
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of galactomannan fragments to help NMR assignment of polysaccharides extracted from lichens†
Louis-Philippe
David
a,
Solenn
Ferron
b,
Bénédicte
Favreau
a,
Oznur
Yeni
c,
Simon
Ollivier
de,
David
Ropartz
de,
Isabelle
Compagnon
 c,
Vincent
Ferrières
*a,
Françoise
Le Dévéhat
*b and
Laurent
Legentil
*a
c,
Vincent
Ferrières
*a,
Françoise
Le Dévéhat
*b and
Laurent
Legentil
*a
aUniv Rennes, Ecole Nationale Supérieure de Chimie de Rennes, CNRS, ISCR – UMR 6226, F-35000 Rennes, France. E-mail: laurent.legentil@ensc-rennes.fr
bUniv Rennes, CNRS, ISCR – UMR 6226, F-35000 Rennes, France
cUniv Lyon, Université Claude Bernard Lyon 1, CNRS, Institut Lumière Matière, F-69622 Villeurbanne, France
dINRAE, UR BIA, F-44316 Nantes, France
eINRAE, BIBS Facility, F-44316 Nantes, France
First published on 21st February 2024
Abstract
The synthesis of six model trisaccharides representative of galactomannans produced by lichens was performed through stereoselective glycosylation. These standards include linear and branched galactomannans bearing either galactofuranosyl or galactopyranosyl entities. The complete assignment of 1H and 13C signals for both forms of synthetically reduced oligosaccharides was performed. The resulting NMR data were used to quickly demonstrate the structural characteristics of minor polysaccharides within different extracts of three representative lichens.
Introduction
Lichens constitute a “self-sustaining ecosystem” where the main partners are photosynthetic organisms (microalgae and/or cyanobacteria) and fungi.1 Such symbiosis relies on the fruitful exchange of metabolites and nutrients between both partners and allows the lichens to grow in all habitats even under harsh conditions like sun-exposed rocks. To date, lichen chemistry has mainly focused on secondary metabolites (more than 1000 compounds have been isolated and identified in the last compendium).2 Such low molecular weight metabolites represent 3–20% of the lichen dry weight while free sugars and polysaccharides represent about 60% of the extractable biomass.3 These polysaccharides contribute to the extreme tolerance of the lichens towards harsh conditions, their fascinating revival ability, their very unique management of water and the maintenance of the complex partnership, including a specific microflora within this long-living microecosystem.4–6 Three main families of polysaccharides could be found among the few lichens that have been studied for their carbohydrate constituents.7,8 β- and α-glucans are common homopolymers found throughout the species while galactomannans are much more heterogeneous. The latter biomolecules are built on a mannan backbone with different decorations depending on the symbiotic partners. For example, polysaccharides from Cetraria islandica9 and Lasallia pustulata10 have a common (1 → 6) linked α-D-mannopyranosyl main chain branched for the first one with β-D-galactopyranose (Galp) at positions 2 and 4, and for the second one with β-D-galactofuranose (Galf) at the same positions (Fig. 1A). The quite rare galactofuranose moiety was also identified within the galactomannans of Umbilicaria,11Usnea,12Gyrophora13 or Roccella.14 For the last lichen, the mannan backbone is made up of α-(1 → 4) linked mannopyranose with galactofuranose at the non-reducing end (Fig. 1B). Recently, such diversity of structures was outlined as a chemotaxonomic character to classify lichens from different species.8 Some of them were also evaluated for their antitumor, antiviral and immunostimulating properties.15 For example, both polysaccharides KI-M-7 and thamnolan, isolated from C. islandica and Thamnolia subuliformis, respectively, showed an increase in the phagocytic activity of granulocytes in the presence of polymers and anticomplementary effects.16,17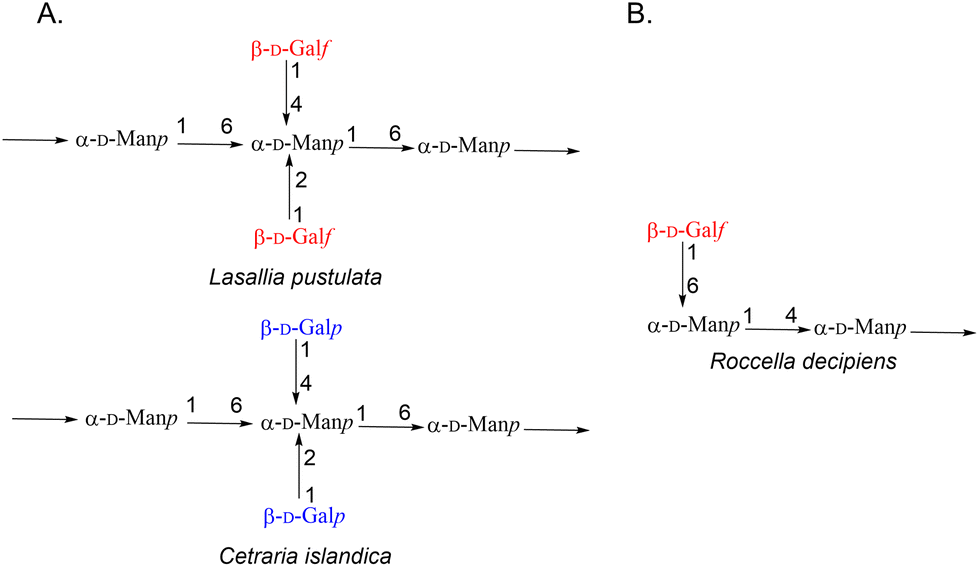 | ||
| Fig. 1 General structures of (A) polysaccharides present in Lasallia, Cetraria and (B) Roccella lichens. | ||
Nevertheless, the studies on such structures remain scarce, due to the difficulty associated with their harvest, extraction and characterisation. The sensitivity of galactofuranosides towards mild hydrolysis conditions also hampers reliable characterisation studies.
Previously, their primary structures were determined after different fractionation steps followed by NMR analysis. The characterisation of the regioisomery relied mainly on 13C data tables that dated from the early 70s.18 Recently, Nifantiev et al. highlighted the difficulty of correlating such 13C chemical shifts with the sequence of the polysaccharide by comparing synthetic galactofuranomannans with the extracellular galactomannan of Aspergillus fumigatus.19 No such systematic comparison by NMR of oligogalactomannans with different extracts of lichens has been reported yet.
