
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of aminoalcohols from substituted alkenes via tungstenooxaziridine catalysis†
Rufai
Madiu
,
Brandon
Dellosso
,
Erin L.
Doran
,
Jenna M.
Doran
,
Ali A.
Pinarci
 ,
Tyler M.
TenHoeve
,
Amari M.
Howard
,
James L.
Stroud
,
Dominic A.
Rivera
,
Dylan A.
Moskovitz
,
Steven J.
Finneran
,
Alyssa N.
Singer
,
Morgan E.
Rossi
and
Gustavo
Moura-Letts
*
,
Tyler M.
TenHoeve
,
Amari M.
Howard
,
James L.
Stroud
,
Dominic A.
Rivera
,
Dylan A.
Moskovitz
,
Steven J.
Finneran
,
Alyssa N.
Singer
,
Morgan E.
Rossi
and
Gustavo
Moura-Letts
*
Department of Chemistry and Biochemistry, Rowan University, 201 Mullica Hill Rd., Glassboro, NJ, USA. E-mail: moura-letts@rowan.edu
First published on 21st February 2024
Abstract
Herein we report the WO2Dipic(H2O) promoted oxyamination of alkenes using sulfonamides as the quantitative source of N. The reaction works for activated and unactivated alkenes in high yields, diastereoselectivities, and stereospecificity. A catalytic cycle involving the formation of tungstenooxaziridine complex 1 as the active catalyst and hydrolysis of tungstenooxazolidine intermediate A as the rate-determining-step has been proposed. Initial kinetic and competition experiments provide evidence for the proposed mechanism.
Introduction
The stereoselective oxyamination of alkenes for the synthesis of aminoalcohols and derivatives remains as one of the most fundamental transformations in organic chemistry to this date.1 Much progress has been achieved towards the stereoselective difunctionalization of alkenes, however there are very few methods that are able to achieve direct oxyamination with high chemo-, regio-, and stereoselectivities.2 This structural motif is prevalent in natural products and commercially available drugs with very important pharmacological profiles3 but also across a large array of chiral ligands, auxiliaries and organocatalysts.4 Therefore, the continuous development of novel methods for the systematic synthesis of aminoalcohols has great significance.5 Since the pioneering work by Sharpless in asymmetric aminohydroxylation,2b multiple strategies have been developed to access aminoalcohols.6 Unfortunately, most suffer from poor regioselectivity or rely on intramolecular methods that require further functional group manipulations. Thus, direct stereoselective oxyamination methods continue to receive much attention in the field of organic chemistry (Fig. 1). Ground-breaking work by Yoon discovered that N-EWG oxaziridines can trigger oxyamination pathways by careful exposure to very specific transition-metal environments.7 Terminal alkenes, when using a copper catalyst, undergo oxyamination with high regioselectivity for oxygen on the least substituted carbon and when using an iron catalyst, oxyamination occurs with high regioselectivity for oxygen on the more substituted carbon.8 | ||
| Fig. 1 Advances in stereoselective oxyamination. | ||
Other efforts have found other transition metals also achieve oxyamination outcomes with general success (Mn, Pd, Ir, Cu, and Se).9 Moreover, efforts in visible-light photocatalysis have also demonstrated synthetically useful efficiencies and generality.10 This new generation of methods offers a large array of catalysts with unique properties, but regioselectivity and poor reactivity across unactivated alkenes remains a problem.
Metallooxaziridines can be obtained from reacting metal-oxides and amino-containing reagents.11 Their chemical properties are analogous to peroxo-complexes that transfer O atoms across alkenes and are comparable to metal oxide complexes known to achieve oxyamination reactions.12 The chemistry of these metallooxaziridines has not been fully exploited; however, we recently reported that zirconooxaziridines are suitable catalysts for the highly stereoselective and stereospecific aziridination of unactivated alkenes.13 Early work by Sharpless provided some insights into the unique properties of these complexes as he found that Mo and W oxaziridines were able to activate π-systems.14
The Moura-Letts laboratory is focused on developing novel methods for the synthesis of complex nitrogen-containing molecules.15 Thus, we envisioned creating a well-defined catalytic system for the oxyamination of alkenes via the formation of metallooxaziridines as the active catalyst for the reaction. To the best of our knowledge, this would be the first report for such a catalytic process.
Results and discussion
Since the discovery of zirconooxaziridine catalysis for the aziridination of alkenes, some results indicated that other chemical pathways could be optimized upon changing the transition-metal in the metallooxaziridine centre. The high chemoselectivity observed for LZrON-Ts was in part due to the group IV transition-metal nature of Zr and the geometry of the respective LUMO across the Zr–N bond. Thus, analogous to CuII and FeIII-promoted chemo- and regioselective diversity in oxyaminations,8 it was hypothesized that high-oxidation state group VI transition metal complexes (LMO2NTs) would provide LUMO's leading to different chemo- and regioselective outcomes. Fortunately, it was discovered that while LWO2NTs provided aziridine in very low yield, it provided aminoalcohol 3a with high selectivity. As observed for FeIII oxyaminations, the reaction chemoselectivity allows the transformation to occur across the N–O bond and, regioselective addition of N-Ts to the least substituted side of the alkene.16The basic premise behind this study was to develop a simple and efficient oxyamination process with high stereoselectivity and stereospecificity for unactivated and activated alkenes (Table 1). Although our efforts for the aziridination of alkenes relied on chloramine T as the source of N, we discovered that further diversification could be introduced with sulfonamides and trichloroisocyanuric acid (TCCA) as an activator/oxidant to provide in situ RSO2NHCl (RSO2-chloramine).17 Initial results using 1-hexene, indicated that metal oxides at 5 mol%, phase-transfer catalyst (PTC) at 7.5 mol%, and excess p-toluenesulfonamide/TCCA in CH3CN/H2O (10/1) promoted oxyamination in low yields, with a clear indication that W was able to provide the expected product with synthetically useful selectivities (entry 3). It was anticipated that dipicolinic acid (Dipic) would provide a more stable complex and it was found that WO2Dipic(H2O) improved the reaction yield to 48% and 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 3a:4a:5a product ratio (entry 5); V or Mo were not as successful at improving the reaction yield (entries 6 and 7). The nature of the PTC was tested, and it was found that TBAI provided a small increase in reaction performance (62% yield, entry 9). The different catalytic steps in this reaction take place in different phases in the reaction mixture. Thus, efficient PTCs are crucial to achieve conversion due to poorly soluble WO2Dipic(H2O) and in situ formed chloramine. Other catalysts failed to improve the reaction yield beyond the efficiency obtained with TBAI (entries 8, 10 and 11).
:1:1 3a:4a:5a product ratio (entry 5); V or Mo were not as successful at improving the reaction yield (entries 6 and 7). The nature of the PTC was tested, and it was found that TBAI provided a small increase in reaction performance (62% yield, entry 9). The different catalytic steps in this reaction take place in different phases in the reaction mixture. Thus, efficient PTCs are crucial to achieve conversion due to poorly soluble WO2Dipic(H2O) and in situ formed chloramine. Other catalysts failed to improve the reaction yield beyond the efficiency obtained with TBAI (entries 8, 10 and 11).
|
|
||||||
|---|---|---|---|---|---|---|
| Entrya,b | Metal catalyst | PTC | Solvent | Temp | Yield (%) |
3a:4a:5a |
|
a Isolated yields.
b TsNH2 (1.5 equiv.) is mixed with TCCA (0.5 equiv.) in CH2Cl2 at 0 °C for 30 min then solvent removed and CH3CN/H2O 0.125M (10:1), metal oxide, additive and alkene are added.
c 5 mol% of metal and 7.5 mol% of PTC.
d WO2Dipic(H2O), Dipic = dipicolinic acid.
e 15 mol%. CTA = cetrimonium, TBA = tetrabutyl n-butylammonium.
|
||||||
| 1 | MoO3c |
CTAB | CH3CN/H2O | rt | 5 | 1:1:1 |
| 2 | V2O5c |
CTAB | CH3CN/H2O | rt | 4 | 1:1:0 |
| 3 | WO3c |
CTAB | CH3CN/H2O | rt | 24 | 4:1:0 |
| 4 | ZrO2c |
CTAB | CH3CN/H2O | rt | 6 | 1:0:5 |
| 5 | WO2Dipicc,d | CTAB | CH3CN/H2O | rt | 48 | 10:1:1 |
| 6 | V2O3Dipicc,d | CTAB | CH3CN/H2O | rt | 10 | 2:1:1 |
| 7 | MoO2Dipicc,d | CTAB | CH3CN/H2O | rt | 8 | 2:1:0 |
| 8 | WO2Dipicc,d | TBAB | CH3CN/H2O | rt | 38 | 10:1:0.5 |
| 9 | WO2Dipicc,d | TBAI | CH3CN/H2O | rt | 62 | 12:1:0.5 |
| 10 | WO2Dipicc,d | TBACl | CH3CN/H2O | rt | 44 | 10:1:1 |
| 11 | WO2Dipicc,d | CTAI | CH3CN/H2O | rt | 39 | 10:1:1 |
| 12 | WO2Dipicd(10 mol%) | TBAIe | CH3CN/H2O | rt | 58 | 10:2:3 |
| 13 | WO 2 Dipic (1 mol%) | TBAI | CH 3 CN/H 2 O | rt | 88 |
16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :0.5:1 :0.5:1
|
| 14 | WO2Dipicd(1 mol%) | TBAI | DCE/H2O | rt | 73 | 16:1:1 |
| 15 | WO2Dipicd(1 mol%) | TBAI | CH2Cl2/H2O | rt | 35 | 16:1:1 |
| 16 | WO2Dipicd(1 mol%) | TBAI | DMF/H2O | rt | 53 | 15:1:2 |
| 17 | WO2Dipicd(1 mol%) | TBAI | CH3CN/H2O | 0 °C | 48 | 16:1:1 |
| 18 | WO2Dipicd(1 mol%) | TBAI | CH3CN/H2O | 60 °C | 32 | 8:1:2 |

