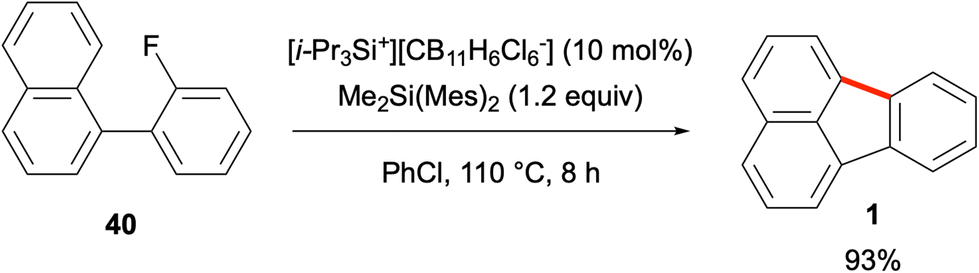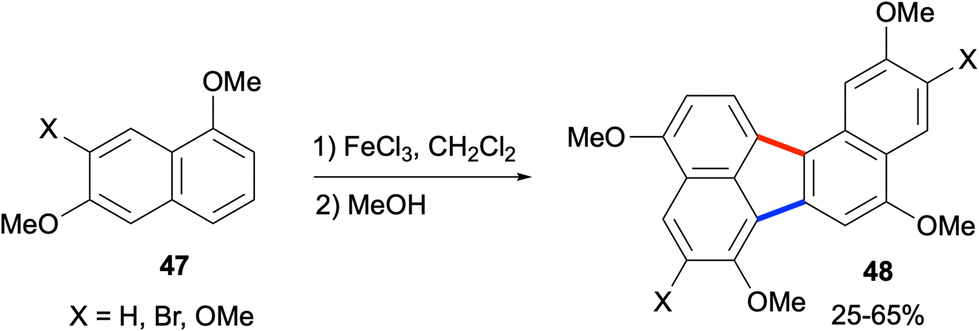DOI:
10.1039/D4OB00083H
(Review Article)
Org. Biomol. Chem., 2024,
22, 2719-2733
Recent advances in the synthesis and applications of fluoranthenes
Received
15th January 2024
, Accepted 4th March 2024
First published on 5th March 2024
Abstract
As an important subclass of polycyclic aromatic hydrocarbons (PAHs), fluoranthenes continue to attract significant attention in synthetic organic chemistry and materials science. In this article, an overview of recent advances in the synthesis of fluoranthene derivatives along with selected applications is provided. First, methods for fluoranthene synthesis with a classification based on strategic bond disconnections are discussed. Then, the total syntheses of natural products featuring the benzo[j]fluoranthene skeleton are covered. Finally, examples of important applications of a variety of fluoranthenes are summarized.
 Yunus Emre Türkmen | Yunus Emre Türkmen received B.Sc. and M.Sc. degrees from the Chemistry Department of Middle East Technical University (METU) in 2005 and 2006, respectively. Afterwards, he worked in the group of Prof. Viresh Rawal at the University of Chicago and received his PhD in 2012. In 2013, he moved to the UK to work as a postdoctoral fellow in the group of Prof. Varinder Aggarwal FRS at the University of Bristol. In 2015, he joined the Chemistry Department of Bilkent University as a faculty member. Some of his research interests are template-directed photochemical cycloaddition reactions, development of synthetic methods towards fluoranthenes, and new catalytic methods for accessing heterocyclic frameworks. He has been a recipient of the BAGEP (Young Scientist Awards Program) award by the Science Academy, Türkiye, in 2019, and the GEBİP (Outstanding Young Scientists) award by the Turkish Academy of Sciences in 2022. Also, he currently serves as an elected member of the International Chemistry Olympiad (IChO) Steering Committee. |
1. Introduction
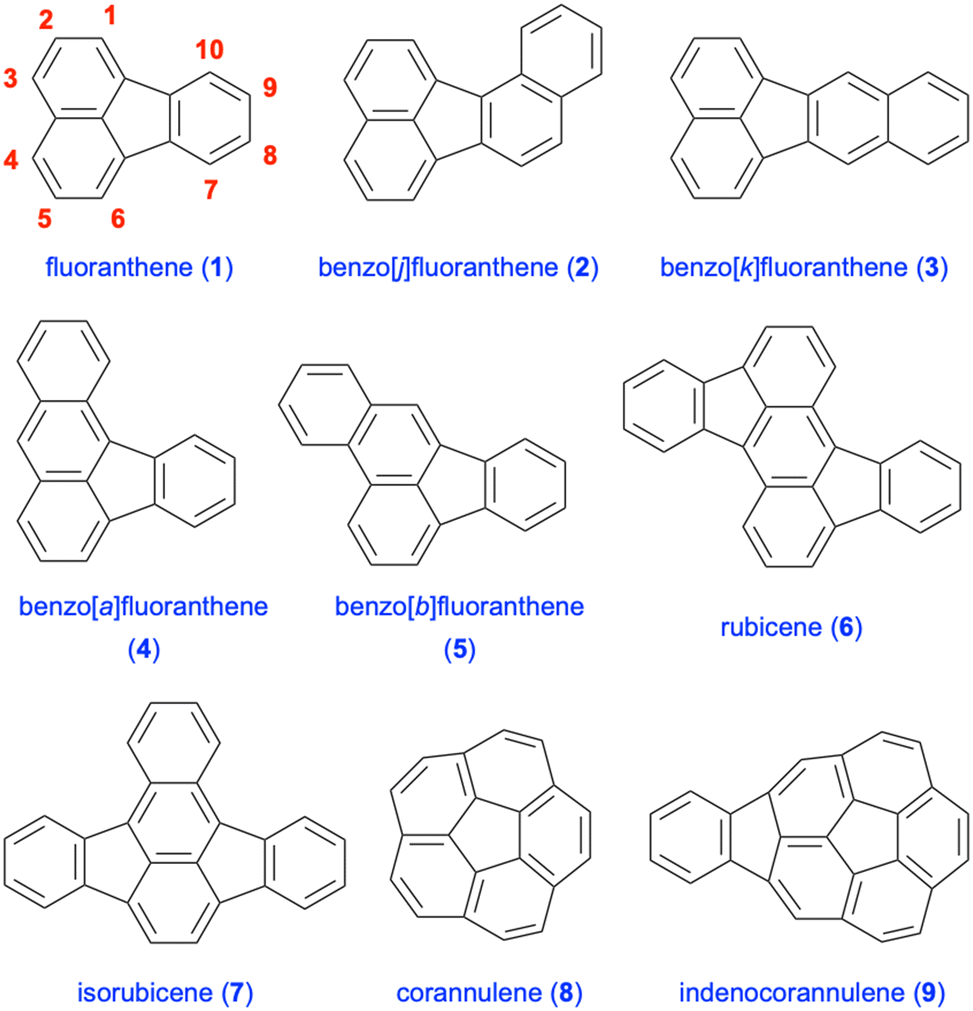
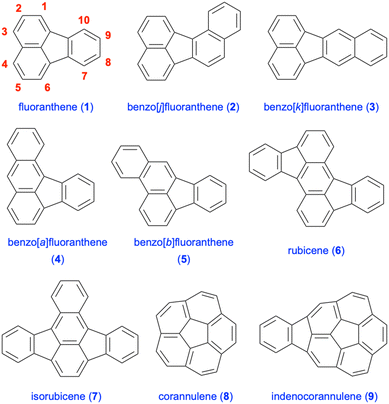
There is continued interest in the design and synthesis of polycyclic aromatic hydrocarbons (PAHs) in the synthetic community due to their attractive fundamental properties and diverse range of applications.1–3 The incorporation of five-membered rings into such polycyclic frameworks leads to the emergence of non-alternant hydrocarbon chemistry,4 in particular, cyclopentannulated PAHs.5,6 An important member of this PAH subclass is fluoranthene (1), which was isolated in 1878 by Fittig and Gebhardt from coal tar, and is a non-alternant, tetracyclic hydrocarbon featuring a combination of naphthalene and benzene rings (Fig. 1).7,8 Fluoranthenes have attracted significant attention because of their interesting photophysical and fluorescence properties,9 and have found a plethora of applications in materials science and organic electronics.6 Among the important fluoranthene-containing PAHs are benzofluoranthenes 2–5, rubicene (6) and isorubicene (7, Fig. 1). Moreover, fluoranthene-containing corannulene (8), indenocorannulene (9) and related bowl-shaped aromatic hydrocarbons have been the focus of immense research as they constitute fragments of fullerenes.10,11
 |
| | Fig. 1 Examples of important PAHs containing the fluoranthene core. | |
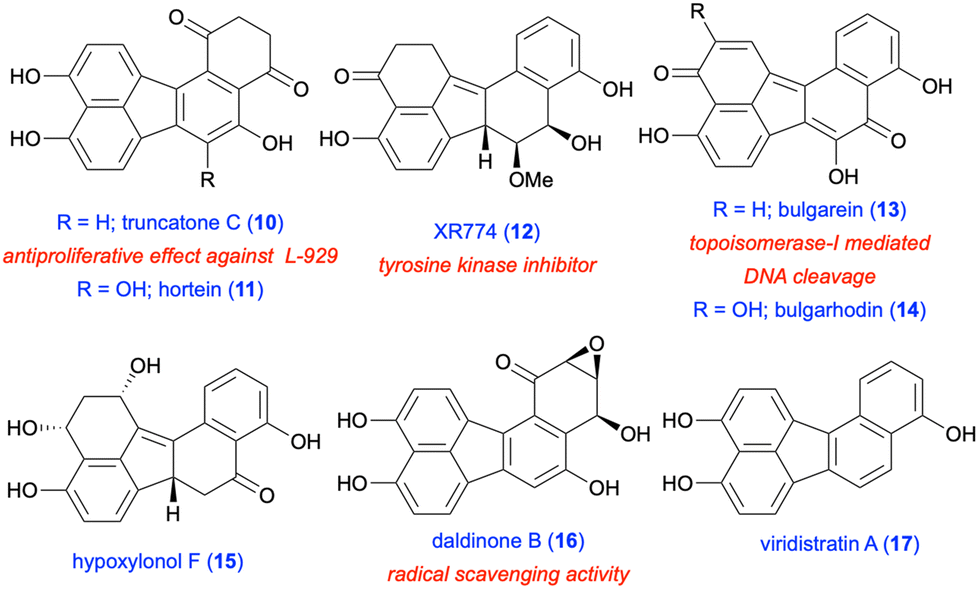
In addition to the examples given above, the structural motif of fluoranthene has been observed in a number of natural products, all of which were isolated from various fungal species.12 The chemistry and biological activities of such fluoranthene-based natural products have recently been reviewed by Podlech and Gutsche.13 All these natural products contain a highly oxygenated benzo[j]fluoranthene skeleton, most of which are dearomatized as a result of the oxidation of one or more of their electron-rich aromatic rings. The presence of the benzo[j]fluoranthene skeleton in these natural products is understandable as their biosyntheses were proposed to involve the oxidative dimerization of 1,8-dihydroxynaphthalene or 1,3,8-trihydroxynaphthalene.13 Among the 33 benzo[j]fluoranthene-derived natural products reported so far,13 only 4 have been synthesized, and these synthetic efforts are summarised in section 3 (vide infra). Selected examples of this class of natural products such as truncatone C (10),14 hortein (11),15 XR774 (12),16 bulgarein (13),17 bulgarhodin (14),17 hypoxylonol F (15),18 daldinone B (16),19,20 and viridistratin A (17)21 are shown in Fig. 2.
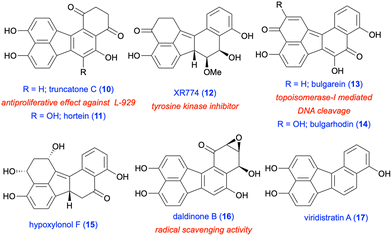 |
| | Fig. 2 Structures of selected natural products with the benzo[j]fluoranthene core. | |
A variety of PAHs including fluoranthene and benzofluoranthenes can be released into the atmosphere via incomplete combustion of fossil fuels, garbage and tobacco, as well as through natural sources such as volcanic eruptions and forest fires.22,23 In this respect, the health and environmental hazards of some polycyclic aromatic hydrocarbons have been the focus of extensive research due to their carcinogenic and mutagenic properties.22,24 Fluoranthene (1), benzo[k]fluoranthene (3) and benzo[b]fluoranthene (5) are some of the PAHs reported in the list of priority pollutants published by the United States Environmental Protection Agency (US-EPA) (Fig. 1).25 Moreover, in the toxic pollutant list published by the same agency, polynuclear aromatic hydrocarbons are listed as a general class of such pollutant compounds.25 The occurrence of 16 PAHs reported in the priority pollutant list of the US-EPA in foodstuffs was reviewed by Zelinkova and Wenzl in 2015.26 In addition, the toxicity of fluoranthene (1) and its biodegradation products to the aquatic environment was reported in 2003.27 In this regard, potential toxicological properties must be considered while designing new fluoranthene analogues, even though there is limited knowledge in the literature on the health and environmental hazards of substituted fluoranthenes and benzofluoranthenes.
This review is intended to give a critical overview of the recent advances in the synthesis and applications of fluoranthenes, rather than being comprehensive. Synthetic strategies towards fluoranthenes with a special emphasis on strategic bond disconnections, total syntheses of benzo[j]fluoranthene-based natural products, and selected applications of fluoranthenes will be discussed. It should be noted that the heterocyclic analogues of fluoranthenes such as azafluoranthenes28 will not be included in this review.
2. Strategies for the synthesis of fluoranthenes
2.1 Construction of the fluoranthene core via coupling of naphthalene and benzene fragments
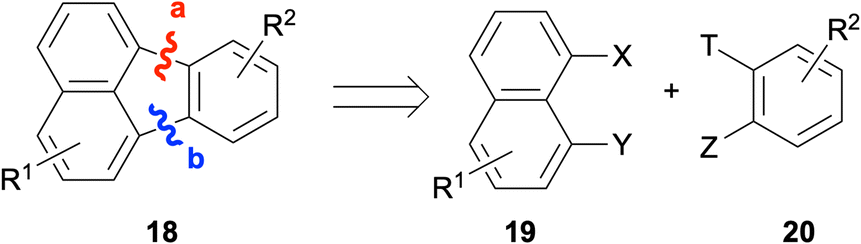
One of the most commonly employed strategies for the construction of fluoranthenes 18 is based on disconnections from bonds a and b, and thus, involves the coupling of a naphthalene fragment 19 and benzene fragment 20 (Scheme 1). While the formation of bonds a and b can be done sequentially in two separate steps, this approach is not very efficient as it requires the isolation of the product in the first step. Instead, the formation of both bonds under the same reaction conditions leads to the synthesis of fluoranthenes 18 directly, and therefore, this domino approach has attracted much attention in this area.
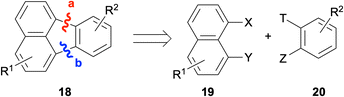 |
| | Scheme 1 Synthesis of fluoranthenes 18via disconnections from bonds a and b. | |
2.1.1 Pd-catalysed reactions.
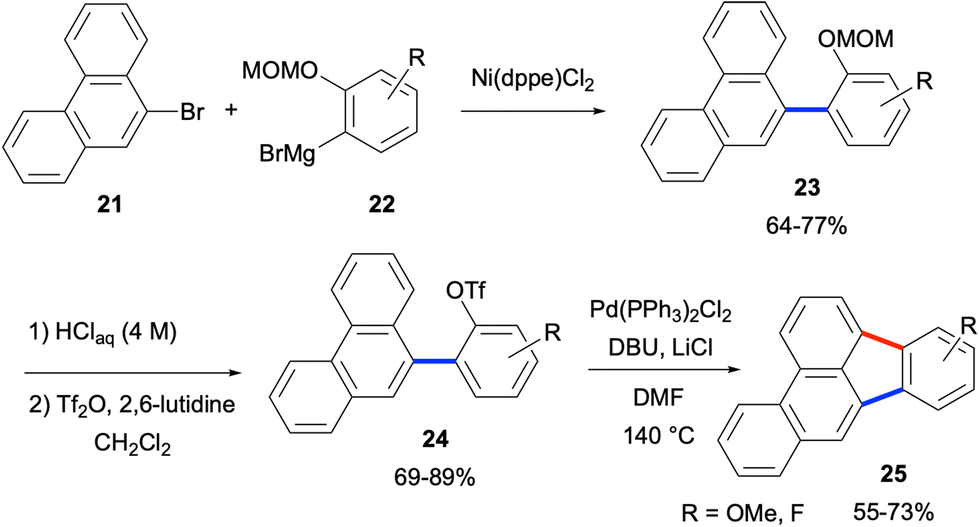
In this category, the initial formation of bond a is generally achieved by a Pd-catalysed cross-coupling reaction such as Suzuki–Miyaura29 or Kumada–Corriu30,31 couplings followed by an intramolecular C–H arylation reaction paving the way for the formation of bond b (Scheme 1). In this respect, the pioneering work of Rice and co-workers in 1993 showcased an effective way of fluoranthene synthesis, an example of which is shown in Scheme 2.32 The initial Ni-catalysed Kumada–Corriu coupling between 9-bromophenanthrene (21) and aryl Grignard reagents 22 gave coupling products 23 in good to high yields (64–77%). The conversion of aryl MOM ethers to aryl triflates 24 (69–89% yields) and their subsequent Pd-catalysed intramolecular C–H arylation reactions afforded benzo[b]fluoranthenes 25 in 55–73% yields (Scheme 2). Notably, it was shown that the same strategy could be applied to the syntheses of a variety of benzofluoranthenes.32 One drawback of this method is the use of MOM- or Me-ethers, the conversion of which to aryl triflates adds two extra steps to the whole synthetic sequence.
