
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Identification of BACE-1 inhibitors through directed C(sp3)–H activation on 5-oxo-pyrrolidine-3-carboxylic acid derivatives†
Lorenzo
Baldini
 ,
Elena
Lenci
,
Cristina
Faggi
and
Andrea
Trabocchi
*
,
Elena
Lenci
,
Cristina
Faggi
and
Andrea
Trabocchi
*
Department of Chemistry “Ugo Schiff”, University of Florence, Via della Lastruccia 13, 50019 Sesto Fiorentino, Florence, Italy. E-mail: andrea.trabocchi@unifi.it
First published on 8th March 2024
Abstract
Convenient synthesis of stereochemically dense 5-oxo-pyrrolidines was obtained from succinic anyhdride and imines by combining the Castagnoli–Cushman reaction with directed Pd-catalyzed C(sp3)–H functionalization, taking advantage of the developing carboxylic group properly derivatized with 8-aminoquinoline as a directing group. These fully substituted 5-oxopyrrolidines were found to be able to inhibit BACE-1 enzyme with sub-micromolar activity, thanks to the interaction of the key aryl appendage introduced by C(sp3)–H activation within BACE-1 S2′ subsite.
Introduction
A convenient synthetic approach for fragment elaboration and decoration of a scaffold in fragment-based approaches1–4 is the site-selective formation of bonds at all growing points of a fragment5 taking advantage of C–H functionalization strategies.6 The site-selective C(sp3)–H functionalisation to form stereocenters is of high value for adding functionalized chemical moieties,7 and in particular late stage aromatic C(sp3)–H functionalization8 can be used to introduce elements that can assist the molecules in passing the BBB.9–12 Among the many different C(sp3)–H arylation methods, Pd-catalyzed C–H activation reactions employing a Directing Group (DG)13 are often used for their high site selectivity, being able to discriminate a particular C–H bond, using distance as the distinctive parameter.14 This strategy represents an optimal tool for the straightforward elaboration of molecular scaffolds and it has been applied to several different N-containing heterocycles.15–18Among the many different N-containing heterocycles, 5-oxopyrrolidines is a privileged structure in medicinal chemistry.19,20 In particular, fully substituted 5-oxopyrrolidines bearing a carbonyl moiety in position 3 are found in many bioactive molecules and natural products involved in the treatment of CNS diseases. Just to give some examples, different Endothelin Receptor Antagonists21 and Glutaminyl-Peptide CycloTransferase-Like (QPCTL) inhibitors,22 used for the treatment of Alzheimer's Disease, contain a fully substituted 5-oxopyrrolidines. Also, this scaffold is present in heliotropamide A and bisavenanthramide B-6 natural products used in traditional Chinese medicine,23 for their acetylcholinesterase inhibitory activity and their neuroprotective effects (Fig. 1).24
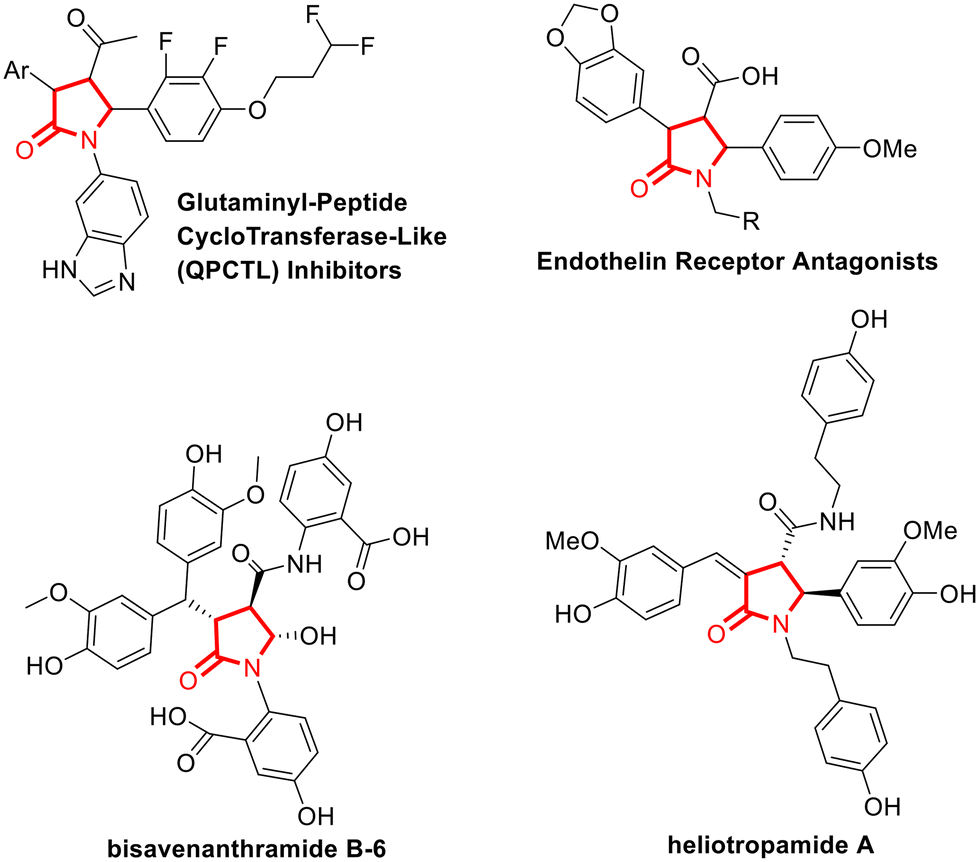 | ||
| Fig. 1 Representative examples of bioactive compounds (top) and natural products (bootom) used in the treatment of CNS diseases, that contains fully substituted 5-oxopyrrolidines bearing a carbonyl moiety in position 3. | ||
Several methods have been developed for the preparation of densely functionalized 5-oxopyrrolidines, including cyclization, [3 + 2] or [4 + 1] cycloaddition,25 cascade reactions, or related transformations.20,26 However, to the best of our knowledge, short and convenient synthesis of fully substituted 5-oxo-pyrrolidines, containing appendages in each available positions and bearing a carbonyl moiety in position 3 were not reported in the literature. In our previous synthetic efforts in the field of CNS drug discovery,27–29 we reported the exploitation of the Castagnoli–Cushman reaction (CCR) to generate C-2 substituted morpholinone scaffolds as BACE-1 inhibitors.28 The CCR is a multicomponent reaction that is capable to give functionalized N-heterocycles depending on the building blocks employed.30–32 In this work, we reasoned to expand the chemical complexity of 5-oxo-pyrrolidine-3-carboxylic acid derivatives resulting from the CCR reaction with a directed C(sp3)–H functionalization to obtain fully substituted 5-oxopyrrolidines. The synthesized compounds were then analyzed for their drug-like properties, considering the range of optimal parameters for CNS drugs, as defined by Wager and coworkers,33 and evaluated for their inhibition activity against different enzymes involved in CNS diseases. In particular, we performed a screening on representative enzymes involved in Alzheimer's Disease (AD), namely BACE-1, an aspartic protease involved in the amyloidogenic pathway by cleaving amyloid precursor protein (APP),34–36 and legumain, also called δ-secretase, a cysteine protease responsible for tau cleaving in AD.37
Results and discussion
The directed C(sp3)–H functionalization of 5-oxo-pyrrolidine-3-carboxamide was developed by taking advantage of the COOH handle resulting from the CCR reaction to install a suitable directing group for Pd-catalysis. In particular, 8-aminoquinoline (8-AQ) was chosen among various directing groups for its capability of chelating the transition metal catalyst and bringing it close to the β-C(sp3)–H bond, thus resulting in the use of distance as chemo- and stereoselective parameters for functionalizing the desired C–H bond. To study the versatility of this novel C(sp3)–H functionalization method, we synthesized 5-oxo-2-phenylpyrrolidine-3-carboxylic acid 2 as a representative substrate of the C–H arylation reaction. Specifically, 5-oxopyrrolidine 2 was obtained in high yield from N-benzylidene-2-methylpropan-2-amine (1) and succinic anhydride, following the reported protocols (Scheme 1).38,39 | ||
| Scheme 1 Synthesis of 8-aminoquinoline-functionalized 5-oxo-2-phenylpyrrolidine-3-carboxamide 3. | ||
Then, the carboxylic acid function resulting from the Castagnoli–Cushman reaction was exploited for inserting the 8-AQ as a directing group through amide coupling employing HOBt and DIC in anhydrous CH2Cl2, to give compound 3 in 67% yield (Scheme 1). As no examples of 8-aminoquinoline-promoted C(sp3)–H arylation of 5-oxopyrrolidine-3-carboxylic acid derivatives are reported in the literature, solvent, substrate concentration and reaction temperature were studied in order to find out best reaction conditions. As reported in Table 1, best conditions for the C–H bond activation step were found to be the employment of 3 equivalents of aryl iodide and 0.2 equivalents of (BnO)2PO2H, heating at 110 °C for 21 h in anhydrous 1,4-dioxane at 0.2 M concentration. The increase of the concentration to 0.9 M and the reduction of reaction temperature to 80 °C did not improve the reaction conversion.
