
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A visible-light-catalyzed sulfonylation reaction of an aryl selenonium salt via an electron donor–acceptor complex†
Yuqing
Wang
,
Liang
Zhao
 ,
Xinyu
Hao
,
Kun
Jin
,
Rong
Zhang
,
Chunying
Duan
and
Yaming
Li
*
,
Xinyu
Hao
,
Kun
Jin
,
Rong
Zhang
,
Chunying
Duan
and
Yaming
Li
*
State Key Laboratory of Fine Chemicals, School of Chemical Engineering, Dalian University of Technology, Dalian 116024, Liaoning, P.R. China. E-mail: ymli@dlut.edu.cn
First published on 26th February 2024
Abstract
An efficient synthesis of sulfone structures through selenonium salts and sodium sulfinates was developed. Under the irradiation of a blue LED lamp, the two substrates generate aryl and sulfonyl radicals through the activation of the intermediate electron donor acceptor (EDA) complex, thereby synthesizing aromatic, heteroaromatic and aliphatic sulfones in medium to good yields. The advantages of this strategy are metal-free, mild conditions and the leaving group is recycled to construct new selenonium salts.
Introduction
The sulfone structure (R–SO2–R) is an important building blocks and is widely used as a synthetic intermediate in fine and bulk chemical fields.1–5 Molecules containing sulfone units play prominent roles in agricultural chemicals, pharmaceutical formulations, and polymers.6–10 Many bioactive molecules and herbicide safety agents also contain this functional group,11–16 including bicalutamide, used for the treatment of prostate cancer; the preferred drug for treating leprosy, dapsone; the migraine drug eletriptan; and the excellent pre-vaccine soil treatment agent pyroxasulfone (Fig. 1). | ||
| Fig. 1 Examples of –SO2-containing drugs. | ||
There are several methods to synthesize sulfones, and the oxidation of sulfides remains a widely applied strategy,17–19 which achieves the conversion of intermediate sulfoxide to hydrocarbons with sulfonyl halides catalyzed by a Lewis or Brønsted acid and offers an alternative approach to sulfone synthesis.20
However, these classic reactions typically exhibit limitations such as harsh reaction conditions and low regioselectivity,21–23 prompting the development of diverse efficient catalyst systems. In 2014, Rao reported the one-step synthesis of symmetric diarylsulfones using K2S2O8 as a sulfonation reagent.24 Furthermore, sulfone is prepared using DABSO [DABCO(SO2)2], potassium pyrosulfite, and thiourea dioxide25–32 as alternative sources of sulfur dioxide. Over the last decades, the synthetic routes to sodium aryl sulfites have been through cross-coupling with various electrophilic reagents, either under transition metal-catalyzed or metal-free conditions. In 2023, Nevado reported a tripartite enantioselective carbo-sulfonylation process of olefins, sodium arenesulfinates, and aryl halides, integrating photoredox and nickel catalysis.33 In 2020, Tang devised a copper-mediated decarboxylative sulfonation reaction between aryl acetic acid and sodium sulfite, offering a novel way of decarboxylative coupling to produce sulfones.34
In 2019, Ritter reported sulfonium salts which allowed for direct access to a vast selection of intricate small-molecule derivatives since there was no requirement for a guiding group to achieve selectivity.35 Subsequently, Ritter broadened the use of sulfonium salts as precursors to include C–O, C–N, and C–CF3 coupling with a variety of functional groups,36–38 which laid a strong foundation for the research of subsequent researchers. In the last few years, the use of electron donor–acceptor (EDA) complex (Scheme 1a) initiation to construct C–S bonds has become increasingly popular39,40 due to the ability of EDA complexes to generate radical ion pairs and take part in a variety of reactions without the need for external photocatalysts. Zhang presented a novel approach in 2019 for sulfone synthesis, involving the formation of EDA complexes between sodium sulfites and aryl halides under a 365 nm wavelength light source (Scheme 1b).41 In 2022, Molander documented the photoactivated sulfonylation of sulfonium salts through an EDA complex approach that employed sulfonium salts as electron-deficient acceptors and sodium sulfite salts as electron-rich donors (Scheme 1c).42 Considering that an EDA complex is a weak noncovalent bonding interaction, the C–Se bond is more active than the C–S bond and less is known about aryl selenonium salts.43 In this paper we report an aryl selenonium salt as an aryl source and a sodium sulfite salt as a sulfur dioxide source to generate sulfones without the involvement of a photosensitizer (Scheme 1d).
