
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOverview of alkyl quercetin lipophenol synthesis and its protective effect against carbonyl stress involved in neurodegeneration†
Léa
Otaegui‡
ab,
Jordan
Lehoux‡
b,
Leo
Martin
b,
Laurent
Givalois
 ac,
Thierry
Durand
b,
Catherine
Desrumaux
ad and
Céline
Crauste
*b
ac,
Thierry
Durand
b,
Catherine
Desrumaux
ad and
Céline
Crauste
*b
aMMDN, Univ Montpellier, INSERM, EPHE, Montpellier, France
bIBMM, Univ Montpellier, CNRS, ENSCM, 34000 Montpellier, France. E-mail: celine.crauste@umontpellier.fr
cLaval University, Department of Neurosciences & Psychiatry, Quebec, Canada
dLIPSTIC LabEx, 21000 Dijon, France
First published on 15th March 2024
Abstract
Oxidative stress and carbonyl stress resulting from the toxicity of small aldehydes are part of the detrimental mechanisms leading to neuronal cell loss involved in the progression of neurodegenerative diseases such as Alzheimer's disease. Polyunsaturated alkylated lipophenols represent a new class of hybrid molecules that combine the health benefits of anti-inflammatory omega-3 fatty acids with the anti-carbonyl and oxidative stress (anti-COS) properties of (poly)phenols in a single pharmacological entity. To investigate the therapeutic potential of quercetin-3-docosahexaenoic acid-7-isopropyl lipophenol in neurodegenerative diseases, three synthetic pathways using chemical or chemo-enzymatic strategies were developed to access milligram or gram scale quantities of this alkyl lipophenol. The protective effect of quercetin-3-DHA-7-iPr against cytotoxic concentrations of acrolein (a carbonyl stressor) was assessed in human SHSY-5Y neuroblastoma cells to underscore its ability to alleviate harmful mechanisms associated with carbonyl stress in the context of neurodegenerative diseases.
Introduction
It is commonly accepted that oxidative stress (i.e., an imbalance between the reactive oxygen species and the antioxidant system or defense of the body) can have detrimental effects on neural tissues and be part of the etiology of neurodegenerative pathologies including Alzheimer's disease (AD).1,2 Oxidative stress (OS) is an early and persistent feature of AD that can be of genetic origin or amplified by environmental factors such as smoking, pollution, or exposure to toxic substances. A direct consequence of OS is the increase in lipid peroxidation, leading to the formation of various aldehydic by-products, including malondialdehyde (MDA), 4-hydroxy-2-nonenal (HNE), and acrolein. Acrolein, the most reactive among them, can also originate from exogenous sources as it is commonly present in the environment as a pollutant. Reactive electrophilic aldehydes are the cause of carbonyl stress toxicity, as they form cellular adducts with nucleophilic proteins, lipids, or nucleic acids, and impair their function. A significant increase in acrolein and other carbonyl stressors was observed in the brain of early Alzheimer's patients,3 associated with toxicity in hippocampal neurons.4 Due to their high neurotoxicity, reactive aldehydes have been identified as neurotoxic mediators of oxidative damage in the progression of AD.5–7A deficiency in polyunsaturated fatty acids (PUFAs), a critical class of fatty acids essential for brain functions, has also been identified as a factor contributing to increased oxidative stress.8 Docosahexaenoic acid (DHA, 22:6 n-3), the most prevalent omega-3 PUFA in neuronal membranes,9 possesses significant free radical scavenging properties and provides protection against peroxidative damage to brain lipids. It also serves as a precursor for the biosynthesis of lipid mediators that regulate inflammatory responses.10 DHA is weakly produced de novo and must be obtained from the diet. In humans, low plasmatic concentrations of DHA have been associated with impaired cognitive function, low hippocampal volumes, and increased amyloid deposition in the brain.11,12 Reduced DHA concentrations have been reported in the brains of AD patients.13 A number of epidemiological studies suggest that dietary DHA consumption is essential for brain development, learning ability and memory, and prevention of cognitive decline.14 However, the success of oral DHA supplementation (a highly oxidizable derivative) depends on individual variability, including that of metabolism.15 Recently, we showed that intranasal administration of nanovectorized DHA reduces tau phosphorylation and restores cognitive functions in two murine models of AD.16 These results pave the way for the development of a new approach for targeting the brain using DHA for the prevention or treatment of AD.
Currently, clinical trials of many drugs targeting AD may have failed because of the single target-based therapeutics (and also the high impenetrability of the blood–brain barrier). In this context, carbonyl and oxidative stresses (COS), in addition to disrupting DHA homeostasis, emerge as interesting targets to combat the progression of AD and serve as the starting point for the development of multi-target pharmacological molecules.
There is currently growing interest in the impact of dietary nutrients on reducing toxic mechanisms leading to the onset of cognitive disorders,17,18 particularly, (poly)phenols,19 well-known antioxidant compounds abundant in fruits and vegetables. Interestingly, depending on their chemical structure, they can also efficiently act as anti-carbonyl stress agents by directly or indirectly trapping reactive aldehydes.20,21 However, the pharmacological development of (poly)phenol drugs often encounters issues related to their high metabolism and low bioavailability. We have previously developed hybrid pharmacological protective molecules called lipophenols, based on the conjugation of PUFAs and (poly)phenols, to overcome PUFA degradation due to oxidation, enhance the bioavailability of (poly)phenols, provide DHA, and target PUFA-rich tissues such as the retina.22 In addition, this approach allows us to leverage the therapeutic characteristics offered by both components of lipophenols (i.e., the (poly)phenol and the PUFA). Alkyl lipophenols of DHA were efficient in providing cellular protection against oxidative and carbonyl stresses involved in age-related macular degeneration (AMD) or genetic Stargardt dystrophy.23–25 The importance of the alkyl (an isopropyl) and the omega-3 chain has been highlighted in cellular models. Such new lipophenolic derivatives were able to protect photoreceptor degeneration induced by light (80% of protection) in a murine model of genetic macular degeneration by IV and oral administration,26,27 thus validating the proof of concept of the pre-clinical interest of those PUFA conjugates.
Considering the close relationship between the COS mechanisms involved in AMD and AD and the essential physiological role of DHA in both retinal and brain functions, alkyl lipophenols of DHA should be evaluated for their potential as pharmacological agents to reduce COS associated with AD. In the present study, we selected quercetin-3-DHA-7-iPr (for quercetin-3-docosahexaenoate-7-isopropyl, Fig. 1), the most efficient anti-COS lipophenol described so far,24 for further investigation to reduce carbonyl stress involved in AD (in vitro first and then using in vivo models in the future). To achieve this goal, new synthetic methodologies for obtaining gram-scale quantities of this alkyl-PUFA lipophenol of quercetin are needed. Indeed, we previously observed that once in vitro screening is validated, the challenge of upscaling the chemical synthesis strategy of lipophenol could impede effective pre-clinical studies involving gram-scale consumption for toxicity evaluation, pharmacokinetic aspects or formulation development. Therefore, new chemical strategies to access quercetin-3-DHA-7-iPr have been developed and compared based on their reproducibility, scalability, the number of steps involved and their potential to contribute to greener chemistry. This work thus highlights the strengths and weaknesses of three new different synthesis strategies. While each pathway leads to the desired molecule, not all meet the criteria required for the in vivo assessment of a therapeutically relevant compound. In the second phase of our study, we shift our focus to the evaluation of the synthesized quercetin-3-DHA-7-iPr in the context of AD.
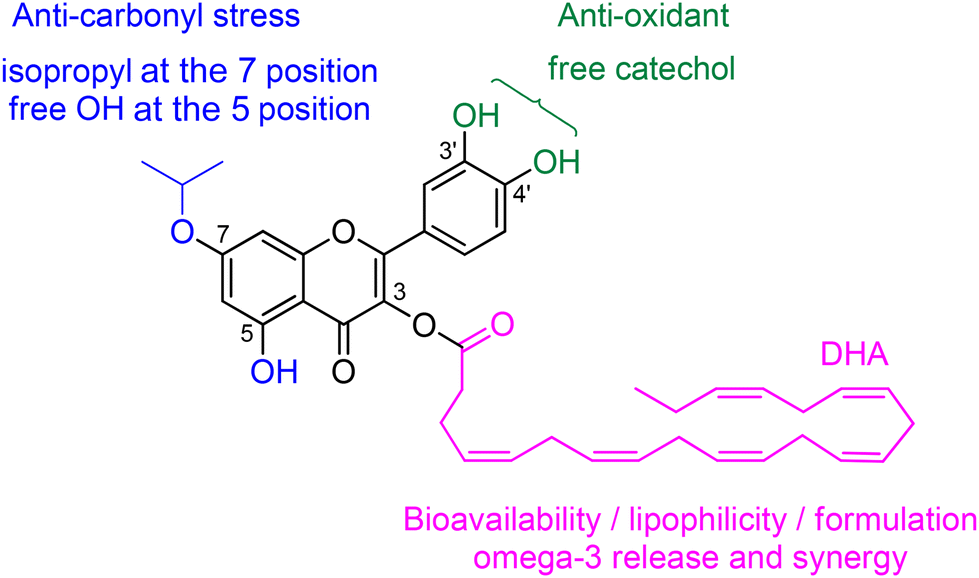 | ||
| Fig. 1 Chemical structure of lipophenol quercetin-3-DHA-7-iPr. | ||
Using acrolein as a potent carbonyl stressor, we developed an efficient cellular assay to investigate the efficacy of this conjugate in protecting against carbonyl stress in human SH-SY5Y neuroblastoma cells. The potential of this conjugate to mitigate the toxic effects of acrolein and the importance of both the isopropyl and the PUFA part in its protective effect were evaluated.