To efficiently perform such screening, we describe herein the synthesis of a library of six trisaccharides that could serve as analytical standards (Fig. 2). Linear β-D-Galf-(1 → 6)-α-D-Manp-(1 → 4)-α-D-Manp1a, linear and branched trisaccharides β-D-Galf-(1 → 4)-α-D-Manp-(1 → 6)-α-D-Manp2a and α-D-Manp-(1 → 6)-[β-D-Galf-(1 → 4)]-α-D-Manp3a were first synthesized as representative galactomannans from Roccella decipiens14 and Lasallia pustulata10 species. Moreover, in order to pursue our efforts in the development of analytical approaches allowing differentiation between galactosyl tautomers,20 we also prepared the trisaccharides bearing Galp residues 1b–3b. This is crucial as Galp-Man motifs were also described in some lichens like Cetraria islandica.9 All standards were obtained as hemiacetals, the main product of hydrolysis of natural galactomannans.
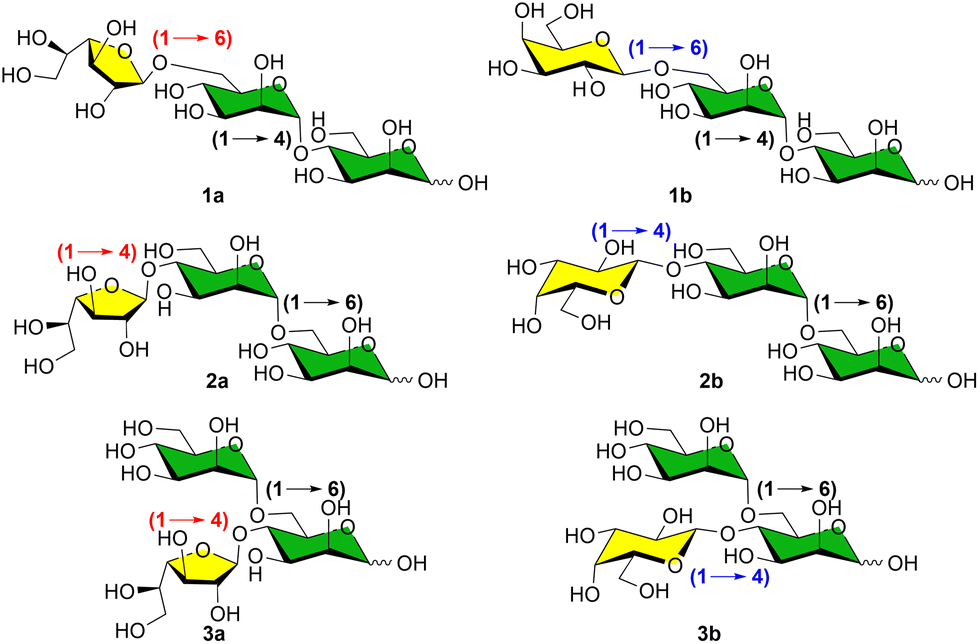 | ||
| Fig. 2 Targeted trisaccharides 1–3 as standards for NMR comparison with extracted fractions from the lichen thalli (series a correspond to Galf-containing trisaccharides and b to Galp-containing ones). | ||
To complete this study, the NMR spectra of all trisaccharides were compared with different fractions of extracts of our representative lichen thalli. We demonstrate here that the presence of biologically relevant galactomannans could be detected in unsuspected fractions, not reported before.
Results and discussion
Synthesis of trisaccharides
The strategy implemented in this study capitalized on recent works on the synthesis of the four possible regioisomers of Galf-Manp and Galp-Manp disaccharides.20–22 In each case, the synthesis of a-family derivatives (Galf family) and b-family compounds (Galp one) followed a similar sequence of reactions.The synthetic cascade to afford linear trisaccharides 1 started from known thioglycosides 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 20 with the established link Gal-(1 → 6)-Manp. They were first converted into the corresponding trichloroacetimidates 6 (Scheme 1). Hydrolysis of 4 assisted by N-iodosaccharin afforded acetals 5, which were reacted with trichloroacetonitrile to give 6. Then, glycosylation of acceptor 7 with the free O-4 position was performed in the presence of a catalytic amount of trimethylsilyl trifluoromethanesulfonate (TMSOTf). Subsequent cleavage of the acetonide group in 8 in the presence of an excess of trifluoroacetic acid in dichloromethane gave 9. This reaction turned out to be sluggish for obtaining 9a and maximum yield was limited to 30%. Addition of dithiothreitol (DTT) allowed the increase of the yield to 41%. As for 9b, a good yield of 66% was obtained without DTT. All ester groups were then removed by Zemplen transesterification.
20 with the established link Gal-(1 → 6)-Manp. They were first converted into the corresponding trichloroacetimidates 6 (Scheme 1). Hydrolysis of 4 assisted by N-iodosaccharin afforded acetals 5, which were reacted with trichloroacetonitrile to give 6. Then, glycosylation of acceptor 7 with the free O-4 position was performed in the presence of a catalytic amount of trimethylsilyl trifluoromethanesulfonate (TMSOTf). Subsequent cleavage of the acetonide group in 8 in the presence of an excess of trifluoroacetic acid in dichloromethane gave 9. This reaction turned out to be sluggish for obtaining 9a and maximum yield was limited to 30%. Addition of dithiothreitol (DTT) allowed the increase of the yield to 41%. As for 9b, a good yield of 66% was obtained without DTT. All ester groups were then removed by Zemplen transesterification.
 | ||
| Scheme 1 Synthesis of trisaccharides 1 based on the Manp-(1,4)-Manp skeleton. Reagents and conditions: (i) N-iodosaccharin, H2O/CH3CN/CH2Cl2; (ii) Cl3CCN, DBU, CH2Cl2; (iii) 5, TMSOTf, CH2Cl2; (iv) TFA, DTT, CH2Cl2; (v) NaOMe, MeOH; (vi) TFA, CH2Cl2; (vii) N-bromosuccinimide, acetone/H2O. | ||
Finally, in order to get as close as possible to structures that would result from controlled hydrolysis of native polysaccharides, the targeted triglycosides 1a and 1b were obtained with excellent yields thanks to the action of the bromonium ion and water on thioglycosides 10.