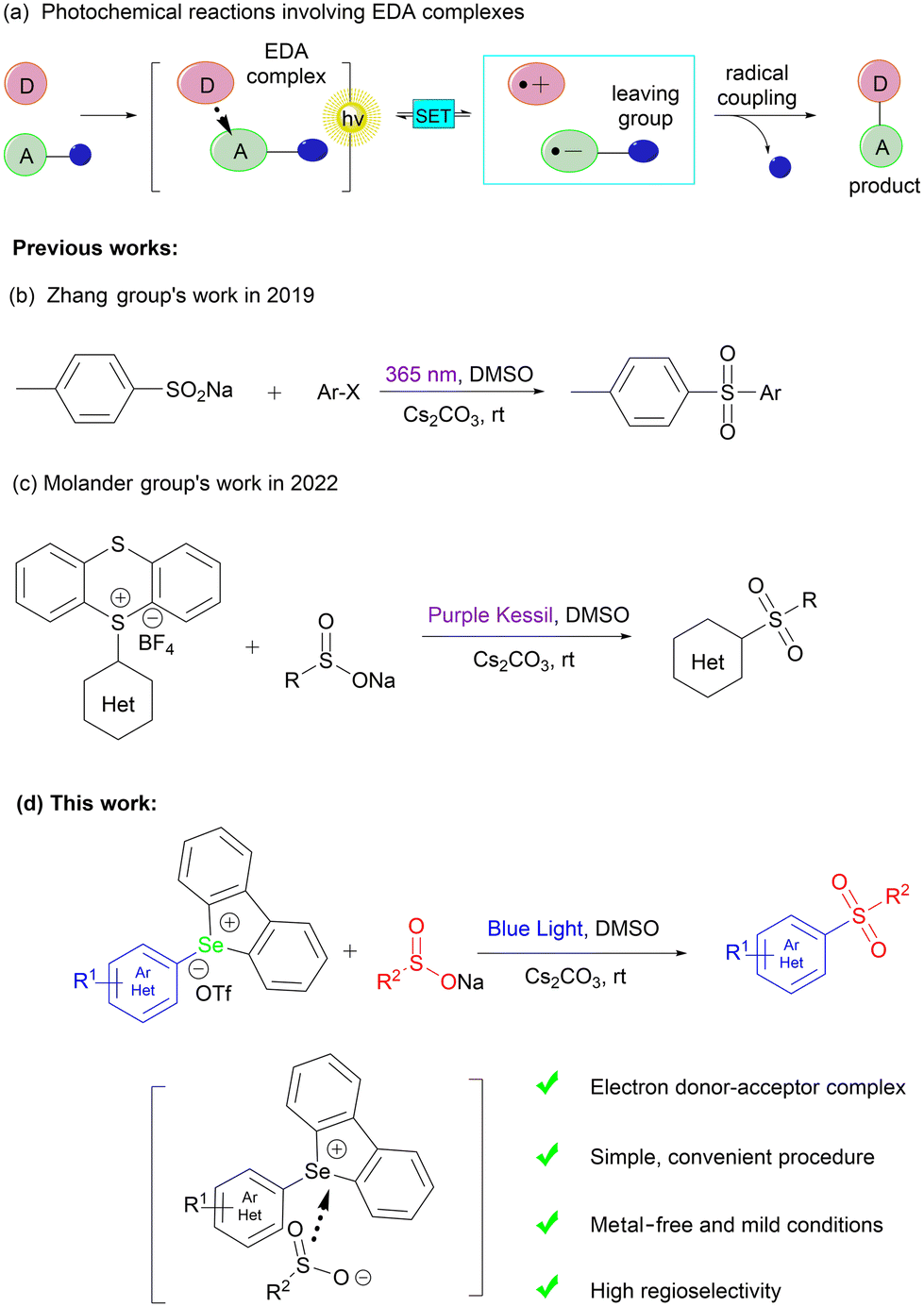 | ||
| Scheme 1 Method for synthesizing sulfone and the mechanism of the EDA complex. | ||
Results and discussion
Initially, an aryl selenonium salt (1a) and sodium benzenesulfinate (2a) were chosen as the model substrates for condition optimization of the visible light-induced C–Se sulfonylation reaction. As shown in Table 1, the base was screened first. The reaction can take place without an additive or catalyst, and the results obtained by using Cs2CO3 are the best (Table 1, entries 1–4). When increasing the amount of Cs2CO3 to 3 or 4 equivalents (Table 1, entries 9 vs. 11 and 12), the yields of the corresponding sulfone increased to 68% or 60%, respectively. Subsequently, various solvents commonly employed in photocatalysis were experimented with, all of which successfully propelled the reaction, and DMSO showed better performance in terms of yields (Table 1, entries 3 vs. 5–8). Adjusting the reaction stoichiometry to six equivalents of sodium benzenesulfinate and one equivalent of selenonium salts, the reaction furnished 3a in 59% yield (Table 1, entries 3 and 9). Control experiments showed that the yield was found to be almost unchanged when [Ru(bpy)3]Cl2 was added (Table 1, entry 10), while no product formation was observed when there was no light irradiation, proving that light was necessary (Table 1, entry 13). Notably, open-to-air or in O2 atmosphere experiments afforded different results to those found under an inert atmosphere, indicating that oxygen affects the transformation (Table 1, entries 13 and 16). Finally, shortening the reaction time to 12 hours led to a low yield of 43%, whereas prolongation of the reaction time to 36 hours resulted in a mere 3% increase in yield (Table 1, entries 14 and 15). Therefore, the optimized conditions were defined as the use of a sodium sulfite salt (6 eq.) and 3 equivalents of Cs2CO3 in DMSO at room temperature under an inert gas atmosphere under blue LED irradiation for 18 hours (Table 2).|
|
||||
|---|---|---|---|---|
| Entry | Base | Solvent | Time (h) | Yielda (%) |
| Reaction conditions: 1a (0.2 mmol), 2a (0.6 mmol), additive (0.4 mmol), and DMSO (0.15 M) at room temperature under blue light irradiation for 18 h.a Isolated yield.b 6 eq. of 2a.c [Ru(bpy)3]Cl2 (5 mol%).d No light or in an O2 environment.e 3 eq. of Cs2CO3.f 4 eq. of Cs2CO3.g Open to air. | ||||
| 1 | None | DMSO | 18 | 27 |
| 2 | K2CO3 | DMSO | 18 | 39 |
| 3 | Cs2CO3 | DMSO | 18 | 49 |
| 4 | CaCO3 | DMSO | 18 | 30 |
| 5 | Cs2CO3 | DMF | 18 | 42 |
| 6 | Cs2CO3 | Acetone | 18 | 31 |
| 7 | Cs2CO3 | DCM | 18 | 38 |
| 8 | Cs2CO3 | MeCN | 18 | 32 |
| 9b, | Cs2CO3 | DMSO | 18 | 59 |
| 10b,c | Cs2CO3 | DMSO | 18 | 58 |
| 11b,e | Cs2CO3 | DMSO | 18 | 68 |
| 12b,f | Cs2CO3 | DMSO | 18 | 60 |
| 13d | Cs2CO3 | DMSO | 18 | nd |
| 14b,e | Cs2CO3 | DMSO | 12 | 43 |