Results and discussion
Chemical synthesis
 | ||
| Scheme 1 Pathways A – chemo-enzymatic synthesis of quercetin-3-DHA-7-iPr. | ||
Then, the monobenzylation of the most reactive OH-3 function of compound 2 was carried out using carefully selected equivalents of benzyl bromide (to consume all of the starting compound 2 and avoid high proportion of di-benzylated 3b – Table S1†).35 The selective alkylation at the 7 position of 3a was performed as the one described for the total synthesis of rhamnetin, the 7-methyl analogue of quercetin.35 The well-known hydrogen bonding between the OH-5 and the carbonyl function reduces the activity at the 5 position, allowing for regioselective alkylation at the 7 position. In contrast to methylation, isopropylation requires a greater amount of reagent to achieve an efficient conversion in mono-isopropylated 4a, due to a lower reactivity of the sterically hindered iodopropane. When one manages to limit dialkylation (4b), the remaining starting material can be easily recovered resulting in an acceptable 53% yield (73% BSMR). Hydrogenation of 4a was then performed using palladium hydroxide under hydrogen to achieve both deprotection of dioxole and benzyl protecting groups with excellent yields. Based on preliminary work (see Fig. S1†), the coupling reaction with DHA was then conducted to minimize the introduction of the fatty acid at the 5 position. Indeed, this position is poorly hydrolysable by the enzyme in the presence of an isopropylated bulky group located at the 7 position. Tri-DHA quercetin 6 was thus isolated in moderate yield (22%) and the absence of DHA at the 5 position was confirmed by the unshielded characteristic 1H NMR signal of the OH-5 position at 12.05 ppm (Scheme 1). A mixture of di-DHA derivatives (uncharacterized) was also obtained (52%) and used as tri-DHA 6 in regioselective enzymatic deacylation. Due to the long C22 carbon chain and the presence of six unsaturated bonds, DHA is not enzymatically cleaved as quickly as a shorter, saturated fatty acid.32 The reaction reaches a plateau after 6–8 days, and neither the addition nor the replacement of the supported lipase improves the final yield. The desired quercetin-3-DHA-7-iPr (7) was recovered using either a tri-DHA (6) or di-DHA mixture. Overall, we have successfully developed a new methodology, leveraging the regioselectivity of the Lipozyme® lipase, to access an alkyl lipophenol of quercetin with a reduced number of steps (Table 1). It is worth noting that this type of strategy could also be an interesting alternative for the synthesis of lipophenols with shorter chains or those featuring saturated fatty acids. Indeed, it is highly likely that the yields of the last two coupling and deacylation steps would yield much better results with a different, shorter, and more linear fatty acid than DHA.
| Pathways | Nbr of steps | Global yield | Reproducibility of the strategy tested | Amount of compound 7 synthesized: | |
|---|---|---|---|---|---|
| each step performed oncea | with batch reproduction | ||||
| a On scale tested, starting from 5 g of quercetin. | |||||
| A “chemo-enzymatic” | 7 | 4.6% | No | 49 mg | nd |
| B1 “butyrate” | 10 | 10.2% | No | 630 mg | nd |
| B2 “acetate” | 8 | 14.6% | Yes | 994 mg | 2.91 g |
 | ||
| Scheme 2 Alkylation scale up issue in the previously reported quercetin-3-DHA-7-iPr pathway.24 | ||
 | ||
| Scheme 3 Synthesis of quercetin-3-DHA-7-iPr through B1 “butyrate” and B2 “acetate” protecting group strategies. | ||
| Synthesized compound | Reaction | Largest scale tested | Yielda | Reproducibilitybn = | Average yield |
|---|---|---|---|---|---|
| a On the largest scale. b Starting material > 1 g. | |||||
| 2 | Catechol protection | 5 g | 81% | 5 | 71.2% |
| 12 | Di-acetylation | 3.2 g | 74% | 6 | 74% |
| 13 | Isopropylation | 3 g | 79% | 9 | 73.4% |
| 14 | Hydrogenation | 2 g | 85% | 3 | 81.6% |
| 15 | TIPS introduction | 2.3 g | 66% | 3 | 77% |
| 10 | De-acetylation | 3.7 g | 94% | 4 | 93% |
| 11 | DHA coupling | 3.1 g | 75% | 4 | 72.5% |
| 7 | TIPS removing | 3 g | 78% | 5 | 80.2% |
In vitro potency of quercetin-3-DHA-7-iPr lipophenol against acrolein toxicity in neuroblastoma cells
The impact of quercetin-3-DHA-7-iPr treatment on cell viability was assessed in human SH-SY5Y neuroblastoma cells with or without exposure to the carbonyl stress inducer acrolein. The activity of quercetin-3-DHA-7-iPr (7) was compared with those of previously synthesized quercetin-7-iPr, quercetin-3-DHA, commercial free DHA acid and quercetin (Fig. 2). As shown in Fig. 3A, the exposure of the cells to quercetin-3-DHA-7-iPr (25 or 50 μM) for 24 h had no impact on cell viability. In contrast, viability did not exceed 50% when cells were exposed to quercetin-7-iPr, DHA, a combination of quercetin-7-iPr + DHA, or quercetin-3-DHA. As shown in Fig. 3B, quercetin-3-DHA-7-iPr (7) exhibited a dose-dependent, strong protective effect against acrolein-induced cell toxicity, and notably, this effect was much higher than the effects of quercetin, quercetin-7-iPr and quercetin-3-DHA at the same concentration, indicating that both the isopropyl group and the DHA moiety of quercetin-3-DHA-7-iPr are necessary to obtain a maximal protective effect under our experimental conditions. Interestingly, the highest concentration of quercetin-3-DHA-7-iPr (7) resulted in higher cell viability than control conditions, showing an increase of around 30%. This may be explained by the fact that serum-free conditions induce oxidative stress, which can be counteracted when lipophenol is added to the culture medium during preincubation. The lack of protection observed during cell treatment with the quercetin-7-iPr + DHA mixture underscores the significance of the covalent lipophenolic structure. | ||
| Fig. 2 Chemical structures of quercetin derivatives compared to that of quercetin-3-DHA-7-iPr (7). | ||
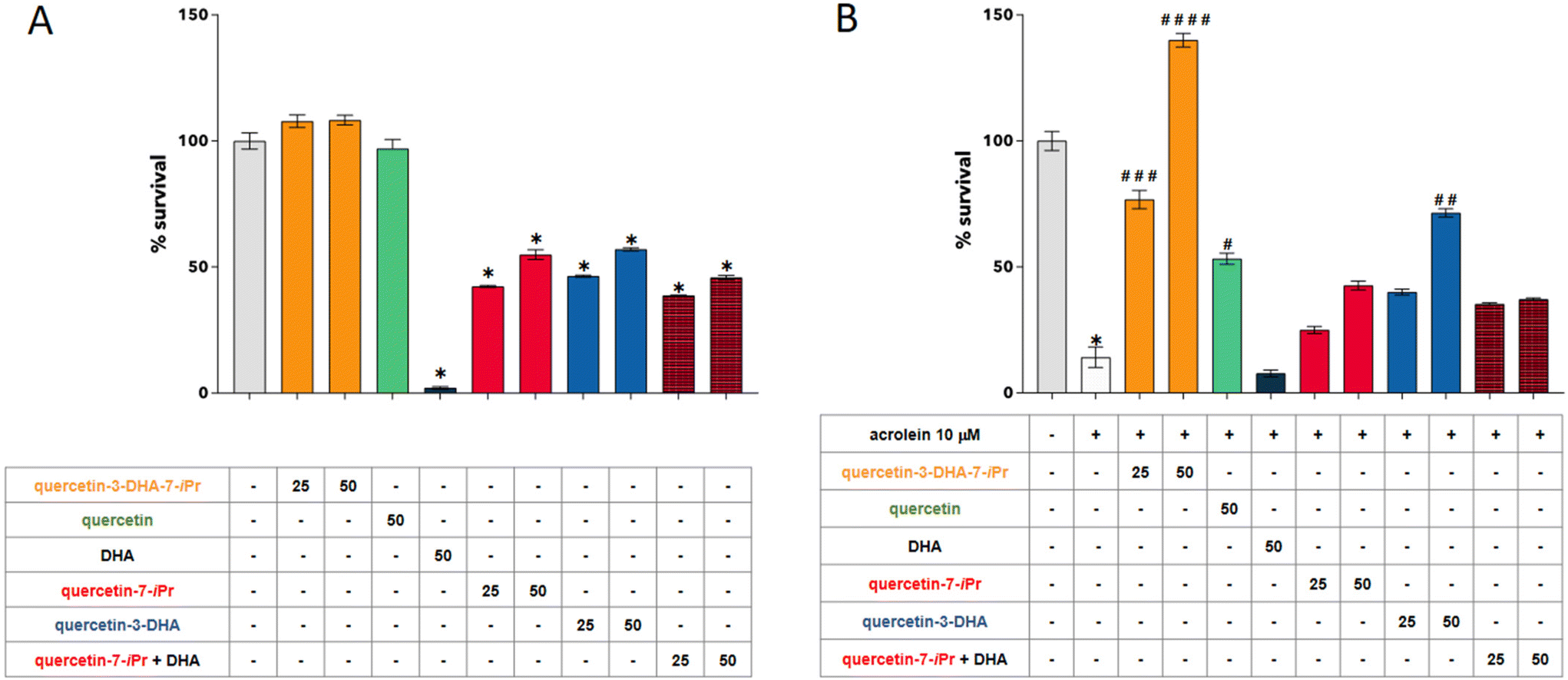 | ||
| Fig. 3 Structure–function relationship of quercetin-3-DHA-7-iPr on neuroblastoma cells: (A) effects of quercetin-3-DHA-7-iPr (25 and 50 μM), quercetin (50 μM), DHA (50 μM), quercetin-7-iPr (25 and 50 μM), quercetin-3-DHA (25 and 50 μM) and quercetin-7-iPr + DHA (25 and 50 μM of each) on cell viability. SH-SY5Y cells were treated for 24 h with 25 and/or 50 μM of each compound in serum-free medium. Cell viability was determined using the MTT assay. The data are expressed as the percentage of control untreated cells and presented as mean ± SEM of two separate experiments; (B) neuroprotective effect of quercetin-3-DHA-7-iPr against acrolein-induced toxicity. The cells were pre-treated for 2 h with quercetin-3-DHA-7-iPr or other tested compounds at 25 and/or 50 μM (quercetin, DHA, quercetin-7-iPr, quercetin-3-DHA and quercetin-7-iPr + DHA) in serum-free medium before the addition of acrolein (10 μM) for 24 h. Cell viability was determined using the MTT assay. The data are expressed as the percentage of control untreated cells and presented as mean ± SEM of two separate experiments. * p < 0.0001 versus the control, # p < 0.05, # # p < 0.01, # # # p < 0.001, and # # # # p < 0.0001 (ANOVA test with Tukey's post-hoc analysis) versus the cells treated with acrolein. | ||
Retinopathies and neurodegenerative diseases have several common features such as the strong involvement of oxidative and carbonyl stresses in their etiology, and the presence of highly selective barriers that limit the diffusion of drugs to the eyes and brain, reducing their therapeutic efficacy. In previous studies conducted in vitro and in vivo, several lipophenols were demonstrated to efficiently protect retinal cells against all-trans retinal-induced carbonyl and oxidative stresses.23–25 Our results are in line with these findings and extend the potential therapeutic benefits of lipophenols to neurodegenerative diseases such as AD. Interestingly, as previously observed with phloroglucinol-IP-DHA on ARPE-19 retinal cells,24 the incorporation of the PUFA segment and the isopropyl moiety into quercetin was found to be essential for providing maximal cellular protection against carbonyl stressor toxicity. This could result from a significant improvement in quercetin lipophilicity and accretion to neuronal cells provided by the DHA graft, and/or from a synergistic effect arising from the alkyl phenol and DHA moieties. The exact mechanism underlying alkyl lipophenol protection against acrolein toxicity will be investigated in the near future, exploring two potential directions: direct aldehyde scavenging to reduce intracellular aldehyde concentration and/or the activation of aldehyde detoxification enzymes through the Nrf2-Keap1 pathway. The therapeutic potential of quercetin-3-DHA-7-iPr will also be addressed in vivo using murine AD models.