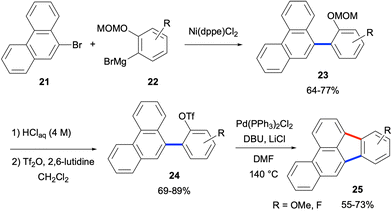 |
| | Scheme 2 Synthesis of fluoranthenes via intramolecular C–H arylation of aryl triflates. | |
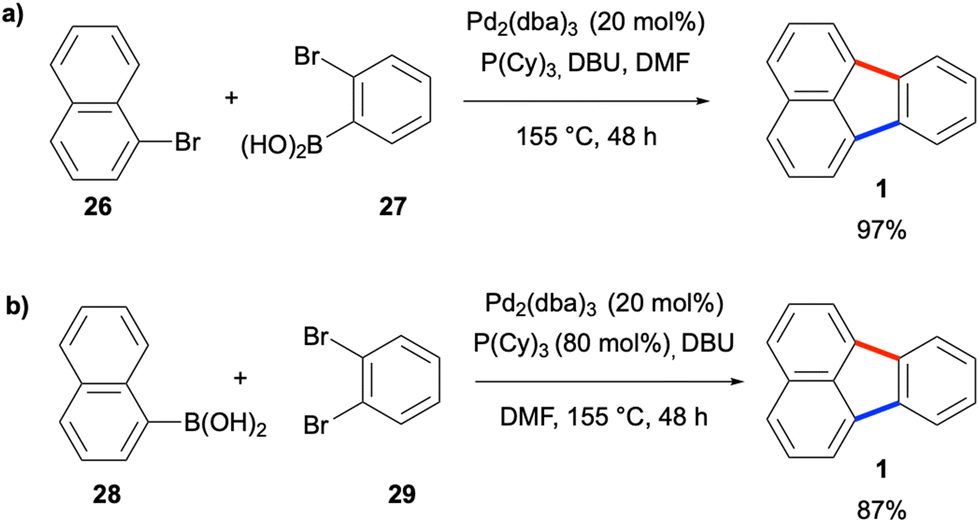
Another early work in this area was reported by de Meijere and co-workers in 2003, where the authors utilised a domino reaction which consists of sequential Pd-catalysed Suzuki–Miyaura and intramolecular C–H arylation reactions for the construction of fluoranthene analogues.33 In the first reaction type developed in this work, bromonaphthalene derivatives such as 26 were coupled with (2-bromophenyl)boronic acid (27) to afford fluoranthenes (Scheme 3a). In a second reaction type, the coupling of naphthalene-1-boronic acid (28) with 1,2-dibromobenzene (29) gave fluoranthene (1) in 87% yield (Scheme 3b). It should be noted that the second step of these transformations was referred to as the Heck-type coupling reaction by the authors. The second method was also successfully applied to the synthesis of indenocorannulene (9) in 40% yield. The major limitations of these two methods are relatively harsh reaction conditions (e.g., high Pd loading and reaction temperature) and a narrow substrate scope.
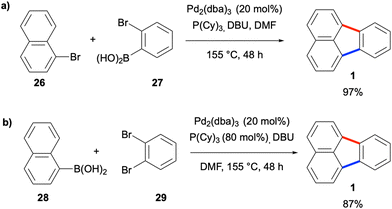 |
| | Scheme 3 Synthesis of fluoranthenes via sequential Suzuki–Miyaura coupling and intramolecular C–H arylation reactions. | |
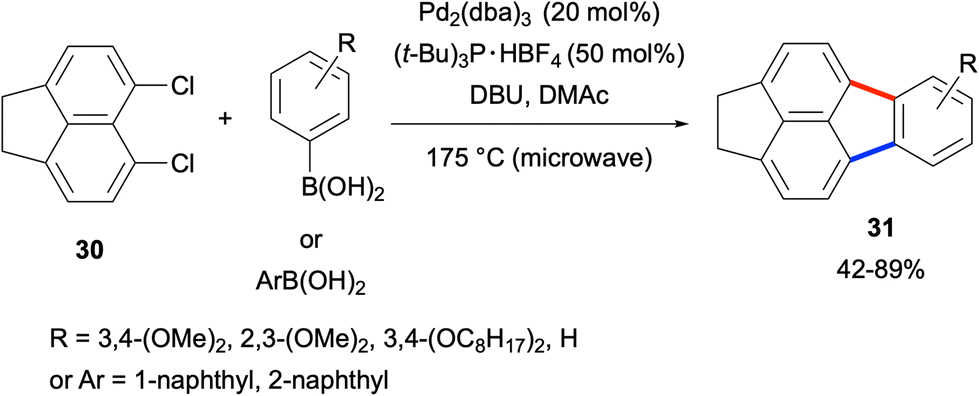
The fluoranthene synthesis methods described above involved the coupling reactions between mono-functionalised naphthalenes and di-functionalised benzene derivatives (Scheme 3). A different approach was undertaken by Quimby and Scott in 2009, where the coupling between the dichloronaphthalene derivative 30 and arylboronic acids was investigated (Scheme 4).34 With the use of 5,6-dichloroacenaphthene (30) as the naphthalene source, fluoranthene derivatives 31 were obtained in 42–89% yields. Similar to the above case, the high Pd loading (40 mol% Pd) and temperature (175 °C) as well as the use of only 30 as the naphthalene component are the limitations of this work.
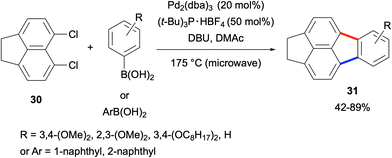 |
| | Scheme 4 Synthesis of fluoranthenes via the coupling of 5,6-dichloroacenaphthene (30) and arylboronic acids. | |
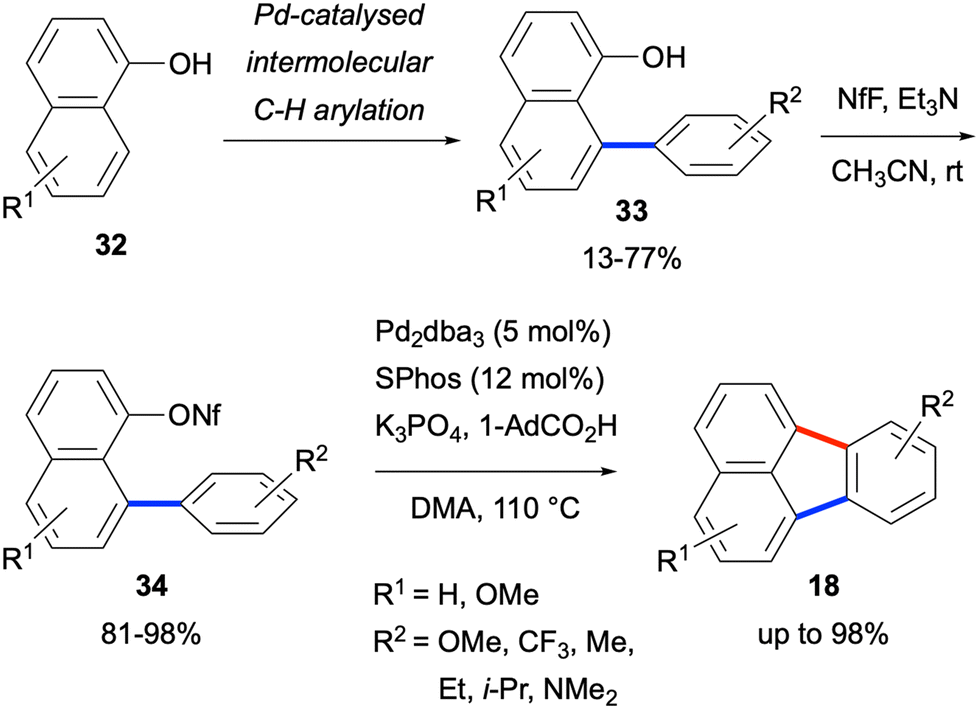
A three-step sequence to access fluoranthenes 18 was developed by Manabe and co-workers in 2016 starting from 1-naphthol derivatives 32 (Scheme 5).35 The sequence involved an initial Pd-catalysed intermolecular C–H arylation reaction of 1-naphthol 32 followed by the nonaflation of the resulting arylation products 33. A final Pd-catalysed intramolecular C–H arylation reaction of 34 resulted in the formation of fluoranthene products 18 in up to 98% yield. While this design led to an increase in the total number of synthetic steps, it gives the overall sequence of a modular character allowing the incorporation of substituents both into the phenyl and naphthalene rings.
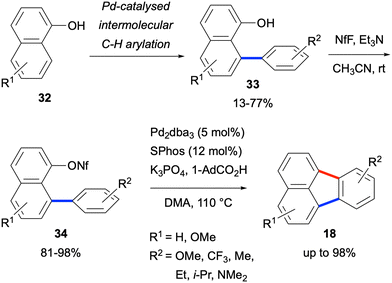 |
| | Scheme 5 Employment of intermolecular and intramolecular C–H arylation reactions to access substituted fluoranthenes. | |
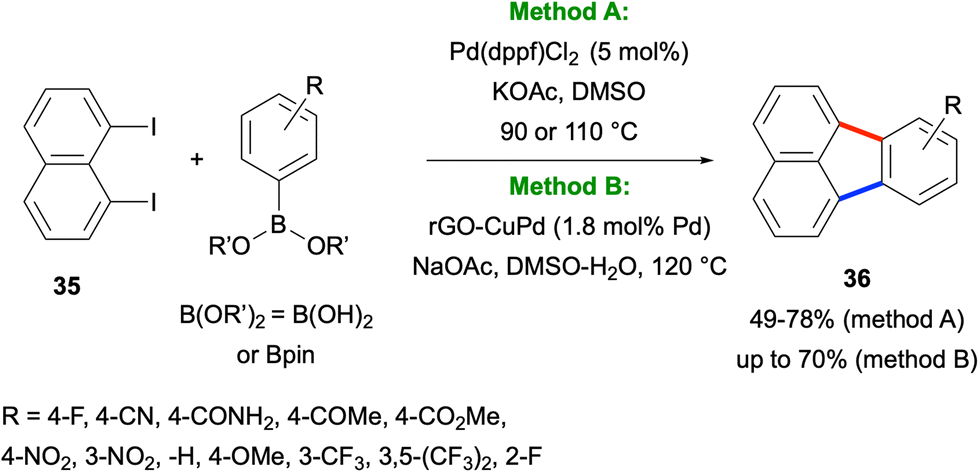
In 2017, Türkmen, Metin and co-workers reported a Pd-catalysed method for the synthesis of fluoranthenes 36 starting from 1,8-diiodonaphthalene (35) and arylboronic acids or pinacol esters (Scheme 6).36 The method works via an initial Suzuki–Miyaura coupling between the two reactants followed by an intramolecular C–H arylation reaction under the same reaction conditions. Importantly, this transformation was found to work successfully under both homogeneous and heterogeneous catalytic conditions. Although it tolerates a broad range of electron-withdrawing and electron-donating substituents on the phenyl ring, the use of only compound 35 as the naphthalene component is a limitation of this method.
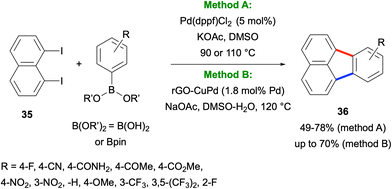 |
| | Scheme 6 Synthesis of fluoranthenes 36 from 1,8-diiodonaphthalene (35) under homogeneous and heterogeneous catalytic conditions. | |
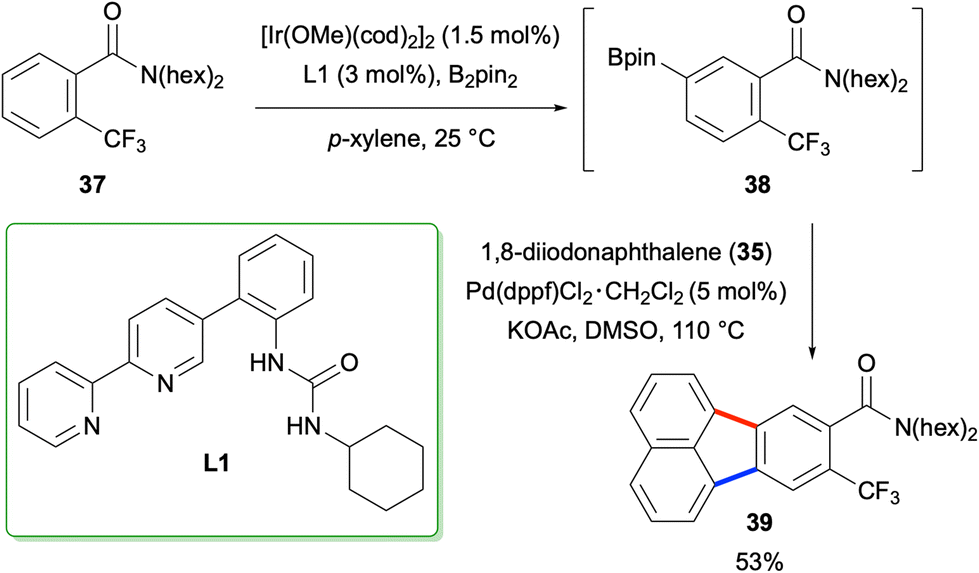
The method described above was employed by Kuninobu and co-workers for the synthesis of fluoranthene 39 in 2019 where the researchers utilised their newly developed meta-selective C–H borylation protocol for the synthesis of arylboronic ester intermediate 38 (Scheme 7).37
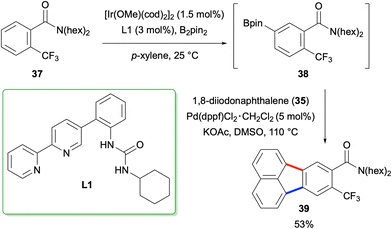 |
| | Scheme 7 Synthesis of fluoranthene 39 starting from amide 37. | |
In addition to the above examples, the Pd-catalysed intramolecular C–H arylation reaction was applied to the synthesis of a variety of fluoranthene-related PAHs including fluorescent dibenzo- and naphtho-fluoranthenes,38 unsymmetrical twistacenes,39 indeno[1,2,3-fg]tetracenes,40 electron-deficient polycyclic aromatic dicarboximides,41 pyrene-bridged acenaphthenes,42 bowl-shaped fullerene fragments,43 and tetra- and pentaindenocorannulenes.44–46 Interestingly, the synthesis of a uniformly 13C-labeled fluoranthene was demonstrated by Gold, Ball and co-workers in 2011 via a Pd-catalysed intramolecular C–H arylation protocol.47
2.1.2 Friedel–Crafts-type reactions.
Despite the importance of Friedel–Crafts reactions in fundamental organic chemistry, the use of aryl cations or their equivalents in such reactions is rather limited due to their unstable nature. In an elegant work by Siegel and co-workers reported in 2011, fluoroarene 40 was found to undergo an efficient Friedel–Crafts-type reaction to give fluoranthene (1) in 93% yield (Scheme 8).48 The reaction proceeds with the use of a sub-stoichiometric amount of triisopropylsilylium hexachlorocarborane (10 mol%) and 1.2 equivalents of dimethyldimesitylsilane at 110 °C. [i-Pr3Si+][CB11H6Cl6−] was proposed to act as an initiator via abstraction of fluoride from fluoroarene 40. After the intramolecular Friedel–Crafts-type cyclisation, the resulting fluoranthene-H+ was proposed to act as a Brønsted acid to protonate dimethyldimesitylsilane to form mesitylene and generate a Me2Si(Mes)+ silylium cation, which in turn would lead to another Friedel–Crafts reaction cycle. The greater strength of the Si–F bond compared to that of the C(aryl)–F bond is the driving force for this transformation. The developed catalytic method was also applied to the synthesis of other fluoranthene analogues, namely benzo[a]fluoranthene (4), rubicene (6), and indenocorannulene (9) in good yields (49–79%).
