (EI, 70 eV): m/z (%) = 463 ([M]+, 100), 462 (78), 461 (43), 460 (13), 459 (13), 436 (5), 385 (9), 232 (11), 231 (26), 230 (12), 217 (5). HRMS (ESI-TOF): calculated for C32H19FN3 ([M + H]+) 464.1563, found 464.1554.
:1). 1H NMR (500 MHz, CDCl3) δ = 8.76 (s, 1H), 8.21 (s, 1H), 8.14 (dd, 3J = 9.2 Hz, 4J = 5.5 Hz, 1H), 7.95 (d, 3J = 4.1 Hz, 1H), 7.73–7.68 (m, 2H), 7.51–7.46 (m, 1H), 7.27–7.23 (m, 1H), 7.22–7.19 (m, 2H), 7.17 (d, 3J = 4.1 Hz, 1H), 7.14 (s, 1H), 6.99–6.95 (m, 2H), 6.89–6.86 (m, 2H), 3.12 (s, 6H), 3.06 (s, 6H). 13C NMR (126 MHz, CDCl3) δ = 160.0 (d, 1J = 246.4 Hz, C), 150.8, 150.7 (C), 146.6 (d, 4J = 6.0 Hz, C), 145.3 (C), 145.1, 144.7 (CH), 144.6 (d, 4J = 2.4 Hz, C), 134.4, 133.9 (C), 130.9 (d, 3J = 8.9 Hz, CH), 129.5 (C), 129.4, 128.9 (CH), 128.8 (d, 3J = 9.2 Hz, C), 126.0, 125.4, 124.8 (C), 120.5 (d, 2J = 26.5 Hz, CH), 120.1, 118.3, 117.5 (C), 114.0, 113.5, 112.3, 110.9 (CH), 110.4 (d, 2J = 23.5 Hz, CH), 107.2 (CH), 40.4, 40.3 (CH3). 19F NMR (282 MHz, CDCl3): δ = −113.6. IR (ATR, cm−1):  , 1523 (s), 1508 (s), 1428 (m), 1360 (s), 1335 (s), 1230 (m), 1191 (s), 1164 (s), 816 (s), 804 (s), 793 (s), 729 (m), 600 (m). MS (EI, 70 eV): m/z (%) = 550 ([M]+, 62), 549 (100), 548 (19), 535 (15), 534 (32), 520 (15), 518 (11), 479 (19), 274 (18), 252 (12). HRMS (EI): calculated for C36H28FN5 ([M]+) 550.2407, found 550.2407.
:1). 1H NMR (300 MHz, CDCl3) δ = 9.40 (s, 1H), 8.77 (d, 3J = 4.7 Hz, 1H), 8.51 (dd, 3J = 9.4 Hz, 4J = 4.5 Hz, 1H), 8.05–7.97 (m, 2H), 7.84 (s, 1H), 7.82–7.75 (m, 2H), 7.71 (d, 3J = 4.7 Hz, 1H), 7.64–7.56 (m, 2H), 7.50–7.43 (m, 2H), 7.40–7.31 (m, 3H). 13C NMR (75 MHz, CDCl3) δ = 164.7 (d, 1J = 256.9 Hz, C), 164.3 (d, 1J = 252.6 Hz, C), 162.0 (d, 1J = 258.1 Hz, C), 156.7 (d, 4J = 5.8 Hz, C), 138.8 (CH), 138.2 (C), 137.4 (d, 4J = 2.0 Hz, C), 137.0, 135.2 (C), 134.7 (CH), 134.4 (C), 130.2 (d, 3J = 8.8 Hz, CH), 130.2 (C), 129.2 (d, 3J = 8.3 Hz. CH), 129.1 (d, 4J = 4.2 Hz, C), 128.7 (d, 3J = 9.8 Hz, C), 128.4 (d, 2J = 26.7 Hz, CH), 122.7 (d, 3J = 9.2 Hz, CH), 122.3 (C), 120.8 (CH), 119.9 (d, 2J = 22.3 Hz, CH), 118.6 (CH), 118.3, 118.3 (C), 117.0 (d, 2J = 22.1 Hz, CH), 116.5 (C), 114.3 (CH), 112.7 (d, 2J = 24.9 Hz, CH). 19F NMR (282 MHz, CDCl3): δ = −102.9, −105.2, −108.5. IR (ATR, cm−1):
, 1523 (s), 1508 (s), 1428 (m), 1360 (s), 1335 (s), 1230 (m), 1191 (s), 1164 (s), 816 (s), 804 (s), 793 (s), 729 (m), 600 (m). MS (EI, 70 eV): m/z (%) = 550 ([M]+, 62), 549 (100), 548 (19), 535 (15), 534 (32), 520 (15), 518 (11), 479 (19), 274 (18), 252 (12). HRMS (EI): calculated for C36H28FN5 ([M]+) 550.2407, found 550.2407.