|
|
|||
|---|---|---|---|
| Entry | Solvent (conc.) | Temp., °C | Yield, % |
| a NMR conversion. | |||
| 1 | Toluene (0.05 M) | 110 | 14 |
| 2 | t-AmylOH (0.05 M) | 110 | 15a |
| 3 | DCE (1 M) | 110 | 36a |
| 4 | HFIP (0.2 M) | 110 | 68 |
| 5 | TFE (0.02 M) | 110 | 59 |
| 6 | 1,4-Dioxane (0.2 M) | 110 | 95 |
| 7 | t-BuOH (0.2 M) | 110 | 29a |
| 8 | 1,4-Dioxane (0.2 M) | 80 | 14a |
| 9 | 1,4-Dioxane (0.9 M) | 80 | 10a |
| 10 | 1,4-Dioxane (0.9 M) | 110 | 73a |
The optimized conditions were applied for conducting a substrate scope study, producing with good to excellent yields 2,4-diaryl-5-oxo-pyrrolidines 4a–j (Scheme 2), with the rare trans 2,4-diaryl substitution pattern. The reaction proved to possess a broad scope for aryl iodide with a high functional group tolerance in the presence of aldehyde or nitro groups. For compound 4g, an X-ray crystallographic analysis was carried out to confirm the relative configuration of the substituents at the scaffold (Scheme 2), indicating the trans 2,3- and cis 3,4-configuration for the major isomer formed during the directed C(sp3)–H bond activation step. Also, gCOSY of 4g allowed to assign the 1H NMR peaks to each proton (Fig. 2).
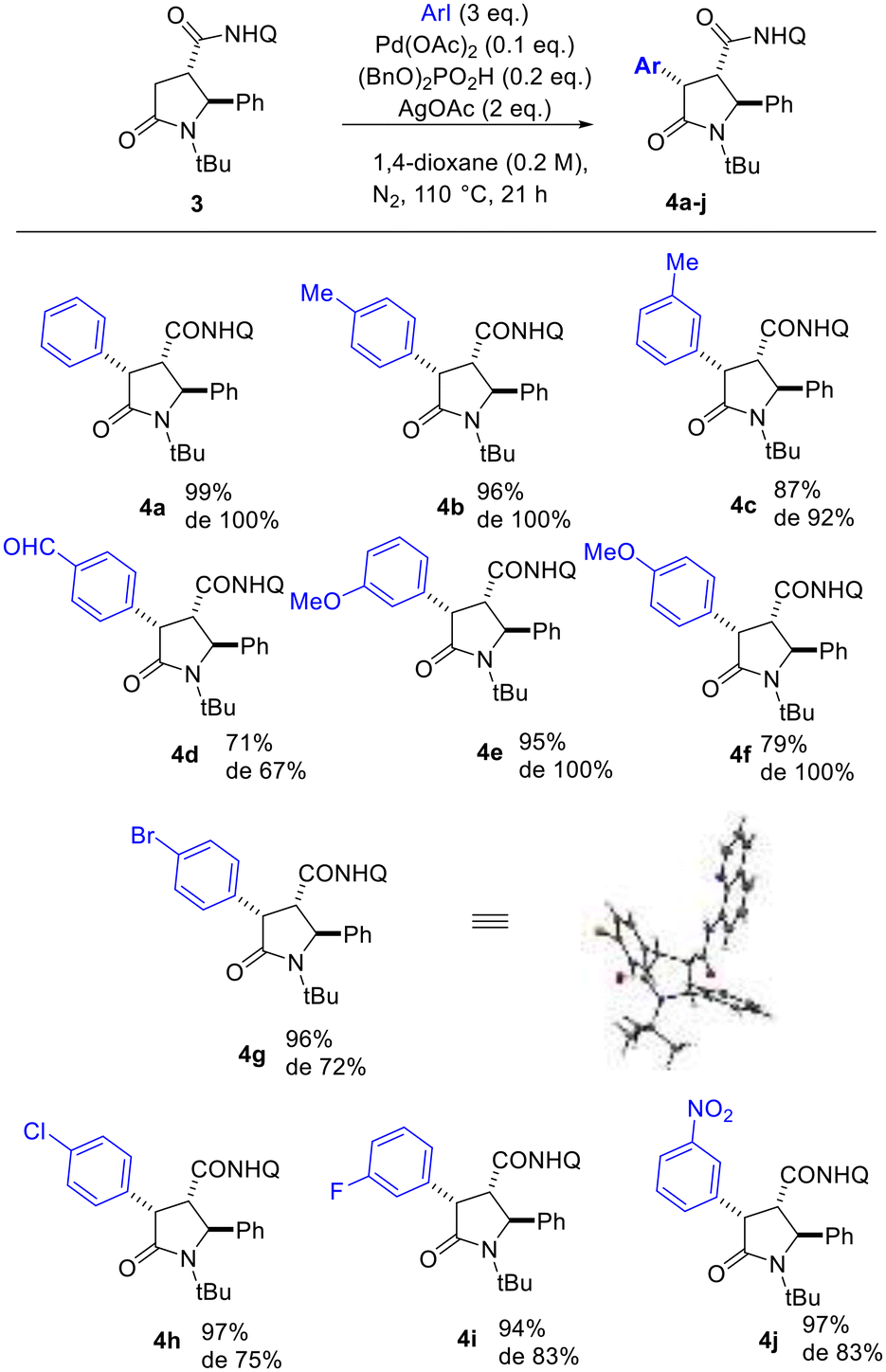 | ||
| Scheme 2 Scope of directed C(sp3)–H functionalization of 5-oxo-2-phenylpyrrolidine-3-carboxamide 3. Diastereoselectivity ratio (de) was determined by the two sets of peaks in the 1H NMR spectrum of the crude mixture attributed to the hydrogen atoms 2-H, 3-H and 4-H of the heterocyclic ring. The X-Ray crystallographic structure of 4g is reported in the Scheme (CCDC: 2165903†). | ||
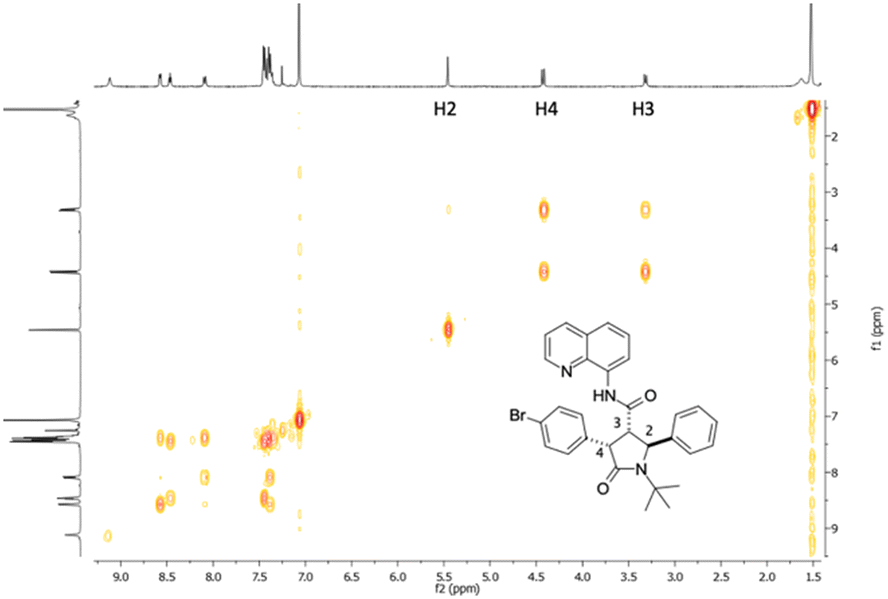 | ||
| Fig. 2 gCOSY NMR plot of compound 4g. | ||
In particular, 4-H was found significantly deshielded in C(sp3)–H functionalized 5-oxo-2-phenylpyrrolidines 4a–j (4.4 ppm with respect to 3.06 ppm found for 3), whereas 3-H and 2-H protons did not shift considerably (3.06 and 5.35 ppm, respectively for compound 3, and around 5.5 and 3.3 ppm for compounds 4a–j).