A very similar approach was performed to synthesize trisaccharides, characterized by a Manp-(1 → 6)-Manp skeleton grafted with a Gal residue at position 4 (Scheme 2). While building block 11a was already described,20 the pyranosidic counterpart 11b had to be synthesized first through glycosylation of thiophenyl 4-hydroxyl mannopyranoside 7 by tetra-O-benzoyl-β-D-galactopyranosyl trichloroacetimidate according to a standard procedure. Then, to gain access to linear compounds 2, the thioglycosides 11 in hand were first transformed into the corresponding trichloroacetimidate donors 13 in two steps. Despite the absence of a participating group at the C-2 position of both donors, glycosylation of 14 yielded the desired Gal-(1 → 4)-Manp-(1 → 6)-Manp trisaccharides 15a and 15b with α-(1 → 6)-linkages only. Removal of the acetonide group was performed this time in hot aqueous acetic acid media to avoid by-product formation. This was followed by deacylation and hydrolysis of the resulting trisaccharides to give the targeted compounds 2a and 2b, respectively.
 | ||
| Scheme 2 Synthesis of the trisaccharides 2 and 3 based on the Manp-(1,6)-Manp skeleton. Reagents and conditions: (i) NBS, H2O/Me2CO; (ii) Cl3CCN, CsCO3, CH2Cl2; (iii) 14, TMSOTf, CH2Cl2; (iv) AcOH, H2O; (v) NaOMe, MeOH; (vi) NIS, CH3CN/H2O; (vii) BzCl, Pyr; (viii) AcCl, MeOH; (ix) 21, TMSOTf, CH2Cl2. | ||
Finally, the branched isomers 3, presenting a Gal entity at the O-4 position of the reducing Man residue, were also prepared from the same disaccharides 11. The first attempt to selectively remove the acetate group from position 6 of mannose in the presence of 2,3-acetonide failed. In order to avoid acetal cleavage, we anticipated that mild conditions were required. Lipase from Candida cylindracea, known to selectively cleave ester at the primary position of mannose derivatives, was unfortunately ineffective probably because of the steric hindrance around this position. We therefore proceeded to the interconversion of the protecting group at OH-2 and OH-3 of thiomannoside 11, from acetonide to benzoyl. Orthogonal acid-catalysed 6-O-deacetylation that preserved the benzoic esters was then performed to access compounds 20. These acceptors were then glycosylated with the mannopyranosyl donor 21 bearing benzoyl groups. Once again, protecting groups were removed according to the standard procedures to give 3a and 3b in 13% and 21% overall yields, respectively, over six steps.
NMR analysis of trisaccharides 1–3 and comparison with extracts from lichen thalli
With targeted trisaccharides in hand, we wanted to confirm the structure of galactomannans in the extracts of representative lichens Lasallia pustulata, Cetraria islandica and Roccella decipiens (Fig. 1). The last lichen does not grow in the Brittany Region (France), hence it was replaced by R. fuciformis, which is easily available. For this purpose, the protocol described by Prieto and coworkers was implemented first on the thallus of L. pustulata.10 Briefly, hot water extraction was performed to provide, after alcoholic precipitation, fraction A2. The remaining insoluble fraction, after cooling, was subjected to alkali extraction to give after filtration, a precipitate (fraction F2) and a filtrate (fraction F1S). We then compared the HSQC maps of water and alkali extracts A2 and F1S, respectively, with the one from the standard trisaccharides 1–3 (see the ESI† for HSQC maps of the corresponding fractions). The main proton and carbon chemical shifts of trisaccharides 1–3 are indexed in the ESI (Tables S1–S6†). Their presence in the natural extract could be easily identified. Indeed, the anomeric position of the mannan core showed a characteristic 1H chemical shift above 4.93 ppm and 13C shift between 100 and 105 ppm, while galactofuranose possesses an anomeric carbon between 106 and 108 ppm, a region where no anomeric carbon of pyranose was found. In addition, the C-4 and C-2 of the furanose ring are above 80 ppm while most carbon atoms from mannose or glucose are found below 78 ppm. Also, the 1H and 13C chemical shifts of the anomeric position of galactopyranose are characteristic (δH below 4.50 ppm and δC between 100 and 103 ppm).Fractions A2 and F1S from L. pustulata surprisingly showed a similar profile with the main anomeric proton at 4.54 ppm that correlates with 13C signal at 102.87 ppm (Fig. 3A). Those signals are characteristics of pustulan, a β-(1 → 6)-glucan, known as the major constituent of the thallus. The presence of an acetyl group (δH 2.17 ppm) is in accordance with the presence of around 15% of acetylated glucose in pustulan. As for the minor constituent of both fractions, 1H and 13C chemical shifts characteristic of galactomannans could be found (Fig. 3A). Interestingly, such a pattern was never reported before on the hot-water extract A2. Therefore, the 1H signal at 5.06 ppm (δC 107.53 ppm) showed a good match with the anomeric proton of trisaccharides 2a and 3a. Interestingly the anomeric proton at 4.93 ppm (δC 99.25 ppm) fits well with signals corresponding to the branched trisaccharides 3a rather than the linear one 2a. This confirmed the prevalence of the branched galactomannan oligomer over Galf-ending mannans. No Galp was identified in the extract which is in accordance with the literature.
 | ||
| Fig. 3 Identification of galactomannans in the natural extract from lichens. HSQC spectrum of the anomeric region of the fractions A2 of L. pustulata (A), R. fuciformis (B) and C. islandica (C) and comparison with trisaccharides 1–3 as standards (D). HSQC correlations are shown in black for lichen extracts; blue for 1a; red for 2a; pink for 2b; green for 3a. Signals in the anomeric region corresponding to putative galactomannans are labelled. | ||
The same trend was observed using the extracts of the Roccella thalli. Again unexpectedly, hot water extract A2 contained galactose in its furanose form (δH 5.00 ppm and δC 107.69 ppm in Fig. 3B). A comparison with standard 1a confirmed the previously reported Galf-(1 → 6)-Manp-(1 → 4) sequence (δH 5.17 ppm and δC 100.68 ppm for mannose anomeric proton). No correlation with the pyranose analogues was identified. Finally, we compared 1–3 with the water-soluble fractions from Cetraria. While a good match was found with standards 2b and 3b, thus confirming the presence of the β-Galp-(1 → 4)-α-Manp motif (δH 4.44 ppm and δC 103.06 ppm in Fig. 3C), some correlation spots were also visible around 107 ppm (δH 5.20 ppm and δC 106.72 ppm). While this chemical shift differed from the one found for β-Galf-(1 → 4)- and β-Galf-(1 → 6)-α-Manp, it is nevertheless representative of the presence of the furanosyl moiety, probably linked in β-(1 → 2) on the mannopyranose core. It is worth noting that this signal could also be found in both Lassalia and Roccella fractions.