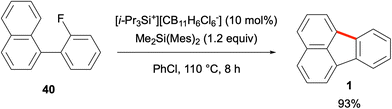 |
| | Scheme 8 Friedel–Crafts-type reaction of fluoroarene 40 to give fluoranthene (1). | |
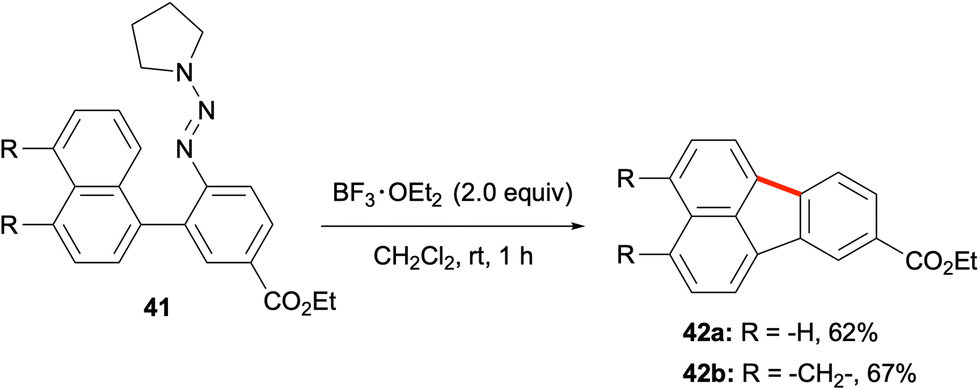
Another method for utilising in situ-generated aryl cations in the synthesis of PAHs including fluoranthenes was developed by Ren and co-workers in 2012 (Scheme 9).49 In this work, aryl triazenes were used as aryl diazonium salt equivalents. The treatment of the aryl triazene reactants with BF3·OEt2 as a Lewis acid was proposed to form the corresponding aryl diazonium, which would subsequently generate an aryl cation intermediate. The intramolecular trapping of this high energy intermediate with another aryl group would give the PAH product. In this respect, the reaction of aryl triazene 41 with BF3·OEt2 in CH2Cl2 at room temperature afforded fluoranthene analogues 42a and 42b in 62% and 67% yields, respectively (Scheme 9). Control experiments with radical trapping agents such as TEMPO, 1,1-diphenylethylene and BHT support the formation of the aryl cation intermediate. While this method holds potential for the synthesis of a variety of fluoranthenes, only two examples of fluoranthenes were reported in this study.
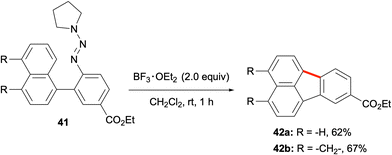 |
| | Scheme 9 Utilization of aryl triazenes as the aryl cation precursor in the synthesis of fluoranthenes 42. | |
The use of aryl diazoniums in the synthesis of fluoranthene analogues starting from aniline derivatives was demonstrated by Cho and co-workers in 2016.50 The reaction of anilines such as 43 with tBuONO leads to the in situ generation of the aryl diazonium intermediate 44 which was proposed to undergo cyclisation to give benzo[b]fluoranthene products 45 in moderate to good yields (Scheme 10; 20–75%). Compounds 46 as deamination side products were proposed to originate from a radical pathway. The method was also applied to the syntheses of benzo[a]fluoranthene (4), fluoranthene (1), and indeno[1,2,3-cd]pyrenes.
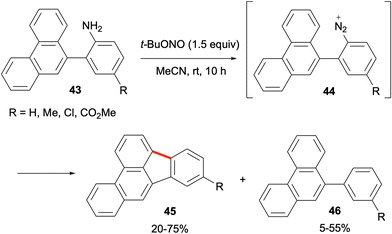 |
| | Scheme 10 Synthesis of benzo[b]fluoranthenes 45 starting from aniline derivatives 43. | |
2.1.3 Synthesis of benzo[j]fluoranthenes under oxidative or reductive couplings.
A final strategy based on the formation of bonds a and b (Scheme 1) for the synthesis of fluoranthenes involves the use of oxidative or reductive conditions for the coupling of two naphthalene-related fragments. In this context, an oxidative dimerization of electron-rich naphthalenes was demonstrated by Bushby and co-workers in 2014 to provide directly benzo[j]fluoranthenes (Scheme 11).51 When naphthalenes 47 were treated with FeCl3 as the oxidant followed by a reductive, non-aqueous work-up with MeOH, benzo[j]fluoranthene products 48 were isolated in moderate yields (25–65%). The reaction was reported not to work when X is iodine or 4-methoxyphenyl.
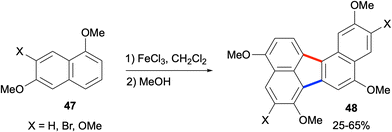 |
| | Scheme 11 Synthesis of benzo[j]fluoranthenes 48via oxidative dimerization of naphthalenes 47. | |
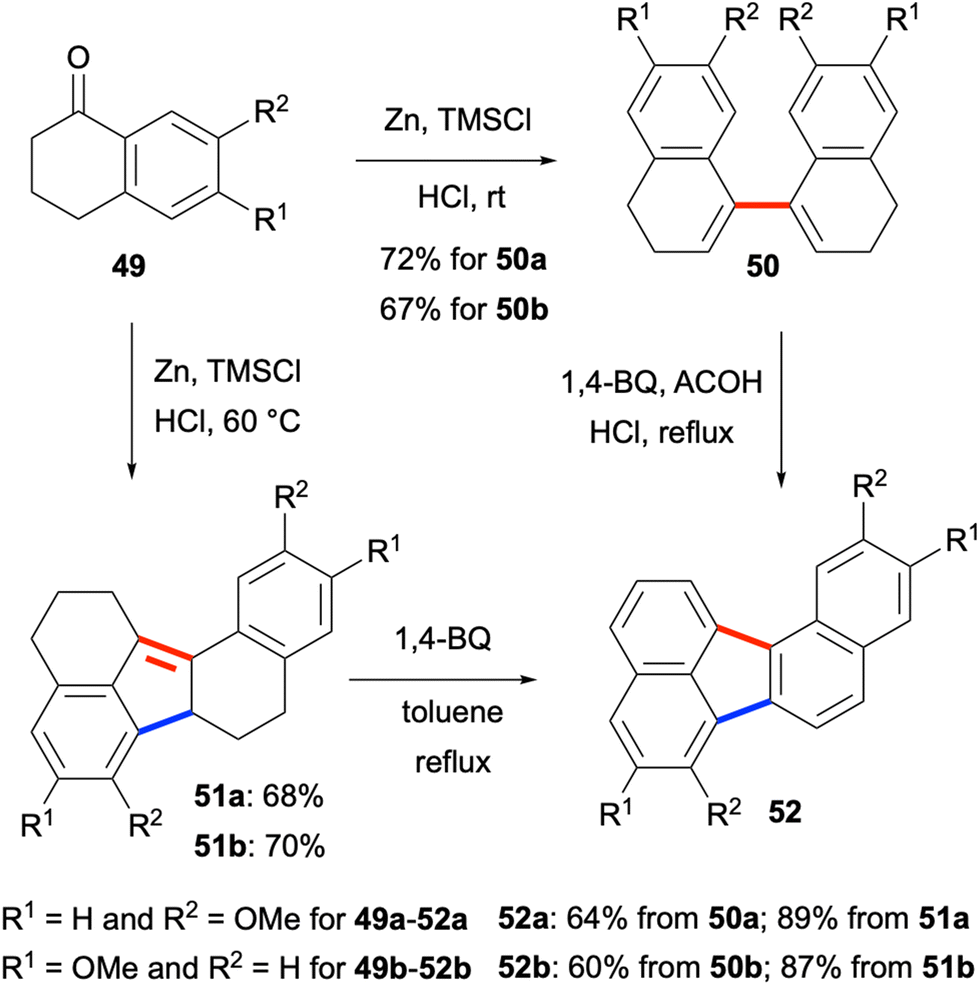
In 2015, methoxytetralones 49 were shown to be converted efficiently to dialkoxybenzo[j]fluoranthenes 52 by Lu, Zhao, Chen and co-workers (Scheme 12).52 Interestingly, the reductive homo-coupling of methoxytetralones 49 when treated with Zn, trimethylsilyl chloride (TMSCl) and HCl gave either bis(dihydronaphthalene) products 50 or hexahydro dialkoxybenzo[j]fluoranthenes 51 selectively depending on the reaction temperature. Oxidation of both compound types with excess 1,4-benzoquinone afforded the final dialkoxybenzo[j]fluoranthenes 52 in good to high yields (Scheme 12). The presence of the alkoxy groups makes these benzo[j]fluoranthenes highly electron rich. Their photophysical properties were investigated in solution (CH2Cl2) and in the solid state as spin-coated films, and they were found to exhibit strong fluorescence with large Stokes shifts in the solid state. Finally, one benzo[j]fluoranthene derivative was demonstrated to exhibit a favourable optical waveguide behaviour. Overall, the two methods mentioned in this category (section 2.1.3) have the ability to provide benzo[j]fluoranthenes in one or two steps in an efficient manner. However, the requirement to use the same naphthalene or tetralone reactants for the homo-coupling reactions is a major limitation.
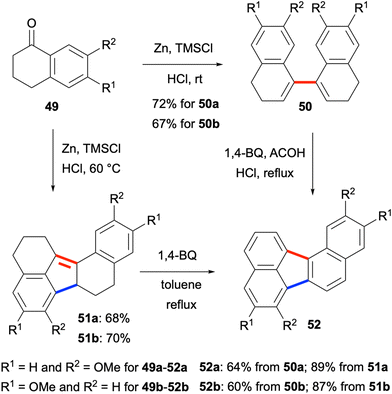 |
| | Scheme 12 Syntheses of fluoranthenes 52 starting from tetralones 49. | |
2.2 Synthesis of fluoranthenes via the de novo construction of benzene or naphthalene rings
In this section, fluoranthene synthesis methods which involve the de novo construction of the benzene ring and to a much lesser extent, the construction of the naphthalene ring will be summarized.
2.2.1 Intermolecular Diels–Alder reactions.
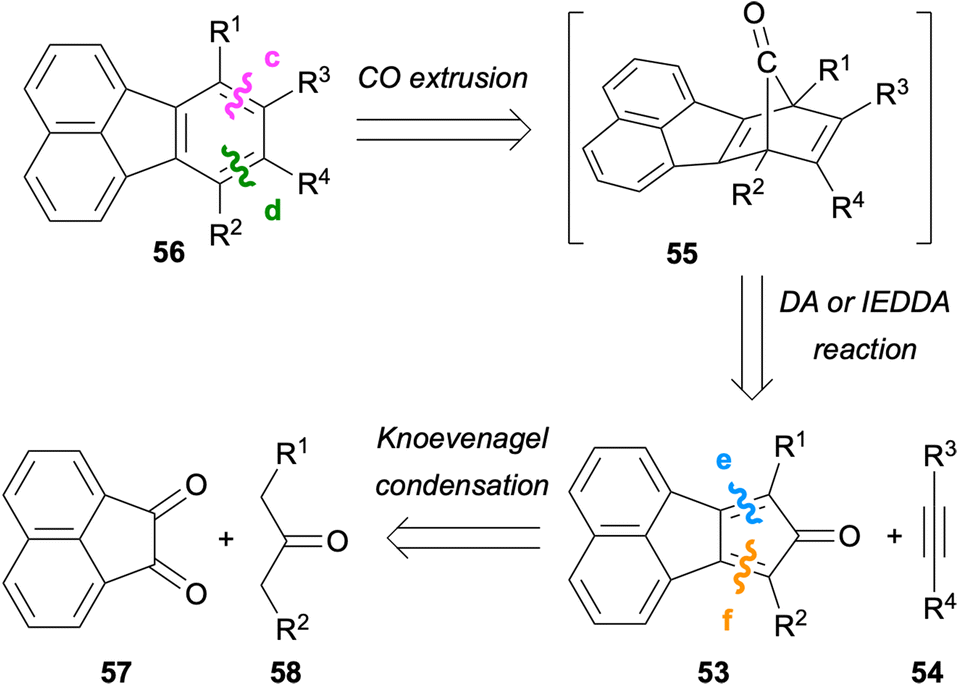
One of the most traditional methods for the synthesis of fluoranthenes involves intermolecular Diels–Alder reactions of naphthalene-fused cyclopentadienone derivatives.7,53 In this design, the Diels–Alder or inverse electron-demand Diels–Alder (IEDDA) reaction between cyclopentadienones 53 and alkynes 54 followed by a spontaneous cheletropic CO extrusion from intermediate 55 provide access to fluoranthenes 56 (Scheme 13). One advantage of this strategy is the easy preparation of cyclopentadienones 53via a double Knoevenagel condensation between diketone 57 and ketones 58. In addition, alkenes can also be used instead of alkynes as dienophiles in these DA or IEDDA reactions, which would give eventually aromatic fluoranthenes after dehydrogenative oxidation.53
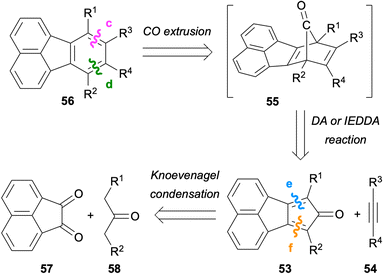 |
| | Scheme 13 Synthesis of fluoranthenes 56via disconnections from bonds c and d. | |
Using this strategy, Wudl and co-workers reported the design and synthesis of 7,8,10-triphenylfluoranthene (59) for applications in OLEDs in 2006 (Scheme 14).54 The double Knoevenagel condensation of acenaphthenequinone (57) and diphenylacetone (60) gave cyclopentadienone derivative 61 in 93% yield. A subsequent IEDDA reaction between 61 and phenylacetylene afforded fluoranthene 59 (66%), which was found to be an efficient blue-light emitter in the solid state.