Conclusion
Unlike chemical strategies for alkylated flavonoids involving the reconstruction of the three-ring skeleton of flavonoids starting from phloroglucinol building blocks,38 we have successfully validated three new chemical strategies for 7-alkylated flavonoids using quercetin as the starting material. With this approach, the challenge was focused on the close reactivity of the different phenolic functions of quercetin. Considering the biological relevance of the isopropyl function in carbonyl stress protection, the isopropylation reaction was carefully optimized to achieve maximum yield and good reproducibility on a gram scale either through a selection of an appropriate equivalent of an alkylated reagent (pathway A) or by utilizing the intra-molecular migration of acetate (pathway B2). The validation of the chemo-enzymatic pathways A highlighted the efficiency of supported lipase regioselective access to alkyl lipophenolic structures. This pathway, suitable for the small-scale synthesis of lipophenolic PUFA conjugates, allows for a reduction in the use of phenolic protecting groups and chemical steps. In the near future, it would be interesting to extrapolate this method to the synthesis of saturated lipophenols. Simultaneously, we have also confirmed the effectiveness of two additional strategies, which are better suited for scale-up synthesis. Notably, a final quantity of around 3 g of quercetin-3-DHA-7-iPr was obtained through pathway B2 (starting from around 15–20 g of quercetin), making it a validated strategy for future pre-clinical studies.The cellular assay conducted in this study validated the protective effect of this isopropyl PUFA lipophenol against acrolein toxicity, a carbonyl stressor implicated in the physiopathology of neurodegenerative diseases, whether originating from environmental exposure or genetic predispositions (via oxidative stress). Given the beneficial role of quercetin lipophenol against carbonyl and reactive oxygen species toxicity, quercetin-3-DHA-7-iPr emerges as an interesting candidate for further in vivo evaluation in mouse models of AD.
According to these in vitro results, the convergence of nutrition and organic chemistry paves the way for innovative solutions in the prevention and/or treatment of neurodegenerative pathologies such as AD. This opens new horizons to investigate deeper therapeutic developments based on lipophenols.
Experimental section
General methods
All solvents were anhydrous reagents from commercial sources. Unless otherwise noted, all chemicals and reagents were obtained commercially and used without purification. The reactions were monitored using TLC on plates that were pre-coated with silica gel 60 (Macherey-Nagel). The reaction components were visualized using a 254 nm UV lamp, stained with acidic p-anisaldehyde or KMNO4 solution followed by gentle heating. Purifications of the synthesized compounds were performed by column chromatography on silica gel 40–63 μm. High-resolution mass spectra (HRMS) were recorded using a Q-Tof micro spectrometer (resolution 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, Waters) or mass spectrometers Synapt G2-S (Waters). Data were obtained by positive or negative electrospray ionization methods between 100 and 1500 Da by direct introduction (ESI, ASAP, FTMS + p APCI ionisation). NMR spectra were recorded at 300, 400 or 500 MHz (1H) and 75, 101 or 126 MHz (13C) using Bruker spectrometers. Chemical shifts are reported in parts per million (ppm, δ) relative to residual deuterated solvent peaks. The NMR spectra were assigned with the help of 2D NMR analyses (COSY, HSQC, and HMBC). The multiplicities reported are as follows: br = broad singlet, m = multiplet, s = singlet, d = doublet, t = triplet, q = quadruplet, quint = quintuplet, h = hextuple, or combinations thereof. For the peak assignments, the following abbreviations were used: Ar = aromatic, CH
000, Waters) or mass spectrometers Synapt G2-S (Waters). Data were obtained by positive or negative electrospray ionization methods between 100 and 1500 Da by direct introduction (ESI, ASAP, FTMS + p APCI ionisation). NMR spectra were recorded at 300, 400 or 500 MHz (1H) and 75, 101 or 126 MHz (13C) using Bruker spectrometers. Chemical shifts are reported in parts per million (ppm, δ) relative to residual deuterated solvent peaks. The NMR spectra were assigned with the help of 2D NMR analyses (COSY, HSQC, and HMBC). The multiplicities reported are as follows: br = broad singlet, m = multiplet, s = singlet, d = doublet, t = triplet, q = quadruplet, quint = quintuplet, h = hextuple, or combinations thereof. For the peak assignments, the following abbreviations were used: Ar = aromatic, CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH = aliphatic alkene, and iP = isopropyl. If the reaction was performed several times, the protocol described is the one performed on the largest scale.
:30); 1H NMR (400 MHz, DMSO-d6): δ 12.37 (s, 1H, OH5), 10.87 (br, 1H, OH7), 9.65 (br, 1H, OH3), 7.84–7.78 (m, 2H, H2′, H6′), 7.58–7.56 (m, 4H, HAr), 7.49–7.42 (m, 6H, HAr), 7.22 (d, 3JH,H = 8.2 Hz, 1H, H5′), 6.47 (d, 4JH,H = 2.0 Hz, 1H, H8), 6.19 (d, 4JH,H = 2.0 Hz, 1H, H6); 13C NMR (75 MHz, DMSO-d6): δ 176.0, 164.1, 160.7, 156.2, 147.6, 146.7, 145.6, 139.4 (2C), 136.4, 129.5 (2C), 128.6 (4C), 125.8 (4C), 125.2, 123.1, 117.1, 108.9, 107.8, 103.1, 98.3, 93.6; HRMS (ESI-TOF) m/z: calcd for C28H17O7 [M − H]−, 465.0974, found 465.0971.
:5); 1H NMR (400 MHz, CDCl3): δ 12.74 (s, 1H, OH5), 7.61–7.59 (m, 4H, HAr), 7.56 (dd, 3JH,H = 8.4 Hz, 4JH,H = 1.8 Hz, 1H, H6′), 7.49 (d, 4JH,H = 1.7 Hz, 1H, H2′), 7.44–7.39 (m, 6H, HAr), 7.28–7.25 (m, 2H, HAr), 7.19–7.12 (m, 3H, HAr), 6.91 (d, 3JH,H = 8.4 Hz, 1H, H5′), 6.37 (d, 4JH,H = 2.2 Hz, 1H, H6 or H8), 6.29 (d, 4JH,H = 2.2 Hz, 1H, H6 or H8), 5.90 (br, 1H, OH7), 5.03 (s, 2H, CH2Bn); 13C NMR (126 MHz, CDCl3): δ 179.0, 162.5, 162.3, 157.1, 157.0, 149.4, 147.4, 139.9 (2C), 137.2, 136.1, 129.5, 129.1, 128.5 (5C), 128.4, 128.3, 126.4 (6C), 124.3, 124.3, 117.9, 109.2, 108.5, 105.9, 99.3, 94.2, 74.7.
:2); 1H NMR (400 MHz, CDCl3): δ 12.68 (s, 1H, OH), 7.62–7.59 (m, 4H, HAr), 7.56 (dd, 3JH,H = 8.4 Hz, 4JH,H = 1.8 Hz, 1H, H6′), 7.50 (d, 4JH,H = 1.7 Hz, 1H, H2′), 7.44–7.34 (m, 12H, HAr), 7.28–7.26 (m, 1H, HAr), 7.21–7.13 (m, 3H, HAr), 6.91 (d, 3JH,H = 8.4 Hz, 1H, H5′), 6.48 (d, 4JH,H = 2.2 Hz, 1H, H6 or H8), 6.44 (d, 4JH,H = 2.2 Hz, 1H, H6 or H8), 5.13 (s, 2H, CH2Bn), 5.04 (s, 2H, CH2Bn); 13C NMR (126 MHz, CDCl3): 178.9, 164.6, 162.2, 156.9 (2C), 149.4, 147.4, 139.9 (2C), 137.4, 136.3, 135.9, 129.5, 129.1, 128.9, 128.5 (8C), 128.3, 127.6, 126.4 (7C), 124.4, 124.2, 118.0, 109.2, 108.5, 106.3, 98.8, 93.2, 74.6, 70.6; HRMS (+ASAP) m/z: calcd for C42H31O7: 647.2070 [M + H]+, found: 647.2067.