Conclusion
Six different oligogalactomannans were synthesized and their HSQC maps were compared with the natural polysaccharides extracted from three different lichens. Thanks to this non-destructive method, the presence of galactomannans could be identified in most fractions, even in the hot water extract which is supposed to contain mostly pustulan. Unexpectedly, the rare hexofuranose form was identified in lichens that are supposed to contain only the pyranose isomers. It confirms that such galactofuranomannans constitute a unique feature of the symbiosis.Experimental
General procedure for hydrolysis of thiomannosides using N-iodosaccharin (A1)
To a 9/1 (v/v) mixture of CH3CN/H2O, thiosaccharide and N-iodosaccharin were successively added. The reaction mixture was stirred at RT and monitored by TLC. After the disappearance of the starting compound, the reaction was quenched by adding triethylamine and concentrated under reduced pressure. The product was partitioned between CH2Cl2 and water, and the organic layer was washed with water. The combined organic layers were dried over MgSO4, filtered and concentrated under reduced pressure. The product was finally chromatographically purified.General procedure for hydrolysis of thiomannosides using N-bromosuccinimide (A2)
To a 9/1 (v/v) mixture of acetone/H2O, thiosaccharide and N-bromosuccinimide were successively added. The reaction mixture was stirred at RT and monitored by TLC. After the disappearance of the starting compound, the reaction medium was diluted with CH2Cl2 and the resulting solution was washed with a saturated aqueous solution of NaHCO3 (3×). The resulting organic phase was dried over MgSO4, filtered and concentrated under reduced pressure. The product was finally chromatographically purified.General procedure for Zemplén transesterification (B)
Into anhydrous MeOH, acylated trisaccharide and a volume of a 0.54 M solution of MeONa in MeOH were successively added. The mixture was stirred overnight at RT and then neutralized with IR 120 (H+-form). After filtration, the solvent was removed under reduced pressure. The residue was suspended in water and the biphasic solution was extracted with CH2Cl2 (3×). The resulting aqueous layer was finally freeze-dried.General procedure for glycosylation (C)
Into anhydrous CH2Cl2, both the acceptor and donor and then activated 4 Å molecular sieves were added. The mixture was cooled to 0 °C and stirred for 15 min before dropwise addition of TMSOTf. The temperature was maintained at 0 °C and the reaction was monitored by TLC (cyclohexane/AcOEt 7/3). When no starting material remained, triethylamine was added and the solution was filtered on a pad of Celite. The resulting filtrate was evaporated under vacuum before flash-chromatography.General procedure for synthesis of trichloroacetimidate (D)
Into anhydrous CH2Cl2, the reducing saccharide, Cs2CO3, and trichloroacetonitrile were successively added. The resulting mixture was stirred at RT until no starting material remained. After filtration, the resulting filtrate was concentrated under reduced pressure and the product was purified by flash-chromatography.β-D-Galactofuranosyl-(1 → 6)-α-D-mannopyranosyl-(1 → 4)-α,β-D-mannopyranose (1a)
To a solution of 10a (47 mg, 0.079 mmol) in a CH3CN/H2O 1:1 mixture (5 mL), N-iodosaccharin (87 mg, 0.28 mmol) was added. The reaction mixture was stirred at RT for 3 h. After the disappearance of the starting compound, the reaction was quenched by adding triethylamine and concentrated under reduced pressure. The resulting crude was partitioned between CH2Cl2 and water, and the aqueous layer was washed with CH2Cl2 (3×). The resulting aqueous phase was freeze-dried and the resulting residue was purified by column chromatography (AcOEt/MeOH/H2O gradient from 8:1:1 up to 6:2:2) to afford 1a (36 mg, 90%) as a white solid. TLC: (BuOH/AcOH/H2O, 6:2:2), Rf = 0.23.
1aα: 1H NMR (D2O, 400 MHz): δ 5.20 (1H, d, J1b,2b 1.5, H-1b), 5.17 (1H, d, J1a,2a 1.8, H-1a), 5.05 (1H, d, J1c,2c 1.8, H-1c), 4.14 (1H, dd, J2c,3c 3.7, J2c,1c 1.8, H-2c), 4.09–4.03 (2H, m, H-2b,H-6b), 4.07 (1H, dd, J3c,4c 6.5, J3c,2c 3.7, H-3c), 4.00 (1H, dd, J4c,3c 6.5, J4c,5c 4.2, H-4c), 3.97 (1H, dd, J3a,4a 9.0, J3a,2a 3.4, H-3a), 3.92–3.76 (8H, m, H-2a, H-4a, H-5a, H-6a, H-3b, H-5b, H-5c), 3.76–3.63 (4H, m, H-4b, H-6′b, H-6c). 13C NMR (D2O, 126 MHz): δ 107.75 (C-1c), 101.74 (C-1b), 93.83 (C-1a), 82.83 (C-4c), 81.05 (C-2c), 76.85 (C-3c), 75.10 (C-4a), 72.56 (C-5b), 71.17 (C-2a), 71.03, 70.87 (C-5a, C-5c), 70.60 (C-3a), 70.27 (C-2b, C-3b), 66.80 (C-6b), 66.62 (C-4b), 62.76 (C-6c), 61.19 (C-6a).
1aβ: 1H NMR (D2O, 500 MHz): δ 5.25 (1H, d, J1b,2b 1.7, H-1b), 5.05 (1H, d, J1c,2c 1.8, H-1c), 4.89 (1H, s, J1a,2a 1.8, H-1a), 4.14 (1H, dd, J2c,3c 3.7, J2c,1c 1.8, H-2c), 4.09–4.03 (2H, m, H-2b, H-6b), 4.07 (1H, dd, J3c,4c 6.5, J3c,2c 3.7, H-3c), 4.00 (1H, dd, J4c,3c 6.5, J4c,5c 4.2, H-4c), 3.92–3.76 (6H, m, H-2a, H-3a, H-6a, H-3b, H-5b, H-5c), 3.76–3.63 (6H, m, H-4a, H-6′a, H-4b, H-6′b, H-6c), 3.53–3.44 (1H, m, H-5a). 13C NMR (D2O, 100 MHz): δ 107.75 (C-1c), 101.74 (C-1b), 93.61 (C-1a), 82.83 (C-4c), 81.05 (C-2c), 76.85 (C-3c), 74.77, 74.69 (C-4a, C-5a), 73.48 (C-3a), 72.56 (C-5b), 71.17 (C-2a), 70.87 (C-5c), 70.27 (C-2b, C-3b), 66.80 (C-6b), 66.62 (C-4b), 62.76 (C-6c), 60.93 (C-6a).