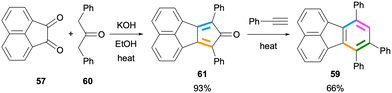 |
| | Scheme 14 Synthesis of fluoranthene 59via an intermolecular Diels–Alder reaction between 61 and phenylacetylene. | |
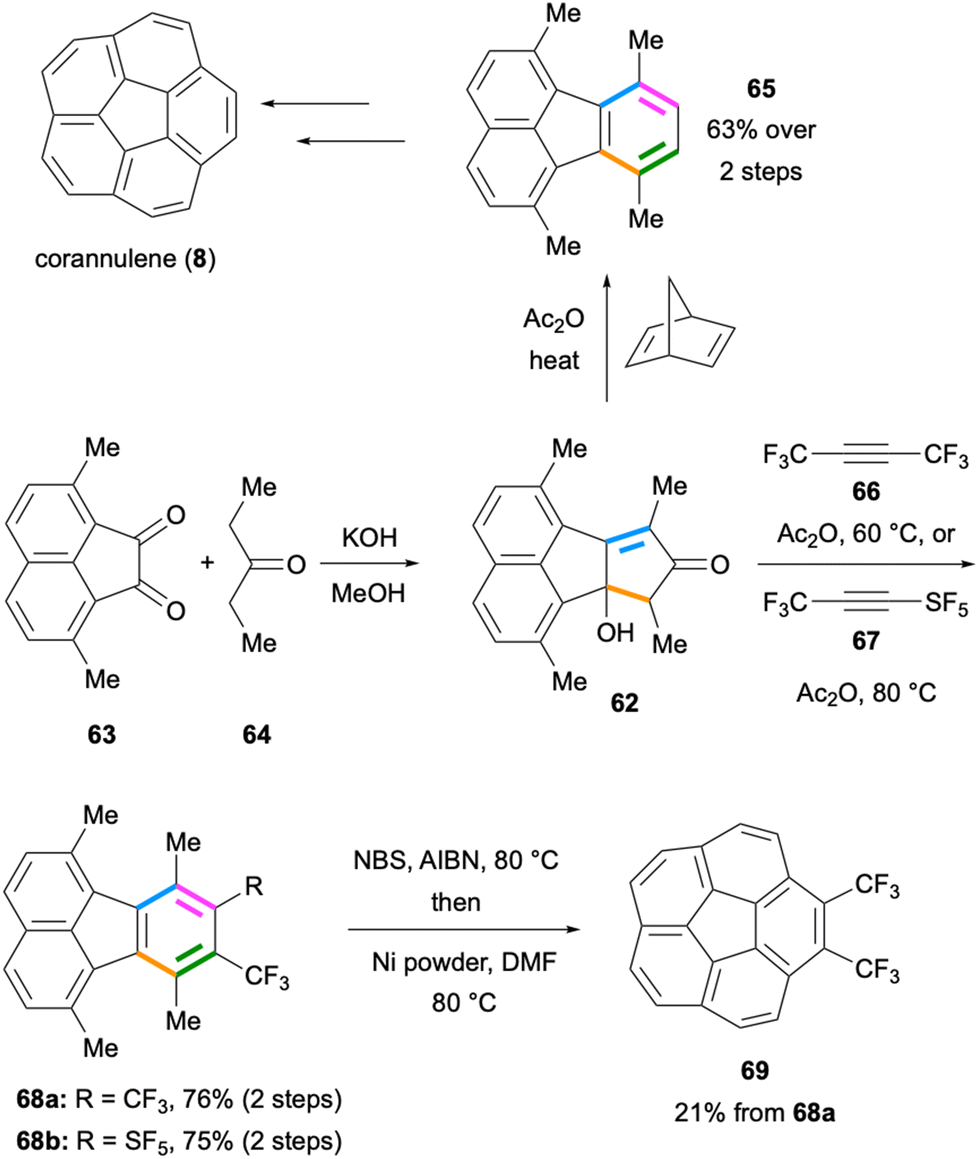
Two important applications of the intermolecular Diels–Alder reaction mentioned above were reported in early 1990s on the synthesis of corannulene (8) independently by the Scott55 and Siegel groups.56,57 In the latter synthesis, the cyclopentadienone precursor 62 was prepared by the double Knoevenagel-type reaction between diketone 63 and 3-pentanone (64) under basic conditions (Scheme 15). The Diels–Alder reaction between the cyclopentadienone formed from 62 and norbornadiene followed by CO extrusion and a retro-Diels–Alder reaction led to the formation of 1,6,7,10-tetramethylfluoranthene (65) which was further converted to corannulene (8) in a few steps. Remarkably, the Siegel group optimised their synthetic route, which enabled the production of corannulene (8) on a kilogram scale.58 A similar Diels–Alder approach was employed by Lentz and co-workers in 2012 for the synthesis of trifluoromethylated corannulenes.59 The Diels–Alder reaction of the in situ-formed cyclopentadienone with hexafluoro-2-butyne (66) gave fluoranthene 68a in 76% yield (Scheme 15). This fluoranthene product was further converted to the bis-trifluoromethylated corannulene analogue 69 in 21% yield. The same cyclopentadienone intermediate was utilised by Duda and Lentz in 2015 in a Diels–Alder reaction with the electron-deficient alkyne 67 as the dienophile (Scheme 15).60 In this reaction, the –SF5-substituted fluoranthene product 68b was isolated in 75% yield. These results underscore the utility of such intermolecular Diels–Alder reactions for the synthesis of electron-deficient fluoranthenes decorated with –CF3 and –SF5 groups. It is also worth noting that both these Diels–Alder reactions give high yields despite the fact that both the diene and dienophile components are electron-deficient.
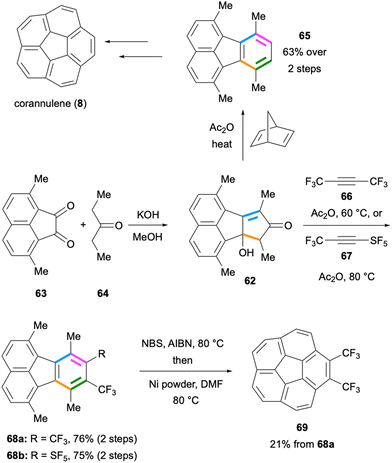 |
| | Scheme 15 Preparation of fluoranthenes 65 and 68via Diels–Alder reactions en route to the synthesis of corannulenes. | |
In addition to the above examples, the intermolecular Diels–Alder reaction discussed in this section has been utilised in the synthesis of substituted fluoranthenes to be used in a broad range of applications such as monomers in the design of covalent organic frameworks (COFs),61 semiconducting materials for thin film organic field-effect transistors (OFETs),62 fluorescent probes for the detection of nitroaromatic compounds including explosive ones,63,64 and as blue fluorescent and electron transport materials in OLEDs.65–67
2.2.2 Intermolecular formal [4 + 2] cycloadditions.
Along with the intermolecular Diels–Alder reactions mentioned in the previous section, the benzene unit of fluoranthenes can also be constructed via formal [4 + 2] cycloadditions through the disconnections of bonds e and f as shown in Scheme 16.
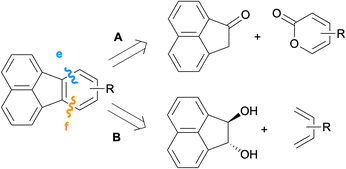 |
| | Scheme 16 Synthesis of fluoranthenes via disconnections from bonds e and f. | |
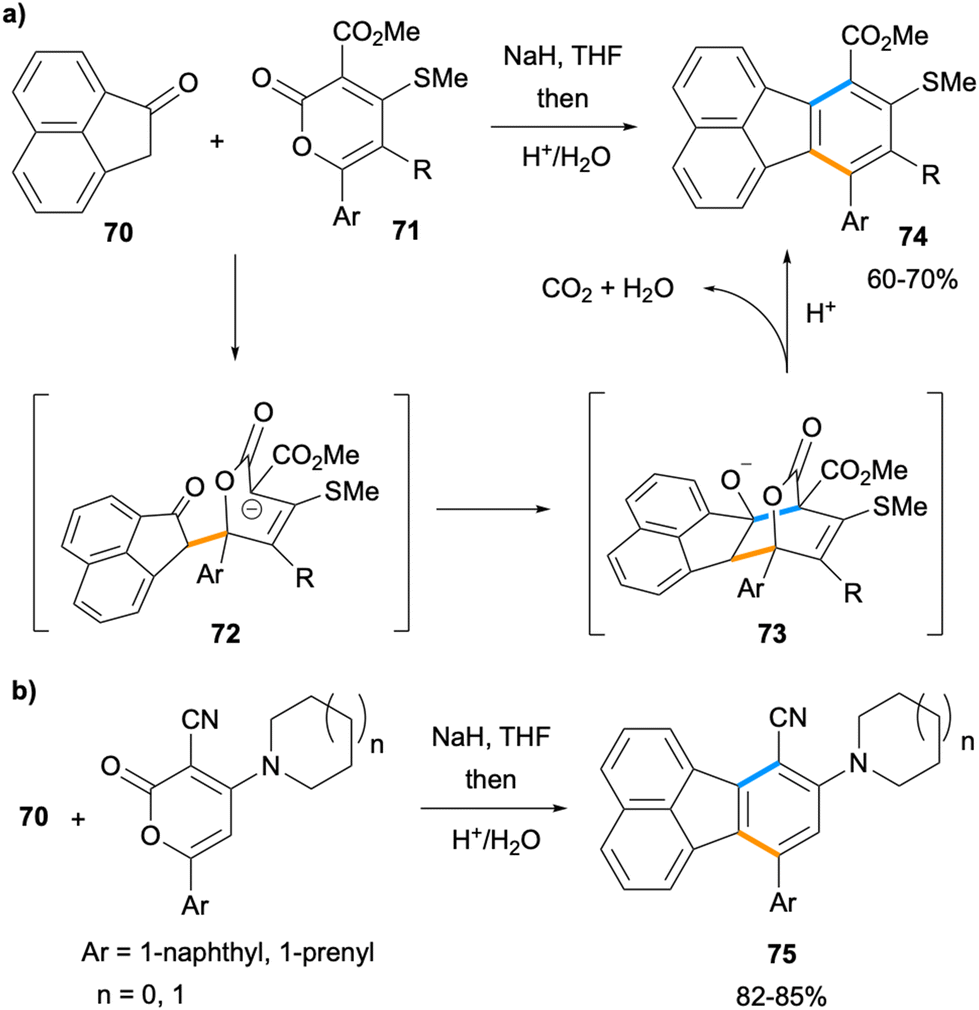
The synthesis of multi-substituted fluoranthenes was shown to be feasible by Goel and co-workers in 2010 by the reactions of ketone 70 with 2-pyrone derivatives (Scheme 17).68 In this method, the enolate of ketone 70 undergoes a stepwise [4 + 2] cycloaddition with 2-pyrone derivatives 71 through the conjugate addition intermediate 72. The acidic treatment of 73 leads to concomitant dehydration and decarboxylation to give the final fluoranthene products 74 in good yields (60–70%). The same approach was applied to the synthesis of donor–acceptor fluoranthenes 75 (82–85% yield) where the donor is an amine such as pyrrolidine or piperidine and the acceptor group is –CN. Compounds 75 turned out to be yellow light-emitting fluoranthene derivatives with emission λmax values of 549–552 nm. In particular, fluoranthene 75a, where the amine is piperidine and Ar is 1-naphthyl, was found to have high thermal stability, and its non-doped organic light emitting device (OLED) was shown to exhibit a low turn-on voltage and good luminance efficiency. It is noteworthy that this method allows the introduction of multiple substituents with electron-donating and electron-withdrawing properties to the fluoranthene core in a single transformation.
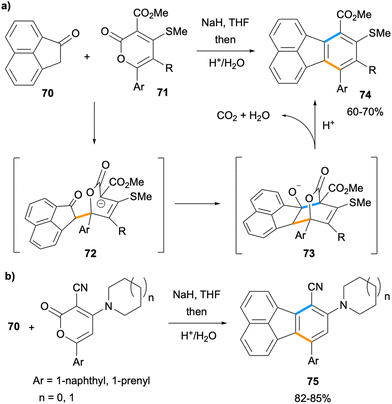 |
| | Scheme 17 Synthesis of fluoranthenes 74 and 75via formal [4 + 2] cycloaddition between ketone 70 and pyrones. | |
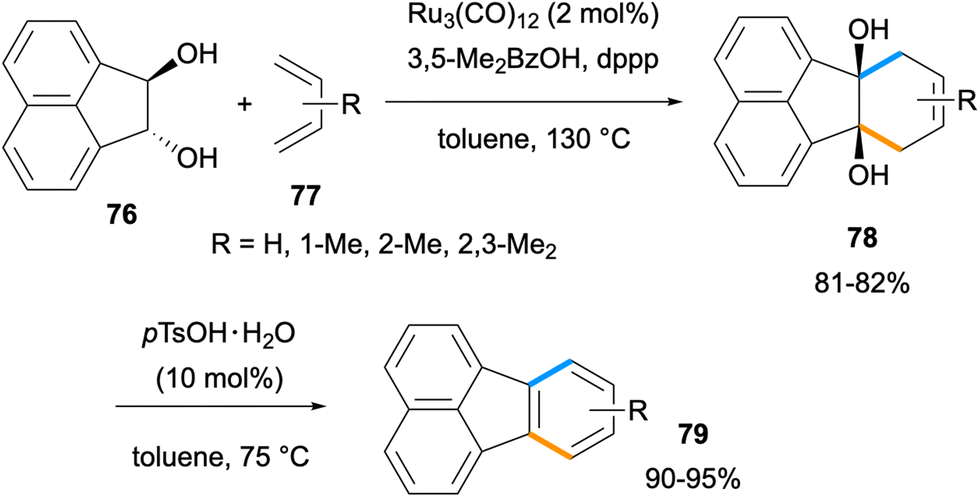
In 2014, Krische and co-workers reported an efficient method for the synthesis of acenes and fluoranthenes utilising a formal [4 + 2] cycloaddition between vicinal diols and dienes.69 The Ru-catalysed benzannulation between acenaphthalene diol 76 with dienes 77 gave cycloadducts 78 in 81–82% yields (Scheme 18). An acid-catalysed dehydration of diols 78 afforded fluoranthenes 79 in high yields (90–95%). A limitation of this study arises from the use of only two acenaphthalene diol derivatives for fluoranthene synthesis.
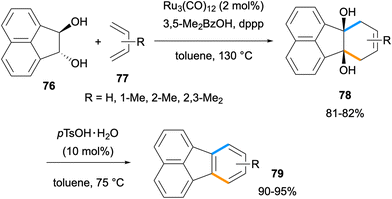 |
| | Scheme 18 Synthesis of fluoranthenes 79via a Ru-catalysed benzannulation of diol 76 with dienes 77. | |
2.2.3 Intramolecular Diels–Alder reactions.
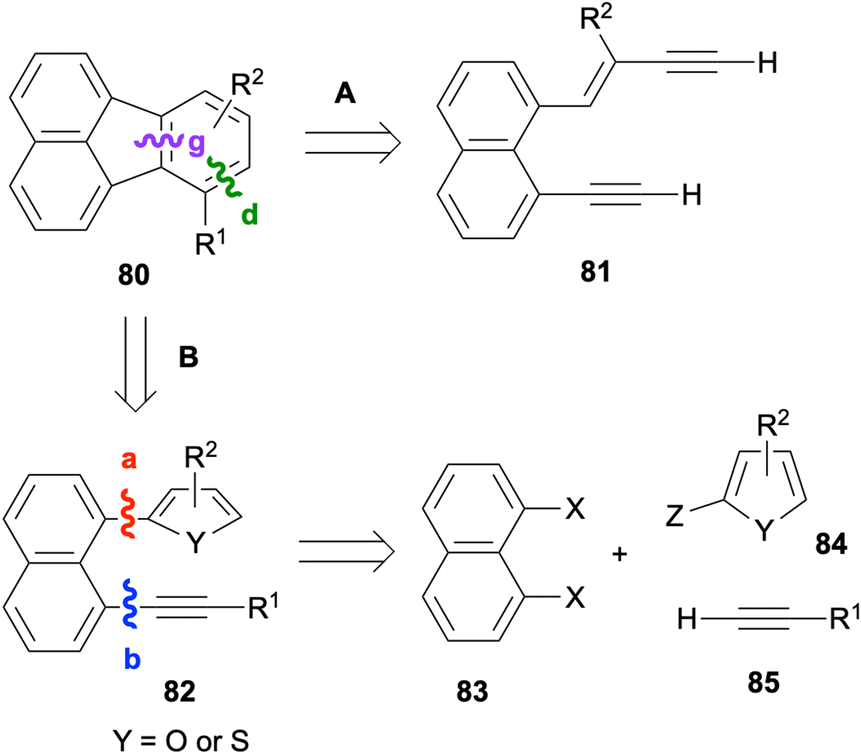
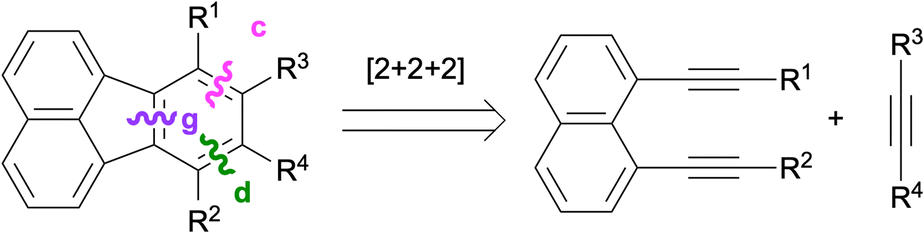
Intramolecular [4 + 2] cycloadditions provide effective ways of constructing the fluoranthene skeleton through the disconnections of bonds d and g (Scheme 19). Earlier examples of this strategy successfully utilised the dehydro-Diels–Alder reactions of 1,8-bis(arylethynyl)naphthalenes for the synthesis of fluoranthenes.70,71 A more recent method based on the dehydro-Diels–Alder reaction of dialkynes 81 is shown in path A in Scheme 19. Alternatively (path B), a regular intramolecular Diels–Alder reaction of 82 can be used for the synthesis of fluoranthenes 80 where naphthalene (83), diene (84) and dienophile (85) components can be assembled in a modular fashion (Scheme 19).