:1); 1H NMR (400 MHz, CDCl3): δ 12.64 (s, 1H, OH), 7.61–7.59 (m, 4H, HAr), 7.56 (dd, 3JH,H = 8.3 Hz, 4JH,H = 1.8 Hz, 1H, H6′), 7.50 (d, 4JH,H = 1.7 Hz, 1H, H2′), 7.44–7.39 (m, 6H, HAr), 7.28–7.26 (m, 2H, HAr), 7.21–7.13 (m, 3H, HAr), 6.91 (d, 3JH,H = 8.3 Hz, 1H, H5′), 6.37 (d, 4JH,H = 2.2 Hz, 1H, H6 or H8), 6.33 (d, 4JH,H = 2.2 Hz, 1H, H6 or H8), 5.04 (s, 2H, CH2Bn), 4.61 (sept, 3JH,H = 6.0 Hz, 1H, CHi-Pr), 1.38 (d, 3JH,H = 6.0 Hz, 6H, CH3i-Pr); 13C NMR (126 MHz, CDCl3): δ 178.9, 164.1, 162.2, 157.0, 156.7, 149.4, 147.4, 140.0 (2C), 137.3, 136.4, 129.5, 129.1, 128.5 (6C), 128.3, 126.4 (7C), 124.2, 117.9, 109.5, 108.5, 105.9, 99.1, 93.6, 74.6, 70.9, 22.1 (2C); HRMS (+ASAP) m/z: calcd for C38H31O7: 599.2070 [M + H]+, found: 599.2074.
:30); 1H NMR (400 MHz, CDCl3): δ 7.60–7.58 (m, 4H, HAr), 7.52 (dd, 3JH,H = 8.3 Hz, 4JH,H = 1.8 Hz, 1H, H6′), 7.48 (d, 4JH,H = 1.7 Hz, 1H, H2′), 7.43–7.38 (m, 6H, HAr), 7.30–7.27 (m, 2H, HAr), 7.16–7.10 (m, 3H, HAr), 6.87 (d, 3JH,H = 8.3 Hz, 1H, H5′), 6.42 (d, 4JH,H = 2.2 Hz, 1H, H6 or H8), 6.33 (d, 4JH,H = 2.2 Hz, 1H, H6 or H8), 5.05 (s, 1H, CH2Bn), 4.61 (sept, 3JH,H = 6.0 Hz, 2H, CHi-Pr), 1.48 (d, 3JH,H = 6.0 Hz, 6H, CH3i-Pr), 1.39 (d, 3JH,H = 6.0 Hz, 6H, CH3i-Pr); 13C NMR (126 MHz, CDCl3): δ 174.0, 162.1, 159.8, 159.0, 153.4, 148.7, 147.2, 140.1 (2C), 139.5, 137.1, 129.4, 129.1, 128.5 (5C), 128.1, 127.9, 126.4 (6C), 125.0, 123.6, 117.6, 110.4, 109.0, 108.3, 100.4, 94.1, 74.3, 72.6, 70.7, 22.1 (4C); HRMS (+ASAP) m/z: calcd for C41H37O7: 641.2539 [M + H]+, found: 641.2537.
:27 mL, v/v), 20% palladium hydroxide (50% w/w, 727 mg) was added. The resulting mixture was stirred at room temperature under an H2 atmosphere for 24 hours. Palladium hydroxide was removed by filtration on Celite and washed with EtOH. The filtrate was concentrated under reduced pressure. Purification of the residue was performed by silica gel column chromatography using DCM/MeOH (95/5) to give compound 5 (810 mg, 2.4 mmol, 98% yield) as a yellow solid. Rf = 0.5 (DCM/MeOH 95:5); 1H NMR (400 MHz, CDCl3): δ 7.75 (d, 4JH,H = 2.1, 1H, H2′), 7.65 (dd, 3JH,H = 8.5 Hz, 4JH,H = 2.1 Hz, 1H, H6′), 6.95 (d, 3JH,H = 8.5 Hz, 1H, H5′), 6.43 (d,3JH,H = 2.2 Hz, 1H, H8 or H6), 6.31 (d,3JH,H = 2.2 Hz, 1H, H8 or H6), 4.62 (sept, 2JH,H = 6.1 Hz, 1H, CHi-Pr), 1.37 (d,3JH,H = 6.1 Hz, 6H, CH3i-Pr); 13C NMR (75 MHz, DMSO-d6): 175.9, 163.2, 160.5, 156.2, 147.8, 147.2, 145.1, 136.0, 121.9, 120.0, 115.5, 115.3, 103.8, 98.4, 92.9, 70.4, 21.7 (2C); HRMS (ESI-TOF) m/z: calcd for C18H15O7 [M − H]− 343.0818, found 343.0816.
:20); 1H NMR (400 MHz, CD3OD): δ 12.05 (s, 1H, OH), 7.71 (dd, 3JH,H = 8.5 Hz, 4JH,H = 2.1 Hz, 1H, H6′), 7.79 (d, 4JH,H = 2.1, 1H, H2′); 7.33 (d, 3JH,H = 8.5 Hz, 1H, H5′), 6.43 (d, 4JH,H = 2.2 Hz, 1H, H6 or H8), 6.36 (d, 4JH,H = 2.2 Hz, 1H, H6 or H8), 5.51–5.27 (m, 36H, CHCHDHA), 4.63 (sept, 3JH,H = 6.0 Hz, 1H, CHi-Pr), 2.89–2.79 (m, 30H, CH2DHA(bis-allylic)), 2.72–2.62 (m, 6H, CO–CH2DHA), 2.54–2.48 (m, 6H, CO–CH2–CH2DHA), 2.10–2.03 (m, 6H, CH3–CH2DHA), 1.38 (d, 3JH,H = 6.0 Hz, 6H, CH3i-Pr), 0.99–0.94 (m, 9H, CH3DHA); 13C NMR (126 MHz, DMSO-d6): δ 175.8, 170.4, 170.2, 170.1, 164.7, 162.1, 157.3, 154.7, 144.5, 142.3, 132.2 (2C), 132.2, 130.2 (3C), 129.9, 128.7(3C), 128.7(3C), 128.4(5C), 128.4, 128.3, 128.2, 128.2, 128.1 (5C), 128.0, 128.0 (2C), 127.9, 127.6, 127.3, 127.3, 127.2, 127.1 (3C), 126.7, 124.0, 124.0, 105.5, 99.7, 94.1, 71.1, 34.1, 34.1, 33.7, 25.8 (14C), 25.7, 22.7(3C), 22.0 (2C), 20.7 (3C), 14.4 (3C); HRMS (FTMS + p APCI) m/z: calcd for C84H107O10 [M − H]− 1275.78588, found 1275.78622.
CH = aliphatic alkene, and iP = isopropyl. If the reaction was performed several times, the protocol described is the one performed on the largest scale.
:30); 1H NMR (400 MHz, DMSO-d6): δ 12.37 (s, 1H, OH5), 10.87 (br, 1H, OH7), 9.65 (br, 1H, OH3), 7.84–7.78 (m, 2H, H2′, H6′), 7.58–7.56 (m, 4H, HAr), 7.49–7.42 (m, 6H, HAr), 7.22 (d, 3JH,H = 8.2 Hz, 1H, H5′), 6.47 (d, 4JH,H = 2.0 Hz, 1H, H8), 6.19 (d, 4JH,H = 2.0 Hz, 1H, H6); 13C NMR (75 MHz, DMSO-d6): δ 176.0, 164.1, 160.7, 156.2, 147.6, 146.7, 145.6, 139.4 (2C), 136.4, 129.5 (2C), 128.6 (4C), 125.8 (4C), 125.2, 123.1, 117.1, 108.9, 107.8, 103.1, 98.3, 93.6; HRMS (ESI-TOF) m/z: calcd for C28H17O7 [M − H]−, 465.0974, found 465.0971.
:5); 1H NMR (400 MHz, CDCl3): δ 12.74 (s, 1H, OH5), 7.61–7.59 (m, 4H, HAr), 7.56 (dd, 3JH,H = 8.4 Hz, 4JH,H = 1.8 Hz, 1H, H6′), 7.49 (d, 4JH,H = 1.7 Hz, 1H, H2′), 7.44–7.39 (m, 6H, HAr), 7.28–7.25 (m, 2H, HAr), 7.19–7.12 (m, 3H, HAr), 6.91 (d, 3JH,H = 8.4 Hz, 1H, H5′), 6.37 (d, 4JH,H = 2.2 Hz, 1H, H6 or H8), 6.29 (d, 4JH,H = 2.2 Hz, 1H, H6 or H8), 5.90 (br, 1H, OH7), 5.03 (s, 2H, CH2Bn); 13C NMR (126 MHz, CDCl3): δ 179.0, 162.5, 162.3, 157.1, 157.0, 149.4, 147.4, 139.9 (2C), 137.2, 136.1, 129.5, 129.1, 128.5 (5C), 128.4, 128.3, 126.4 (6C), 124.3, 124.3, 117.9, 109.2, 108.5, 105.9, 99.3, 94.2, 74.7.
:2); 1H NMR (400 MHz, CDCl3): δ 12.68 (s, 1H, OH), 7.62–7.59 (m, 4H, HAr), 7.56 (dd, 3JH,H = 8.4 Hz, 4JH,H = 1.8 Hz, 1H, H6′), 7.50 (d, 4JH,H = 1.7 Hz, 1H, H2′), 7.44–7.34 (m, 12H, HAr), 7.28–7.26 (m, 1H, HAr), 7.21–7.13 (m, 3H, HAr), 6.91 (d, 3JH,H = 8.4 Hz, 1H, H5′), 6.48 (d, 4JH,H = 2.2 Hz, 1H, H6 or H8), 6.44 (d, 4JH,H = 2.2 Hz, 1H, H6 or H8), 5.13 (s, 2H, CH2Bn), 5.04 (s, 2H, CH2Bn); 13C NMR (126 MHz, CDCl3): 178.9, 164.6, 162.2, 156.9 (2C), 149.4, 147.4, 139.9 (2C), 137.4, 136.3, 135.9, 129.5, 129.1, 128.9, 128.5 (8C), 128.3, 127.6, 126.4 (7C), 124.4, 124.2, 118.0, 109.2, 108.5, 106.3, 98.8, 93.2, 74.6, 70.6; HRMS (+ASAP) m/z: calcd for C42H31O7: 647.2070 [M + H]+, found: 647.2067.