:1). 1H NMR (300 MHz, CDCl3) δ = 9.40 (s, 1H), 8.77 (d, 3J = 4.7 Hz, 1H), 8.51 (dd, 3J = 9.4 Hz, 4J = 4.5 Hz, 1H), 8.05–7.97 (m, 2H), 7.84 (s, 1H), 7.82–7.75 (m, 2H), 7.71 (d, 3J = 4.7 Hz, 1H), 7.64–7.56 (m, 2H), 7.50–7.43 (m, 2H), 7.40–7.31 (m, 3H). 13C NMR (75 MHz, CDCl3) δ = 164.7 (d, 1J = 256.9 Hz, C), 164.3 (d, 1J = 252.6 Hz, C), 162.0 (d, 1J = 258.1 Hz, C), 156.7 (d, 4J = 5.8 Hz, C), 138.8 (CH), 138.2 (C), 137.4 (d, 4J = 2.0 Hz, C), 137.0, 135.2 (C), 134.7 (CH), 134.4 (C), 130.2 (d, 3J = 8.8 Hz, CH), 130.2 (C), 129.2 (d, 3J = 8.3 Hz. CH), 129.1 (d, 4J = 4.2 Hz, C), 128.7 (d, 3J = 9.8 Hz, C), 128.4 (d, 2J = 26.7 Hz, CH), 122.7 (d, 3J = 9.2 Hz, CH), 122.3 (C), 120.8 (CH), 119.9 (d, 2J = 22.3 Hz, CH), 118.6 (CH), 118.3, 118.3 (C), 117.0 (d, 2J = 22.1 Hz, CH), 116.5 (C), 114.3 (CH), 112.7 (d, 2J = 24.9 Hz, CH). 19F NMR (282 MHz, CDCl3): δ = −102.9, −105.2, −108.5. IR (ATR, cm−1):  , 1461 (m), 1339 (m), 1224 (s), 1158 (m), 828 (s), 802 (s), 779 (m), 725 (m), 587 (m), 560 (s), 540 (s), 505 (s). MS (EI, 70 eV): m/z (%) = 499 ([M]+, 100), 498 (61), 497 (31), 459 (43), 458 (86), 457 (13), 250 (44), 249 (20), 248 (17), 239 (18), 229 (24). HRMS (ESI-TOF): calculated for C32H17F3N3 ([M + H]+) 500.1375, found 500.1377.
, 1461 (m), 1339 (m), 1224 (s), 1158 (m), 828 (s), 802 (s), 779 (m), 725 (m), 587 (m), 560 (s), 540 (s), 505 (s). MS (EI, 70 eV): m/z (%) = 499 ([M]+, 100), 498 (61), 497 (31), 459 (43), 458 (86), 457 (13), 250 (44), 249 (20), 248 (17), 239 (18), 229 (24). HRMS (ESI-TOF): calculated for C32H17F3N3 ([M + H]+) 500.1375, found 500.1377.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the State of Mecklenburg-Vorpommern is gratefully acknowledged.References
- H. Balli and M. Zeller, Neue Heteroarene: Synthese und spektrale Daten von Indolizino[6,5,4,3-aij]chinolin («Ullazin») und einigen Derivaten, Helv. Chim. Acta, 1983, 66, 2135–2139 CrossRef CAS.