HRMS (ESI): calcd for C18H32O16Na [M + Na]+ 527.1588, found 527.1585.
β-D-Galactopyranosyl-(1 → 6)-α-D-mannopyranosyl-(1 → 4)-α,β-D-mannopyranose (1b)
To a solution of 10b (60 mg, 0.1 mmol) in a 9:1 acetone/H2O mixture (6 mL), N-bromosuccinimide (21 mg, 0.12 mmol) was added. The reaction mixture was stirred at RT for 3 h. After the disappearance of the starting compound, the reaction was quenched by adding triethylamine and concentrated under reduced pressure. The resulting crude was partitioned between CH2Cl2 and water, and the aqueous layer was washed with CH2Cl2 (3×). The resulting aqueous phase was freeze-dried and the resulting residue was purified by column chromatography (AcOEt/MeOH/H2O gradient from 8:1:1 up to 6:2:2) to afford 1b (50 mg, 99%) as a white solid. TLC: (AcOEt/MeOH/H2O, 6:2:2), Rf = 0.12.
1bα: 1H NMR (D2O, 400 MHz): δ 5.22 (1H, d, J1b,2b 1.8, H-1b), 5.17 (1H, d, J1a,2a 1.9, H-1a), 4.45 (1H, d, J1c,2c 7.8, H-1c), 4.22 (1H, d, J6b,6′b 9.5, H-6b), 4.07 (1H, dd, J2b,3b 3.3, J2b,1b 1.8, H-2b), 3.97 (1H, dd, J3a,4a 9.0, J3a,2a 3.5, H-3a), 3.94–3.85 (6H, m, H-2a, H-5a, H-6a, H-5b, H-6′b, H-4c), 3.84–3.69 (7H, m, H-4a, H-6′a, H-3b, H-4b, H-5c, H-6c), 3.67 (1H, dd, J3c,2c 10.0, J3c,4c 3.4, H-3c), 3.57 (1H, dd, J2c,3c 10.0, J2c,1c 7.8, H-2c). 13C NMR (D2O, 100 MHz): δ 103.36 (C-1c), 101.71 (C-1b), 93.78 (C-1a), 75.14 (C-5c), 74.94 (C-4a), 72.62, 72.59 (C-5b, C-3c), 71.13 (C-2a), 70.90 (C-5a), 70.81 (C-2c), 70.61 (C-3a), 70.21 (C-2b, C-3b), 68.66 (C-6b, C-4c), 66.33 (C-4b), 61.16, 61.01 (C-6a, C-6c).
1bβ: 1H NMR (D2O, 400 MHz): δ 5.22 (1H, d, J1b,2b 1.8, H-1b), 4.89 (1H, d, J1a,2a 1.0, H-1a), 4.45 (1H, d, J1c,2c 7.8, H-1c), 4.22 (1H, d, J6b,6′b 9.5, H-6b), 4.07 (1H, dd, J2b,3b 3.3, J2b,1b 1.8, H-2b), 3.97 (1H, dd, J3a,4a 9.0, J3a,2a 3.5, H-3a), 3.94–3.85 (5H, m, H-2a, H-6a, H-5b, H-6′b, H-4c), 3.84–3.69 (7H, m, H-4a, H-6′a, H-3b, H-4b, H-5c, H-6c), 3.67 (1H, dd, J3c,2c 10.0, J3c,4c 3.4, H-3c), 3.57 (1H, dd, J2c,3c 10.0, J2c,1c 7.8, H-2c), 3.47 (1H, ddd, J5a,4a 9.6, J5a,6′a 6.1, J5a,6a 2.2, H-5a). 13C NMR (D2O, 100 MHz): δ 103.33 (C-1c), 101.65 (C-1b), 93.57 (C-1a), 75.14 (C-5c), 74.58 (C-5a), 71.48 (C-4a), 72.62, 72.59 (C-5b, C-3c), 71.70 (C-2a), 70.81 (C-2c), 70.61 (C-3a), 70.21 (C-2b, C-3b), 68.66 (C-6b, C-4c), 66.33 (C-4b), 61.16, 61.01 (C-6a, C-6c).
HRMS (ESI): calcd for C18H32O16Na [M + Na]+ 527.1588, found 527.1582.
β-D-Galactofuranosyl-(1 → 4)-α-D-mannopyranosyl-(1 → 6)-α,β-D-mannopyranose (2a)
To a solution of 17a (40 mg, 0.07 mmol) in an acetone/H2O 9:1 mixture (6 mL), N-bromosuccinimide (36 mg, 0.2 mmol) was added. The reaction mixture was stirred at RT for 3 h. After the disappearance of the starting compound, the reaction was quenched by adding triethylamine and concentrated under reduced pressure. The resulting crude was partitioned between CH2Cl2 and water, and the aqueous layer was washed with CH2Cl2 (3×). The resulting aqueous phase was freeze-dried and the resulting residue was purified by column chromatography (AcOEt/MeOH/H2O gradient from 8:1:1 up to 6:2:2) to afford 2a (20 mg, 59%) as a white solid.
2aα: 1H NMR (D2O, 500 MHz): δ 5.17 (1H, d, J1a,2a 1.9, H-1a), 5.08 (1H, d, J1c,2c 2.3, H-1c), 4.72 (1H, d, J1b,2b 1.1, H-1b), 4.17 (1H, dd, J6a,6′a 11.1, J6a,5a 2.0, H-6a), 4.12–4.07 (4H, m, H-2b, H-2c, H-3c, H-4c), 3.99–3.91 (3H, m, H-2a, H-5a, H-6b), 3.89–3.77 (4H, m, H-3a, H-6′a, H-6′b, H-5c), 3.77–3.59 (5H, m, H-4a, H-3b, H-4b, H-6c), 3.52–3.46 (1H, m, H-5b). 13C NMR (D2O, 126 MHz): δ 107.91 (C-1c), 100.44 (C-1b), 94.09 (C-1a), 82.62 (C-2c), 80.91 (C-4c), 75.91 (C-3c), 75.14 (C-4b), 75.04 (C-5b), 71.44 (C-3b), 71.24 (C-5a), 70.58 (C-2a), 70.48 (C-3a), 70.23 (C-2b), 70.12 (C-5c), 68.60 (C-6a), 66.67 (C-4a), 62.67 (C-6c), 60.43 (C-6b).