To further improve the reaction performance, catalyst loading was increased to 10 mol% but a complex mixture and lowered chemo- and regioselectivity due to catalyst decomposition was observed (entry 12); however, when loading was reduced to 1 mol% the yield and product ratio increased to 88% and 16:0.5:1 respectively (entry 13). The reaction solvent was also examined, and CH3CN/H2O remained to be optimal. Coordinating solvents (CH3CN) are known to accelerate N-Ts transfer reactions and the role of H2O as a ligand in the proposed process was anticipated. The reaction at different temperatures also failed to provide similarly high yields and selectivities (entries 17 and 18).
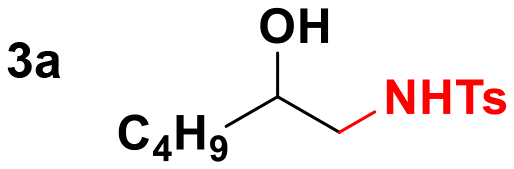
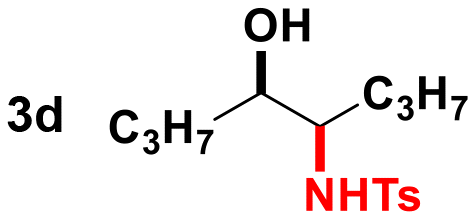
Given the results observed for the oxyamination of 1-hexene, this study focused on addressing the generality across different types of unactivated alkenes (Table 2).18 1-Octene provided the corresponding aminoalcohol in equally high yield and similar observable rate (91% and 16 h, entry 2). Activation of alkenes, depending upon the nature of the N-transfer reagent, often suffers from poor diastereospecificity due to the formation of nitrene reactive intermediates that lead to stereochemical erosion.19 Analogous to the aziridination reaction, high stereospecificity was expected due to the fast formation of metalloheterocyclic intermediates (intermediate A) through a biradical transition state.
|
|
||||
|---|---|---|---|---|
| Entry | Alkene | Productc | Yielda,b (%) | d.r |
|
a Conditions: TsNH2 (1.5 equiv.) is mixed with TCCA (0.5 equiv.) in CH2Cl2 at 0 °C for 30 min then solvent removed and CH3CN/H2O 0.125M (10:1), metal oxide, additive and alkene are added, and reaction stirred at rt for 16 h or until full conversion by TLC.
b Isolated yields.
c Reaction was purified by standard silica gel chromatography.
d 6:1 mixture of 3c and 4c.
|
||||
| 1 |