- J. H. Delcamp, A. Yella, T. W. Holcombe, M. K. Nazeeruddin and M. Grätzel, The molecular engineering of organic sensitizers for solar-cell applications, Angew. Chem., Int. Ed., 2013, 52, 376–380 CrossRef CAS PubMed.
- (a) J. Feng, Y. Jiao, W. Ma, M. K. Nazeeruddin, M. Grätzel and S. Meng, First Principles Design of Dye Molecules with Ullazine Donor for Dye Sensitized Solar Cells, J. Phys. Chem. C, 2013, 117, 3772–3778 CrossRef CAS; (b) Q. Le Bao, S. Thogiti, G. Koyyada and J. H. Kim, Synthesis and photovoltaic performance of novel ullazine-based organic dyes for dye-sensitized solar cells, Jpn. J. Appl. Phys., 2019, 58, 12011 CrossRef; (c) C. Cebrián, Ullazine-based materials: towards novel opportunities in organic electronics, J. Mater. Chem. C, 2018, 6, 11943–11950 RSC.
- (a) Q. Ge, B. Li and B. Wang, Synthesis of substituted benzoijimidazo-2,1,5-dequinolizine by rhodium(III)-catalyzed multiple C-H activation and annulations, Org. Biomol. Chem., 2016, 14, 1814–1821 RSC; (b) Y. Guo, L. Zhang, C. Li, M. Jin, Y. Zhang, J. Ye, Y. Chen, X. Wu and X. Liu, BN/BO-Ullazines and Bis-BO-Ullazines: Effect of BO Doping on Aromaticity and Optoelectronic Properties, J. Org. Chem., 2021, 86, 12507–12516 CrossRef CAS PubMed; (c) C. Li, Y. Liu, Z. Sun, J. Zhang, M. Liu, C. Zhang, Q. Zhang, H. Wang and X. Liu, Synthesis, Characterization, and Properties of Bis-BN Ullazines, Org. Lett., 2018, 20, 2806–2810 CrossRef CAS PubMed; (d) P. Pierrat, S. Hesse, C. Cebrián and P. C. Gros, Controlling charge-transfer properties through a microwave-assisted mono- or bis-annulation of dialkynyl-N-(het)arylpyrroles, Org. Biomol. Chem., 2017, 15, 8568–8575 RSC; (e) S. Janke, S. Boldt, P. Nakielski, A. Villinger, P. Ehlers and P. Langer, Synthesis and Properties of 5-Azaullazines, J. Org. Chem., 2023, 88, 10470–10482 CrossRef CAS PubMed.
- S. Boldt, S. Parpart, A. Villinger, P. Ehlers and P. Langer, Synthesis and Properties of Aza-ullazines, Angew. Chem., Int. Ed., 2017, 56, 4575–4578 CrossRef CAS PubMed.
- (a) G. Zhang, P. Gautam and J. M. W. Chan, Symmetrical and unsymmetrical fluorine-rich ullazines via controlled cycloaromatizations, Org. Chem. Front., 2020, 7, 787–795 RSC; (b) H. Qiao, Y. Deng, R. Peng, G. Wang, J. Yuan and S. Tan, Effect of π-spacers and anchoring groups on the photovoltaic performances of ullazine-based dyes, RSC Adv., 2016, 6, 70046–70055 RSC; (c) D. Wan, X. Li, R. Jiang, B. Feng, J. Lan, R. Wang and J. You, Palladium-Catalyzed Annulation of Internal Alkynes: Direct Access to π-Conjugated Ullazines, Org. Lett., 2016, 18, 2876–2879 CrossRef CAS PubMed; (d) S. Mathew, N. A. Astani, B. F. E. Curchod, J. H. Delcamp, M. Marszalek, J. Frey, U. Rothlisberger, M. K. Nazeeruddin and M. Grätzel, Synthesis, characterization and ab initio investigation of a panchromatic ullazine–porphyrin photosensitizer for dye-sensitized solar cells, J. Mater. Chem. A, 2016, 4, 2332–2339 RSC; (e) K. Kanno, Y. Liu, A. Iesato, K. Nakajima and T. Takahashi, Chromium-mediated synthesis of polycyclic aromatic compounds from halobiaryls, Org. Lett., 2005, 7, 5453–5456 CrossRef CAS PubMed.