2aβ: 1H NMR (D2O, 500 MHz): δ 5.08 (1H, d, J1c,2c 2.3, H-1c), 4.91 (1H, d, J1a,2a 1.0, H-1a), 4.73 (1H, d, J1b,2b 1.1, H-1b), 4.20 (1H, dd, J6a,6′a 11.3, J6a,5a 2.1, H-6a), 4.12–4.07 (4H, m, H-2b, H-2c, H-3c, H-4c), 3.99–3.91 (2H, m, H-2a, H-6b), 3.89–3.77 (5H, m, H-3a, H-5a, H-6′a, H-6′b, H-5c), 3.77–3.59 (5H, m, H-4a, H-3b, H-4b, H-6c), 3.56–3.50 (1H, m, H-5b). 13C NMR (D2O, 126 MHz): δ 107.91 (C-1c), 100.47 (C-1b), 93.72 (C-1a), 82.62 (C-2c), 80.91 (C-4c), 75.91 (C-3c), 74.90 (C-5b), 72.94 (C-4b), 71.44 (C-3b), 71.16 (C-5a), 70.58 (C-2a), 70.48 (C-3a), 70.23 (C-2b), 70.12 (C-5c), 68.56 (C-6a), 66.43 (C-4a), 62.67 (C-6c), 61.04 (C-6b).
HRMS (ESI): calcd for C18H32O16Na [M + Na]+ 527.1588, found 527.15891.
β-D-Galactofuranosyl-(1 → 4)-α-D-mannopyranosyl-(1 → 6)-α,β-D-mannopyranose (2b)
To a solution of 17b (12 mg, 0.02 mmol) in a CH3CN/H2O 1:1 mixture (5 mL), N-iodosaccharine (14 mg, 0.045 mmol) was added. The reaction mixture was stirred at RT for 3 h. After the disappearance of the starting compound, the reaction was quenched by adding triethylamine and concentrated under reduced pressure. The resulting crude was partitioned between CH2Cl2 and water, and the aqueous layer was washed with CH2Cl2 (3×). The resulting aqueous phase was freeze-dried and the resulting residue was purified by column chromatography (AcOEt/MeOH/H2O gradient from 8:1:1 up to 6:2:2) to afford 2b (6 mg, 60%) as a white solid.
2bα: 1H NMR (D2O, 500 MHz): δ 5.17 (1H, d, J1a,2a 1.7, H-1a), 4.91 (1H, d, J1b,2b 1.7, H-1b), 4.45 (1H, d, J1c,2c 7.8, H-1c), 4.06 (1H, dd, J2b,3b 3.4, J2b,1b 1.7, H-2b), 4.00–3.91 (6H, m, H-2a, H-5a, H-6a, H-3b, H-4c, H-6c), 3.91–3.79 (4H, m, H-3a, H-4b, H-5b, H-6′c), 3.78–3.72 (5H, m, H-4a, H-6′a, H-6b, H-5c), 3.68 (1H, dd, J3c,2c 9.9, J3c,4c 3.3, H-3c), 3.55 (1H, dd, J2c,3c 9.9, J2c,1c 7.8, H-2c). 13C NMR (D2O, 126 MHz): 103.02 (C-1c), 99.33 (C-1b), 94.17 (C-1a), 76.40 (C-4b), 75.37 (C-5c), 72.50 (C-3c), 71.28 (C-5b), 70.95 (C-2c), 70.63 (C-5a), 70.58 (C-2a), 70.41 (C-3a), 69.41 (C-2b), 69.32 (C-3b), 68.59 (C-4c), 66.66 (C-4a), 65.91 (C-6a), 61.10 (C-6b), 60.23 (C-6c).
2bβ: 1H NMR (D2O, 500 MHz): δ 4.92 (1H, d, J1b,2b 1.8, H-1b), 4.90 (1H, d, J1a,2a 1.0, H-1a), 4.45 (1H, d, J1c,2c 7.8, H-1c), 4.06 (1H, dd, J2b,3b 3.4, J2b,1b 1.8, H-2b), 4.00–3.91 (6H, m, H-2a, H-3a, H-6a, H-3b, H-4c, H-6c), 3.91–3.79 (3H, m, H-4b, H-5b, H-6′c), 3.78–3.72 (4H, m, H-6′a, H-6b, H-5c), 3.68 (1H, dd, J3c,2c 9.9, J3c,4c 3.3, H-3c), 3.67–3.64 (1H, m, H-4a), 3.58–3.49 (1H, m, H-5a), 3.55 (1H, dd, J2c,3c 9.9, J2c,1c 7.8, H-2c). 13C NMR (D2O, 125 MHz): 103.02 (C-1c), 99.39 (C-1b), 93.81 (C-1a), 76.40 (C-4b), 75.37 (C-5c), 74.25 (C-5a), 73.14 (C-2a), 72.50 (C-3c), 71.28 (C-5b), 71.14 (C-3a), 70.95 (C-2c), 69.38 (C-2b), 69.30 (C-3b), 68.59 (C-4c), 66.45 (C-4a), 65.99 (C-6a), 61.10 (C-6b), 60.23 (C-6c).
HRMS (ESI): calcd for C18H32O16Na [M + Na]+ 527.1588, found 527.1583.
[β-D-Galactofuranosyl-(1 → 4)]-α-D-mannopyranosyl-(1 → 6)-α,β-D-mannopyranose (3a)
To a solution of 23a (20 mg, 0.03 mmol) in a CH3CN/H2O 9:1 mixture (5 mL), N-iodosaccharine (16 mg, 0.05 mmol) was added. The reaction mixture was stirred at RT for 2 h. After the disappearance of the starting compound, the reaction was quenched by adding triethylamine and concentrated under reduced pressure. The resulting crude was partitioned between CH2Cl2 and water, and the aqueous layer was washed with CH2Cl2 (3×). After freeze-drying of the aqueous phase, the target trisaccharide 3a was chromatographically purified on silica gel (AcOEt/MeOH/H2O gradient from 8:1:1 up to 6:2:2) and isolated (8 mg, 50%) as a white solid.