:1); 1H NMR (400 MHz, CDCl3): δ 12.64 (s, 1H, OH), 7.61–7.59 (m, 4H, HAr), 7.56 (dd, 3JH,H = 8.3 Hz, 4JH,H = 1.8 Hz, 1H, H6′), 7.50 (d, 4JH,H = 1.7 Hz, 1H, H2′), 7.44–7.39 (m, 6H, HAr), 7.28–7.26 (m, 2H, HAr), 7.21–7.13 (m, 3H, HAr), 6.91 (d, 3JH,H = 8.3 Hz, 1H, H5′), 6.37 (d, 4JH,H = 2.2 Hz, 1H, H6 or H8), 6.33 (d, 4JH,H = 2.2 Hz, 1H, H6 or H8), 5.04 (s, 2H, CH2Bn), 4.61 (sept, 3JH,H = 6.0 Hz, 1H, CHi-Pr), 1.38 (d, 3JH,H = 6.0 Hz, 6H, CH3i-Pr); 13C NMR (126 MHz, CDCl3): δ 178.9, 164.1, 162.2, 157.0, 156.7, 149.4, 147.4, 140.0 (2C), 137.3, 136.4, 129.5, 129.1, 128.5 (6C), 128.3, 126.4 (7C), 124.2, 117.9, 109.5, 108.5, 105.9, 99.1, 93.6, 74.6, 70.9, 22.1 (2C); HRMS (+ASAP) m/z: calcd for C38H31O7: 599.2070 [M + H]+, found: 599.2074.
:30); 1H NMR (400 MHz, CDCl3): δ 7.60–7.58 (m, 4H, HAr), 7.52 (dd, 3JH,H = 8.3 Hz, 4JH,H = 1.8 Hz, 1H, H6′), 7.48 (d, 4JH,H = 1.7 Hz, 1H, H2′), 7.43–7.38 (m, 6H, HAr), 7.30–7.27 (m, 2H, HAr), 7.16–7.10 (m, 3H, HAr), 6.87 (d, 3JH,H = 8.3 Hz, 1H, H5′), 6.42 (d, 4JH,H = 2.2 Hz, 1H, H6 or H8), 6.33 (d, 4JH,H = 2.2 Hz, 1H, H6 or H8), 5.05 (s, 1H, CH2Bn), 4.61 (sept, 3JH,H = 6.0 Hz, 2H, CHi-Pr), 1.48 (d, 3JH,H = 6.0 Hz, 6H, CH3i-Pr), 1.39 (d, 3JH,H = 6.0 Hz, 6H, CH3i-Pr); 13C NMR (126 MHz, CDCl3): δ 174.0, 162.1, 159.8, 159.0, 153.4, 148.7, 147.2, 140.1 (2C), 139.5, 137.1, 129.4, 129.1, 128.5 (5C), 128.1, 127.9, 126.4 (6C), 125.0, 123.6, 117.6, 110.4, 109.0, 108.3, 100.4, 94.1, 74.3, 72.6, 70.7, 22.1 (4C); HRMS (+ASAP) m/z: calcd for C41H37O7: 641.2539 [M + H]+, found: 641.2537.
:27 mL, v/v), 20% palladium hydroxide (50% w/w, 727 mg) was added. The resulting mixture was stirred at room temperature under an H2 atmosphere for 24 hours. Palladium hydroxide was removed by filtration on Celite and washed with EtOH. The filtrate was concentrated under reduced pressure. Purification of the residue was performed by silica gel column chromatography using DCM/MeOH (95/5) to give compound 5 (810 mg, 2.4 mmol, 98% yield) as a yellow solid. Rf = 0.5 (DCM/MeOH 95:5); 1H NMR (400 MHz, CDCl3): δ 7.75 (d, 4JH,H = 2.1, 1H, H2′), 7.65 (dd, 3JH,H = 8.5 Hz, 4JH,H = 2.1 Hz, 1H, H6′), 6.95 (d, 3JH,H = 8.5 Hz, 1H, H5′), 6.43 (d,3JH,H = 2.2 Hz, 1H, H8 or H6), 6.31 (d,3JH,H = 2.2 Hz, 1H, H8 or H6), 4.62 (sept, 2JH,H = 6.1 Hz, 1H, CHi-Pr), 1.37 (d,3JH,H = 6.1 Hz, 6H, CH3i-Pr); 13C NMR (75 MHz, DMSO-d6): 175.9, 163.2, 160.5, 156.2, 147.8, 147.2, 145.1, 136.0, 121.9, 120.0, 115.5, 115.3, 103.8, 98.4, 92.9, 70.4, 21.7 (2C); HRMS (ESI-TOF) m/z: calcd for C18H15O7 [M − H]− 343.0818, found 343.0816.
:20); 1H NMR (400 MHz, CD3OD): δ 12.05 (s, 1H, OH), 7.71 (dd, 3JH,H = 8.5 Hz, 4JH,H = 2.1 Hz, 1H, H6′), 7.79 (d, 4JH,H = 2.1, 1H, H2′); 7.33 (d, 3JH,H = 8.5 Hz, 1H, H5′), 6.43 (d, 4JH,H = 2.2 Hz, 1H, H6 or H8), 6.36 (d, 4JH,H = 2.2 Hz, 1H, H6 or H8), 5.51–5.27 (m, 36H, CHCHDHA), 4.63 (sept, 3JH,H = 6.0 Hz, 1H, CHi-Pr), 2.89–2.79 (m, 30H, CH2DHA(bis-allylic)), 2.72–2.62 (m, 6H, CO–CH2DHA), 2.54–2.48 (m, 6H, CO–CH2–CH2DHA), 2.10–2.03 (m, 6H, CH3–CH2DHA), 1.38 (d, 3JH,H = 6.0 Hz, 6H, CH3i-Pr), 0.99–0.94 (m, 9H, CH3DHA); 13C NMR (126 MHz, DMSO-d6): δ 175.8, 170.4, 170.2, 170.1, 164.7, 162.1, 157.3, 154.7, 144.5, 142.3, 132.2 (2C), 132.2, 130.2 (3C), 129.9, 128.7(3C), 128.7(3C), 128.4(5C), 128.4, 128.3, 128.2, 128.2, 128.1 (5C), 128.0, 128.0 (2C), 127.9, 127.6, 127.3, 127.3, 127.2, 127.1 (3C), 126.7, 124.0, 124.0, 105.5, 99.7, 94.1, 71.1, 34.1, 34.1, 33.7, 25.8 (14C), 25.7, 22.7(3C), 22.0 (2C), 20.7 (3C), 14.4 (3C); HRMS (FTMS + p APCI) m/z: calcd for C84H107O10 [M − H]− 1275.78588, found 1275.78622.
From pathway A. To a solution of compound 6 (100 mg, 0.08 mmol) (or the mixture of quercetin di-DHA-iPr (198 mg)) in tert-butylmethylether (13.4 mL), n-BuOH (0.50 mL) and supported Lipozyme® from Mucor miehei (1
:1 w/w, 200 mg) were added. The resulting mixture was stirred at 40 °C for 8 days, after which the supported enzyme was filtrated and rinsed several times with DCM. The filtrate was concentrated under reduced pressure. Purification of the residue was performed by silica gel column chromatography (solid deposit) using a pentane/EtOAc gradient of 95/5 to 90/10 to give the desired product 7 (13 mg, 0.02 mmol, 26% from 6; 36 mg, 0.05 mmol, 27% from the di-DHA mixture, and 19% from 5) as a yellow solid.
From pathway B1 and B2. The synthesis was realized starting from 11 (3.08 g, 3.14 mmol) as described by Moine et al.24 and yielded 7 (1.61 g, 2.45 mmol, 78%) as a yellow solid. Rf = 0.5 (DCM/MeOH 95
:5); 1H NMR (400 MHz, CDCl3): δ 12.13 (s, 1H, OH5), 7.36–7.34 (m, 2H, H6′, H2′); 6.95 (d, 3JH,H = 9.0 Hz, 1H, H5′), 6.41 (d, 4JH,H = 2.1 Hz, 1H, H6 or H8), 6.34 (d, 4JH,H = 2.1 Hz, 1H, H6 or H8), 5.89 (br, 2H, OH3′,4′), 5.47–5.41 (m, 12H, CHCHDHA), 4.62 (sept, 3JH,H = 6.0 Hz, 1H, CHi-Pr), 2.94–2.76 (m, 10H, CH2DHA(bis allylic)), 2.74–2.63 (m, 2H, CO–CH2DHA), 2.57–2.46 (m, 2H, CO–CH2–CH2DHA), 2.12–2.01 (m, 2H, CH3–CH2DHA), 1.38 (d, 3JH,H = 6.0 Hz, 6H, CH3i-Pr), 0.99–0.93 (m, 3H, CH3DHA); 13C NMR (126 MHz, CDCl3): δ 175.9, 171.1, 164.6, 161.9, 157.2, 156.7, 147.5, 143.8, 132.2, 130.8, 130.0, 128.7, 128.6, 128.5 (2C), 128.2 (2C), 128.1, 128.0, 127.5, 127.2, 122.2, 121.9, 115.4, 115.2, 105.2, 99.6, 94.1, 71.1, 34.0, 25.8 (4C), 25.7, 22.7, 22.0 (2C), 20.7, 14.4; HRMS (ESI+) m/z calcd for C40H46O8 [M + H]+ 655.3265, found 655.3273.