|
|
88 | — |
| 2 |

|

|
91 | — |
| 3 |

|

|
82d | 18:1 |
| 4 |

|
|
84 | 20:1 |
| 5 |

|

|
88 | 20:1 |
| 6 |

|

|
93 | 20:1 |
| 7 |

|
|
91 | 18:1 |
| 8 |
|

|
95 | 20:1 |
| 9 |
|

|
92 | 19:1 |

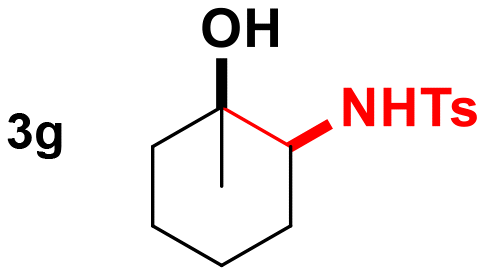
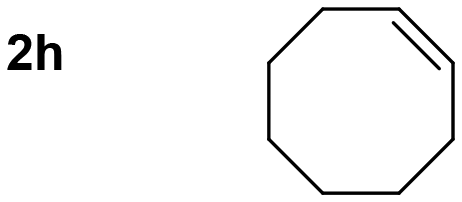
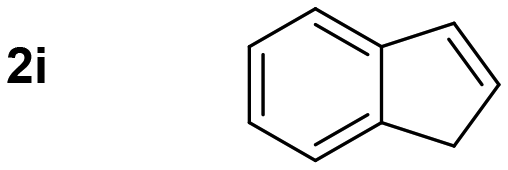
Thus, we tested 1,2-disubstituted alkenes and we found that (E)-2-octene, and symmetric (E)-4-octene reacted to produce the corresponding aminoalcohols in high yields and diastereoselectivities without visible stereochemical erosion (entries 3 and 4). Correspondingly, (Z)-4-octene provided the product in good yield, high diastereoselectivity and a comparable observable rate (88% and 18 h, entry 5). These results provide indication of a highly stereospecific delivery of both groups across the alkene, the high syn-selectivity correlates well with a tungstenooxaziridine syn-addition across the N–O bond. Further exploration of the scope revealed that cyclohexene, 1-methylcyclohexene, cyclooctene, and indene provided the corresponding aminoalcohols in high yields and slightly slower rates despite the increased substitution (18–22 h, entries 6–9).20 The reaction proved to be successful across a variety of unactivated alkenes with high stereoselectivity and stereospecificity. The focus then turned towards addressing the generality across styrenes with activating and deactivating substituents (Table 3).
|
|
|||
|---|---|---|---|
| Entry | Alkene | Productc | Yielda,b (%) |
|
a Conditions: TsNH2 (1.5 equiv.) is mixed with TCCA (0.5 equiv.) in CH2Cl2 at 0 °C for 30 min then solvent removed and CH3CN/H2O 0.125M (10:1), metal oxide, additive and alkene are added, and reaction stirred at rt for 16 h or until full conversion by TLC.
b Isolated yields.
c Reaction was purified by standard silica gel chromatography.
d 10:1 mixture of 3ad (22:1 d.r.) and 4ad.
|
|||
| 1 |

|

|
91 |
| 2 | 2k R = 4-methyl | 3k R = 4-methyl | 88 |
| 3 | 2l R = 3-methyl | 3l R = 3-methyl | 89 |
| 4 | 2m R = 2-methyl | 3m R = 2-methyl | 86 |
| 5 | 2n R = 4-tbutyl | 3n R = 4-tbutyl | 90 |
| 6 | 2o R = 2,4,6-trimethyl | 3o R = 2,4,6-trimethyl | 86 |
| 7 | 2p R = 4-methoxy | 3p R = 4-methoxy | 88 |
| 8 | 2q R = 4-phenyl | 3q R = 4-phenyl | 84 |
| 9 | 2r R = 2-naphthyl | 3r R = 2-naphthyl | 80 |
| 10 | 2s R = 4-fluoro | 3s R = 4-fluoro | 82 |
| 11 | 2t R = 4-chloro | 3t R = 4-chloro | 80 |
| 12 | 2u R = 4-iodo | 3u R = 4-iodo | 86 |
| 13 | 2v R = 3-chloro | 3v R = 3-chloro | 89 |
| 14 | 2w R = 2-chloro | 3w R = 2-chloro | 84 |
| 15 | 2x R = 2,6-dichloro | 3x R = 2,6-dichloro | 91 |
| 16 | 2y R = 4-trifluoromethyl | 3y R = 4-trifluoromethyl | 86 |
| 17 | 2z R = 4-cyano | 2z R = 4-cyano | 80 |
| 18 | 2aa R = 4-nitro | 3aa R = 4-nitro | 82 |
| 19 |