The diastereoselectivity ratio was determined by 1H NMR, specifically evaluating the 2-H signal appearing as a singlet for the major isomer and as an upfield doublet for the minor one (see Fig. 3 for 4j), resulting in good to high de values depending on the installed aryl moiety (Scheme 2). To give an insight to the conversion of the C(sp3)–H bond functionalization reaction and the epimerization degree at C-3 at different times of the reaction mixture, we carried out a kinetic study by NMR employing 1-iodo-3-nitrobenzene (Fig. 3) and 4-iodobenzaldehyde as aryl iodides (see ESI, Fig. S1 and S2†). The reaction temperature was kept fixed at 110 °C to allow for the reaction to proceed to completion (Table 1), and at given times (1, 2, 4, 8, 16, 21 hours) aliquots were taken from the reaction mixture and analyzed by 1H NMR to assess % conversion and epimerization. The results indicated reaction completion at 16 to 21 h and epimerization starting after 4 h reacting, although not increasing significantly during the reaction time.
 | ||
| Fig. 3 Kinetic study by NMR of the C(sp3)–H functionalization for the synthesis of 4j. The expansions of 1H NMR spectra recorded at different times (1, 2, 4, 6, 8, 16, 21 h) of the reaction mixture, employing 1-iodo-3-nitrobenzene as aryl iodide, highlight the reaction conversion and the epimerization degree at the NMR signals of 2-H proton. | ||
The proposed mechanism for the diastereoselective β-C(sp3)–H bond activation reaction involves the coordination of the Pd by the directing group via ligand exchange to form the intermediate species II, followed by insertion to generate the palladacycle III (Fig. 4). The oxidative addition of the aryl species in IV enables the following C–H functionalization to form the arylated specie V and subsequent reductive elimination, resulting to the release of Pd catalyst I and to the diastereostereoselective attachment of the aryl group with the relative cis configuration with respect to the amide bond bearing the DG.
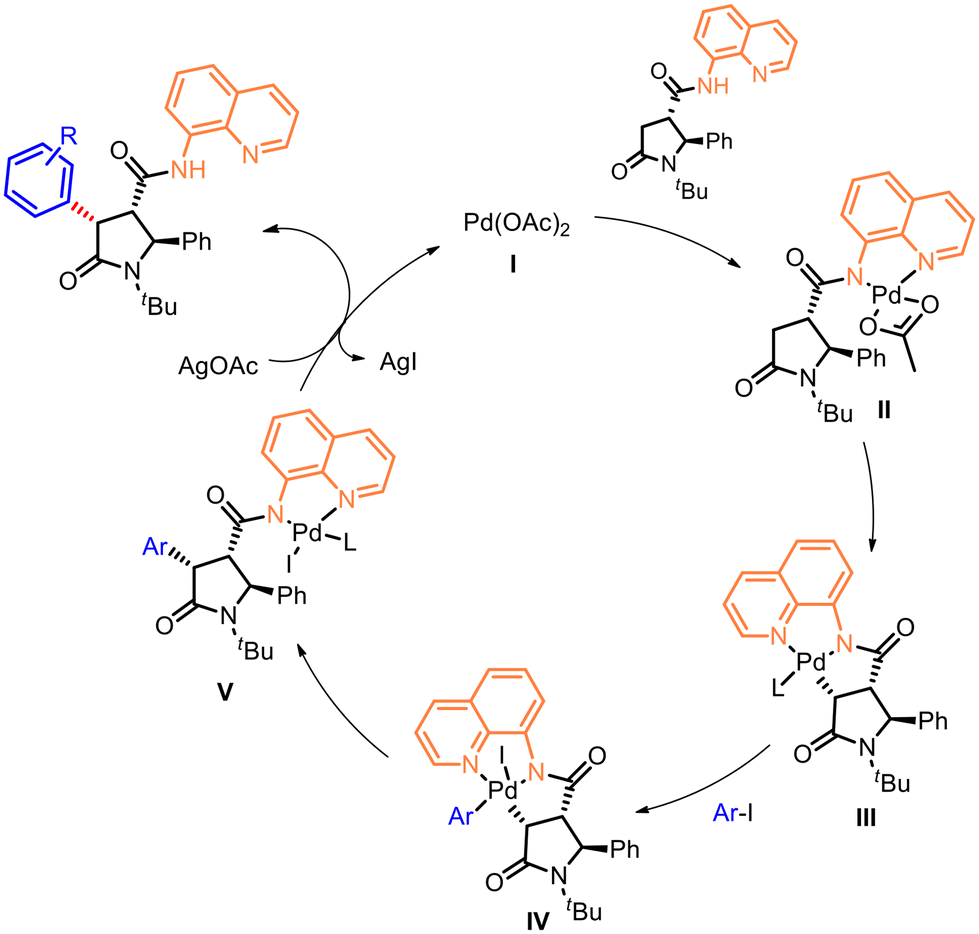 | ||
| Fig. 4 Proposed mechanism for the directed C–H aryl functionalization at C-4 of 5-oxopyrrolidines. | ||
Finally, a representative directing group removal was carried out under standard H2O2/LiOH conditions to allow for potential additional functionalization of the carboxylic group of the 5-oxopyrrolidines. Thus, the 8-AQ directing group was removed from compounds 4e and 4g through a two step-procedure, which foresees the Boc protection of the nitrogen atom of the carboxamide group, followed by oxidative cleavage employing H2O2 and LiOH (Scheme 3). Such procedure allowed to obtain the corresponding carboxylic acids 5a and 5b with an overall yield of 66 and 76%, respectively.