:4); 1H NMR (500 MHz, CDCl3): δ 7.60–7.55 (m, 4H, HAr), 7.44–7.38 (m, 7H, HAr), 7.36 (d, 4JH,H = 1.7 Hz, 1H, H2′), 7.28 (d, 4JH,H = 2.2 Hz, 1H, H8), 6.97 (d, 3JH,H = 8.3 Hz, 1H, H5′), 6.83 (d, 4JH,H = 2.2 Hz, 1H, H6), 2.71 (t, 3JH,H = 7.5 Hz, 2H, CH2butyrate), 2.57 (t, 3JH,H = 7.3 Hz, 2H, CH2butyrate), 2.57 (t, 3JH,H = 7.4 Hz, 2H, CH2butyrate), 1.87–1.74 (m, 4H, 2 CH2butyrate), 1.72 (q, 3JH,H = 7.4 Hz, 2H, CH2butyrate), 1.05 (t, 3JH,H = 7.4 Hz, 3H, CH3butyrate), 1.05 (t, 3JH,H = 7.4 Hz, 3H, CH3butyrate), 0.94 (t, 3JH,H = 7.4 Hz, 3H, CH3butyrate); 13C NMR (126 MHz, CDCl3): δ 172.1, 170.8, 170.7, 170.2, 157.0, 155.2, 154.3, 150.6, 149.9, 147.7, 139.7 (2C), 133.5, 129.6 (2C), 128.5 (4C), 126.4 (4C), 123.7, 123.3, 118.3, 114.9, 113.9, 108.9, 108.8, 108.6, 36.3, 36.1, 35.9, 18.4 (2C), 18.1, 13.8, 13.7, 13.7; HRMS (+ESI) m/z: calcd for C40H37O10: 677.2381 [M + H]+, found: 677.2568.
:2); 1H NMR (500 MHz, CDCl3): δ 7.94 (br, 1H, OH) 7.60–7.55 (m, 4H, HAr), 7.43–7.33 (m, 8H, HAr), 6.91 (d, 3JH,H = 8.7 Hz, 1H, H5′), 6.56 (d, 4JH,H = 2.3 Hz, 1H, H8), 6.43 (d, 4JH,H = 2.3 Hz, 1H, H6), 2.68 (t, 3JH,H = 7.4 Hz, 2H, CH2butyrate), 2.56 (t, 3JH,H = 7.4 Hz, 2H, CH2butyrate), 1.77 (q, 3JH,H = 7.4 Hz, 2H, CH2butyrate), 1.69 (q, 3JH,H = 7.4 Hz, 2H, CH2butyrate), 0.91 (t, 3JH,H = 7.4 Hz, 3H, CH3butyrate), 0.99 (t, 3JH,H = 7.4 Hz, 3H, CH3butyrate); 13C NMR (126 MHz, CDCl3): δ 172.9, 171.9, 170.5, 161.5, 157.8, 155.0, 150.7, 149.8, 147.7, 139.8 (2C), 132.7, 129.6 (2C), 128.5 (4C), 126.4 (4C), 123.8, 123.4, 118.3, 110.5, 109.3, 108.7, 108.6, 101.4, 36.1, 36.0, 18.3, 18.1, 13.8, 13.6; HRMS (+ESI) m/z: calcd for C36H31O9: 607.1963 [M + H]+, found: 607.1952.
From pathway B1 (in 5 steps from 9). Compound 9 (5.0 g, 8.24 mmol, 1 equiv.) was dissolved in acetone (400 mL) under N2. K2CO3 (2.28 g, 16.5 mmol, 2 equiv.) and diisopropyl sulfate (2.73 mL, 16.5 mmol, 2 equiv.) were added to the solution and the mixture was stirred at 40 °C overnight. The mixture was then filtered on Celite and the filtrate was mostly evaporated and dissolved in 150 mL DCM. The organic layer was washed with brine, dried over MgSO4 and the solvents were removed under reduce pressure to afford the crude product as a yellow oil. The crude was then purified through silica gel column chromatography with a pentane/EtOAc gradient of 90/10 to 80/20 as the eluent. The fractions containing the desired product and the major by-products were gathered and solvents were evaporated to afford 4.26 g of a white solid. The solid was dissolved in pyridine (20 mL) under N2. Butyric anhydride (1.8 mL, 11 mmol) was added and the mixture was stirred at room temperature overnight. The mixture was then quenched with 100 mL distilled water and extracted with 2 × 100 mL EtOAc. The organic layers were gathered, washed with 1 M HCl (100 mL), water (100 mL) and brine, and dried over MgSO4. Solvents were removed under reduced pressure to afford the crude product as yellow oil. The crude was then purified through silica gel column chromatography with a pentane/EtOAc gradient of 90/10 to 80/20 as the eluent to yield an unseparable mixture of compounds S1 and tributyrate by-product 8 as a white solid (3.91 g), in 57/43 proportion estimated using 1H NMR integration of the H6/8 signal. S1: Rf = 0.4 (cyclohexane/EtOAc 80
:20); a mixture of compound S1 and by-product 8 (3.1 g) was dissolved in THF/EtOH (77/33 mL) and Pd(OH)2 (20 wt%, 620 mg) was added. The mixture was stirred vigorously under an H2 atmosphere for 24 hours. The mixture was then filtered on Celite and the solvents were evaporated to afford the crude product as a green paste. The crude was then purified through silica gel column chromatography with a DCM/MeOH gradient of 100/0 to 96/4 as the eluent to yield an inseparable mixture of the desired product S2 and its tributyrated by-product (1.9 g) as a yellow solid, in 58/42 proportion estimated using 1H NMR integration of the H5′ signal. S2: Rf = 0.25 (DCM/MeOH, 96:4); the previously mentioned mixture of S2 (1.9 g) was dissolved in dry THF (50 mL). Distilled Et3N (2.19 mL, 15.7 mmol) and TIPSOTf (2.37 mL, 8.63 mmol) were added and the mixture was stirred at room temperature for 45 minutes. The mixture was then quenched with 50 mL distilled water and extracted with EtOAc (2 × 50 mL). The gathered organic phases were washed with 1 M HCl (50 mL) and brine, dried over MgSO4 and the solvents were removed under reduced pressure to afford the crude product as yellow oil. The crude was then purified through silica gel column chromatography with a pentane/EtOAc gradient of 80/20 as the eluent to yield an inseparable mixture of compounds S3 and its tributyrated by-product as yellow oil (2.77 g), in 61/39 proportion estimated using 1H NMR integration of the H6/8 signal. S3: Rf = 0.4 (cyclohexane/EtOAc, 60:40); the previously mentioned mixture of S3 (2.77 g) was dissolved in DCM/MeOH (28/15 mL) under N2 and cooled to 0 °C. NH3 (7 N in MeOH, 6 mL, 41.8 mmol, 12 equiv.) was added and the mixture was stirred at room temperature overnight. The mixture was then diluted with 100 mL DCM. The organic layer was washed with 1 M HCl (50 mL) and brine, dried over MgSO4 and the solvents were removed under reduced pressure to afford the crude product as brown oil. The crude was then purified through silica gel column chromatography with a pentane/EtOAc gradient of 90/10 as the eluent to yield the pure desired product 10 (1.24 g, 1.89 mmol, 25% over 5 steps) as a brown solid.
From pathway B2 (in 1 step from 15):. The synthesis was realized starting from 15 (3.66 g, 4.94 mmol) as described by Moine et al.24 and yielded 10 (3.06 g, 4.65 mmol, 95%) as a brownish solid. Rf = 0.4 (cyclohexane/EtOAc, 95
:5); 1H NMR (400 MHz, CDCl3): δ 11.71 (s, 1H, OH5), 7.83 (d, 4JH,H = 2.3 Hz, 1H, H2′), 7.66 (dd, 3JH,H = 8.6 Hz, 4JH,H = 2.3 Hz, 1H, H6′), 6.95 (d, 3JH,H = 8.6 Hz, 1H, H5′), 6.56 (s, 1H, OH3), 6.39 (d, 4JH,H = 2.1 Hz, 1H, H8), 6.34 (d, 4JH,H = 2.1 Hz, 1H, H6), 4.62 (hept, 3JH,H = 6.0 Hz, 1H, CHi-Pr), 1.39 (d, 3JH,H = 6.0 Hz, 6H, 2 CH3i-Pr), 1.38–1.29 (m, 6H, 6 CHTIPS), 1.18–1.11 (m, 36H, 12 CH3TIPS); 13C NMR (126 MHz, CDCl3) δ 175.2, 164.2, 161.0, 156.9, 149.4, 147.2, 145.7, 135.7, 123.5, 120.9, 120.0, 119.5, 103.7, 98.8, 93.7, 71.0, 22.1 (2C), 18.2 (6C), 18.1 (6C), 13.3 (3C), 13.1 (3C); HRMS (ESI-TOF) m/z calcd for C36H55O7Si2 [M − H]− 655.3486, found 655.3487.
:50); 1H NMR (400 MHz, CDCl3): δ 12.19 (s, 1H, OH5), 7.39 (d, 4JH,H = 2.3 Hz, 1H, H2′), 7.33 (dd, 3JH,H = 8.5 Hz, 4JH,H = 2.2 Hz, 1H, H6′), 6.90 (d, 3JH,H = 8.5 Hz, 1H, H5′), 6.36 (d, 4JH,H = 2.2 Hz, 1H, H8), 6.34 (d, 4JH,H = 2.2 Hz, 1H, H6), 5.51–5.24 (m, 12H, CHCHDHA) 4.61 (hept, 3JH,H = 6.0 Hz, 1H, CHi-Pr), 2.89–2.78 (m, 10H, CH2DHA(bis allylic)), 2.69 (t, 3JH,H = 7.6 Hz, 2H, CO–CH2DHA), 2.51 (q, 3JH,H = 7.3 Hz, 2H, CO–CH2–CH2DHA), 2.07 (p, 3JH,H = 7.3 Hz, 2H, CH3–CH2DHA), 1.39 (d, 3JH,H = 6.0 Hz, 6H, 2 CH3i-Pr), 1.36–1.29 (m, 6H, 6 CHTIPS), 1.16–1.10 (m, 36H, 12 CH3TIPS), 0.96 (t, 3JH,H = 7.5 Hz, 3H, CH3DHA); 13C NMR (101 MHz, CDCl3): δ 175.9, 170.4, 164.4, 162.2, 157.2, 156.4, 150.5, 147.2, 132.2, 131.2, 129.8, 128.7, 128.5, 128.4 (2C), 128.3, 128.2, 128.2, 128.0, 127.7, 127.2, 122.2, 122.1, 119.9, 119.9, 105.3, 99.2, 94.0, 71.0, 33.9, 25.8 (4C), 25.7, 22.8, 22.0 (2C), 20.7, 18.1 (6C), 18.1 (6C), 14.4, 13.3 (3C), 13.2 (3C); HRMS (ASAP-TOF) calcd for C58H87O8Si2 [M + H]+ 967.5939, found 967.5927.