|

|
91 |
| 20 |

|

|
88 |
| 21 |
|
|
84d |

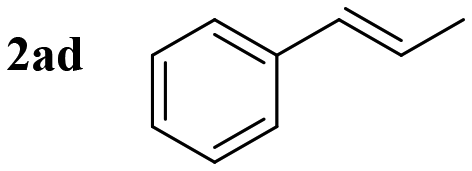
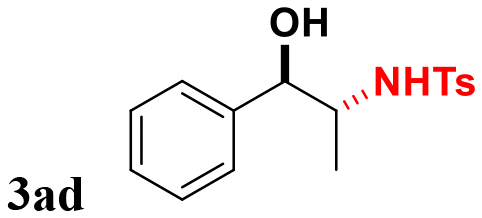
Styrene (entry 1) and styrenes with various alkyl electron-donating-groups (EDGs) were successful at providing the expected aminoalcohols in high yields and at faster observable rates (4–8 h, entries 2–6). Moreover, 4-methoxy styrene worked in similarly high yield and faster observable rate (3 h, entry 7). Other substitution patterns (biphenyl and napthyl, entries 8 and 9) were also very productive for this transformation. The electronic properties around the ring were then changed; and 4-fluoro, 4-chloro, and 4-iodo provided aminoalcohol 3 in similar yields without apparent loss in observable reaction rates (4–8 h, entries 10–12). Moreover, 3-chloro, 2-chloro, 2,6-dichloro, and 4-trifluoromethyl worked in similar high yields but slower rates (12–14 h, entries 13–16). Surprisingly, 4-cyano and 4-nitro worked in slightly lower yields but there was no noticeable decrease in reaction rates (5 and 6 h, entries 17 and 18). The small differences in reaction efficiencies across styrenes with different substitution patterns support a concerted mechanism. We were also interested in addressing the reaction selectivity across disubstituted styrenes. It was discovered that α-methyl-styrene, α-methyl-toluene, and β-methyl-styrene provided aminoalcohol 3 in high yields and without activation of the allylic methyl group (entries 19–21).
Scope of sulfonamides would allow the introduction of more complex functionality across aminoalcohol 3 (Table 4). It was found that sulfonamides with different nitro groups around the ring reacted to form the respective N-transfer reagent and then provided 3 in great yields (entries 1–3). Moreover, substituted heterocyclic sulfonamides reacted well and the respective aminoalcohols were also obtained in high yields (entries 4–6). Substituted benzenesulfonamides (4-Br, 4-F, 2-Me) and methylsulfonamide also provided 3 in great yields and optimal stereoselectivities (entries 7–10).
|
|
|||
|---|---|---|---|
| Entry | Sulfonamide | Productc | Yielda,b(%) |
|
a Conditions: RNH2 (1.5 equiv.) is mixed with TCCA (0.5 equiv.) in CH2Cl2 at 0 °C for 30 min then solvent removed and CH3CN/H2O 0.125M (10:1), metal oxide, additive and t-butylstyrene are added and reaction stirred at rt for 16 h or until full conversion by TLC.
b Isolated yields.
c Reaction was purified by standard silica gel chromatography.
|
|||
| 1 |