 | ||
| Scheme 3 Representative two-steps removal of 8-AQ directing groups. Reagents and conditions: (a) (Boc)2O, DMAP, MeCN, 25 to 50 °C, 2 h, then r.t., 18 h; (b) H2O2, LiOH, 0 °C, 20 min, then 25 °C, 18 h. | ||
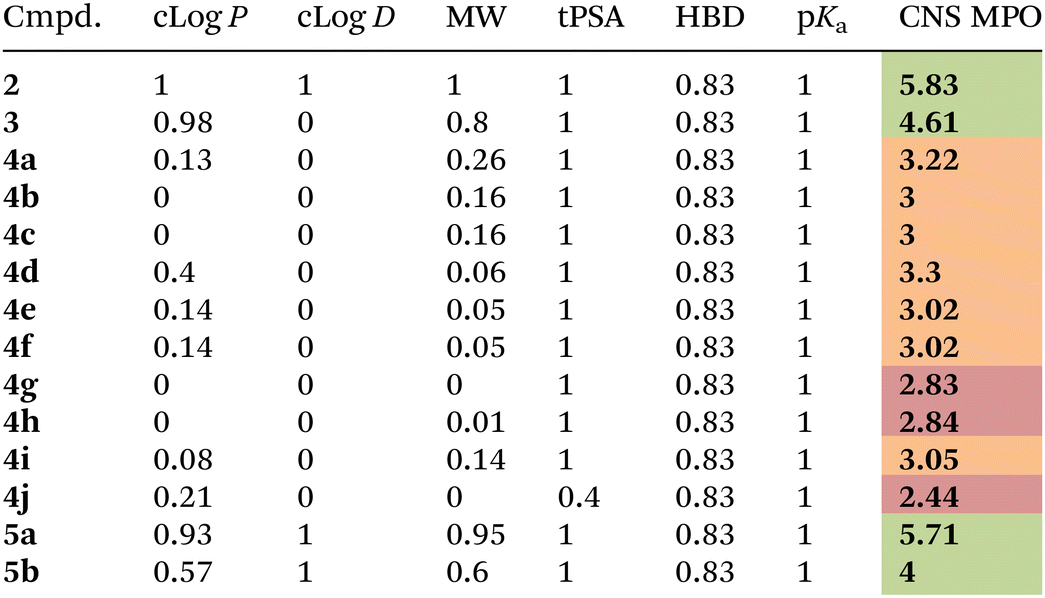
Compounds 4a–j and their corresponding unsubstituted precursors 3 and 2 were assessed for their physicochemical properties in order to establish their valence as potential compounds for central nervous system drug discovery. As reported by Wager and coworkers,33 a strict relationship between several physicochemical properties and favorable in vitro attributes for the CNS drugs (including high permeability, low P-gp efflux liability, low metabolic clearance, and high safety) is observed. In particular, the CNS MPO (CNS MultiParameter Optimization) method provides a single score (spanning from 0 to 6), that weights and condenses the penalty of six different physicochemical properties that are not in the desirable range. Particularly, the desirable properties for a drug to be able to cross the blood brain barrier (BBB) are: (a) calculated partition coefficient (clog![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P) less than 3; (b) distribution coefficient at pH = 7.4 (clogD) less than 2; (c) molecular weight (MW) less than 360 Da; (d) topological polar surface area (tPSA) between 40 and 90; (e) number of hydrogen bond donors (HBD) less than 0.5; (f) pKa less than 8. Thus, cLogP, tPSA, HBD and MW values were calculated using the web-based public tool SwissADME,40 whereas pKa and cLogD values were calculated with ACD Labs Software v 6.041 (Table 2). Then, the CNS MPO method was applied to measure penalty values for each physicochemical parameter (see ESI†) and the overall CNS MPO score was found. As shown in Table 2, the good scores (higher > 4) observed for the final compounds 5a–5b confirmed that the C–H arylated method did not alter the desirable physicochemical properties required for the CNS drugs and revealed the need of removing the 8-AQ directing group, as the corresponding compounds 4e and 4g possess CNS scores around 3, with cLogD, cLogP and MW values outside the desirable ranges. To demonstrate the utility of these fragments in CNS drug discovery, we performed a screening of this library on representative enzymes involved in neurodegenerative diseases, in particular BACE-1 and legumain. While no active compounds were found towards legumain, two molecules (4g and 5b) showed >50% inhibition towards BACE-1 at 10 μM concentration.
P) less than 3; (b) distribution coefficient at pH = 7.4 (clogD) less than 2; (c) molecular weight (MW) less than 360 Da; (d) topological polar surface area (tPSA) between 40 and 90; (e) number of hydrogen bond donors (HBD) less than 0.5; (f) pKa less than 8. Thus, cLogP, tPSA, HBD and MW values were calculated using the web-based public tool SwissADME,40 whereas pKa and cLogD values were calculated with ACD Labs Software v 6.041 (Table 2). Then, the CNS MPO method was applied to measure penalty values for each physicochemical parameter (see ESI†) and the overall CNS MPO score was found. As shown in Table 2, the good scores (higher > 4) observed for the final compounds 5a–5b confirmed that the C–H arylated method did not alter the desirable physicochemical properties required for the CNS drugs and revealed the need of removing the 8-AQ directing group, as the corresponding compounds 4e and 4g possess CNS scores around 3, with cLogD, cLogP and MW values outside the desirable ranges. To demonstrate the utility of these fragments in CNS drug discovery, we performed a screening of this library on representative enzymes involved in neurodegenerative diseases, in particular BACE-1 and legumain. While no active compounds were found towards legumain, two molecules (4g and 5b) showed >50% inhibition towards BACE-1 at 10 μM concentration.
|
A dose–response measurement for 4g and 5b in the range 0.01–100 μM of inhibitor concentration was performed, confirming the inhibition profile of the compounds with an IC50 value of 340 nM and 853 nM, respectively (Fig. 5, top and Fig. S3 at ESI†). These compounds both contain the newly installed 4-Br-phenyl moiety, suggesting the importance of this moiety as a new vector interacting with the enzyme, as the corresponding parent compounds 2 and 3 were found inactive. This result was confirmed by molecular docking studies carried out with Autodock 4.0 and low energy ligand–protein AMMP energy minimization, where an interaction between the 4-bromophenyl moiety of most active compound 4g and Tyr198 of hydrophobic S2′ subsite in the catalytic cleft of BACE-1 enzyme was observed (Fig. 5, bottom).
 | ||
| Fig. 5 Top: hit compounds identified as BACE-1 inhibitors (4g and 5b) in comparison with the parent corresponding 5-oxo-pyrrolidines 2 and 3. Bottom: (2S,3S,4S)-4g docked into the active site of BACE1 (PDB: 5CLM), highlighting protein residues (purple) that form key interactions: Gly230 vs. 8-AQ, Tyr198 vs. 4-Br-phenyl ring. Nonpolar hydrogen atoms are omitted for clarity. | ||
Conclusion
In conclusion, in view of developing a short synthesis for the generation and growth of sp3-rich fragments, we set up a straightforward synthetic pathway which combines the use of a DOS-compatible reaction like CCR with the 8-AQ directed Pd-catalyzed β-C(sp3)–H bond activation reaction. This protocol allows for the generation of densely functionalized 5-oxopyrrolidines possessing three stereocenters. The reaction conditions of the C–H arylation were optimized and the generality of the process was demonstrated for a range of aryl iodides bearing different substituents, resulting in high yields and good to high diastereoselectivity. Preliminary enzyme inhibition assays of newly synthesised compounds towards BACE-1 allowed to identify two molecules both bearing the key 4-Br-phenyl moiety required for inhibition potency at submicromolar concentration, resulting in the identification of new hit compounds useful for subsequent elaboration of such new chemical entities as BACE-1 inhibitors. Finding potent ligands for BACE-1 is still a challenging task in drug discovery, particularly for the requirement of potent inhibitors of this enzyme while passing efficiently the blood–brain barrier (BBB), so the identification of a new moiety capable of interacting strongly within the catalytic cleft of the enzyme is a powerful tool for fragment-based approaches, which have been particularly useful for BACE-1 drug discovery,5–7 followed by optimization of such fragment hits using fragment-linking.42,43Experimental
General
Analytical grade solvents and commercially available reagents were used without further purification. Glutaric acid was recrystallized from CH2Cl2/Et2O. Reactions requiring an inert atmosphere were carried out under a nitrogen atmosphere. CH2Cl2, 1,2-DCE, and 1,4-dioxane were freshly distilled over CaH2. THF, toluene, and xylene were dried by distillation over Na/benzophenone. tBuOH was dried over Na2SO4. 1H-NMR and 13C-NMR spectra were recorded on Varian Mercury 400 (1H: 400 MHz, 13C: 100 MHz), Varian Inova (1H: 400 MHz, 13C: 100 MHz) or Varian Mercury 200 (1H: 200 MHz, 13C: 50 MHz) spectrometer. The chemical shifts (δ) and coupling constants (J) are expressed in parts per million (ppm) and hertz (Hz), respectively. Flash column chromatography (FCC) purifications were performed manually using glass columns with Merck silica gel (40–63 μm) or automatically using the Biotage Isolera system and SNAP silica cartridges. TLC analyses were performed on Merck silica gel 60 F254 plates. Elemental analyses were performed on a Thermoscientific FlashSmart Elemental Analyzer. ESI-MS spectra were recorded on a Thermo Scientific LCQ fleet ion-trap double quadrupole mass spectrometer using electrospray (ES+) ionization techniques. A VWR® Ultrasonic cleaner (frequency 45 kHz, power 80 W) was used for reactions conducted under ultrasound irradiation.N-Benzylidene-2-methylpropan-2-amine (1)
Following the procedure reported by Stefani et al.,38 in a dry flask, tert-butyl amine (116 mg, 1.59 mmol) and benzaldehyde (168 mg, 1.59 mmol) were dissolved in absolute EtOH (2.65 mL), and silica (477 mg) was added. The mixture was reacted in an inert atmosphere under ultrasound irradiation at room temperature for 2 h. Silica was filtered, and the solvent was removed under reduced pressure to provide the title compound 1, that was used without further purification (256 mg, quantitative yield). 1H NMR (200 MHz, CDCl3) δ 8.29 (s, 1H, HC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 7.76 (dd, J = 6.6, 3.0 Hz, 2H, HCAr), 7.41 (m 3H, HCAr), 1.31 (s, 9H, C(CH3)3). 13C NMR (50 MHz, CDCl3) δ 154.1 (CN), 129.1 (CAr), 127.5 (2C, CAr), 126.9 (2C, CAr), 56.2 (C(CH3)3), 28.7 (3C, CH3).