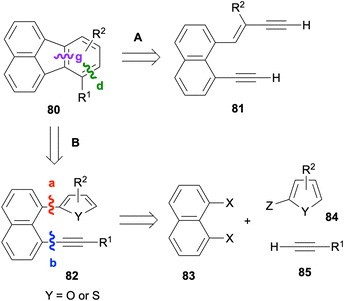 |
| | Scheme 19 Synthesis of fluoranthenes 80via disconnections from bonds d and g. | |
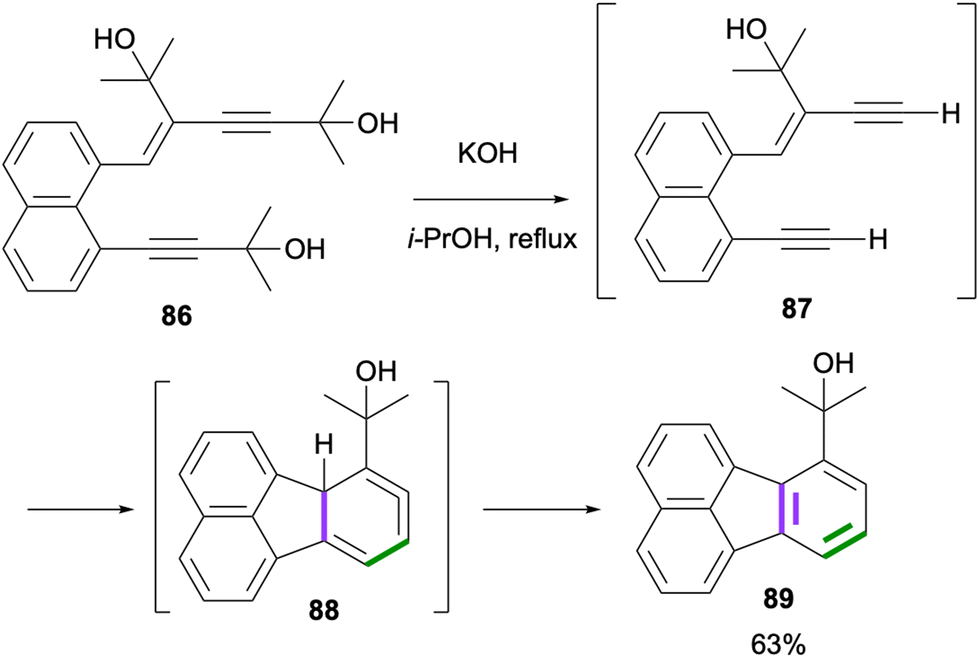
A dehydro-Diels–Alder reaction towards the synthesis of fluoranthenes was reported by Echavarren and co-workers in 1998.72 The initial deprotection of the alkyne groups of 86 under basic conditions gave 87, which underwent a dehydro-Diels–Alder reaction under the same conditions to afford the fluoranthene product 89 in 63% yield through the intermediacy of cyclic allene 88 (Scheme 20). A similar reaction was performed for the synthesis of unsubstituted fluoranthene (1) in 65% yield. The major drawback of this method is the specialized nature of the substrates leading to a limited reaction scope.
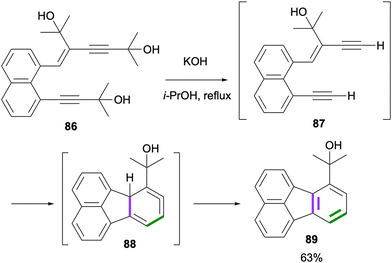 |
| | Scheme 20 Synthesis of fluoranthene 89via a dehydro-Diels–Alder reaction. | |
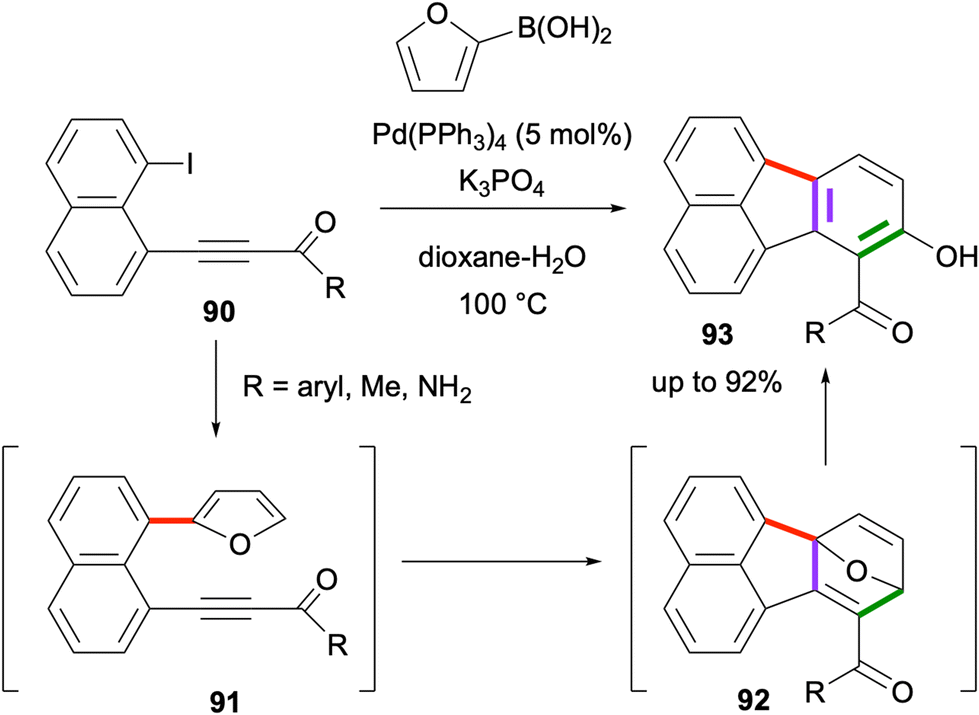
An effective domino reaction sequence towards the synthesis of hydroxyfluoranthenes was developed in 2022 by Türkmen and co-workers.73 In this domino sequence, the reaction of 1-alkynyl-8-iodonaphthalene derivatives 90 with furan-2-boronic acid under Pd catalysis initially gives the Suzuki–Miyaura coupling product 91 (Scheme 21). Under the same reaction conditions, this intermediate was found to undergo an intramolecular Diels–Alder reaction to form cycloadduct 92, which spontaneously gives the final hydroxyfluoranthene product 93via an aromatization-driven ring opening reaction. This three-step domino sequence gives good to high yields (up to 92%) when the alkyne is electron-deficient, whereas the reaction requires a higher temperature or gives a lower yield when arylacetylenes are used instead of alkynones as the dienophile moiety. Finally, the intramolecular Diels–Alder reaction was found not to proceed when furan was replaced with a benzofuran ring.74
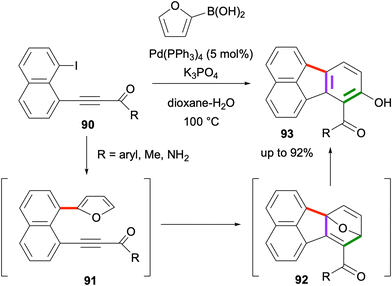 |
| | Scheme 21 Synthesis of hydroxyfluoranthenes 93via a three-step domino reaction sequence. | |
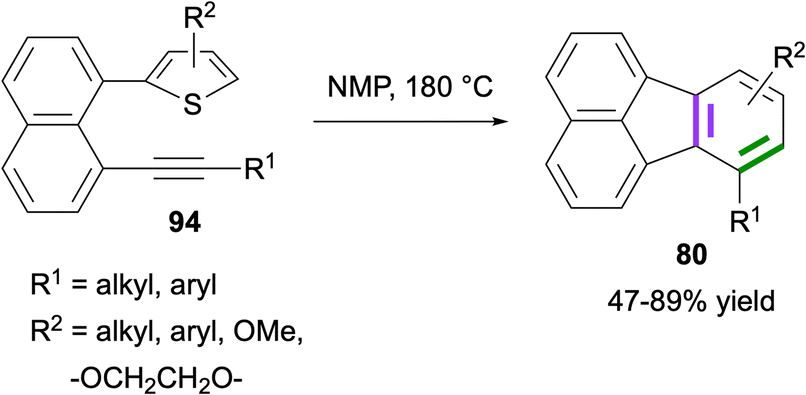
In 2023, Zhou and co-workers reported the synthesis of fluoranthenes 80via an intramolecular Diels–Alder reaction of 94 followed by subsequent spontaneous sulfur extrusion (Scheme 22).75 The reaction was found to have a broad substrate scope, and fluoranthenes 80 were isolated in 47–89% yields. It is important to note that while the X-ray structures of two fluoranthene products were included in the article, no other spectroscopic characterization data were reported for the products.
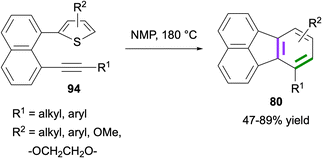 |
| | Scheme 22 Synthesis of fluoranthenes 80 starting from 94. | |
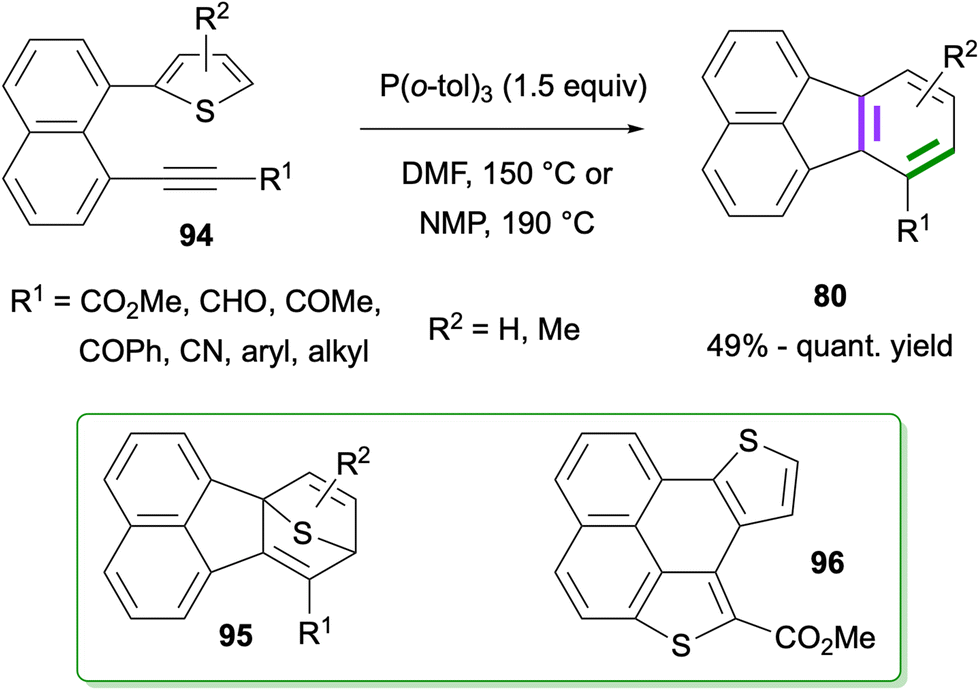
Recently, a similar intramolecular Diels–Alder reaction between thiophene and alkyne groups tethered by a naphthalene ring at the peri-positions was reported by Okitsu, Yakura and co-workers (Scheme 23).76 An initial intramolecular Diels–Alder reaction of 94 at 150 or 190 °C was proposed to give intermediate 95, which further afforded fluoranthenes 80 in good to excellent yields (49%-quant.) upon extrusion of sulfur. Importantly, the observation on the formation of side product 96 prompted the researchers to use P(o-tol)3 as a sulfur scavenger which indeed led to an increase in the reaction yield by reacting with the generated sulfur to give S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) P(o-tol)3. A broad range of functional groups on the alkyne as well as the –Me substituent at different positions of the thiophene ring were found to tolerate the reaction giving fluoranthenes 80 in high yields.
P(o-tol)3. A broad range of functional groups on the alkyne as well as the –Me substituent at different positions of the thiophene ring were found to tolerate the reaction giving fluoranthenes 80 in high yields.
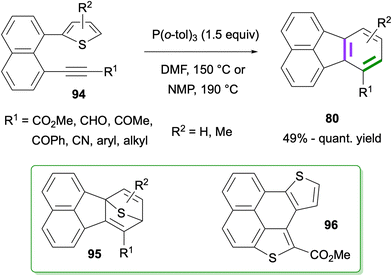 |
| | Scheme 23 Synthesis of fluoranthenes 80via an intramolecular Diels–Alder reaction followed by sulfur extrusion facilitated by the use of P(o-tol)3 as a sulfur scavenger. | |
2.2.4 [2 + 2 + 2] cycloadditions.
Another cycloaddition reaction which proved to be useful for fluoranthene synthesis is a transition metal-catalysed [2 + 2 + 2] reaction. This type of cycloaddition paves the way for the formation of bonds c, d and g in a single step leading to fluoranthenes in an efficient manner (Scheme 24).
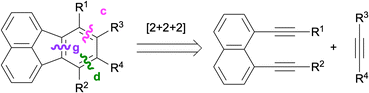 |
| | Scheme 24 Synthesis of fluoranthenes via disconnections from bonds c, d and g. | |
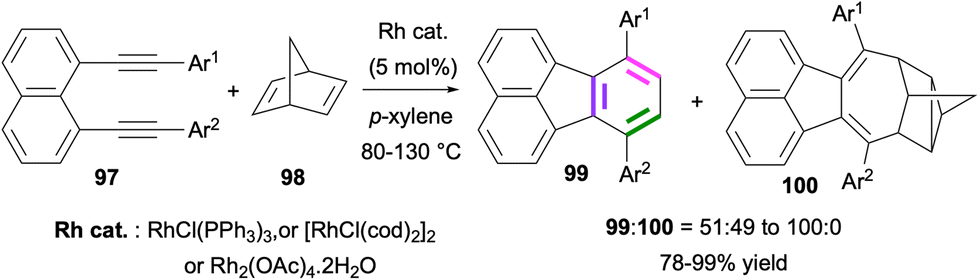
In 2005, Siegel and co-workers reported a Rh-catalysed [2 + 2 + 2] cycloaddition for the synthesis of fluoranthenes.77 The reaction of 1,8-dialkynylnaphthalenes 97 with norbornadiene (98) under Rh catalysis provided access to fluoranthene products 99 along with 100 as an unusual side product with moderate to high selectivity (Scheme 25). The formation of fluoranthenes 99 can be rationalized by an initial [2 + 2 + 2] cycloaddition followed by a spontaneous thermal elimination of cyclopentadiene. The structure of a derivative of 100 was confirmed by single-crystal X-ray analysis. This reaction was further extended by Siegel and co-workers in 2006 to the use of alkynes as [2 + 2 + 2] cycloaddition partners instead of norbornadiene for the synthesis of fluoranthenes and indenocorannulenes.78
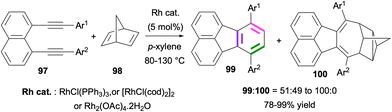 |
| | Scheme 25 Synthesis of fluoranthenes 99via Rh-catalysed [2 + 2 + 2] cycloadditions of dialkynes 97 with norbornadiene (98). | |
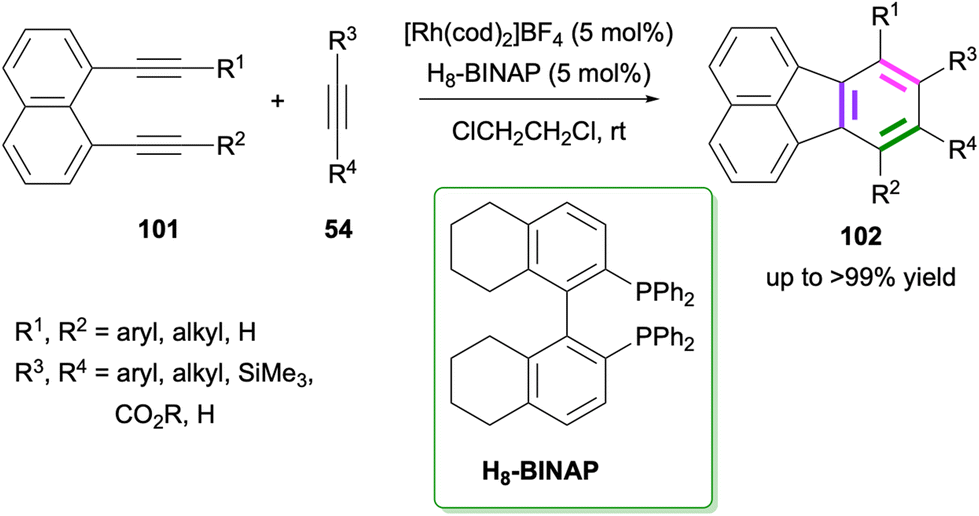
Another Rh-catalysed [2 + 2 + 2] cycloaddition between 1,8-dialkynylnaphthalenes 101 and alkynes 54 to give fluoranthenes 102 was reported by Nagashima, Tanaka and co-workers in 2023 (Scheme 26).79 The reaction works successfully with 5 mol% catalyst loading at room temperature affording di-, tri- and tetra-substituted fluoranthenes in up to >99% yield. Importantly, the reaction tolerates the presence of internal and terminal alkynes on both the diyne and alkyne reaction partners. Nitrile and isocyanates were also shown to be competent reaction partners instead of alkynes affording azafluoranthene products under the optimised reaction conditions. Experimental mechanistic studies and DFT calculations revealed that attractive C–H⋯π and π⋯π non-covalent interactions between the ligand and aromatic rings of the substrates facilitate the reaction by stabilising the transition state.