3aα: 1H NMR (D2O, 500 MHz): δ 5.17 (1H, d, J1a,2a 1.8, H-1a), 5.11 (1H, d, J1c,2c 2.2, H-1c), 4.93 (1H, d, J1b,2b 1.8, H-1b), 4.13–4.08 (3H, m, H-2c, H-3c, H-4c), 4.05–3.93 (5H, m, H-2a, H-3a, H-5a, H-6a, H-2b), 3.92–3.81 (4H, m, H-4a, H-3b, H-6b, H-5c), 3.80–3.71 (2H, m, H-6′a, H-6′b), 3.71–3.63 (4H, m, H-4b, H-5b, H-6c). 13C NMR (D2O, 126 MHz): δ 107.77 (C-1c), 100.34 (C-1b), 93.88 (C-1a), 82.66 (C-2c), 81.04 (C-4c), 76.06 (C-3c), 75.25 (C-4a), 72.92 (C-5b), 70.50, 70.49 (C-5c, C-2a), 70.44 (C-3b), 69.86 (C-5a), 69.65 (C-2b), 68.76 (C-3a), 66.62 (C-4b), 66.15 (C-6a), 62.68 (C-6c), 60.84 (C-6b).
3aβ: 1H NMR (D2O, 500 MHz): δ 5.11 (1H, d, J1c,2c 2.2, H-1c), 4.94 (1H, d, J1b,2b 1.8, H-1b), 4.90 (1H, s, H-1a), 4.13–4.08 (3H, m, H-2c, H-3c, H-4c), 4.05–3.93 (4H, m, H-2a, H-3a, H-6a, H-2b), 3.92–3.81 (3H, m, H-3b, H-6b, H-5c), 3.80–3.71 (3H, m, H-4a, H-6′a, H-6′b), 3.71–3.63 (4H, m, H-4b, H-5b, H-6c), 3.64–3.59 (1H, m, H-5a). 13C NMR (D2O, 126 MHz): δ 107.73 (C-1c), 100.30 (C-1b), 93.77 (C-1a), 82.75 (C-2c), 81.00 (C-4c), 76.06 (C-3c), 74.95 (C-4a), 73.38 (C-5a), 72.92 (C-5b), 70.50, 70.49 (C-5c, C-2a), 70.44 (C-3b), 69.65 (C-2b), 68.76 (C-3a), 66.62 (C-4b), 66.06 (C-6a), 62.68 (C-6c), 60.87 (C-6b).
HRMS (ESI): calcd for C18H32O16Na [M + Na]+ 527.1588, found 527.1590.
[β-D-Galactopyranosyl-(1 → 4)]-α-D-mannopyranosyl-(1 → 6)-α,β-D-mannopyranose (3b)
To a solution of 23b (33 mg, 0.06 mmol) in a CH3CN/H2O 9:1 mixture (5 mL), N-iodosaccharin (26 mg, 0.08 mmol) was added. The reaction mixture was stirred at RT for 2 h. After the disappearance of the starting compound, the reaction was quenched by adding triethylamine and concentrated under reduced pressure. The resulting crude was partitioned between CH2Cl2 and water, and the aqueous layer was washed with CH2Cl2 (3×). After freeze-drying of the aqueous phase, the target trisaccharide 3b was chromatographically purified on silica gel (AcOEt/MeOH/H2O gradient from 8:1:1 up to 6:2:2) and isolated (25 mg, 83%) as a white solid.
3bα: 1H NMR (D2O, 500 MHz): δ 5.17 (1H, s, H-1a), 4.93 (1H, s, H-1b), 4.44 (1H, d, J1c,2c 8.1, H-1c), 4.10–3.97 (4H, m, H-2a, H-5a, H-6a, H-2b), 3.96–3.87 (4H, m, H-3a, H4a, H-6b, H-4c), 3.87–3.70 (7H, m, H-6′a, H-3b, H-5b, H-6′b, H-5c, H-6c), 3.70–3.64 (2H, m, H-4b, H-3c), 3.58–3.51 (1H, m, H-2c).
13C NMR (D2O, 126 MHz): δ 102.99 (C-1c), 100.22 (C-1b), 93.80 (C-1a), 76.32 (C-4a), 75.49 (C-5c), 72.89 (C-5b), 72.45 (C-3c), 70.89 (C-2c), 70.52 (C-3b), 70.03 (C-2b), 69.84 (C-2a), 69.62 (C-5a), 68.92 (C-3a), 68.61 (C-4c), 66.61 (C-4b), 65.99 (C-6a), 61.13 (C-6c), 60.95 (C-6b).
3bβ: 1H NMR (D2O, 500 MHz): δ 4.94 (1H, s, H-1b), 4.91 (1H, s, H-1a), 4.44 (1H, d, J1c,2c 8.1, H-1c), 4.10–3.97 (3H, m, H-2a, H-6a, H-2b), 3.96–3.87 (3H, m, H-3a, H-6b, H-4c), 3.87–3.70 (8H, m, H-4a, H-6′a, H-3b, H-5b, H-6′b, H-5c, H-6c), 3.70–3.64 (2H, m, H-4b, H-3c), 3.58–3.50 (2H, m, H-5a, H-2c). 13C NMR (D2O, 126 MHz): δ 102.99 (C-1c), 100.17 (C-1b), 93.67 (C-1a), 75.92 (C-4a), 75.49 (C-5c), 73.45 (C-5a), 72.84 (C-5b), 72.45 (C-3c), 70.90 (C-2c), 70.50 (C-3b), 70.03 (C-2b), 69.81 (C-2a), 68.92 (C-3a), 68.61 (C-4c), 66.65 (C-4b), 65.91 (C-6a), 61.13 (C-6c), 60.95 (C-6b).
HRMS (ESI): calcd for C18H32O16Na [M + Na]+ 527.1588, found 527.1586.