| 15b,e | Cs2CO3 | DMSO | 36 | 71 |
| 16b,g | Cs2CO3 | DMSO | 18 | 38 |

| a All reactions were performed in a Schlenk tube with 1 (1.0 eq., 0.2 mmol), sodium sulfite (6 eq.), and Cs2CO3 (3 eq.) in DMSO (1.34 mL) at room temperature under an inert gas atmosphere under blue LED irradiation for 18 h. |
|---|

|
With the optimized reaction conditions in hand, we investigated the scope of this transformation. Various sodium aryl sulfites were first evaluated, and substrates with electron-donating (3b–3e) or electron-withdrawing groups (3f–3j) at the para position of the benzene ring exhibited favourable reactions with yields ranging from 50% to 77%. The chlorine group at the ortho- or meta-position could also exhibit reactivity to give the sulfone products (3l and 3m). The yields of the corresponding sulfones obtained from biphenyl and polycyclic aromatic hydrocarbon were 42% (3k) and 26% (3o), respectively. Medically relevant heterocyclic sodium sulfites are also suitable electron donors, providing the corresponding pyridyl (3q) and thiophenyl (3p) derivatives in moderate yields. The sulfonation technique was expanded to include alkyl sulfinic acids in order to achieve the desired methyl sulfone 3r and cyclopropyl sulfone 3s. Ultimately, as anticipated, the presence of the steric effect restricted the accessibility of 3n in a slightly diminished yield (33%). We also attempted trifluoromethyl sulfone, but no target product (3t) was detected. In addition, the sulfonylation of 4-chlorophenyl, 4-ethylphenyl and 2-methoxypyridyl selenonium salts was carried out, respectively (Table 3), and the corresponding sulfones 3ca and 3da were obtained in 52% and 63% yields. Unfortunately, the performance of the electron-deficient pyridine heterocycle selenonium salt 1b was poor. In order to elucidate the mechanism of this sulfonylation reaction, UV-vis and radical trapping studies were performed (Scheme 2). The UV-vis spectra of each component and the reaction mixture were measured in DMSO to investigate the formation of the intermediate EDA complex, with the selenonium salt (Scheme 2a, black square) exhibiting absorption bands in the visible region alongside 2a (Scheme 2a, red circle), and a combination of 1a and 2a (Scheme 2a, green triangle) demonstrated the formation of new molecular aggregates in the basal state along with the deepening of the color. When Cs2CO3 was added to the mixture (Scheme 2a, blue triangle), significant color changes and red shifts in UV absorption were observed, indicating the formation of EDA complexes. Under the reaction conditions, radical trapping experiments were conducted utilizing TEMPO (2,2,6,6-tetramethyl-1-piperidinyloxyl radical) and 1,1-diphenylethene. To our delight, both aryl and sulfonyl radicals were captured, affording 6 in 32% yield, while also the product 8 was detected by APCI Tof-MS. Free radical trapping agents inhibited their complete transformation into 3a. It is shown that the sulfonylation reaction proceeds mainly via the radical pathway (see the ESI† for more experimental details) (Scheme 2b).