N), 7.76 (dd, J = 6.6, 3.0 Hz, 2H, HCAr), 7.41 (m 3H, HCAr), 1.31 (s, 9H, C(CH3)3). 13C NMR (50 MHz, CDCl3) δ 154.1 (CN), 129.1 (CAr), 127.5 (2C, CAr), 126.9 (2C, CAr), 56.2 (C(CH3)3), 28.7 (3C, CH3).
(2S*,3S*)-1-(tert-Butyl)-5-oxo-2-phenylpyrrolidine-3-carboxylic acid (2)
In a dry round bottom flask charged with a magnetic stir bar, imine 1 (1.85 g, 11.5 mmol, 1 eq.) was dissolved in 12 mL of dry xylene (1 M), succinic anhydride (1.15 g, 1 eq.) was added and the reaction mixture was refluxed overnight. The reaction mixture was then cooled to room temperature and the solvent was removed under reduced pressure. The oily crude was dissolved in a mixture of Et2O and a saturated aqueous solution of NaHCO3, and the two phases were separated. The aqueous phase was then acidified to pH = 1 and extracted three times with CH2Cl2. The organic phase was collected, washed with Brine, and concentrated in vacuo. Pure 2 was obtained in 83% yield (2.5 g, 9.57 mmol) after flash chromatography (EtOAc/n-hexane (2:1), Rf = 0.05). 1H NMR (400 MHz, CDCl3) δ 10.84 (s, 1H, CO2H), 7.40–7.25 (m, 5H, HCAr), 5.29 (s, 1H, HC2), 2.95 (dd, J = 17.1, 9.2 Hz, 1H, HC4), 2.78 (dd, J = 21.7, 13.4 Hz, 2H, HC3 + H′C4), 1.34 (s, 9H, CH3). 13C NMR (100 MHz, CDCl3) δ 176.2 (COOH), 174.9 (NCO), 142.3 (CAr), 129.1 (CAr), 128.0 (CAr), 125.3 (2C, CAr), 64.7 (C(CH3)3), 55.7 (C2), 46.0 (C3), 33.8 (C4), 27.9 (3C, CH3). MS (ESI) m/z (%) = 260.10 (100, [M − H]−). Anal. calcd for C15H19NO3: C, 68.94; H, 7.33; N, 5.36. Found: C, 69.08; H, 7.36; N, 5.33.
(2S*,3S*)-1-(tert-Butyl)-5-oxo-2-phenyl-N-(quinolin-8-yl)pyrrolidine-3-carboxamide (3)
Compound 2 (2.31 g, 8.86 mmol, 1 eq.), HOBt·H2O (1.44 g, 10.6 mmol, 1.2 eq.) and 44 mL of dry CH2Cl2 (0.2 M) were added to a dry round bottom and stirred for 5 minutes under inert atmosphere. Then, DIC (1.34 g, 10.6 mmol, 1.2 eq.) was added, and the reaction mixture was stirred for other 5 minutes before adding 8-aminoquinoline (1.41 g, 9.75 mmol, 1.1 eq.). The mixture was left stirring at room temperature for 72 h under inert atmosphere, then it was diluted with CH2Cl2, and a saturated aqueous NaHCO3 solution was added and extracted three times with CH2Cl2. The organic phase was dried over Na2SO4, and the solvent was removed under reduced pressure. The crude product was chromatographed (Et2O/n-hexane (2:1), Rf = 0.29) to provide pure 3 (2.3 g, 67% yield). 1H NMR (400 MHz, CDCl3) δ 9.85 (s, 1H, OCNH), 8.80–8.68 (m, 2H, HCAr), 8.15 (dd, J = 8.3, 1.6 Hz, 1H, HCAr), 7.58–7.47 (m, 2H, HCAr), 7.48–7.35 (m, 3H, HCAr), 7.35–7.30 (m, 3H, HCAr), 5.35 (s, 1H, HC2), 3.06 (m, 2H, HC4 + HC3), 2.75 (dd, J = 16.4, 1.5 Hz, 1H, H′C4), 1.40 (s, 9H, CH3). 13C NMR (100 MHz, CDCl3) δ 173.5 (CONH), 171.1 (CONtBu), 148.1 (CAr), 143.6 (CAr), 138.1 (CAr), 136.4 (CAr), 134.0 (CAr), 129.1 (CAr), 127.9 (CAr), 127.8 (CAr), 127.3 (CAr), 125.4 (2C, CAr), 121.9 (CAr), 121.6 (CAr), 116.7 (CAr), 64.7 (C(CH3)3), 55.5 (C2), 48.4 (C3), 41.8 (C4), 28.0 (3C, CH3). MS (ESI) m/z (%) = 388.16 (100, [M + H]+). Anal. calcd for C24H25N3O2: C, 74.39; H, 6.50; N, 10.84. Found: C, 74.51; H, 6.53; N, 10.56.
General procedure for the C(sp3)–H bond arylation reaction
A dry microwave reaction vial was charged with compound 3 (1 eq.), dibenzyl phosphate (0.2 eq.), AgOAc (2 eq.), ArI (3 eq.), Pd(OAc)2 (0.1 eq.), and vacuum/N2 cycles were performed (n.b. When aryl iodides were liquid, they were added immediately after vacuum/N2 cycles). Dry 1,4-dioxane was added, and the reaction mixture was reacted at 110 °C in a pre-heated oil bath under rapid stirring for 21 h under inert atmosphere. After cooling to room temperature, the reaction mixture was diluted with EtOAc, filtered over Celite and concentrated in vacuo. The crude product was purified by FCC (Et2O/n-hexane) to provide the pure arylated product.(2S*,3S*,4S*)-1-(tert-Butyl)-5-oxo-2,4-diphenyl-N-(quinolin-8-yl)pyrrolidine-3-carboxamide (4a)
Compound 4a was obtained following general procedure for the C(sp3)–H bond arylation reaction (60 mg, 99% yield, Et2O/n-hexane (2:1), Rf = 0.31). 1H NMR (400 MHz, CDCl3) δ 9.15 (s, 1H, OCNH), 8.58 (dd, J = 4.1, 1.3 Hz, 1H, HCAr), 8.46 (dd, J = 6.0, 3.0 Hz, 1H, HCAr), 8.07 (d, J = 8.1 Hz, 1H, HCAr), 7.48–7.32 (m, 8H, HCAr), 7.21 (d, J = 7.4 Hz, 2H, HCAr), 6.98 (t, J = 7.7 Hz, 2H, HCAr), 6.78 (t, J = 7.4 Hz, 1H, HCAr), 5.48 (s, 1H, HC2), 4.49 (d, J = 9.5 Hz, 1H, HC4), 3.37 (d, J = 9.4 Hz, 1H, HC3), 1.55 (s, 9H, CH3). 13C NMR (100 MHz, CDCl3) δ 173.4 (CONH), 169.4 (CONtBu), 147.5 (CAr), 143.0 (CAr), 137.8 (CAr), 136.3 (CAr), 135.2 (CAr), 133.5 (CAr), 132.1 (CAr), 129.6 (2C, CAr), 129.2 (2C, CAr), 128.1 (2C, CAr), 127.9 (CAr), 127.9 (CAr), 127.2 (CAr), 126.9 (CAr), 125.7 (2C, CAr), 121.6 (CAr), 121.4 (CAr), 62.2 (C(CH3)3), 56.3 (C2), 56.0 (C4), 50.5 (C3), 28.1 (3C, CH3). MS (ESI) m/z (%) = 486.27 (100, [M + Na]+). Anal. calcd for C30H29N3O2: C, 77.73; H, 6.31; N, 9.06. Found: C, 77.95; H, 6.38; N, 8.99.