|

|
93 |
| 2 |

|
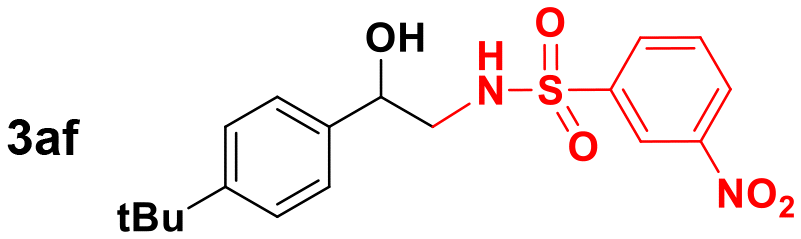
|
89 |
| 3 |

|
|
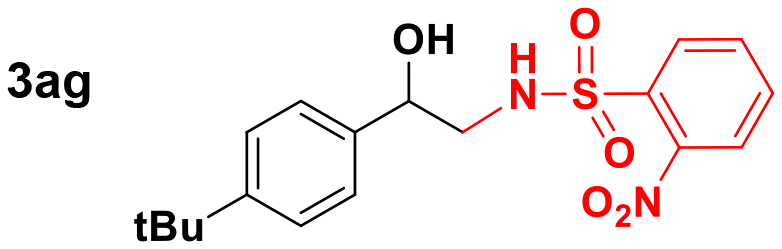
84 |
| 4 |

|

|
82 |
| 5 |
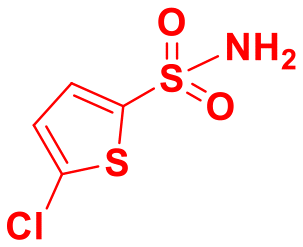
|

|
86 |
| 6 |
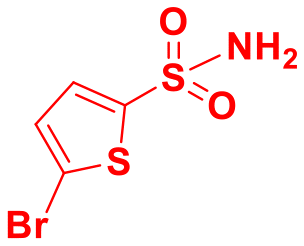
|

|
88 |
| 7 |

|

|
80 |
| 8 |

|

|
84 |
| 9 |
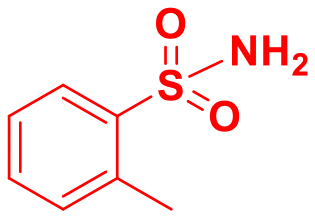
|

|
91 |
| 10 |

|

|
84 |

The foundational knowledge discovered for the zirconooxaziridine-mediated aziridination of alkenes along with the available studies on metal-oxide promoted alkene difunctionalization helped design the mechanistic studies for this reaction. The main questions to be addressed were the mode of N-Ts group transfer, competing pathways and the mode of OH transfer across the alkene and metal-centre.
Based on the observed chemoselectivity and high stereoselectivity, we anticipated a potentially fast tungstenooxaziridine addition across the alkene with formation of short-lived biradical species through homolytic cleavage of the N–O bond in the transition state.21 Experiments with deuterated substrates showed that 2j1-d and 2p1-d reacted to provide 3j1-d and 3p1-d without any deuterium scrambling, thus both experiments confirming that a highly concerted syn-addition is likely to be the predominant pathway. We also wanted to address if 3 formed through hydrolysis of aziridine 5 under the reaction conditions, and we found that 5j does not provide 3j under the reaction conditions. Other control experiments showed that WO2Dipic(H2O) is crucial for reaction conversion, thus no halogen-mediated activation pathway is taking place, and that the reaction can also work under stoichiometric amounts of 1 (low conversion, details on ESI†).
Thus, we propose a catalytic cycle in which 1 forms within minutes from reacting WO2Dipic(H2O) with Ts-chloramine, obtained in-situ from reacting p-toluenesulfonamide and TCCA (Fig. 2). Complex 1 can also be efficiently isolated by reacting WO2Dipic(H2O) and p-toluenesulfonamide/TCCA or chloramine T in CHCl3 or MeOH, and its presence can be detected in the catalytic reaction mixture spectroscopically.22 Ligand exchange allows for alkene coordination in such a way, so the HOMO of styrene is aligned with the N–O bond α* (LUMO) of complex 1. This is then followed by fast syn-addition across the alkene to provide tungstenooxazolidine intermediate A. The intermediate then undergoes slow, rate-determining-step hydrolysis followed by incorporation of a second water molecule to provide intermediate B. The water molecule in A exchanges across the weakly coordinating N-Ts group and the resulting intermediate B is then poised to trigger fast proton transfer to release 3 and regenerate precatalyst WO2Dipic(H2O).23
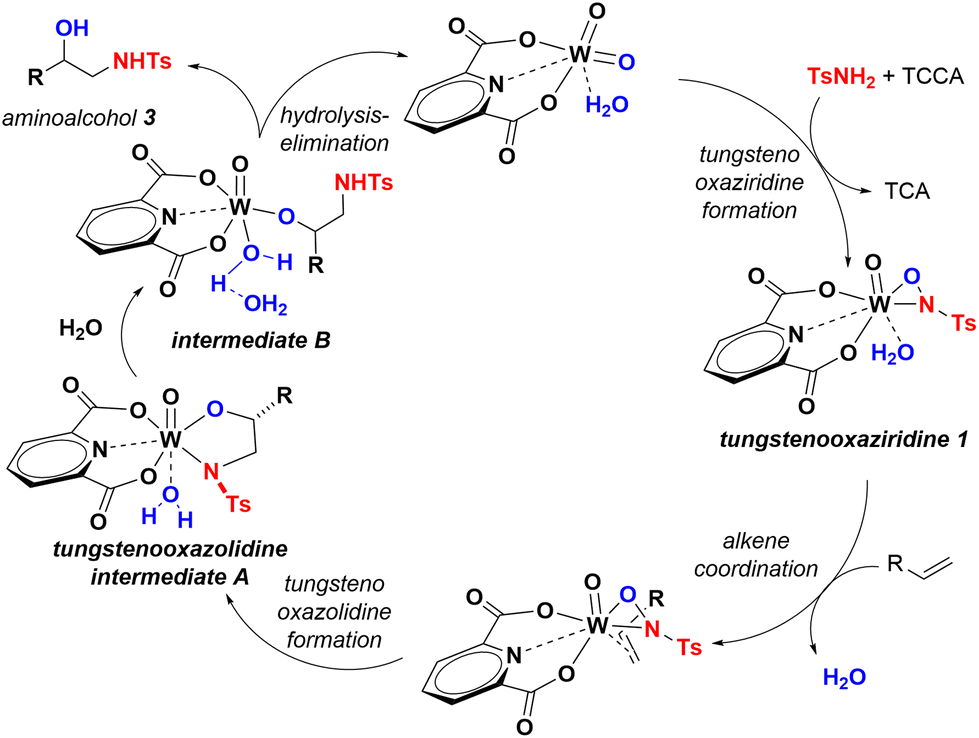 | ||
| Fig. 2 Proposed catalytic cycle. | ||
Further analysis of the reaction kinetics revealed that hydrolysis of intermediate A is irreversible with a secondary kinetic solvent isotope effect (KSIE, KH2O/KD2O = 0.83, details in ESI†). The inverse KSIE obtained through pseudo-first-order kinetics validates a slow, rate-determining hydrolytic step. Experimental verification was obtained by further deuterium-labeling competition studies (Fig. 3). The results showed no secondary KIE when reacting a 1:1 mixture of 2j/2j2-d2 (kH/kD = 1.02) and 2l/2j1-d (kH/kD = 1.01). These results are in agreement with a fast, highly concerted tungstenooxaziridine addition across the alkene step and a slow hydrolytic rate-determining-step. The proposed catalytic cycle is in further agreement with a Hammett correlation study employing substituted styrenes (2j,k,n,p,s-u), which shows ρ-values of −1.87 for EDG-styrenes and 0.75 for EWG-styrenes. Concave Hammett plots are often observed on highly concerted cycloadditions with developing biradical characters in their transition-states, thus confirming the proposed catalytic cycle.
 | ||
| Fig. 3 Deuterium-labeling competition studies. | ||
Conclusions
In summary, this work managed the discovery of a stereoselective tungstenooxaziridine-mediated catalytic oxyamination of alkenes. The reaction works with high efficiency and stereoselectivity for alkenes with diverse substitution patterns and for styrenes with a variety of functional groups. The proposed catalytic cycle involves the formation of active catalyst tungstenooxaziridine 1 that then delivers the N-Ts and O groups through the formation of tungstenooxazolidine intermediate A, followed by rate-determining-step hydrolysis to provide aminoalcohol 3. Further experiments to better understand and fully characterize all mechanism intermediates are ongoing and a follow up manuscript is in preparation.Author contributions