|
 | ||
| Scheme 2 Control experiments. | ||
Subsequently, we compared the photoinduced sulfonation reactions of selenonium salts with those of sulfonium salts under different light sources. Under the irradiation of purple light, sulfonium salts exhibit greater reactivity than selenium salts (Scheme 2c, entry 1). However, under blue light irradiation, the reactivity of selenonium salts is slightly better (Scheme 2c, entry 2). When the maximum absorption wavelength is 520 nm (Scheme 2c, entry 3), the selenonium salts still exhibit good reactivity, suggesting a variance in the maximum absorption levels of selenonium and sulfonium salts. Finally, different competitive experiments were carried out (Scheme 2d). An evaluation of the reactivity of sulfinates 2a and 2s gave the corresponding products 3a and 3s in 52% and 13% yields, respectively. This suggested that activation by EDA complexes is more inclined to generate aryl sulfones. Moreover, the electron-rich product 3d is more inclined to be generated than the electron-deficient product 3i. This may be due to the sulfonyl radicals with electron-donating groups being more likely to collide with aryl radicals to form covalent bonds.
Based on our experiments, a possible mechanism was proposed as follows (Scheme 3). The electron-rich sulfinate anion 2 and the electron-deficient selenonium salt 1 form the EDA complex A in the ground state, which is then activated by blue-light irradiation and undergoes a single electron transfer (SET) within the complex to produce the sulfonyl radical D, affording the intermediate radical anion B. The dibenzoselenophenol, acting as a suitable leaving group, triggers an irreversible fragmentation event and generates aryl radical C. Subsequent selective coupling of the sulfonyl and aryl radicals generates the sulfone product 3.
 | ||
| Scheme 3 A proposed reaction mechanism. | ||
Conclusions
In conclusion, we have developed a new and efficient method of sulfone synthesis to generate aliphatic sulfones or aromatic sulfones in medium to good yields by a photocatalytic method using aryl selenonium salts and sodium sulfite as substrates in the absence of exogenous metal complexes or photosensitizers. As supported by mechanistic studies, the sulfonation reaction involves the formation of an EDA complex, followed by electron transfer within the complex under visible light irradiation, generating aryl radicals and sulfonyl radicals and selective coupling.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC) (Project No. 22078045 and 21176039).References
- J. Corpas, S.-H. Kim-Lee, P. Mauleón, R. G. Arrayás and J. C. Carretero, Chem. Soc. Rev., 2022, 51, 6774–6823 RSC.