(2S*,3S*,4S*)-1-(tert-Butyl)-5-oxo-2-phenyl-N-(quinolin-8-yl)-4-(p-tolyl)pyrrolidine-3-carboxamide (4b)
Compound 4b was obtained following general procedure for the C(sp3)–H bond arylation reaction (59 mg, 96% yield, Et2O/n-hexane (2:1), Rf = 0.42). 1H NMR (400 MHz, CDCl3) δ 9.12 (s, 1H, OCNH), 8.58 (dd, J = 4.2, 1.4 Hz, 1H, HCAr), 8.47 (dd, J = 6.8, 2.1 Hz, 1H, HCAr), 8.08 (d, J = 8.0 Hz, 1H, HCAr), 7.50–7.31 (m, 8H, HCAr), 7.06 (d, J = 8.0 Hz, 2H, HCAr), 6.74 (d, J = 7.9 Hz, 2H, HCAr), 5.48 (s, 1H, HC2), 4.44 (d, J = 9.4 Hz, 1H, HC4), 3.33 (d, J = 9.4 Hz, 1H, HC3), 1.82 (s, 3H, OCH3), 1.54 (s, 9H, CH3). 13C NMR (100 MHz, CDCl3) δ 173.6 (CONH), 169.5 (CONtBu), 147.3 (CAr), 143.0 (CAr), 137.1 (CAr), 136.4 (CAr), 133.6 (CAr), 132.0 (CAr), 131.1 (CAr), 129.4 (2C, CAr), 129.2 (2C, CAr), 128.8 (2C, CAr), 127.8 (CAr), 127.6 (CAr), 127.2 (CAr), 125.7 (2C, CAr), 121.5 (CAr), 121.2 (CAr), 116.6 (CAr), 62.1 (C(CH3)3), 56.3 (C2), 55.9 (C4), 50.1 (C3), 28.1 (3C, CH3), 20.6. MS (ESI) m/z (%) = 500.27 (100, [M + Na]+). Anal. calcd for C31H31N3O2: C, 77.96; H, 6.54; N, 8.80. Found: C, 78.14; H, 6.59; N, 8.73.
(2S*,3S*,4S*)-1-(tert-Butyl)-5-oxo-2-phenyl-N-(quinolin-8-yl)-4-(m-tolyl)pyrrolidine-3-carboxamide (4c)
Compound 4c was obtained following general procedure for the C(sp3)–H bond arylation reaction (53 mg, 87% yield, Et2O/n-hexane (2:1), Rf = 0.41). 1H NMR (400 MHz, CDCl3) δ 9.08 (s, 1H, OCNH), 8.56 (d, J = 3.3 Hz, 1H, HCAr), 8.48 (dd, J = 6.4, 2.3 Hz, 1H, HCAr), 8.06 (d, J = 8.1 Hz, 1H, HCAr), 7.50–7.31 (m, 8H, HCAr), 6.97 (d, J = 7.2 Hz, 2H, HCAr), 6.82 (t, J = 7.7 Hz, 1H, HCAr), 6.49 (d, J = 7.5 Hz, 1H, HCAr), 5.49 (s, 1H, HC2), 4.45 (d, J = 9.5 Hz, 1H, HC4), 3.33 (d, J = 9.5 Hz, 1H, HC3), 1.91 (s, 3H, OCH3), 1.55 (s, 9H, CH3). 13C NMR (100 MHz, CDCl3) δ 173.4 (CONH), 169.4 (CONtBu), 147.5 (CAr), 143.0 (CAr), 137.5 (CAr), 136.1 (CAr), 134.9 (CAr), 133.7 (CAr), 130.2 (CAr), 129.2 (2C, CAr), 128.0 (CAr), 127.8 (CAr), 127.6 (CAr), 127.5 (CAr), 127.1 (CAr), 126.5 (CAr), 125.7 (2C, CAr), 121.4 (CAr), 121.3 (CAr), 116.1 (CAr), 110.4 (CAr), 62.0 (C(CH3)3), 56.3 (C2), 55.9 (C4), 50.5 (C3), 28.1 (3C, CH3), 20.9 (CH3). MS (ESI) m/z (%) = 478.23 (100, [M + H]+). Anal. calcd for C31H31N3O2: C, 77.96; H, 6.54; N, 8.80. Found: C, 78.13; H, 6.61; N, 8.73.
(2S*,3S*,4S*)-1-(tert-Butyl)-4-(4-formylphenyl)-5-oxo-2-phenyl-N-(quinolin-8-yl)pyrrolidine-3-carboxamide (4d)
Compound 4d was obtained following general procedure for the C(sp3)–H bond arylation reaction (45 mg, 71% yield, Et2O/n-hexane (2:1), Rf = 0.33). 1H NMR (400 MHz, CDCl3) δ 9.54 (s, 1H, CHO), 9.12 (s, 1H, OCNH), 8.54 (d, J = 4.0 Hz, 1H, HCAr), 8.50–8.38 (m, 1H, HCAr), 8.04 (d, J = 8.2 Hz, 1H, HCAr), 7.46 (t, J = 7.7 Hz, 4H, HCAr), 7.43–7.31 (m, 8H, HCAr), 5.48 (s, 1H, HC2), 4.55 (d, J = 9.3 Hz, 1H, HC4), 3.37 (d, J = 9.3 Hz, 1H, HC3), 1.55 (s, 9H, CH3). 13C NMR (100 MHz, CDCl3) δ 191.5 (CHO), 172.4 (CONH), 168.8 (CONtBu), 147.8 (CAr), 142.5 (CAr), 142.4 (CAr), 137.9 (CAr), 136.1 (CAr), 135.0 (CAr), 133.4 (CAr), 130.5 (2C, CAr), 129.5 (2C, CAr), 129.3 (2C, CAr), 128.1 (CAr), 127.6 (CAr), 127.0 (CAr), 125.6 (2C, CAr), 121.9 (CAr), 121.5 (CAr), 116.2 (CAr), 62.3 (C(CH3)3), 56.3 (C2), 56.2 (C4), 50.4 (C3), 28.1 (3C, CH3). MS (ESI) m/z (%) = 514.28 (100, [M + Na]+). Anal. calcd for C31H29N3O3: C, 75.74; H, 5.95; N, 8.55. Found: C, 75.91; H, 6.99; N, 8.51.
(2S*,3S*,4S*)-1-(tert-Butyl)-4-(3-methoxyphenyl)-5-oxo-2-phenyl-N-(quinolin-8-yl)pyrrolidine-3-carboxamide (4e)
Compound 4e was obtained following general procedure for the C(sp3)–H bond arylation reaction (60 mg, 95% yield, Et2O/n-hexane (2:1), Rf = 0.36). 1H NMR (400 MHz, CDCl3) δ 9.10 (s, 1H, OCNH), 8.60–8.52 (m, 1H, HCAr), 8.49 (dd, J = 6.0, 3.0 Hz, 1H, HCAr), 8.06 (dd, J = 8.2, 1.3 Hz, 1H, HCAr), 7.48–7.32 (m, 8H, HCAr), 6.90–6.67 (m, 3H, HCAr), 6.30–6.18 (m, 1H, HCAr), 5.50 (s, 1H, HC2), 4.45 (d, J = 9.5 Hz, 1H, HC4), 3.49 (s, 3H, OCH3), 3.40–3.26 (m, 1H, HC3), 1.53 (s, 9H, CH3). 13C NMR (100 MHz, CDCl3) δ 173.2 (CONH), 169.3 (CONtBu), 159.3 (CAr), 147.6 (CAr), 142.9 (CAr), 136.5 (CAr), 133.7 (CAr), 130.7 (CAr), 129.7 (CAr), 129.2 (2C, CAr), 129.1 (CAr), 127.9 (CAr), 127.5 (CAr), 127.0 (CAr), 125.7 (2C, CAr), 122.9 (CAr), 121.8 (CAr), 121.5 (CAr), 121.3 (CAr), 114.7 (CAr), 113.0 (CAr), 62.1 (C(CH3)3), 56.2 (C2), 56.0 (C4), 54.8 (OCH3), 50.5 (C3), 28.0 (3C, CH3). MS (ESI) m/z (%) = 516.18 (100, [M + Na]+). Anal. calcd for C31H31N3O3: C, 75.43; H, 6.33; N, 8.51. Found: C, 75.60; H, 6.35; N, 8.47.