General procedure for lichen extraction
All lichen samples were collected in France. Cetraria islandica, Lasallia pustulata and Roccella fuciformis were collected respectively in the Alpes (Haute-Savoie (74), August 2009), in Sainte Urcize (Cantal (15), June 2005) and near the plage des Chevrets (St Malo, Ille et Vilaine (35), June 2005). Vouchers were stored in the Herbarium of the lab under the codes (JB/09/106), (JB/19/227) and (JB/05/58), respectively.According to Pereyra,10 the thalli of lichens were separated from the support (soil and rock) debris, washed with water (3×) and dried overnight at 40 °C. 15 g of thalli were boiled for 30 min at 100 °C (3×) and filtered through a cheesecloth. After cooling, the soluble fraction was centrifuged (2000g, 15 min). The supernatant was precipitated with EtOH (v/v) and filtered through a Büchner filter. Fractions A2 (>10 kDa) were obtained after ultrafiltration using an Amicon device with a regenerated cellulose filter (10 kDa, Merck Milipore), a concentration step under vacuum and a lyophilization step. They were stored in the dark at room temperature. The insoluble fraction after hot water extraction was treated with NaOH 1 M (3 × 200 mL) at 25 °C followed by centrifugation (2000g, 15 min). The supernatant was precipitated with EtOH (v/v) and filtered through a Büchner filter. Fractions F1S (>10 kDa) were obtained after ultrafiltration using the Amicon device with a regenerated cellulose filter (10 kDa, Merck Milipore), a concentration step under vacuum and a lyophilization step.
Author contributions
V. F., L. L., I. C. and D. R. contributed to the conceptualisation and acquisition of the financial support. B. F., L.-P. D. and L. L. synthesized all oligosaccharides and intermediates. L. L., S. F. and F. L. D. participated in the interpretation of the 2D HSQC maps of the lichen extracts. L. L. and V. F. shared the preparation of the initial draft. All contributors participated in the approval of the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from Agence Nationale de la Recherche (project ALGAIMS ANR-18-CE29-0006-02, https://algaims-35.webself.net/accueil) is gratefully acknowledged. Part of this work was performed using the PRISM core facility (Biogenouest, Univ Rennes, Univ Angers, INRAE, CNRS, France). Special thanks to Joel Esnault for the pictures of the lichens.References
- D. L. Hawksworth and M. Grube, New Phytol., 2020, 227, 1281–1283 CrossRef.
- K. Müller, Appl. Microbiol. Biotechnol., 2001, 56, 9–16 CrossRef.
- P. Le Pogam, G. Herbette and J. Boustie, in Recent Advances in Lichenology, ed. D. K. Upreti, P. K. Divakar, V. Shukla and R. Bajpai, Springer India, 2015 Search PubMed.
- F. Gasulla, E. M. del Campo, L. M. Casano and A. Guéra, Plants, 2021, 10, 807 CrossRef CAS.
- T. Spribille, G. Tagirdzhanova, S. Goyette, V. Tuovinen, R. Case and W. F. Zandberg, FEMS Microbiol. Lett., 2020, 367, fnaa023 CrossRef CAS PubMed.
- M. González-Hourcade, E. M. del Campo, M. R. Braga, A. Salgado and L. M. Casano, Environ. Microbiol., 2020, 22, 3096–3111 CrossRef PubMed.
- P. A. J. Gorin and E. Barreto-Bergter, in The Polysaccharides, ed. G. O. Aspinall, Academic Press, 1983, pp. 365–409, DOI:10.1016/B978-0-12-065602-8.50011-X.
- E. S. Olafsdottir and K. Ingólfsdottir, Planta Med., 2001, 67, 199–208 CrossRef CAS PubMed.
- P. A. J. Gorin and M. Iacomini, Carbohydr. Res., 1984, 128, 119–132 CrossRef CAS.
- M. T. Pereyra, A. Prieto, M. Bernabé and J. A. Leal, Lichenologist, 2003, 35, 177–185 CrossRef.
- E. R. Carbonero, F. R. Smiderle, A. H. P. Gracher, C. G. Mellinger, G. Torri, T. Ahti, P. A. J. Gorin and M. Iacomini, Carbohydr. Polym., 2006, 63, 13–18 CrossRef CAS.
- P. A. J. Gorin and M. Iacomini, Carbohydr. Res., 1985, 142, 253–267 CrossRef CAS.
- Y. Sone, M. Isoda-Johmura and A. Misaki, Biosci., Biotechnol., Biochem., 1996, 60, 213–215 CrossRef CAS PubMed.
- E. R. Carbonero, L. M. C. Cordeiro, C. G. Mellinger, G. L. Sassaki, E. Stocker-Wörgötter, P. A. J. Gorin and M. Iacomini, Carbohydr. Res., 2005, 340, 1699–1705 CrossRef CAS.
- G. Shrestha, L. L. St. Clair and K. L. O'Neill, Phytother. Res., 2015, 29, 317–322 CrossRef CAS.
- E. S. Olafsdottir, S. Omarsdottir, B. S. Paulsen, K. Jurcic and H. Wagner, Phytomedicine, 1999, 6, 273–279 CrossRef CAS.
- K. Ingolfsdottir, K. Jurcic, B. Fischer and H. Wagner, Planta Med., 1994, 60, 527–531 CrossRef CAS.
- L. D. Hall and L. F. Johnson, J. Chem. Soc. D, 1969, 509–510, 10.1039/C29690000509.
- V. B. Krylov, D. A. Argunov, A. S. Solovev, M. I. Petruk, A. G. Gerbst, A. S. Dmitrenok, A. S. Shashkov, J. P. Latgé and N. E. Nifantiev, Org. Biomol. Chem., 2018, 16, 1188–1199 RSC.
- B. Favreau, O. Yeni, S. Ollivier, J. Boustie, F. Le Dévéhat, J.-P. Guégan, M. Fanuel, H. Rogniaux, R. Brédy, I. Compagnon, D. Ropartz, L. Legentil and V. Ferrières, J. Org. Chem., 2021, 86, 6390–6405 CrossRef CAS PubMed.
- O. Yeni, S. Ollivier, B. Moge, D. Ropartz, H. Rogniaux, L. Legentil, V. Ferrières and I. Compagnon, J. Am. Chem. Soc., 2023, 145, 15180–15187 CrossRef CAS PubMed.
- S. Ollivier, L. Legentil, O. Yeni, L.-P. David, V. Ferrières, I. Compagnon, H. Rogniaux and D. Ropartz, J. Am. Soc. Mass Spectrom., 2023, 34, 627–639 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and related spectroscopic data for all intermediaries 4–23, 1H and 13C indexation table of trisaccharides 1–3, 2D HSQC maps of lichen extracts, and 1H and 13C NMR spectra of all intermediates and final products. See DOI: https://doi.org/10.1039/d4ob00047a |
| This journal is © The Royal Society of Chemistry 2024 |