:30); 1H NMR (400 MHz, CDCl3): δ 12.21 (s, 1H, OH), 7.61–7.56 (m, 4H, HAr), 7.45 (dd, 3JH,H = 8.3 Hz, 4JH,H = 1.8 Hz, 1H, HAr), 7.43–7.38 (m, 7H, HAr), 7.00 (d, 3JH,H = 8.3 Hz, 1H, HAr), 6.82 (d, 4JH,H = 2.0 Hz, 1H, HAr), 6.58 (d, 4JH,H = 2.0 Hz, 1H, HAr), 2.36 (s, 3H, CH3Ac), 2.33 (s, 3H, CH3Ac); 13C NMR (101 MHz, CDCl3): δ 176.4, 168.4, 167.9, 161.8, 157.2, 156.3, 156.0, 150.3, 147.9, 139.6 (2C), 131.5, 129.6 (2C), 128.6 (4C), 126.4 (4C), 124.0, 123.0, 118.5, 109.0, 108.8, 108.5, 105.5, 101.2, 21.3, 20.7; HRMS (ESI-TOF) m/z calcd for C32H23O9 [M + H]+ 551.1342, found 551.1342.
:30); 1H NMR (400 MHz, CDCl3): δ 7.64–7.52 (m, 4H, HAr), 7.45–7.32 (m, 8H, HAr), 6.97 (d, 3JH,H = 8.2 Hz, 1H, HAr), 6.77 (d, 4JH,H = 2.4 Hz, 1H, HAr), 6.57 (d, 4JH,H = 2.4 Hz, 1H, HAr), 4.62 (hept, 3JH,H = 5.9 Hz, 1H, CHi-Pr), 2.42 (s, 3H, CH3Ac), 2.31 (s, 3H, CH3Ac), 1.39 (d, 3JH,H = 6.0 Hz, 6H, 2 CH3i-Pr); 13C NMR (75 MHz, CDCl3): δ 170.3, 169.8, 168.3, 162.3, 158.3, 154.7, 150.8, 149.7, 147.8, 139.8 (2C), 133.2, 129.6 (2C), 128.5 (4C), 126.4 (4C), 123.6, 123.3, 118.3, 110.7, 109.7, 108.8, 108.4, 100.0, 71.4, 21.9, 21.3, 20.9; HRMS (ESI-TOF) m/z: calcd for C35H29O9 [M + H]+ 593.1812, found 593.1801.
:4); 1H NMR (400 MHz, DMSO-d6): δ 9.65 (br, 2H, 2 OH), 7.34 (d, 4JH,H = 2.3 Hz, 1H, HAr), 7.29 (dd, 3JH,H = 8.4, 4JH,H = 2.3 Hz, 1H, HAr), 7.21 (d, 4JH,H = 2.4 Hz, 1H, HAr), 6.92 (d, 3JH,H = 8.4 Hz, 1H, HAr), 6.76 (d, 4JH,H = 2.4 Hz, 1H, HAr), 4.86 (hept, 3JH,H = 5.9 Hz, 1H, CHi-Pr), 2.32 (s, 3H, CH3Ac), 2.29 (s, 3H, CH3Ac), 1.33 (d, 3JH,H = 6.0 Hz, 6H, 2 CH3i-Pr); 13C NMR (101 MHz, DMSO-d6): δ 168.9, 168.8, 167.9, 161.7, 157.5, 154.3, 150.0, 149.0, 145.4, 131.7, 120.3, 119.7, 115.9, 115.0, 109.6, 109.4, 100.2, 71.0, 21.5 (2C), 20.9, 20.4; HRMS (ESI-TOF) m/z: calcd for C22H19O9 [M − H]− 427.1029, found 427.1031.
:30); 1H NMR (300 MHz, CDCl3) δ 7.36 (d, 4JH,H = 2.3 Hz, 1H, H2′), 7.30 (dd, 3JH,H = 8.5, 4JH,H = 2.3 Hz, 1H, H6′), 6.91 (d, 3JH,H = 8.4 Hz, 1H, H5′), 6.74 (d, 4JH,H = 2.5 Hz, 1H, H8), 6.56 (d, 4JH,H = 2.5 Hz, 1H, H6), 4.61 (hept, 3JH,H = 6.0 Hz, 1H, CHi-Pr), 2.42 (s, 3H, CH3Ac), 2.32 (s, 3H, CH3Ac), 1.39 (d, 3JH,H = 6.0 Hz, 6H, 2 CH3i-Pr), 1.38–1.27 (m, 6H, 6 CHTIPS), 1.13 (d, 3JH,H = 7.3 Hz, 36H, 12 CH3TIPS); 13C NMR (101 MHz, CDCl3) δ 170.4, 169.8, 168.2, 162.2, 158.3, 154.9, 150.8, 150.2, 147.1, 133.2, 122.3, 121.6, 120.0, 119.9, 110.7, 109.4, 100.1, 71.3, 21.9 (2C), 21.3, 20.8, 18.1 (6C), 18.1 (6C), 13.3 (3C), 13.2 (3C); HRMS (ESI-TOF) m/z: calcd for C40H61O9Si2 [M + H]+ 741.3854, found 741.3849.
Cell culture experiments
SHSY-5Y cells (purchased from ATCC) were used at passage 15. They were cultured in DMEM/F12 medium supplemented with 10% FBS and 1% penicillin–streptomycin. For the experiments, the cells were plated and allowed to differentiate in 96-well plates at a density of 40000 cells per well in DMEM/F12 medium supplemented with 3% FBS, 1% penicillin–streptomycin and 3.36 mM retinoic acid. After 72 h, they were rinsed with DPBS and pre-treated with various concentrations of DHA, quercetin, quercetin-7-iPr or lipophenolic compounds for 2 h in serum-free DMEM/F12 medium before the addition of 10μM acrolein. Cell viability was determined after a 24-hour incubation using the MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide, Sigma-Aldrich) test. Briefly, MTT was dissolved at 0.5 mg mL−1 in PBS and added to the cells for 1 h. Then, the medium was discarded, replaced with DMSO, and the plate was placed on an orbital shaker for 1 h at room temperature to dissolve formazan crystals. A microplate reader (Multiskan Sky, Thermo Scientific) was used to measure the absorbance at 540 nm. The data are expressed as the percentage of control untreated cells and presented as mean ± SEM of two separate experiments. Statistical comparisons were performed using the ANOVA test with Tukey's post-hoc analysis.
Author contributions
LO, LM and JL carried out the chemical syntheses and analysed the products. LO conducted and evaluated all biological assays. JL, CD and CC wrote the manuscript. TD and LG supervised the project. CD and CC initiated, supervised the project and acquired financial support for the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the University of Montpellier, the ANR (LipoPheRet - ANR-18-CE18-0017 and LabEx LIPSTIC - ANR-11-LABX-0021), the Région Bourgogne Franche-Comté, and the Fonds Européen de Développement Régional. CNRS and INSERM are thanked for their support.References
- S. Mani, R. Dubey, I.-C. Lai, M. A. Babu, S. Tyagi, G. Swargiary, D. Mody, M. Singh, S. Agarwal, D. Iqbal, S. Kumar, M. Hamed, P. Sachdeva, A. G. Almutary, H. M. Albadrani, S. Ojha, S. K. Singh and N. K. Jha, Oxidative Stress and Natural Antioxidants: Back and Forth in the Neurological Mechanisms of Alzheimer's Disease, J. Alzheimer's Dis., 2023, 96, 877–912 CAS.
- M. Perluigi, F. Di Domenico and D. A. Butterfield, Oxidative damage in neurodegeneration: roles in the pathogenesis and progression of Alzheimer disease, Physiol. Rev., 2024, 104, 103–197 CrossRef CAS PubMed.
- T. I. Williams, B. C. Lynn, W. R. Markesbery and M. A. Lovell, Increased levels of 4-hydroxynonenal and acrolein, neurotoxic markers of lipid peroxidation, in the brain in Mild Cognitive Impairment and early Alzheimer's disease, Neurobiol. Aging, 2006, 27, 1094–1099 CrossRef CAS PubMed.
- M. A. Lovell, C. Xie and W. R. Markesbery, Acrolein is increased in Alzheimer's disease brain and is toxic to primary hippocampal cultures, Neurobiol. Aging, 2001, 22, 187–194 CrossRef CAS PubMed.
- M. J. Picklo, T. J. Montine, V. Amarnath and M. D. Neely, Carbonyl toxicology and Alzheimer's disease, Toxicol. Appl. Pharmacol., 2002, 184, 187–197 CrossRef CAS PubMed.
- Q. Liu, A. K. Raina, M. A. Smith, L. M. Sayre and G. Perry, Hydroxynonenal, toxic carbonyls, and Alzheimer disease, Mol. Aspects Med., 2003, 24, 305–313 CrossRef CAS PubMed.
- M. Singh, T. N. Dang, M. Arseneault and C. Ramassamy, Role of by-products of lipid oxidation in Alzheimer's disease brain: a focus on acrolein, J. Alzheimer's Dis., 2010, 21, 741–756 CAS.
- S. M. Innis, Dietary (n-3) Fatty Acids and Brain Development12, J. Nutr., 2007, 137, 855–859 CrossRef CAS PubMed.