R. M. and B. D. contributed equally for most of the experiments (∼60%). E. L. D. (20%) and J. M. D. (10%) performed significant parts of the experiments as well. A. A. P and T. T made significant contributions at the discovery stage. D. A. R., A. M. H., J. L. S., D. A. M., S. J. F., A. N. S., and M. E. R. participated in compound characterization, and SI preparation. G. M. L. wrote the draft and supervised the research. All authors discussed the results and prepared the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is based upon work supported by the National Science Foundation CAREER and MRI awards. Under grant numbers CHE-1752085 and CHE-1827938.References
- (a) C. Wata and T. Hashimoto, Organoiodine-Catalyzed Enantioselective Intermolecular Oxyamination of Alkenes, J. Am. Chem. Soc., 2021, 143, 1745–1751 CrossRef CAS PubMed; (b) H. Lei, J. H. Conway, C. C. Cook and T. Rovis, Ligand Controlled Ir-Catalyzed Regiodivergent Oxyamination of Unactivated Alkenes, J. Am. Chem. Soc., 2019, 141, 11864–11869 CrossRef CAS PubMed; (c) I. Cho, C. K. Prier, Z.-J. Jia, E. K. Zhang, T. Goerbe and F. H. Arnold, Enantioselective Aminohydroxylation of Styrenyl Olefins Catalyzed by an Engineered Hemoprotein, Angew. Chem., Int. Ed., 2019, 58, 3138–3142 CrossRef CAS PubMed; (d) H.-C. Shen, Y.-F. Wu, Y. Zhang, L.-F. Fan, Z.-Y. Han and L.-Z. Gong, Palladium-Catalyzed Asymmetric Aminohydroxylation of 1,3-Dienes, Angew. Chem., Int. Ed., 2018, 57, 2372–2376 CrossRef CAS PubMed; (e) M. M. Heravi, T. B. Lashaki, B. Fattahi and V. Zadsirjan, Application of Asymmetric Sharpless Aminohydroxylation in Total Synthesis of Natural Products and Some Synthetic Complex Bioactive Molecules, RSC Adv., 2018, 8, 6634–6659 RSC.
- (a) A. Gontala, H. Huh and S. K. Woo, Photoredox-Catalyzed Synthesis of Beta-Amino Alcohols: Hydroxymethylation of Imines with alpha-Silyl Ether as Hydroxymethyl Radical Precursor, Org. Lett., 2023, 25, 21–23 CrossRef CAS PubMed; (b) K. B. Sharpless, A. O. Chong and K. Oshima, Osmium-Catalyzed Vcinial Oxyamination of Olefins by Chloramine-T, J. Org. Chem., 1976, 41, 177–179 CrossRef CAS; (c) G. Li, H.-T. Chang and K. B. Sharpless, Catalytic Asymmetric Aminohydroxylation (AA) of Olefins, Angew. Chem., Int. Ed. Engl., 1996, 35, 451–454 CrossRef CAS; (d) T. J. Donohoe, P. D. Johnson, M. Helliwell and M. Keenan, The Regioselective Aminohydroxylation of Allylic Carbamates, Chem. Commun., 2001, 2078–2079 RSC; (e) T. J. Donohoe, P. D. Johnson, A. Cowley and M. Keenan, The Tethered Aminohydroxylation (TA) of Cyclic Allylic Carbamates, J. Am. Chem. Soc., 2002, 124, 12934–12935 CrossRef CAS PubMed; (f) P. H. Fuller, J.-W. Kim and S. R. Chemler, Copper Catalyzed Enantioselective Intramolecular Aminohydroxylation of Alkenes, J. Am. Chem. Soc., 2008, 130, 17638–17639 CrossRef CAS PubMed.
- (a) N. U. Sata and N. Fusetani, Amininols A and B, New Bicyclic Aminoalcohols from anUnidentifed Tunicate of the Family Polyclinidae, Tetrahedron Lett., 2000, 41, 489–492 CrossRef CAS; (b) E. Abraham, S. G. Davies, N. L. Millican, R. L. Nicholson, P. M. Roberts and A. D. Smith, Asymmetric Synthesis of Vicinal Aminoalcohols: Xestoaminol C, Sphinganine and Sphingosine, Org. Biomol. Chem., 2008, 6, 1655 RSC; (c) E. D. Calder, A. M. Zaed and A. Sutherland, Preparation of anti-Vicinal Aminoalcohols: Asymmetric Synthesis of D-Erythro-Sphinganine, (+)-Spisulosine, and D-Ribo-Phytosphingosine, J. Org. Chem., 2013, 78, 7223–7233 CrossRef CAS PubMed; (d) J. N. Park, S. Y. Ko and H. Y. Koh, Synthetic Applications of the Cyclic Iminocarbonate Rearrangement: Enantioselective Synthesis of Chloramphenicol and 4_epi-Cytoxazone, Tetrahedron Lett., 2000, 41, 5553–5556 CrossRef CAS.