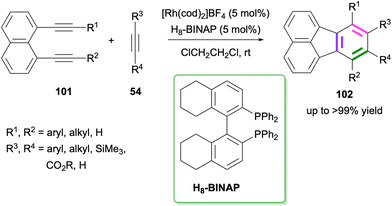 |
| | Scheme 26 Synthesis of fluoranthenes 102via Rh-catalysed [2 + 2 + 2] cycloadditions of dialkynes 101 with alkynes 54. | |
In relation to the methods covered in this section, an I2-mediated cyclisation of 1,8-dialkynylnaphthalenes was developed by Wang and co-workers in 2011 leading to fluoranthenes via the formation of bonds c, d and g, as shown in Scheme 24.80
2.2.5 Hydroarylation reactions.
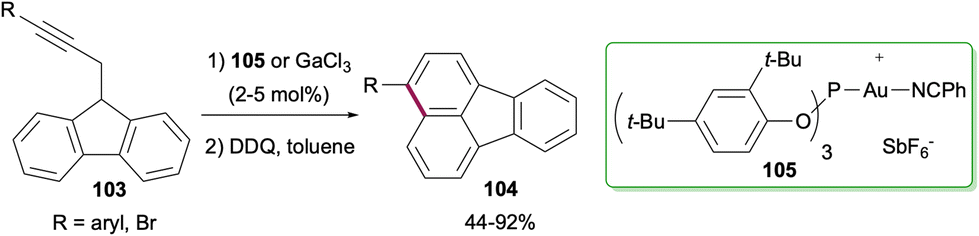
Synthesis of fluoranthenes through the construction of the naphthalene ring is much less common compared to the methods that proceed via the construction of the phenyl ring. One such attempt was reported by Echavarren and co-workers in 2011 where an intramolecular hydroarylation reaction of propargylic fluorenes 103 afforded fluoranthenes 104 having aryl or Br substituents on the naphthalene ring in good to high yields (44–92%; Scheme 27).81 This transformation was found to be catalysed effectively either by GaCl3 or the electrophilic Au(I) catalyst 105.
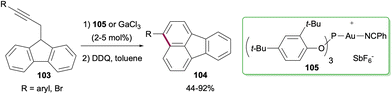 |
| | Scheme 27 Synthesis of fluoranthenes 104via the construction of the naphthalene ring. | |
3. Total synthesis of natural products with the benzo[j]fluoranthene core
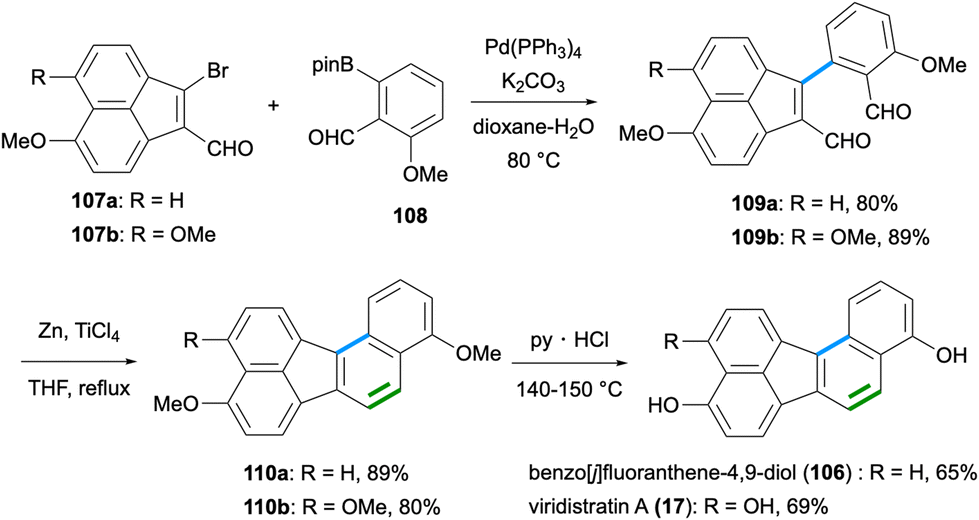
The synthesis of benzo[j]fluoranthene-4,9-diol (106),82 which also constitutes the first total synthesis of a fluoranthene-based natural product, was reported by Dallavalle and co-workers in 2013 (Scheme 28).83 Later, the name viridistratin D was proposed for this natural product by Podlech and Gutsche.13 In this total synthesis,83 a Pd-catalysed Suzuki–Miyaura reaction between vinyl bromide 107a and aryl pinacol boronic ester 108 gave the cross-coupling product 109a in 80% yield. A subsequent intramolecular McMurry coupling between the two aldehydes of 109a mediated by TiCl4 and Zn afforded the benzo[j]fluoranthene product 110a in 89% yield. A final deprotection of the methyl ethers by pyridinium chloride at high temperature gave the fungal natural product 106 in good yield (65%). It should be noted that the same synthetic sequence starting from 107b was applied by the Dallavalle group to the synthesis of the benzo[j]fluoranthene analogue 17,83 which later turned out to be a natural product itself and was named viridistratin A after its isolation from Annulohypoxylon viridistratum in 2020 (Scheme 28).21
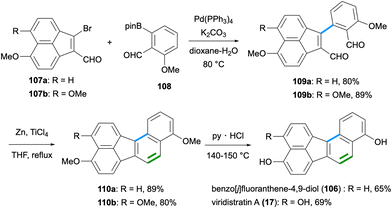 |
| | Scheme 28 Total synthesis of benzo[j]fluoranthene-4,9-diol (106) and viridistratin A (17). | |
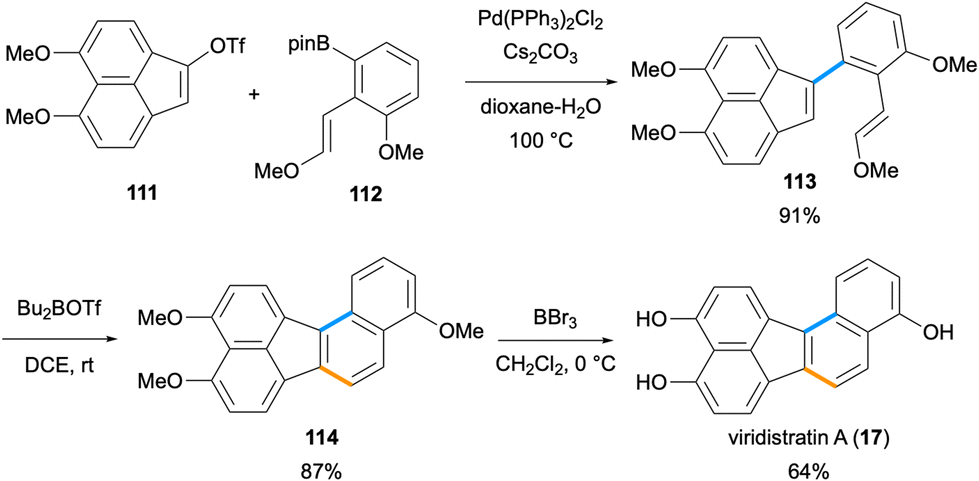
Recently, the second total synthesis of viridistratin A (17) has been accomplished by Yun and co-workers (Scheme 29).84 The authors developed a Lewis acid-catalysed Prins cyclisation for the construction of benzo[a]fluorene, benzo[c]fluorene and benzo[j]fluoranthene ring systems. In the synthetic route to viridistratin A (17), the cyclisation precursor enol ether 113 was synthesized via a Suzuki–Miyaura cross-coupling between acenaphthylene triflate 111 and pinacol boronate 112 in 91% yield. The key Prins cyclisation was conducted with the use of Bu2BOTf as the Lewis acid catalyst to give the benzo[j]fluoranthene derivative 114 in high yield (87%). A global deprotection of the three methyl ethers by BBr3 resulted in the formation of the natural product viridistratin A (17) in 64% yield. The overall synthesis proceeded in ten steps (longest linear sequence) starting from 1,8-dimethoxynaphthalene in 22% overall yield.84
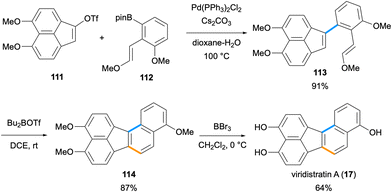 |
| | Scheme 29 Total synthesis of viridistratin A (17) via Prins cyclisation as the key step. | |
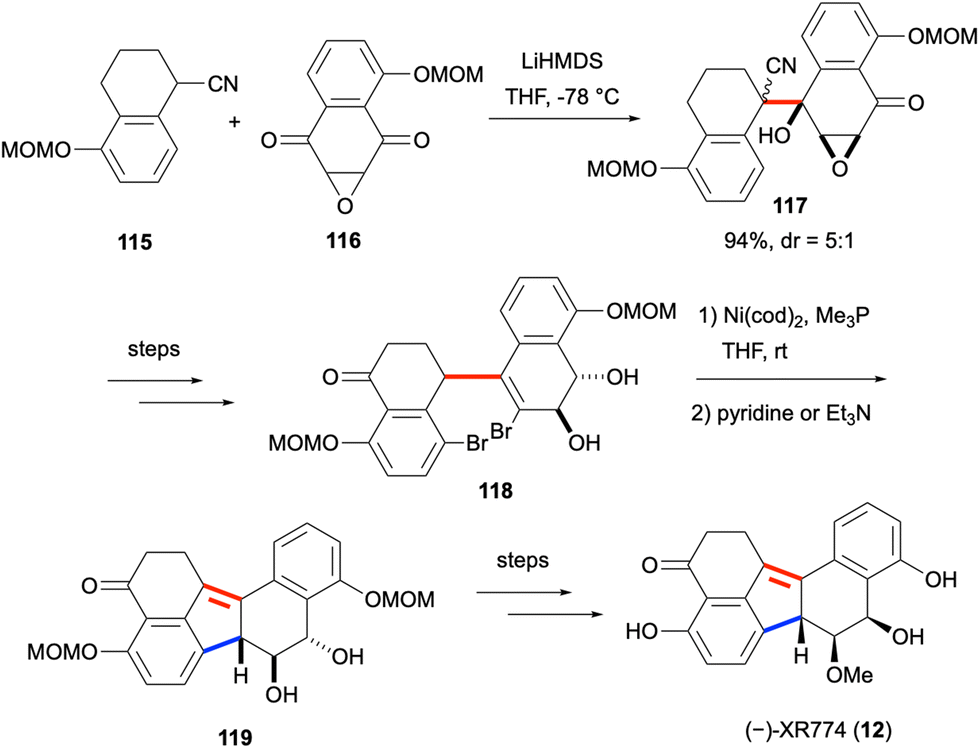
XR774 (12) is a fungal natural product with a dearomatized benzo[j]fluoranthene core isolated in 2001 from Cladosporium cf. cladosporioides, and was found to be an effective tyrosine kinase inhibitor.16,85 With its three contiguous stereogenic centers, it is one of the chiral benzo[j]fluoranthene-based natural products. The total synthesis of this natural product was accomplished in 2018 by Hosokawa and co-workers, and the two key steps of the synthesis are shown in Scheme 30.86 Compounds 115 and 116 were prepared from commercially available starting materials in three and two steps, respectively. The α-deprotonation of the nitrile of 115 with LiHMDS, and its subsequent attack on one of the ketone groups of 116 gave 117 in 94% yield and with a diastereomeric ratio (dr) of 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. This diastereomeric mixture was converted to 118 in eight steps. The reductive coupling of the aryl bromide and vinyl bromide groups of 118 with the use of four equivalents of Ni(cod)2 followed by a base-mediated alkene isomerization afforded 119 featuring a dearomatized benzo[j]fluoranthene skeleton. Compound 119 could be converted to (±)-XR774 (12) in two steps. Moreover, the synthesis of enantiomerically pure (−)-XR774 (12) was shown to be possible via the resolution of one of the racemic intermediates, which also paved the way for the determination of the absolute configuration of the natural product.
:1. This diastereomeric mixture was converted to 118 in eight steps. The reductive coupling of the aryl bromide and vinyl bromide groups of 118 with the use of four equivalents of Ni(cod)2 followed by a base-mediated alkene isomerization afforded 119 featuring a dearomatized benzo[j]fluoranthene skeleton. Compound 119 could be converted to (±)-XR774 (12) in two steps. Moreover, the synthesis of enantiomerically pure (−)-XR774 (12) was shown to be possible via the resolution of one of the racemic intermediates, which also paved the way for the determination of the absolute configuration of the natural product.
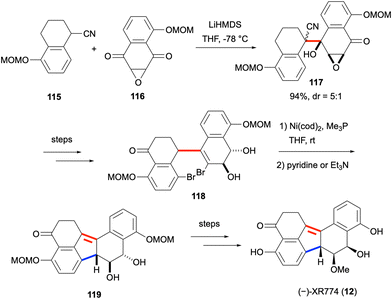 |
| | Scheme 30 Total synthesis of (−)-XR774 (12). | |
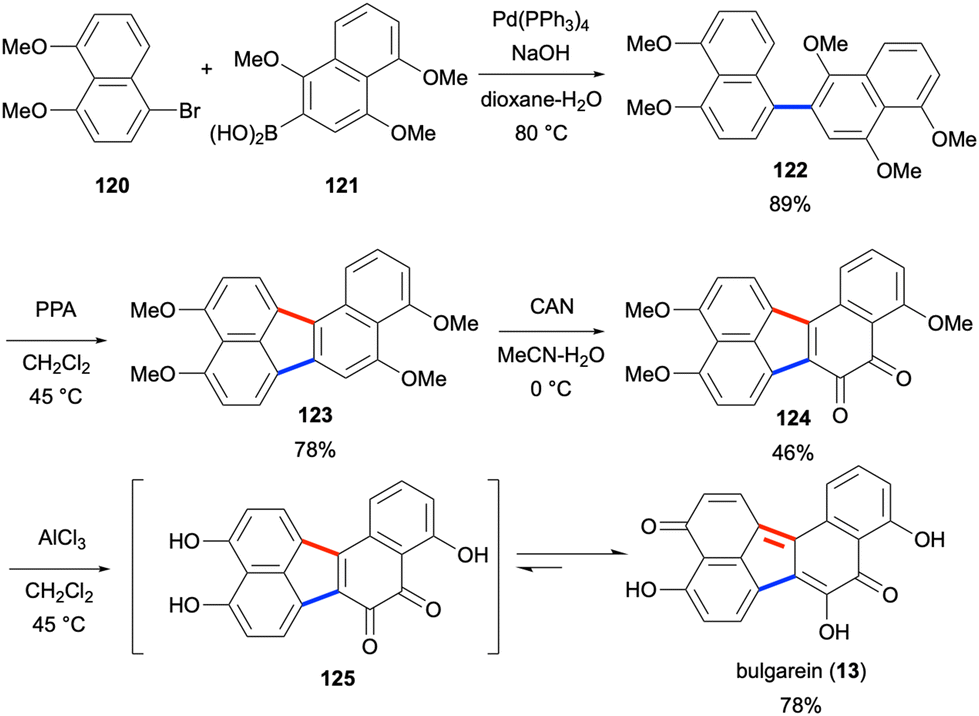
Bulgarein (13) is another highly oxygenated fungal natural product with the benzo[j]fluoranthene core, which was isolated in 1976 from Bulgaria inquinans.17 In 1993, bulgarein (13) was shown to induce mammalian topoisomerase I-mediated DNA cleavage, which was comparable to the activity of camptothecin.87 Despite earlier attempts towards the synthesis of bulgarein (13),88 the first total synthesis was reported in 2022 by Swieca and Spiteller.89 In this eight-step synthesis (longest linear sequence), the two naphthalene units 120 and 121 were attached via a Suzuki–Miyaura coupling to give binaphthalene product 122 in 89% yield (Scheme 31). The cyclisation reaction of 122 mediated by polyphosphoric acid (PPA) led to the formation of benzo[j]fluoranthene product 123 in 78% yield. Oxidation of this electron-rich PAH derivative by ceric ammonium nitrate (CAN) afforded the ortho-quinone 124 in moderate yield (46%). Global demethylation by the use of AlCl3 afforded bulgarein (13) in 78% yield, presumably through the intermediacy of 125.