- R. J. S. Lacombe, M. E. Smith, K. Perlman, G. Turecki, N. Mechawar and R. P. Bazinet, Quantitative and carbon isotope ratio analysis of fatty acids isolated from human brain hemispheres, J. Neurochem., 2023, 164, 44–56 CrossRef CAS PubMed.
- N. G. Bazan, V. L. Marcheselli and K. Cole-Edwards, Brain response to injury and neurodegeneration: endogenous neuroprotective signaling, Ann. N. Y. Acad. Sci., 2005, 1053, 137–147 CAS.
- H. N. Yassine, Q. Feng, I. Azizkhanian, V. Rawat, K. Castor, A. N. Fonteh, M. G. Harrington, L. Zheng, B. R. Reed, C. DeCarli, W. J. Jagust and H. C. Chui, Association of Serum Docosahexaenoic Acid With Cerebral Amyloidosis, JAMA Neurol., 2016, 73, 1208–1216 CrossRef PubMed.
- M. Haution-Bitker, T. Gilbert, A. Vignoles, C. Lecardonnel, S. Watelet, E. Blond, J. Drai and M. Bonnefoy, Associations between Plasmatic Polyunsaturated Fatty Acids Concentrations and Cognitive Status and Decline in Neurocognitive Disorders, J. Nutr., Health Aging, 2018, 22, 718–725 CrossRef CAS PubMed.
- R. P. Bazinet and S. Layé, Polyunsaturated fatty acids and their metabolites in brain function and disease, Nat. Rev. Neurosci., 2014, 15, 771–785 CrossRef CAS PubMed.
- I. M. Dighriri, A. M. Alsubaie, F. M. Hakami, D. M. Hamithi, M. M. Alshekh, F. A. Khobrani, F. E. Dalak, A. A. Hakami, E. H. Alsueaadi, L. S. Alsaawi, S. F. Alshammari, A. S. Alqahtani, I. A. Alawi, A. A. Aljuaid and M. Q. Tawhari, Effects of Omega-3 Polyunsaturated Fatty Acids on Brain Functions: A Systematic Review, Cureus, 2022, 14, e30091 Search PubMed.
- B. Troesch, M. Eggersdorfer, A. Laviano, Y. Rolland, A. D. Smith, I. Warnke, A. Weimann and P. C. Calder, Expert Opinion on Benefits of Long-Chain Omega-3 Fatty Acids (DHA and EPA) in Aging and Clinical Nutrition, Nutrients, 2020, 12, 2555 CrossRef CAS PubMed.
- C. Zussy, R. John, T. Urgin, L. Otaegui, C. Vigor, N. Acar, G. Canet, M. Vitalis, F. Morin, E. Planel, C. Oger, T. Durand, S. L. Rajshree, L. Givalois, P. V. Devarajan and C. Desrumaux, Intranasal Administration of Nanovectorized Docosahexaenoic Acid (DHA) Improves Cognitive Function in Two Complementary Mouse Models of Alzheimer's Disease, Antioxidants, 2022, 11, 838 CrossRef CAS PubMed.
- C. H. Maclean, A. M. Issa, S. J. Newberry, W. A. Mojica, S. C. Morton, R. H. Garland, L. G. Hilton, S. B. Traina and P. G. Shekelle, Effects of omega-3 fatty acids on cognitive function with aging, dementia, and neurological diseases, Evid. Rep. Technol. Assess, 2005, 1–3 Search PubMed.
- S. Śliwińska and M. Jeziorek, The role of nutrition in Alzheimer's disease, Rocz. Panstw. Zakl. Hig., 2021, 72, 29–39 Search PubMed.
- S. Quideau, D. Deffieux, C. Douat-Casassus and L. Pouységu, Plant polyphenols: chemical properties, biological activities, and synthesis, Angew. Chem., Int. Ed., 2011, 50, 586–621 CrossRef CAS PubMed.
- Q. Zhu, Z. P. Zheng, K. W. Cheng, J. J. Wu, S. Zhang, Y. S. Tang, K. H. Sze, J. Chen, F. Chen and M. Wang, Natural polyphenols as direct trapping agents of lipid peroxidation-derived acrolein and 4-hydroxy-trans-2-nonenal, Chem. Res. Toxicol., 2009, 22, 1721–1727 Search PubMed.
- X. Shao, H. Chen, Y. Zhu, R. Sedighi, C.-T. Ho and S. Sang, Essential Structural Requirements and Additive Effects for Flavonoids to Scavenge Methylglyoxal, J. Agric. Food Chem., 2014, 62, 3202–3210 CrossRef CAS PubMed.
- C. Crauste, M. Rosell, T. Durand and J. Vercauteren, Omega-3 polyunsaturated lipophenols, how and why?, Biochimie, 2016, 120, 62–74 CrossRef CAS PubMed.
- C. Crauste, C. Vigor, P. Brabet, M. Picq, M. Lagarde, C. Hamel, T. Durand and J. Vercauteren, Synthesis and Evaluation of Polyunsaturated Fatty Acid-Phenol Conjugates as Anti-Carbonyl-Stress Lipophenols, Eur. J. Org. Chem., 2014, 4548–4561 CrossRef CAS.
- E. Moine, M. Boukhallat, D. Cia, N. Jacquemot, L. Guillou, T. Durand, J. Vercauteren, P. Brabet and C. Crauste, New lipophenols prevent carbonyl and oxidative stresses involved in macular degeneration, Free Radicals Biol. Med., 2021, 162, 367–382 CrossRef CAS PubMed.
- A. Cubizolle, D. Cia, E. Moine, N. Jacquemot, L. Guillou, M. Rosell, C. Angebault-Prouteau, G. Lenaers, I. Meunier, J. Vercauteren, T. Durand, C. Crauste and P. Brabet, Isopropyl-phloroglucinol-DHA protects outer retinal cells against lethal dose of all-trans-retinal, J. Cell. Mol. Med., 2020, 24, 5057–5069 CrossRef CAS PubMed.
- N. Taveau, A. Cubizolle, L. Guillou, N. Pinquier, E. Moine, D. Cia, V. Kalatzis, J. Vercauteren, T. Durand, C. Crauste and P. Brabet, Preclinical pharmacology of a lipophenol in a mouse model of light-induced retinopathy, Exp. Mol. Med., 2020, 52, 1090–1101 CrossRef CAS PubMed.
- M. Vincent, L. Simon, P. Brabet, P. Legrand, C. Dorandeu, J. L. K. Him, T. Durand, C. Crauste and S. Begu, Formulation and Evaluation of SNEDDS Loaded with Original Lipophenol for the Oral Route to Prevent Dry AMD and Stragardt's Disease, Pharmaceutics, 2022, 14, 1029 CrossRef CAS PubMed.
- W. Y. Oh, P. Ambigaipalan and F. Shahidi, Preparation of Quercetin Esters and Their Antioxidant Activity, J. Agric. Food Chem., 2019, 67, 10653–10659 CrossRef CAS PubMed.
- W. Y. Oh, P. Ambigaipalan and F. Shahidi, Quercetin and its ester derivatives inhibit oxidation of food, LDL and DNA, Food Chem., 2021, 364, 130394 CrossRef CAS PubMed.
- P. Torres, A. Poveda, J. Jimenez-Barbero, A. Ballesteros and F. J. Plou, Regioselective Lipase-Catalyzed Synthesis of 3-O-Acyl Derivatives of Resveratrol and Study of Their Antioxidant Properties, J. Agric. Food Chem., 2010, 58, 807–813 CrossRef CAS PubMed.
- L. Chebil, J. Anthoni, C. Humeau, C. Gerardin, J.-M. Engasser and M. Ghoul, Enzymatic Acylation of Flavonoids: Effect of the Nature of the Substrate, Origin of Lipase, and Operating Conditions on Conversion Yield and Regioselectivity, J. Agric. Food Chem., 2007, 55, 9496–9502 CrossRef CAS PubMed.
- A. Patti, M. Piattelli and G. Nicolosi, Use of Mucor miehei lipase in the preparation of long chain 3-O-acylcatechins, J. Mol. Catal. B: Enzym., 2000, 10, 577–582 CrossRef CAS.
- H. Peng and F. Shahidi, Quercetin Fatty Acid Monoesters (C2:0–C18:0): Enzymatic Preparation and Antioxidant Activity, J. Agric. Food Chem., 2022, 70, 14073–14083 CrossRef CAS PubMed.
- E. Moine, P. Brabet, L. Guillou, T. Durand, J. Vercauteren and C. Crauste, New Lipophenol Antioxidants Reduce Oxidative Damage in Retina Pigment Epithelial Cells, Antioxidants, 2018, 7, 197 CrossRef PubMed.
- M. Bouktaib, S. Lebrun, A. Atmani and C. Rolando, Hemisynthesis of all the O-monomethylated analogues of quercetin including the major metabolites, through selective protection of phenolic functions, Tetrahedron, 2002, 58, 10001–10009 CrossRef CAS.
- M. Li, X. Han and B. Yu, Facile Synthesis of Flavonoid 7-O-Glycosides, J. Org. Chem., 2003, 68, 6842–6845 CrossRef CAS PubMed.
- J. Liu, J. Fu, W. Li, Y. Zou, Z. Huang, J. Xu, S. Peng and Y. Zhang, Utilization of the inherent nucleophile for regioselective O-acylation of polyphenols via an intermolecular cooperative transesterification, Tetrahedron, 2016, 72, 4103–4110 CrossRef CAS.
- S.-H. Jung, H.-Y. Heo, J.-W. Choe, J. Kim and K. Lee, Anti-Melanogenic Properties of Velutin and Its Analogs, Molecules, 2021, 26, 3033 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional information: all 1H and 13C NMR spectra of pure compounds, 1H NMR spectra and chemical structures of S1, S2, and S3 isolated as mixtures, Table S1, and Fig. S1 and S2. See DOI: https://doi.org/10.1039/d4ob00066h |
| ‡ These authors contributed equally to this work as first authors. |
| This journal is © The Royal Society of Chemistry 2024 |