- (a) A. V. Malkov, M. A. Kabeshov, M. Bella, O. Kysilka, D. A. Malyshev, K. Pluháčková and P. Kočovský, Vicinal Aminoalcohols as Organocatalysts in Asymmetric Cross-Aldol Reaction of Ketones: Application in the Synthesis of Convolutamydine A., Org. Lett., 2007, 9, 5473–5476 CrossRef CAS PubMed; (b) M. M. Heravi, V. Zadsirjan and B. Farajpour, Applications of Oxazolidinones as Chiral Auxiliaries in the Asymmetric Alkylation Reaction Applied to Total Synthesis, RSC Adv., 2016, 6, 30498–30551 RSC; (c) G. Desimoni, G. Faita and K. A. Jørgensen, C2-Symmetrical Chiral Bis(Oxazoline) Ligands in Asymmetric Catalysis, Chem. Rev., 2006, 106, 3561–3651 CrossRef CAS PubMed.
- (a) K. S. Williamson, D. J. Michaelis and T. P. Yoon, Advances in the Chemistry of Oxaziridines, Chem. Rev., 2014, 114, 8016–8036 CrossRef CAS PubMed; (b) C.-Y. Huang and A. G. Doyle, The Chemistry of Transition Metals with Three-Membered Ring Heterocycles, Chem. Rev., 2014, 114, 8153–8198 CrossRef CAS PubMed.
- (a) E. M. Mumford, B. N. Hemric and S. E. Denmark, Catalytic, Enantioselective Syn-Oxyamination of Alkenes, J. Am. Chem. Soc., 2021, 143, 13408–13417 CrossRef CAS PubMed; (b) G. Yin, X. Mu and G. Liu, Palladium(II)-Catalyzed Oxidative Difunctionalization of Alkenes: Bond Forming at a High-Valent Palladium Center, Acc. Chem. Res., 2016, 49, 2413–2423 CrossRef CAS PubMed; (c) S. Dong, X. Liu, Y. Zhu, P. He, L. Lin and X. Feng, Organocatalytic Oxyamination of Azlactones: Kinetic Resolution of Oxaziridines and Asymmetric Synthesis of Oxazolin-4-ones, J. Am. Chem. Soc., 2013, 135, 10026–10029 CrossRef CAS PubMed; (d) M. C. Paderes, J. B. Keister and S. R. Chemler, Mechanistic Analysis and Optimization of the Copper-Catalyzed Enantioselective Intramolecular Alkene Aminooxygenation, J. Org. Chem., 2013, 78, 506–515 CrossRef CAS PubMed; (e) D. J. Michaelis, C. J. Shaffer and T. P. Yoon, Copper(II)-Catalyzed Aminohydroxylation of Olefins, J. Am. Chem. Soc., 2007, 129, 1866–1867 CrossRef CAS PubMed.
- (a) K. S. Williamson and T. P. Yoon, Iron Catalyzed Asymmetric Oxyamination of Olefins, J. Am. Chem. Soc., 2012, 134, 12370–12373 CrossRef CAS PubMed; (b) D. J. Michaelis, M. A. Ischay and T. P. Yoon, Activation of N-Sulfonyl Oxaziridines Using Copper(II) Catalysis: Aminohydroxylation of Styrenes and 1,3-Dienes, J. Am. Chem. Soc., 2008, 130, 6610–6615 CrossRef CAS PubMed; (c) K. M. Partridge, M. E. Anzovino and T. P. Yoon, Cycloadditions, of N-Sulfonyl Nitrones Generated by Lewis Acid Catalyzed Rearrangement of Oxaziridines, J. Am. Chem. Soc., 2008, 130, 2920–2921 CrossRef CAS PubMed.
- K. S. Williamson and T. P. Yoon, Iron-Catalyzed Aminohydroxylation of Olefins, J. Am. Chem. Soc., 2010, 132, 4570–4571 CrossRef CAS PubMed.
- (a) M. Nishimura, S. Minakata, T. Takahashi, Y. Oderaotoshi and M. Komatsu, Asymmetric N1 Unit Transfer to Olefins with a Chiral Nitridomanganese Complex: Novel Stereoselective Pathways to Aziridines and Oxazolines, J. Org. Chem., 2002, 67, 2101–2110 CrossRef CAS PubMed; (b) S. Hajra, S. M. S. Akhtar and S. M. Aziz, Catalytic Asymmetric Aminolactonization of 1,2-Disubstituted Alkenoic Acid Esters, Chem. Commun., 2014, 50, 6913–6916 RSC; (c) R. Zhu and S. L. Buchwald, Copper-Catalyzed Oxytrifluoromethylation of Unactivated Alkenes, J. Am. Chem. Soc., 2015, 137, 8069–8077 CrossRef CAS PubMed; (d) H.-C. Shen, Y.-F. Wu, Y. Zhang, L.-F. Fan, Z.-Y. Han and L.-Z. Gong, Palladium-Catalyzed Asymmetric Aminohydroxylation of 1,3-Dienes, Angew. Chem., Int. Ed., 2018, 57, 2372–2376 CrossRef CAS PubMed; (e) S. Kim, D. Kim, S. Y. Hong and S. Chang, Tuning Orbital Symmetry of Iridium Nitrenoid Enables Catalytic Diastereo- and Enantioselective Alkene Difunctionalization, J. Am. Chem. Soc., 2021, 143, 3993–4004 CrossRef CAS PubMed.
- N. L. Reed, M. I. Herman, V. P. Miltchev and T. P. Yoon, Photocatalytic Oxyamination of Alkenes: Copper(II) Salts as Terminal Oxidants in Photoredox Catalysis, Org. Lett., 2018, 20, 7345–7350 CrossRef CAS PubMed.
- D. A. Muccigrosso, S. E. Jacobson, P. A. Apgar and F. Mares, Group 6 Metallooxaziridines: Preparation, Characterization, and Reaction with Cyclohexanone, J. Am. Chem. Soc., 1978, 100, 7063–7065 CrossRef CAS.
- J. A. Connor and E. A. V. Ebsworth, Peroxy Compounds of Transition Metals, Adv. Inorg. Chem. Radiochem., 1964, 6, 279–381 CrossRef CAS.
- A. Pinarci, N. Daniecki, T. M. TenHoeve, B. Dellosso, R. Madiu, L. Mejia, S. E. Bektas and G. Moura-Letts, Synthesis of N-Tosylaziridines from Substituted Alkenes via Zirconooxaziridine Catalysis, Chem. Commun., 2022, 58, 4909–4912 RSC.