(2S*,3S*,4S*)-1-(tert-Butyl)-4-(4-methoxyphenyl)-5-oxo-2-phenyl-N-(quinolin-8-yl)pyrrolidine-3-carboxamide (4f)
Compound 4f was obtained following general procedure for the C(sp3)–H bond arylation reaction (50 mg, 79% yield, Et2O/n-hexane (2:1), Rf = 0.38). 1H NMR (400 MHz, CDCl3) δ 9.14 (s, 1H, OCNH), 8.58 (d, J = 4.0 Hz, 1H, HCAr), 8.52–8.45 (m, 1H, HCAr), 8.07 (s, 1H, HCAr), 7.48–7.31 (m, 8H, HCAr), 7.10 (d, J = 8.5 Hz, 2H, HCAr), 6.50 (d, J = 8.5 Hz, 2H, HCAr), 5.47 (s, 1H, HC2), 4.44 (d, J = 9.4 Hz, 1H, HC4), 3.37 (s, 3H, OCH3), 3.32 (d, J = 9.2 Hz, 1H, HC3), 1.53 (s, 9H, CH3). 13C NMR (100 MHz, CDCl3) δ 173.6 (CONH), 169.5 (CONtBu), 158.4 (CAr), 147.5 (CAr), 143.0 (CAr), 137.8 (CAr), 136.3 (CAr), 133.7 (CAr), 130.7 (2C, CAr), 129.2 (2C, CAr), 127.9 (CAr), 127.6 (CAr), 127.1 (CAr), 125.7 (2C, CAr), 121.6 (CAr), 121.3 (CAr), 116.4 (CAr), 113.7 (2C, CAr), 108.5 (CAr), 62.1 (C(CH3)3), 56.3 (C2), 55.9 (C4), 54.8 (OCH3), 49.7 (C3), 28.1 (3C, CH3). MS (ESI) m/z (%) = 516.30 (100, [M + Na]+). Anal. calcd for C31H31N3O3: C, 75.43; H, 6.33; N, 8.51. Found: C, 75.61; H, 6.36; N, 8.47.
(2S*,3S*,4S*)-4-(4-Bromophenyl)-1-(tert-butyl)-5-oxo-2-phenyl-N-(quinolin-8-yl)pyrrolidine-3-carboxamide (4g)
Compound 4g was obtained following general procedure for the C(sp3)–H bond arylation reaction (67 mg, 96% yield, EtOAc/n-hexane (1:3), Rf = 0.38). 1H NMR (400 MHz, CDCl3) δ 9.15 (s, 1H, OCNH), 8.58 (dd, J = 4.2, 1.5 Hz, 1H, HCAr), 8.47 (t, J = 4.5 Hz, 1H, HCAr), 8.10 (d, J = 8.1 Hz, 1H, HCAr), 7.48–7.30 (m, 8H, HCAr), 7.07 (s, 4H, HCAr) 5.46 (s, 1H, HC2), 4.43 (d, J = 9.3 Hz, 1H, HC4), 3.33 (d, J = 9.2 Hz, 1H, HC3), 1.53 (s, 9H, CH3). 13C NMR (100 MHz, CDCl3) δ 172.8 (CONH), 169.1 (CONtBu), 147.8 (CAr), 142.7 (CAr), 136.2 (CAr), 134.2 (CAr), 131.4 (2C, CAr), 131.2 (2C, CAr), 129.3 (2C, CAr), 128.0 (CAr), 127.7 (CAr), 127.1 (CAr), 125.7 (2C, CAr), 121.9 (CAr), 121.6 (CAr), 121.2 (CAr), 110.5 (CAr), 110.0 (CAr), 108.5 (CAr), 62.2 (C(CH3)3), 56.2 (C2), 56.1 (C4), 49.8 (C3), 28.1 (3C, CH3). MS (ESI) m/z (%) = 564.21 (100, [M + Na]+). Anal. calcd for C30H28BrN3O2: C, 66.42; H, 5.20; N, 7.75. Found: C, 66.59; H, 5.30; N, 7.68.
(2S*,3S*,4S*)-1-(tert-Butyl)-4-(4-chlorophenyl)-5-oxo-2-phenyl-N-(quinolin-8-yl)pyrrolidine-3-carboxamide (4h)
Compound 4h was obtained following general procedure for the C(sp3)–H bond arylation reaction (63 mg, 97% yield, Et2O/n-hexane (2:1), Rf = 0.31). 1H NMR (400 MHz, CDCl3) δ 9.14 (s, 1H, OCNH), 8.57 (dd, J = 4.2, 1.5 Hz, 1H, HCAr), 8.48 (t, J = 4.5 Hz, 1H, HCAr), 8.08 (dd, J = 8.3, 1.5 Hz, 1H, HCAr), 7.48–7.32 (m, 8H, HCAr), 7.14 (d, J = 8.4 Hz, 2H, HCAr), 6.93 (d, J = 8.4 Hz, 2H, HCAr), 5.47 (s, 1H, HC2), 4.45 (d, J = 9.3 Hz, 1H, HC4), 3.33 (d, J = 9.3 Hz, 1H, HC3), 1.54 (s, 9H, CH3). 13C NMR (100 MHz, CDCl3) δ 172.9 (CONH), 169.1 (CONtBu), 147.8 (CAr), 142.6 (CAr), 137.9 (CAr), 136.1 (CAr), 133.6 (CAr), 133.5 (CAr), 132.9 (CAr), 131.0 (2C, CAr), 129.2 (2C, CAr), 128.2 (2C, CAr), 128.0 (CAr), 127.6 (CAr), 127.0 (CAr), 125.6 (2C, CAr), 121.8 (CAr), 121.5 (CAr), 116.3 (CAr), 62.2 (C(CH3)3), 56.2 (C2), 56.0 (C4), 49.7 (C3), 28.0 (3C, CH3). MS (ESI) m/z (%) = 520.28 (100, [M + Na]+). Anal. calcd for C30H28ClN3O2: C, 72.35; H, 5.67; N, 8.44. Found: C, 72.54; H, 5.69; N, 8.38.
(2S*,3S*,4S*)-1-(tert-Butyl)-4-(3-fluorophenyl)-5-oxo-2-phenyl-N-(quinolin-8-yl)pyrrolidine-3-carboxamide (4i)
Compound 4i was obtained following general procedure for the C(sp3)–H bond arylation reaction (68 mg, 94% yield, Et2O/n-hexane (2:1), Rf = 0.40). 1H NMR (400 MHz, CDCl3) δ 9.16 (s, 1H, OCNH), 8.60 (dd, J = 4.2, 1.5 Hz, 1H, HCAr), 8.48 (dd, J = 5.6, 3.3 Hz, 1H, HCAr), 8.07 (d, J = 8.2 Hz, 1H, HCAr), 7.49–7.31 (m, 8H, HCAr), 6.99 (dd, J = 7.6, 4.7 Hz, 2H, HCAr), 6.90 (td, J = 8.1, 6.1 Hz, 1H, HCAr), 6.48–6.40 (m, 1H, HCAr), 5.48 (s, 1H, HC2), 4.48 (d, J = 9.4 Hz, 1H, HC4), 3.35 (d, J = 9.4 Hz, 1H, HC3), 1.54 (s, 9H, CH3). 13C NMR (100 MHz, CDCl3) δ 172.7 (CONH), 169.0 (CONtBu), 163.7 (CAr), 161.2 (CAr), 147.7 (CAr), 142.7 (CAr), 137.6 (d, J = 7.7 Hz, 1C, CAr), 136.2 (CAr), 133.5 (CAr), 129.5 (d, J = 8.4 Hz, 1C, CAr), 129.2 (2C, CAr), 128.0 (CAr), 127.6 (CAr), 127.1 (CAr), 125.6 (2C, CAr), 125.2 (d, J = 2.8 Hz, 1C, CAr), 121.7 (CAr), 121.4 (CAr), 116.8 (d, J = 22.2 Hz, 1C, CAr), 116.2 (CAr), 113.8 (d, J = 21.2 Hz, CAr), 62.1 (C(CH3)3), 56.1 (C2), 56.0 (C4), 50.0 (C3), 28.0 (3C, CH3). MS (ESI) m/z (%) = 504.27 (100, [M + Na]+). Anal. calcd for C30H28FN3O2: C, 74.82; H, 5.86; N, 8.73. Found: C, 75.21; H, 5.82; N, 8.68.