- (a) E. S. Larsen, G. Ahumada, P. R. Sultane and C. W. Bielawski, Stereoelectronically-induced allosteric binding: shape complementarity promotes positive cooperativity in fullerene/buckybowl complexes, Chem. Commun., 2022, 58, 6498–6501 RSC; (b) S. Li, Y. Sun, X. Li, O. Smaga, S. Koniarz, M. Stępień and P. J. Chmielewski, 1,3-Dipolar cycloaddition of polycyclic azomethine ylide to norcorroles: towards dibenzoullazine-fused derivatives, Chem. Commun., 2022, 58, 6510–6513 RSC.
- J. Zhou, W. Yang, B. Wang and H. Ren, Friedel-Crafts arylation for the formation of C(sp2)–C(sp2) bonds: a route to unsymmetrical and functionalized polycyclic aromatic hydrocarbons from aryl triazenes, Angew. Chem., Int. Ed., 2012, 51, 12293–12297 CrossRef CAS PubMed.
- D. Wang, Y. Liu, L. Wang, H. Cheng, Y. Zhang and G. Gao, Synthesis of π-extended dibenzo[d,k]ullazines by a palladium-catalyzed double annulation using arynes, Chin. Chem. Lett., 2021, 32, 1407–1410 CrossRef CAS.
- R. Berger, M. Wagner, X. Feng and K. Müllen, Polycyclic aromatic azomethine ylides: a unique entry to extended polycyclic heteroaromatics, Chem. Sci., 2015, 6, 436–441 RSC.
- S. Ito, Y. Tokimaru and K. Nozaki, Isoquinolino[4,3,2-de]phenanthridine: synthesis and its use in 1,3-dipolar cycloadditions to form nitrogen-containing polyaromatic hydrocarbons, Chem. Commun., 2015, 51, 221–224 RSC.
- M. Richter, M. Borkowski, Y. Fu, E. Dmitrieva, A. Popov, J. Ma, T. Marszalek, W. Pisula and X. Feng, Synthesis and Self-Assembly Behavior of Double Ullazine-Based Polycyclic Aromatic Hydrocarbons, Org. Mater., 2021, 03, 198–203 CrossRef CAS.
- S. Ito, Y. Tokimaru and K. Nozaki, Benzene-Fused Azacorannulene Bearing an Internal Nitrogen Atom, Angew. Chem., Int. Ed., 2015, 54, 7256–7260 CrossRef CAS PubMed.
- J. Hager, S. Kang, P. J. Chmielewski, T. Lis, D. Kim and M. Stępień, Acenaphthylene-fused ullazines: fluorescent π-extended monopyrroles with tunable electronic gaps, Org. Chem. Front., 2022, 9, 3179–3185 RSC.
- D. Miao, C. Aumaitre and J.-F. Morin, Photochemical synthesis of π-extended ullazine derivatives as new electron donors for efficient conjugated D–A polymers, J. Mater. Chem. C, 2019, 7, 3015–3024 RSC.
- L. S. Povarov, α,β-Unsaturated Ethers and their analogues in reactions of diene synthesis, Russ. Chem. Rev., 1967, 36, 656–670 CrossRef.
- (a) J. Clerigué, M. T. Ramos and J. C. Menéndez, Enantioselective catalytic Povarov reactions, Org. Biomol. Chem., 2022, 20, 1550–1581 RSC; (b) E. A. Kuznetsova, A. V. Smolobochkin, T. S. Rizbayeva, A. S. Gazizov, J. K. Voronina, O. A. Lodochnikova, D. P. Gerasimova, A. B. Dobrynin, V. V. Syakaev, D. N. Shurpik, I. I. Stoikov, A. R. Burilov, M. A. Pudovik and O. G. Sinyashin, Diastereoselective intramolecular cyclization/Povarov reaction cascade for the one-pot synthesis of polycyclic quinolines, Org. Biomol. Chem., 2022, 20, 5515–5519 RSC.