- X. Chang, X. Chen, S. Zhang, S. Lu, Y. Zhao, D. Zhang, L. Yang, Y. Ma and P. Sun, Environ. Chem. Lett., 2023, 21, 681–687 CrossRef CAS.
- Z.-J. Wang, S. Zheng, J. K. Matsui, Z. Lu and G. A. Molander, Chem. Sci., 2019, 10, 4389–4393 RSC.
- A. L. Moure, R. Gómez Arrayás and J. C. Carretero, Chem. Commun., 2011, 47, 6701–6703 RSC.
- Q. Yan, G. Xiao, Y. Wang, G. Zi, Z. Zhang and G. Hou, J. Am. Chem. Soc., 2019, 141, 1749–1756 CrossRef CAS PubMed.
- M. Nambo, Y. Maekawa and C. M. Crudden, ACS Catal., 2022, 12, 3013–3032 CrossRef CAS.
- Z. Wei, Z. Lou, C. Ni, W. Zhang and J. Hu, Chem. Commun., 2022, 58, 10024–10027 RSC.
- Y. Zhao, S. Li, Y. Fan, C. Chen, X. Dong, R. Wang and Y.-Y. Jiang, Org. Chem. Front., 2023, 10, 5923–5932 RSC.
- M. Staniszewska, A. Sobiepanek, M. Gizińska, E. Peña-Cabrera, I. J. Arroyo-Córdoba, M. Kazek, Ł. Kuryk, M. Wieczorek, M. Koronkiewicz, T. Kobiela and Z. Ochal, Eur. J. Med. Chem., 2020, 191, 112139 CrossRef CAS PubMed.
- Z. Zhou, Q. Liu, Z. Huang and Y. Zhao, Org. Lett., 2022, 24, 4433–4437 CrossRef CAS PubMed.
- X. Jia, C. Huang, X. Zhang and Z. Lian, Org. Chem. Front., 2021, 8, 5310–5315 RSC.
- Y.-J. Jhang, C.-Y. Chang, Y.-H. Lin, C.-C. Lee and Y.-K. Wu, Tetrahedron Lett., 2021, 73, 153060 CrossRef CAS.
- Y. Wang, W. Jiang and C. Huo, J. Org. Chem., 2017, 82, 10628–10634 CrossRef CAS PubMed.
- M. Baunach, L. Ding, K. Willing and C. Hertweck, Angew. Chem., Int. Ed., 2015, 54, 13279–13283 CrossRef CAS PubMed.
- N. von Wolff, J. Char, X. Frogneux and T. Cantat, Angew. Chem., Int. Ed., 2017, 56, 5616–5619 CrossRef CAS PubMed.
- R. Kumar and I. N. N. Namboothiri, Org. Lett., 2011, 13, 4016–4019 CrossRef CAS PubMed.
- Y. Li, L. Liu, D. Shan, F. Liang, S. Wang, L. Yu, J.-Q. Liu, Q. Wang, X. Shao and D. Zhu, ACS Catal., 2023, 13, 13474–13483 CrossRef CAS.
- X. Xu, L. Yan, S. Wang, P. Wang, A. X. Yang, X. Li, H. Lu and Z.-Y. Cao, Org. Biomol. Chem., 2021, 19, 8691–8695 RSC.
- Z. Cheng, P. Sun, A. Tang, W. Jin and C. Liu, Org. Lett., 2019, 21, 8925–8929 CrossRef CAS PubMed.
- L. Tang, K. Du, B. Yu and L. He, Chin. Chem. Lett., 2020, 31, 2991–2992 CrossRef CAS.
- T. J. Blacklock, P. Sohar, J. W. Butcher, T. Lamanec and E. J. J. Grabowski, J. Org. Chem., 1993, 58, 1672–1679 CrossRef CAS.
- K. Bahrami, M. M. Khodaei and M. Sheikh Arabi, J. Org. Chem., 2010, 75, 6208–6213 CrossRef CAS PubMed.