- (a) L. S. Liebeskind, K. B. Sharpless, R. D. Wilson and J. A. Ibers, The First d0 Metallooxaziridines. Amination of Olefins, J. Am. Chem. Soc., 1978, 100, 7061–7063 CrossRef CAS; (b) S. K. Dutta, D. B. McConville, W. J. Younfs and M. Chaudhury, Reactivity of Mo-Ot Terminal Bonds toward Substrates having Simultaneous Proton- and Electron-Donor Properties, Inorg. Chem., 1997, 36, 2517–2522 CrossRef CAS.
- (a) P. Das, D. W. Almond, L. N. Tumbelty, B. E. Austin and G. Moura-Letts, From Heterocycles to Carbacycles: Synthesis of Carbocyclic Nucleoside Analogues from Enals and Hydroxylamines, Org. Lett., 2020, 22, 5491 CrossRef CAS PubMed; (b) G. J. Haun, A. N. Paneque, D. Almond, B. E. Austin and G. Moura-Letts, Synthesis of Chromenoisoxazolidines from Substituted Salicylic Nitrones via Visible-Light Photocatalysis, Org. Lett., 2019, 21(5), 1388 CrossRef CAS PubMed; (c) D. J. Quinn, L. N. Tumbelty, E. M. Moscarello, A. N. Paneque, A. H. Zinsky, M. Russ, G. J. Haun and G. M. Moura-Letts, One-Pot Synthesis of Vinylisoxazolidines from Simple Hydroxylamines and Conjugated Carbonyls, Tetrahedron Lett., 2017, 58(50), 4682–4686 CrossRef CAS; (d) P. Trieu, W. H. Filkin, A. Pinarci, E. M. Tobias, R. Madiu, B. Dellosso, J. Roldan, P. Das, B. E. Austimn and G. Moura-Letts, Synthesis, of Isoxazolidines from Substituted Vinylnitrones andConjugated Carbonyls via Visible-Light Photocatalysis, ChemPhotoChem, 2023, 3, e202200277 CrossRef.
- D. J. Michaelis, K. S. Williamson and T. P. Yoon, Oxaziridine-Mediated Enantioselective Aminohydroxylation of Styrenes Catalyzed by Copper(II) Bis(oxazoline) Complexes, Tetrahedron, 2009, 65, 5118–5124 CrossRef CAS PubMed.
- D. A. Rogers, A. T. Bensalah, A. T. Espinosa, J. L. Hoerr, F. H. Refai, A. K. Pitzel, J. J. Alvarado and A. A. Lamar, Amplification of Trichloroisocyanuric Acid (TCCA) Reactivity for Chlorination of Arenes and Heteroarenes via Catalytic Organic Dye Activation, Org. Lett., 2019, 21, 4229–4233 CrossRef CAS PubMed . Sulfonamides and TCCA (trichloroisocyanuric acid) produce Ts-chloramines with full conversion at 0 °C.
-
General protocol for synthesis of aminoalcohol
3
: In a 16 mL vial packed with a magnetic stirrer, sulfonamide (0.75 mmol, 1.5 equiv.) and of TTCA (0.25 mmol, 0.5 equiv.) were mixed in CH2Cl2 (4 mL) at 0 °C for 30 min. The solvent was then removed under vacuum and the residue was reconstituted with CH3CN/H2O (4 mL, 10:1) and then WO2Dipic(H2O) (0.005 mmol, 1 mol%), TBAI (0.0375 mmol, 7.5 mol%) and alkene (0.5 mmol, 1 equiv.) were added and the resulting mixture was allowed to stir for 16 h or until disappearance of alkene by TLC. The crude was then filtered by a 1:1 silica gel/celite pad and the resulting crude was then purified by silica gel chromatography to provide the corresponding aminoalcohol.
- P. Muller and C. Fruit, Enantioselective Catalytic Aziridination and Asymmetric Nitrene Insertions into CH Bonds, Chem. Rev., 2003, 103, 2905–2920 CrossRef PubMed.
- The relative stereochemistry for aminoalcohols 3c–3i was determined by NMR analysis of the coupling constants between the newly formed stereocenters.
- K. Yamaguchi, H. Isober, M. Shoji, T. Kawakami and K. Miyagawa, The Nature of the Chemical Bonds of High-Valent Transition-Metal Oxo (M=O) and Peroxo (MOO) Compounds: A Historical Perspective of the Metal Oxyl-Radical Character by the Classical to Quantum Computations, Molecules, 2023, 7119 CrossRef CAS PubMed . Review on stereospecificity of biradical forming reactions.
- Efforts to duplicate the reaction efficiencies under stoichiometric conditions provided lower yields, but reactions in CDCl3 at one equiv. of 1 displayed the characteristic resonances for the Ts group in 1.
- H. Sugimoto, A. Mikami, K. Kai, P. K. Sajith, Y. Shiota, K. Yoshizawa, K. Asano, T. Suzuki and S. Itoh, Cis-1,2-Aminohydroxylation of Alkenes Involving a Catalytic Cycle of Osmium(III) and Osmium(V) Centers: Os(O)(NHTs) Active Oxidant with a Macrocyclic Tetradentate Ligand, Inorg. Chem., 2015, 54, 7073–7082 CrossRef CAS PubMed . Intermediate A was measured by MS and 1H-NMR in a stoichiometric reaction in the absence of water.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization data for tungstenooxaziridine and known and unknown aminoalcohols is included. See DOI: https://doi.org/10.1039/d4ob00022f |
| This journal is © The Royal Society of Chemistry 2024 |