- B. Chakraborty, A. Kar, R. Chanda and U. Jana, Application of the Povarov Reaction in Biaryls under Iron Catalysis for the General Synthesis of Dibenzo[a,c]Acridines, J. Org. Chem., 2020, 85, 9281–9289 CrossRef CAS PubMed.
- F. Spruner von Mertz, R. Molenda, S. Boldt, A. Villinger, P. Ehlers and P. Langer, Synthesis and Properties of Diphenylbenzo[j]naphtho[2,1,8-def][2,7]phenanthrolines, Chem. – Eur. J., 2023, 29, e202204011 CrossRef CAS PubMed.
- R. Molenda, S. Boldt, A. Villinger, P. Ehlers and P. Langer, Synthesis of 2-Azapyrenes and Their Photophysical and Electrochemical Properties, J. Org. Chem., 2020, 85, 12823–12842 CrossRef CAS PubMed.
- S. Parpart, S. Boldt, P. Ehlers and P. Langer, Synthesis of Unsymmetrical Aza-Ullazines by Intramolecular Alkynyl-Carbonyl Metathesis, Org. Lett., 2018, 20, 122–125 CrossRef CAS PubMed.
- A. M. Brouwer, Standards for photoluminescence quantum yield measurements in solution (IUPAC Technical Report), Pure Appl. Chem., 2011, 83, 2213–2228 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, J. Williams, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev. C.01, Wallingford, CT, 2016 Search PubMed.
- (a) D. Bischof, M. W. Tripp, P. E. Hofmann, C.-H. Ip, S. I. Ivlev, M. Gerhard, U. Koert and G. Witte, Regioselective Fluorination of Acenes: Tailoring of Molecular Electronic Levels and Solid-State Properties, Chem. – Eur. J., 2022, 28, e202103653 CrossRef CAS PubMed; (b) J. Holec, B. Cogliati, J. Lawrence, A. Berdonces-Layunta, P. Herrero, Y. Nagata, M. Banasiewicz, B. Kozankiewicz, M. Corso, D. G. de Oteyza, A. Jancarik and A. Gourdon, A Large Starphene Comprising Pentacene Branches, Angew. Chem., Int. Ed., 2021, 60, 7752–7758 CrossRef CAS PubMed; (c) O. Francesconi, M. Martinucci, L. Badii, C. Nativi and S. Roelens, A Biomimetic Synthetic Receptor Selectively Recognising Fucose in Water, Chem. – Eur. J., 2018, 24, 6828–6836 CrossRef CAS PubMed.
- M.-L. Tan, S. Tong, S.-K. Hou, J. You and M.-X. Wang, Copper-Catalyzed N,N-Diarylation, of Amides for the Construction of 9,10-Dihydroacridine Structure and Applications in the Synthesis of Diverse Nitrogen-Embedded Polyacenes, Org. Lett., 2020, 22, 5417–5422 CrossRef CAS PubMed.
- J. Liu, C. Chen and C. Fang, Polarity-Dependent Twisted Intramolecular Charge Transfer in Diethylamino Coumarin Revealed by Ultrafast Spectroscopy, Chemosensors, 2022, 10, 411 CrossRef CAS.
- P. v. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. van Eikema Hommes, Nucleus-Independent Chemical Shifts: A Simple and Efficient Aromaticity Probe, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS PubMed.
- E. Paenurk and R. Gershoni-Poranne, Simple and efficient visualization of aromaticity: bond currents calculated from NICS values, Phys. Chem. Chem. Phys., 2022, 24, 8631–8644 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Single crystal X-ray data, 1H-, 19F, 13C-NMR spectra of isolated compounds; computational details. CCDC 2292306. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ob00091a |
| This journal is © The Royal Society of Chemistry 2024 |