- S. Répichet, C. Le Roux, P. Hernandez, J. Dubac and J.-R. Desmurs, J. Org. Chem., 1999, 64, 6479–6482 CrossRef.
- Y. Yang, Z. Chen and Y. Rao, Chem. Commun., 2014, 50, 15037–15040 RSC.
- A. S. Deeming, C. J. Russell, A. J. Hennessy and M. C. Willis, Org. Lett., 2014, 16, 150–153 CrossRef CAS PubMed.
- B. N. Rocke, K. B. Bahnck, M. Herr, S. Lavergne, V. Mascitti, C. Perreault, J. Polivkova and A. Shavnya, Org. Lett., 2014, 16, 154–157 CrossRef CAS PubMed.
- A. S. Deeming, C. J. Russell and M. C. Willis, Angew. Chem., Int. Ed., 2016, 55, 747–750 CrossRef CAS PubMed.
- D. Zheng, Y. An, Z. Li and J. Wu, Angew. Chem., Int. Ed., 2014, 53, 2451–2454 CrossRef CAS PubMed.
- J. Zhang, J. Wu, X. Chang, P. Wang, J. Xia and J. Wu, Org. Chem. Front., 2022, 9, 917–922 RSC.
- Y. Liu, Q.-L. Wang, Z. Chen, H. Li, B.-Q. Xiong, P.-L. Zhang and K.-W. Tang, Chem. Commun., 2020, 56, 3011–3014 RSC.
- J. C. Hethcox, H. C. Johnson, J. Kim, X. Wang, L. Cheng, Y. Cao, M. Tan, D. A. DiRocco and Y. Ji, Angew. Chem., Int. Ed., 2023, 62, e202217623 CrossRef CAS PubMed.
- Y. Li, J.-B. Liu, F.-S. He and J. Wu, Chin. J. Chem., 2020, 38, 361–366 CrossRef CAS.
- X. Du, I. Cheng-Sánchez and C. Nevado, J. Am. Chem. Soc., 2023, 145, 12532–12540 CrossRef CAS PubMed.
- Y. Wu, J. Chen, L. Li, K. Wen, X. Yao, J. Pang, T. Wu and X. Tang, Org. Lett., 2020, 22, 7164–7168 CrossRef CAS PubMed.
- F. Berger, M. B. Plutschack, J. Riegger, W. Yu, S. Speicher, M. Ho, N. Frank and T. Ritter, Nature, 2019, 567, 223–228 CrossRef CAS PubMed.
- R. Sang, S. E. Korkis, W. Su, F. Ye, P. S. Engl, F. Berger and T. Ritter, Angew. Chem., Int. Ed., 2019, 58, 16161–16166 CrossRef CAS PubMed.
- P. S. Engl, A. P. Häring, F. Berger, G. Berger, A. Pérez-Bitrián and T. Ritter, J. Am. Chem. Soc., 2019, 141, 13346–13351 CrossRef CAS PubMed.
- F. Ye, F. Berger, H. Jia, J. Ford, A. Wortman, J. Börgel, C. Genicot and T. Ritter, Angew. Chem., Int. Ed., 2019, 58, 14615–14619 CrossRef CAS PubMed.
- L. Lin, Z. Yang, J. Liu, J. Wang, J. Zheng, J.-L. Li, X. Zhang, X.-W. Liu, H. Jiang and J. Li, Green Chem., 2021, 23, 5467–5473 RSC.
- X.-X. Zhang, H. Zheng, Y.-K. Mei, Y. Liu, Y.-Y. Liu, D.-W. Ji, B. Wan and Q.-A. Chen, Chem. Sci., 2023, 14, 11170–11179 RSC.
- L. Chen, J. Liang, Z.-y. Chen, J. Chen, M. Yan and X.-j. Zhang, Adv. Synth. Catal., 2019, 361, 956–960 CrossRef CAS.
- A. Granados, M. J. Cabrera-Afonso, M. Escolano, S. O. Badir and G. A. Molander, Chem. Catal., 2022, 2, 898–907 CrossRef CAS PubMed.
- Q. Wang, X. Hao, K. Jin, R. Zhang, C. Duan and Y. Li, Org. Biomol. Chem., 2022, 20, 4427–4430 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ob00060a |
| This journal is © The Royal Society of Chemistry 2024 |