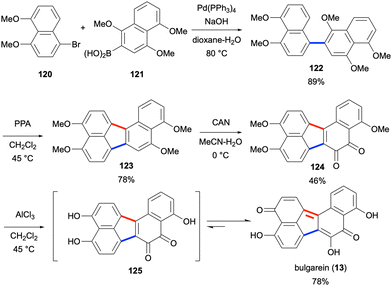 |
| | Scheme 31 Total synthesis of bulgarein (13). | |
4. Selected applications of fluoranthenes
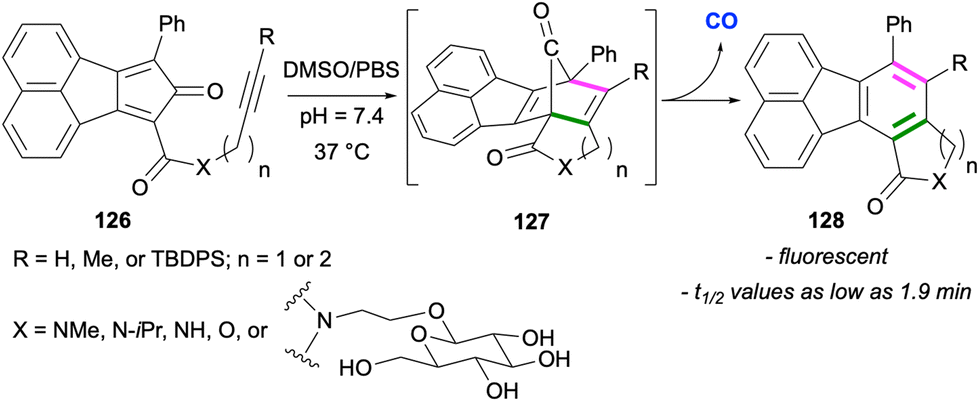
In a series of elegant applications, Wang and co-workers utilised intramolecular and intermolecular Diels–Alder reactions leading to fluoranthenes for controlled release of carbon monoxide, which is an endogenous signalling molecule.90–92 In a study reported in 2016, the researchers designed compounds 126 as CO prodrugs, which are capable of undergoing an intramolecular Diels–Alder reaction under near physiological conditions (Scheme 32).90 In this design, the cyclopentadienone derivatives 126 were observed to be stable for weeks at room temperature in the solid state. However, when subjected to near physiological conditions (pH 7.4, 37 °C) in DMSO/PBS, the Diels–Alder reaction between the cyclopentadienone moiety and the internal alkyne gave intermediate 127, which spontaneously releases CO via a cheletropic reaction. The rate of the formation of the fluoranthene products 128 and thus the rate of CO release was determined for all substrates, and the minimum half-life (t1/2) value was determined as 1.9 min for the substrate in which X = NMe, R = H and n = 1. It was also demonstrated that the introduction of a glucose unit as a side chain led to an increase in water solubility. It should be mentioned that the fluorescence nature of fluoranthene end products 128 made the real time monitoring of CO release possible. The potential of this approach in therapeutic applications was showcased in a colitis animal (mice) model and cell culture anti-inflammatory essays.
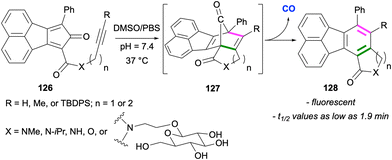 |
| | Scheme 32 An intramolecular Diels–Alder reaction that releases CO under physiological conditions. | |
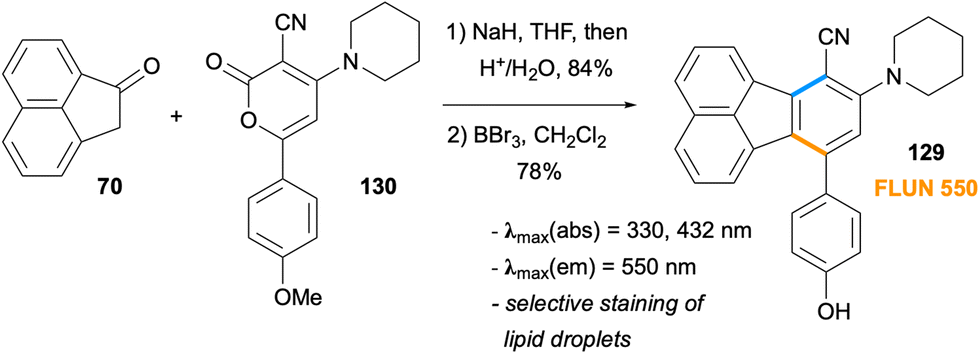
In 2014, a set of fluoranthene derivatives were examined by Goel, Mitra and co-workers with the aim of discovering new stains for lipid droplets.93 Among the molecules tested, compound 129, named FLUN 550, was selected based on cellular uptake studies (Scheme 33). The synthesis of this fluoranthene derivative was achieved by a formal [4 + 2] cycloaddition between the enolate of ketone 70 and 2-pyrone derivative 130 followed by a demethylation by BBr3. Fluoranthene 129 with the piperidine ring as the donor and –CN group as the acceptor units was found to exhibit an emission λmax of 550 nm, a large Stokes shift (220 nm), and fluorescence quantum yields of 35% in DMSO and 3.8% in water. Detailed studies revealed FLUN 550 (129) to be a cell permeable and highly selective staining agent for lipid droplets in in vitro live cell imaging in a variety of cell types and single-celled parasites, as well as in in vivo imaging of soil nematode C. elegans. Another analytical application of fluoranthenes was reported by Wang and co-workers in 2022 where three fluoranthene dyes were used as fluorescent probes for the determination of water content in methanol.94
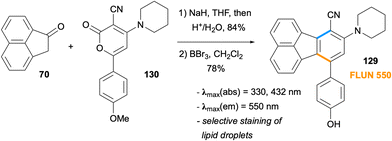 |
| | Scheme 33 Development of FLUN 550 (129) as a selective staining agent for lipid droplets. | |
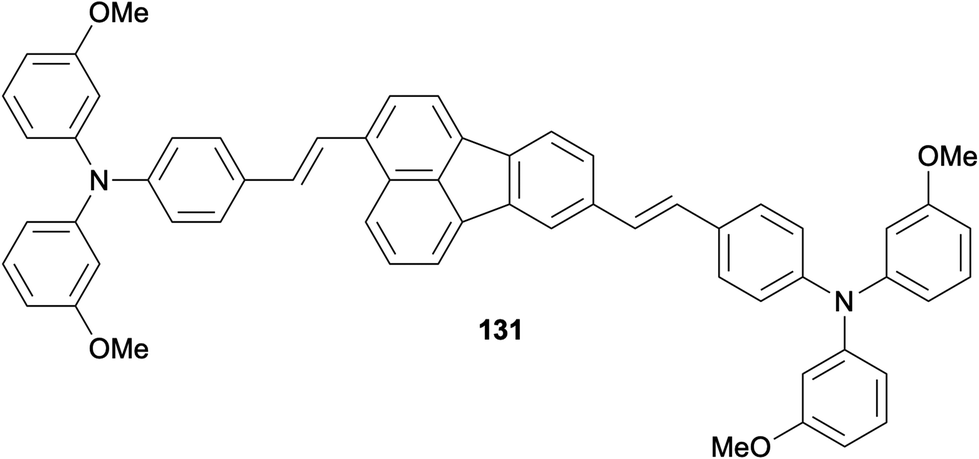
In an attempt to find alternatives to the commonly used spirobifluorenes and poly(triarylamine)s as organic hole-transporting materials (HTMs), Zhu, Li and co-workers examined a series of fluoranthene derivatives as dopant-free HTMs in perovskite solar cells.95 In this work, the researchers carried out a systematic investigation of the structure–property relationship on the designed fluoranthene derivatives. Studies revealed fluoranthene 131 to have the optimal properties among the tested compounds, which gave a power conversion efficiency of 19.3% in a perovskite solar cell as a dopant-free organic HTM (Fig. 3).
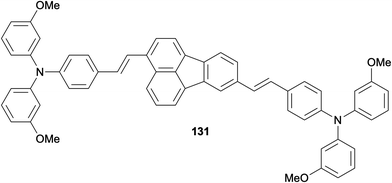 |
| | Fig. 3 Fluoranthene derivative 131 used as a hole-transporting material in perovskite solar cells. | |
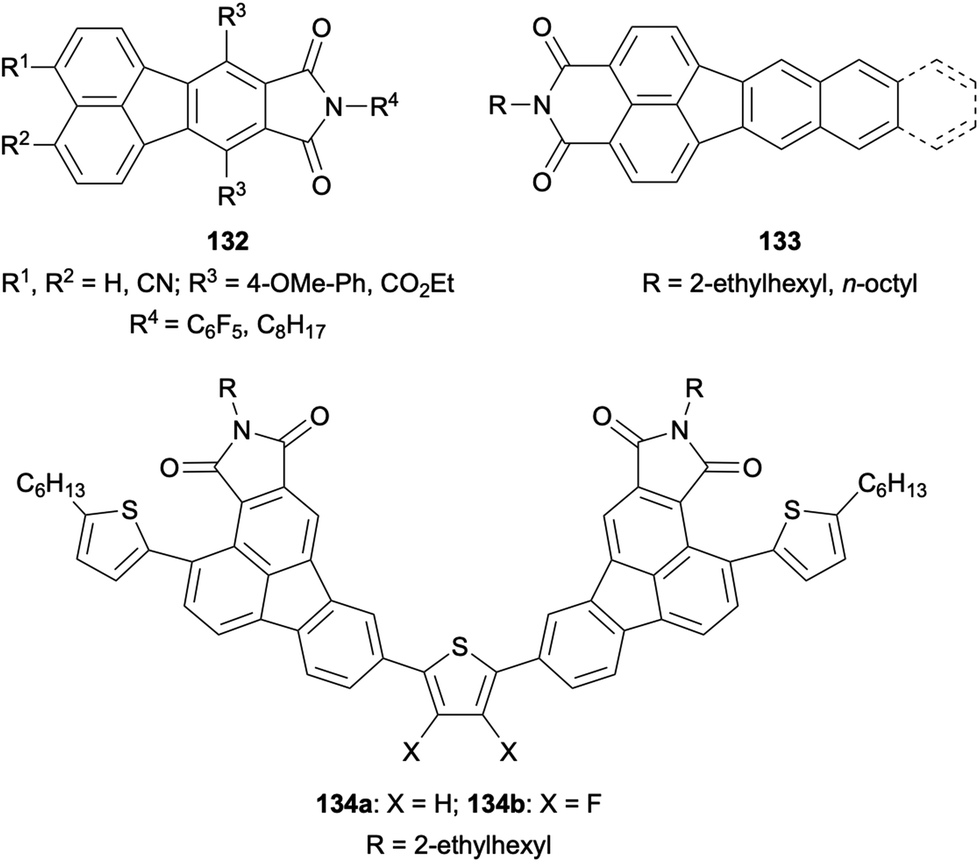
Finally, a variety of fluoranthene-fused imides were synthesized and investigated due to their interesting photophysical properties. For instance, 8,9-fluoranthene imides 132 were reported by Pei and co-workers in 2010 (Fig. 4).96 The photophysical and electrochemical properties of these imides were investigated in detail, and it was shown that their LUMO energy levels can be tuned through the careful control of the nature and position of the substituents. In 2017, the optical and electronic properties of π-extended 3,4-fluoranthene imides 133 with push–pull structures were examined by Kawase and co-workers.97 Recently, thiophene-bridged 2,3-fluoranthene imides such as 134 were designed by Li and co-workers as organic semiconductors (Fig. 4).98 All fluoranthene-fused imides developed in this study were shown to exhibit aggregation-induced emission, with compound 134a having the highest photoluminescence quantum yield (5.9%) in the solid state as a powder. Interestingly, compound 134a was found to behave as a p-type semiconductor, whereas the introduction of fluorines into the thiophene ring in 134b switches its behaviour into an n-type semiconductor.
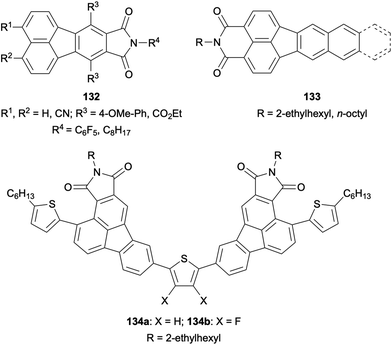 |
| | Fig. 4 Structures of fluoranthene-fused imides 132–134. | |
5. Conclusion and outlook
In this review, recent developments in the synthesis and applications of fluoranthenes, particularly in the past two decades, are discussed. First, in section 2, synthetic strategies towards the synthesis of fluoranthenes and related polycyclic aromatic hydrocarbons are classified in a systematic manner based on strategic bond disconnections. In the subsequent section, the recent syntheses of benzo[j]fluoranthene-based natural products are summarized. Finally, in section 3, selected examples of applications of fluoranthenes in a variety of areas such as the design of prodrugs, fluorescent probes, hole-transporting materials and organic semiconductors are provided.
The majority of the strategies developed for the construction of fluoranthenes enable the presence of substituents on the benzene ring of fluoranthenes rather than the naphthalene portion. As a result, a broad range of fluoranthenes with multiple substituents on the benzene ring can be synthesized with the available methods, whereas diversification on the naphthalene unit is rather limited. This limitation can be overcome by the introduction of novel synthetic methods that involve the de novo construction of the naphthalene moiety of fluoranthenes with a variety of substituents at the desired positions. The development of methods for the direct introduction of various substituents on the naphthalene unit of fluoranthenes may offer an alternative solution to this problem. For instance, in a study reported by Itami and co-workers in 2012, 3-arylfluoranthenes were synthesised directly via a Pd-catalysed intermolecular C–H arylation reaction, which takes place on the naphthalene ring.99 Given the remarkable recent advances in the area of C–H activation reactions, it is envisaged that fluoranthenes with diverse substituents on the naphthalene unit can be easily accessible. In relation to the above point, another major current limitation is the scarcity of methods for the preparation of substituted 1,8-dihalonaphthalenes or their equivalents. Therefore, there is a definite need for the development of new methods that will allow the synthesis of substituted 1,8-dihalonaphthalene analogues to be used in the synthetic methods described in sections 2.1.1, 2.2.3 and 2.2.4. Finally, the fact that only four natural products with the benzo[j]fluoranthene skeleton have been synthesized so far points to a broad range of opportunities in this area. In particular, the total syntheses of such benzo[j]fluoranthene-based natural products have the potential to lead to a diverse array of synthetic analogues which will set the ground for detailed structure–activity relationship studies.
Note added in proof
During the evaluation of the revision of this manuscript, Salem, Doucet and co-workers reported the synthesis of fluoranthenes starting from 1,8-dibromonaphthalene via mono or double C–H functionalisation reactions.100
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The author acknowledges financial support from the GEBIP Award of the Turkish Academy of Sciences.
References
-
R. G. Harvey, Polycyclic Aromatic Hydrocarbons, Wiley-VCH, New York, 1997 Search PubMed.
- X. Feng, W. Pisula and K. Müllen, Pure Appl. Chem., 2009, 81, 2203 CrossRef CAS.
-
Polyarenes I, ed. J. S. Siegel and Y.-T. Wu, Springer-Verlag, Berlin, Heidelberg, 2014 Search PubMed.
- A. Konishi and M. Yasuda, Chem. Lett., 2021, 50, 195 CrossRef CAS.
- K. N. Plunkett, Synlett, 2013, 898 CrossRef CAS.
- Y.-H. Liu and D. F. Perepichka, J. Mater. Chem. C, 2021, 9, 12448 RSC.
- S. H. Tucker and M. Whalley, Chem. Rev., 1952, 50, 483 CrossRef CAS.
-
R. Schmidt, K. Griesbaum, A. Behr, D. Biedenkapp, H.-W. Voges, D. Garbe, C. Paetz, G. Collin, D. Mayer and H. Höke, Hydrocarbons, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag, 2015 Search PubMed.
- H. Güsten and G. Heinrich, J. Photochem., 1982, 18, 9 CrossRef.
- Y.-T. Wu and J. S. Siegel, Chem. Rev., 2006, 106, 4843 CrossRef CAS PubMed.
-
Y.-T. Wu and J. S. Siegel, Synthesis, Structure, and Physical Properties of Aromatic Molecular-Bowl Hydrocarbons, in Polyaranes I, ed. J. S. Siegel and Y.-T. Wu, Springer-Verlag, Berlin, Heidelberg, 2014, pp. 63–120 Search PubMed.
- M. Gill, Nat. Prod. Rep., 2003, 20, 615 RSC.
- J. Podlech and M. Gutsche, J. Nat. Prod., 2023, 86, 1632 CrossRef CAS.
- E. Sudarman, E. Kuhnert, K. D. Hyde, E. B. Sir, F. Surup and M. Stadler, Tetrahedron, 2016, 72, 6450 CrossRef CAS.
- G. Brauers, R. Ebel, R. Edrada, V. Wray, A. Berg, U. Gräfe and P. Proksch, J. Nat. Prod., 2001, 64, 651 CrossRef CAS PubMed.
- S. K. Wrigley, A. M. Ainsworth, D. A. Kau, S. M. Martin, S. Bahl, J. S. Tang, D. J. Hardick, P. Rawlins, R. Sadheghi and M. Moore, J. Antibiot., 2001, 54, 479 CrossRef CAS PubMed.
- R. L. Edwards and H. J. Lockett, J. Chem. Soc., Perkin Trans. 1, 1976, 2149 RSC.
- M. Fukai, M. Tsukada, K. Miki, T. Suzuki, T. Sugita, K. Kinoshita, K. Takahashi, M. Shiro and K. Koyama, J. Nat. Prod., 2012, 75, 22 CrossRef CAS PubMed.
- D. N. Quang, T. Hashimoto, M. Tanaka, M. Baumgartner, M. Stadler and Y. Asakawa, J. Nat. Prod., 2002, 65, 1869 CrossRef CAS PubMed.
- L. Du, J. B. King and R. H. Cichewicz, J. Nat. Prod., 2014, 77, 2454 CrossRef CAS PubMed.
- K. Becker, A.-C. Wessel, J. J. Luangsa-ard and M. Stadler, Biomolecules, 2020, 10, 805 CrossRef CAS PubMed.
- K.-H. Kim, S. A. Jahan, E. Kabir and R. J. C. Brown, Environ. Int., 2013, 60, 71 CrossRef CAS PubMed.
- Y. Zhang and S. Tao, Atmos. Environ., 2009, 43, 812 CrossRef CAS.
- C.-E. Boström, P. Gerde, A. Hanberg, B. Jenström, C. Johansson, T. Kyrklund, A. Rannug, M. Törnqvist, K. Victorin and R. Westerholm, Environ. Health Perspect., 2002, 110, 451 Search PubMed.
-
https://www.epa.gov/eg/toxic-and-priority-pollutants-under-clean-water-act
, last accessed on February 14 2024.
- Z. Zelinkova and T. Wenzl, Polycycl. Aromat. Compd., 2015, 35, 248 CrossRef CAS PubMed.
- E. Šepič, M. Bricelj and H. Leskovšek, Chemosphere, 2003, 52, 1125 CrossRef PubMed.
- T. Sawano, K. Takamura, T. Yoshikawa, K. Murata, M. Koga, R. Yamada, T. Saito, K. Tabata, Y. Ishii, W. Kashihara, T. Nishihara, K. Tanabe, T. Suzuki and R. Takeuchi, Org. Biomol. Chem., 2023, 21, 323 RSC.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS.
- K. Tamao, K. Sumitani and M. Kumada, J. Am. Chem. Soc., 1972, 94, 4374 CrossRef CAS.
- R. J. P. Corriu and J. P. Masse, J. Chem. Soc., Chem. Commun., 1972, 144 RSC.
- J. E. Rice and Z.-W. Cai, J. Org. Chem., 1993, 58, 1415 CrossRef CAS.
- H. A. Wegner, L. T. Scott and A. de Meijere, J. Org. Chem., 2003, 68, 883 CrossRef CAS PubMed.
- J. M. Quimby and L. T. Scott, Adv. Synth. Catal., 2009, 351, 1009 CrossRef CAS.
- M. Yamaguchi, M. Higuchi, K. Tazawa and K. Manabe, J. Org. Chem., 2016, 81, 3967 CrossRef CAS PubMed.
- S. Pal, Ö. Metin and Y. E. Türkmen, ACS Omega, 2017, 2, 8689 CrossRef CAS PubMed.
- J. Wang, T. Torigoe and Y. Kuninobu, Org. Lett., 2019, 21, 1342 CrossRef CAS PubMed.
- G. S. Mohammad-Pour, R. T. Ly, D. C. Fairchild, A. Burnstine-Townley, D. A. Vazquez-Molina, K. D. Trieu, A. D. Campiglia, J. K. Harper and F. J. Uribe-Romo, J. Org. Chem., 2018, 83, 8036 CrossRef CAS PubMed.
- S. Chen, J. Xiao, X. Zhang, X. Shen, X. Liu, F. Shen, Y. Yi and Y. Song, Dyes Pigm., 2016, 134, 9 CrossRef CAS.
- X. Gu, W. A. Luhman, E. Yagodkin, R. J. Holmes and C. J. Douglas, Org. Lett., 2012, 14, 1390 CrossRef CAS PubMed.
- S. Seifert, D. Schmidt, K. Shoyama and F. Würthner, Angew. Chem., Int. Ed., 2017, 56, 7595 CrossRef CAS PubMed.
- J. Polkaehn, P. Ehlers, A. Villinger and P. Langer, Org. Biomol. Chem., 2023, 21, 9669 RSC.
- L. Wang and P. B. Shevlin, Org. Lett., 2000, 2, 3703 CrossRef CAS PubMed.
- E. A. Jackson, B. D. Steinberg, M. Bancu, A. Wakamiya and L. T. Scott, J. Am. Chem. Soc., 2007, 129, 484 CrossRef CAS PubMed.
- S. Lampart, L. M. Roch, A. K. Dutta, Y. Wang, R. Warshamanage, A. D. Finke, A. Linden, K. K. Baldridge and J. S. Siegel, Angew. Chem., Int. Ed., 2016, 55, 14648 CrossRef CAS PubMed.
- W. Hagui, H. Doucet and J.-F. Soulé, Chem, 2019, 5, 2006 CAS.
- Z. Zhang, R. Sangaiah, A. Gold and L. M. Ball, Org. Biomol. Chem., 2011, 9, 5431 RSC.
- O. Allemann, S. Duttwyler, P. Romanato, K. K. Baldridge and J. S. Siegel, Science, 2011, 332, 574 CrossRef CAS PubMed.
- J. Zhou, W. Yang, B. Wang and H. Ren, Angew. Chem., Int. Ed., 2012, 51, 12293 CrossRef CAS PubMed.
- Y. Choi, T. Chatterjee, J. Kim, J. S. Kim and E. J. Cho, Org. Biomol. Chem., 2016, 14, 6804 RSC.
- D. J. Tate, M. Abdelbasit, C. A. Kilner, H. J. Shepherd, S. L. Warriner and R. J. Bushby, Tetrahedron, 2014, 70, 67 CrossRef CAS.
- X.-J. Li, M. Li, W. Yao, H.-Y. Lu, Y. Zhao and C.-F. Chen, RSC Adv., 2015, 5, 18609 RSC.
- C. F. H. Allen and J. A. VanAllan, J. Org. Chem., 1952, 17, 845 CrossRef CAS.
- R. C. Chiechi, R. J. Tseng, F. Marchioni, Y. Yang and F. Wudl, Adv. Mater., 2006, 18, 325 CrossRef CAS.
- L. T. Scott, M. M. Hashemi, D. T. Meyer and H. B. Warren, J. Am. Chem. Soc., 1991, 113, 7082 CrossRef CAS.
- A. Borchardt, A. Fuchicello, K. V. Kilway, K. K. Baldridge and J. S. Siegel, J. Am. Chem. Soc., 1992, 114, 1921 CrossRef CAS.
- A. Borchardt, K. Hardcastle, P. Gantzel and J. S. Siegel, Tetrahedron Lett., 1993, 34, 273 CrossRef CAS.
- A. M. Butterfield, B. Gilomen and J. S. Siegel, Org. Process Res. Dev., 2012, 16, 664 CrossRef CAS.
- B. M. Schmidt, S. Seki, B. Topolinski, K. Ohkubo, S. Fukuzumi, H. Sakurai and D. Lentz, Angew. Chem., Int. Ed., 2012, 51, 11385 CrossRef CAS PubMed.
- B. Duda and D. Lentz, Org. Biomol. Chem., 2015, 13, 5625 RSC.
- C. M. Thompson, G. Occhialini, G. T. McCandless, S. B. Alahakoon, V. Cameron, S. O. Nielsen and R. A. Smaldone, J. Am. Chem. Soc., 2017, 139, 10506 CrossRef CAS PubMed.
- Q. Yan, Y. Zhou, B.-B. Ni, Y. Ma, J. Wang, J. Pei and Y. Cao, J. Org. Chem., 2008, 73, 5328 CrossRef CAS PubMed.
- N. Venkatramaiah, S. Kumar and S. Patil, Chem. Commun., 2012, 48, 5007 RSC.
- Y. Chandrasekaran, N. Venkatramaiah and S. Patil, Chem. – Eur. J., 2016, 22, 5288 CrossRef CAS PubMed.
- S. Kumar and S. Patil, New J. Chem., 2015, 39, 6351 RSC.
- S. Kumar and S. Patil, J. Phys. Chem. C, 2015, 119, 19297 CrossRef CAS.
- S. Kumar, D. Kumar, Y. Patil and S. Patil, J. Mater. Chem. C, 2016, 4, 193 RSC.
- A. Goel, V. Kumar, S. Chaurasia, M. Rawat, R. Prasad and R. S. Anand, J. Org. Chem., 2010, 75, 3656 CrossRef CAS PubMed.
- L. M. Geary, T.-Y. Chen, T. P. Montgomery and M. J. Krische, J. Am. Chem. Soc., 2014, 136, 5920 CrossRef CAS PubMed.
- B. Bossenbroek, D. C. Sanders, H. M. Curry and H. Shechter, J. Am. Chem. Soc., 1969, 91, 371 CrossRef CAS.
- H. A. Staab and J. Ipaktschi, Chem. Ber., 1971, 104, 1170 CrossRef CAS.
- J. J. González, A. Francesch, D. J. Cárdenas and A. M. Echavarren, J. Org. Chem., 1998, 63, 2854 CrossRef.
- D. Ahmadli, Y. Sahin, E. Calikyilmaz, O. Şahin and Y. E. Türkmen, J. Org. Chem., 2022, 87, 6336 CrossRef CAS PubMed.
- E. Calikyilmaz, G. Karaoglu, M. Demir, O. Şahin, B. Ulgut, A. Akdag and Y. E. Türkmen, Chem. Commun., 2024, 60, 550 RSC.
- S. Xie, W. Chen, S. Liu, H. Zong, B. Ming and G. Zhou, Chin. Chem. Lett., 2023, 34, 107642 CrossRef CAS.
- T. Okitsu, Y. Shinohara, H. Luo, M. Hatano and T. Yakura, Chem. – Asian J., 2024, e202301031 CrossRef CAS PubMed.
- Y.-T. Wu, A. Linden and J. S. Siegel, Org. Lett., 2005, 7, 4353 CrossRef CAS PubMed.
- Y.-T. Wu, T. Hayama, K. K. Baldridge, A. Linden and J. S. Siegel, J. Am. Chem. Soc., 2006, 128, 6870 CrossRef CAS PubMed.
- R. Abe, Y. Nagashima, J. Tanaka and K. Tanaka, ACS Catal., 2023, 13, 1604 CrossRef CAS.
- X. Chen, P. Lu and Y. Wang, Chem. – Eur. J., 2011, 17, 8105 CrossRef CAS PubMed.
- S. Pascual, C. Bour, P. de Mendoza and A. M. Echavarren, Beilstein J. Org. Chem., 2011, 7, 1520 CrossRef CAS PubMed.
- Y. L. Zhang, J. Zhang, N. Jiang, Y. H. Lu, L. Wang, S. H. Xu, W. Wang, G. F. Zhang, Q. Xu, H. M. Ge, J. Ma, Y. C. Song and R. X. Tan, J. Am. Chem. Soc., 2011, 133, 5931 CrossRef CAS PubMed.
- S. Lahore, U. Narkhede, L. Merlini and S. Dallavalle, J. Org. Chem., 2013, 78, 10860 CrossRef CAS PubMed.
- M. Jeong, M. Yoon, L. H. Nguyen, S. Kim, J. Han, C. S. Tran, J. Kim, J. Jo, Y.-S. Jung, J.-W. Yoo and H. Yun, Adv. Synth. Catal., 2024, 366, 390 CrossRef CAS.
- R. Sadeghi, P. Depledge, P. Rawlins, N. Dhanjal, A. Manic, S. Wrigley, B. Foxwell and M. Moore, Int. Immonopharmacol., 2001, 1, 33 CrossRef CAS PubMed.
- K. Tatsuta, D. Sekine, S. Hayama, Y. Kataoka, S. Hayashi and S. Hosokawa, J. Org. Chem., 2018, 83, 7010 CrossRef CAS PubMed.
- N. Fujii, Y. Yamashita, Y. Saitoh and H. Nakano, J. Biol. Chem., 1993, 268, 13160 CrossRef CAS PubMed.
-
(a)
D. Ahmadli and Y. E. Turkmen, 261st ACS National Meeting, American Chemical Society, 2021;
(b)
D. Ahmadli, Total synthesis of biologically active fungal natural products daldiquinone and bulgarein, and intramolecular Diels–Alder reactions for fluoranthene synthesis, M.Sc. Thesis, Bilkent University, Ankara, Turkey, 2021 Search PubMed.
- P. Swieca and P. Spiteller, Eur. J. Org. Chem., 2022, e202101576 CrossRef CAS.
- X. Ji, C. Zhou, K. Ji, R. E. Aghoghovbia, Z. Pan, V. Chittavong, B. Ke and B. Wang, Angew. Chem., Int. Ed., 2016, 55, 15846 CrossRef CAS PubMed.
- Y. Zheng, X. Ji, B. Yu, K. Ji, D. Gallo, E. Csizmadia, M. Zhu, M. R. Choudhury, L. K. C. De La Cruz, V. Chittavong, Z. Pan, Z. Yuan, L. E. Otterbein and B. Wang, Nat. Chem., 2018, 10, 787 CrossRef CAS.
- L. K. C. De La Cruz, S. L. Benoit, Z. Pan, B. Yu, R. J. Maier, X. Ji and B. Wang, Org. Lett., 2018, 20, 897 CrossRef CAS PubMed.
- A. Goel, A. Sharma, M. Kathuria, A. Bhattacharjee, A. Verma, P. R. Mishra, A. Nazir and K. Mitra, Org. Lett., 2014, 16, 756 CrossRef CAS PubMed.
- Y.-C. Liu, G.-D. Lu, J.-H. Zhou, J.-W. Rong, H.-Y. Liu and H.-Y. Wang, RSC Adv., 2022, 12, 7405 RSC.
- X. Sun, F. Wu, C. Zhong, L. Zhu and Z. Li, Chem. Sci., 2019, 10, 6899 RSC.
- L. Ding, H.-Z. Ying, Y. Zhou, T. Lei and J. Pei, Org. Lett., 2010, 12, 5522 CrossRef CAS PubMed.
- I. Kawajiri, M. Nagahara, H. Ishikawa, Y. Yamamoto, J. Nishida, C. Kitamura and T. Kawase, Can. J. Chem., 2017, 95, 371 CrossRef CAS.
- X. Sun, M.-Y. Liao, X. Yu, Y.-S. Wu, C. Zhong, C.-C. Chueh, Z. Li and Z. Li, Chem. Sci., 2022, 13, 996 RSC.
- K. Kawasumi, K. Mochida, T. Kajino, Y. Segawa and K. Itami, Org. Lett., 2012, 14, 418 CrossRef CAS PubMed.
- N. Ketata, L. Liu, R. B. Salem and H. Doucet, Beilstein J. Org. Chem., 2024, 20, 427 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab
ab