(2S*,3S*,4S*)-1-(tert-Butyl)-4-(3-nitrophenyl)-5-oxo-2-phenyl-N-(quinolin-8-yl)pyrrolidine-3-carboxamide (4j)
Compound 4j was obtained following general procedure for the C(sp3)–H bond arylation reaction (67 mg, 97% yield, Et2O/n-hexane (2:1), Rf = 0.27). 1H NMR (400 MHz, CDCl3) δ 9.12 (s, 1H, OCNH), 8.49 (dd, J = 4.2, 1.5 Hz, 1H, HCAr), 8.43 (dd, J = 5.8, 3.1 Hz, 1H, HCAr), 8.13 (s, 1H, HCAr), 8.05 (dd, J = 8.3, 1.5 Hz, 1H, HCAr), 7.57 (dd, J = 10.7, 4.8 Hz, 2H, HCAr), 7.50–7.44 (m, 2H, HCAr), 7.44–7.37 (m, 5H, HCAr), 7.35 (dd, J = 8.3, 4.2 Hz, 1H, HCAr), 7.08 (t, J = 8.0 Hz, 1H, HCAr), 5.49 (s, 1H, HC2), 4.60 (d, J = 9.3 Hz, 1H, HC4), 3.40 (d, J = 9.3 Hz, 1H, HC3), 1.55 (s, 9H, CH3). 13C NMR (100 MHz, CDCl3) δ 172.0 (CONH), 168.7 (CONtBu), 147.9 (CAr), 147.7 (CAr), 142.4 (CAr), 137.8 (CAr), 137.5 (CAr), 136.2 (CAr), 135.7 (CAr), 133.2 (CAr), 129.4 (2C, CAr), 129.0 (CAr), 128.2 (CAr), 127.6 (CAr), 127.0 (CAr), 125.6 (2C, CAr), 125.1 (CAr), 121.9 (2C, CAr), 121.7 (CAr), 116.2 (CAr), 62.2 (C(CH3)3), 56.3 (C2), 56.1 (C4), 50.0 (C3), 28.1 (3C, CH3). MS (ESI) m/z (%) = 531.23 (100, [M + Na]+). Anal. calcd for C30H28N4O4: C, 70.85; H, 5.55; N, 11.02. Found: C, 71.08; H, 5.62; N, 10.93.
(2S*,3S*,4S*)-1-(tert-Butyl)-4-(3-methoxyphenyl)-5-oxo-2-phenylpyrrolidine-3-carboxylic acid (5a)
Compound 4e (209 mg, 0.423 mmol) was solubilized in MeCN (2.2 mL), then (Boc)2O (277 mg, 1.27 mmol) and DMAP (5 mg, 0.042 mmol) were added. The reaction solution was warmed to 50 °C and stirred for 2 hours. Then, it was cooled to room temperature, stirred for additional 18 hours, and the solvent was removed in vacuo. The crude Boc-derivative was solubilized in THF/H2O (3:1) (4.2 mL), then LiOH·H2O (106 mg, 2.52 mmol) and 30% H2O2 solution (480 μL, 1.90 mmol) were successively added at 0 °C. The reaction mixture was stirred for 20 minutes at 0 °C, then warmed to room temperature and stirred for additional 18 hours. Et2O was added to the reaction mixture, the aqueous phase was acidified to pH = 2 with 6M HCl and extracted three times with EtOAc. The collected organic phase was washed with brine, dried over Na2SO4, and the solvent was removed under reduced pressure to afford pure carboxylic derivative 5a (136 mg, 66% yield). 1H NMR (400 MHz, CDCl3) δ 7.41 (t, J = 7.2 Hz, 2H, HCAr), 7.34 (dd, J = 13.2, 7.2 Hz, 3H, HCAr), 7.14 (t, J = 7.8 Hz, 1H, HCAr), 6.78–6.72 (m, 3H, HCAr), 5.13 (s, 1H, HC2), 4.23 (d, J = 9.3 Hz, 1H, HC4), 3.71 (s, 3H, OCH3), 3.16 (d, J = 9.3 Hz, 1H, HC3), 1.39 (s, 9H, CH3). 13C NMR (100 MHz, CDCl3) δ 175.7 (COOH), 173.5 (CONtBu), 159.2 (CAr), 142.2 (CAr), 136.6 (CAr), 129.2 (2C, CAr), 128.1 (CAr), 125.7 (2C, CAr), 121.7 (CAr), 114.7 (CAr), 113.3 (CAr), 108.5 (CAr), 62.4 (C(CH3)3), 56.0 (C2), 55.0 (C4), 53.9 (OCH3), 49.4 (C3), 27.9 (3C, CH3). MS (ESI) m/z (%) = 733.00 (100, [2M − H]−), 366.17 (55, [M − H]−). Anal. calcd for C22H25NO4: C, 71.91; H, 6.86; N, 3.81. Found: C, 72.12; H, 6.90; N, 3.77.
(2S*,3S*,4S*)-1-(tert-Butyl)-4-(4-bromo)-5-oxo-2-phenylpyrrolidine-3-carboxylic acid (5b)
Compound 5b was obtained in 76% yield as described for 5a starting from 4g (209 mg, 0.423 mmol) as a 2:1 mixture of unseparable epimers. 1H NMR (400 MHz, CDCl3) δ 7.45–7.28 (m, 7H, HCAr), 7.14 (d, J = 8.5 Hz, 2H minor, HCAr), 7.08 (d, J = 8.4 Hz, 2H major, HCAr), 5.13 (s, 1H major + 1H minor, HC2), 4.26 (d, J = 9.2 Hz, 1H major, HC4), 3.96 (d, J = 7.1 Hz, 1H minor, HC4), 3.20 (d, J = 9.3 Hz, 1H major, HC3), 3.06–2.99 (m, 1H minor, HC3), 1.42 (s, 9H major, CH3), 1.34 (s, 9H minor, CH3). 13C NMR (100 MHz, CDCl3) δ 174.8 (major, COOH), 173.3 (minor, COOH), 172.7 (2C, major + minor, CONtBu), 144.2 (minor, CAr), 141.9 (major, CAr), 136.7 (minor, CAr), 134.2 (minor, CAr), 131.8 (2C, minor, CAr), 131.4 (2C, major, CAr), 131.3 (2C, major, CAr), 129.8 (2C, minor, CAr), 129.3 (2C, major, CAr), 129.1 (2C, minor, CAr), 128.3 (major, CAr), 128.1 (minor, CAr), 125.8 (2C, minor, CAr), 125.6 (2C, major, CAr), 121.6 (major, CAr), 121.4 (minor, CAr), 63.1 (minor, ![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) (CH3)3), 62.3 (major, (CH3)3), 56.7 (minor, C2), 56.1 (major, C2), 53.7 (major, C4), 53.5 (minor, C4), 51.2 (minor, C3), 48.6 (major, C3), 28.2 (3C, minor, CH3), 28.0 (3C, major, CH3). MS (ESI) m/z (%) = 438.10 (100, [M + Na]+). Anal. calcd for C21H22BrNO3: C, 60.59; H, 5.33; N, 3.36. Found: C, 60.98; H, 5.40; N, 3.28.
(CH3)3), 62.3 (major, (CH3)3), 56.7 (minor, C2), 56.1 (major, C2), 53.7 (major, C4), 53.5 (minor, C4), 51.2 (minor, C3), 48.6 (major, C3), 28.2 (3C, minor, CH3), 28.0 (3C, major, CH3). MS (ESI) m/z (%) = 438.10 (100, [M + Na]+). Anal. calcd for C21H22BrNO3: C, 60.59; H, 5.33; N, 3.36. Found: C, 60.98; H, 5.40; N, 3.28.
X-Ray data collection
Single crystals were mounted in a loop and intensity data collected at 100° K with a Bruker Apex-II CCD diffractometer, using a Cu–Kα (λ = 1.54184 Å) radiation. Data were collected with the Bruker APEX2 program, integrated and reduced with the Bruker SAINT software. The integrated intensities, measured using the φ and ω scan mode, were corrected for Lorentz and polarization effects. The substantial redundancy in data allows empirical absorption corrections to be applied using SADABS-2016/2 (Bruker AXS area detector scaling and absorption correction).40 Structures were solved by direct methods of SIR201941 and refined using the full-matrix least squares on F2 provided by SHELXL-2014/6.42 The non-hydrogen atoms were refined anisotropically whereas hydrogen atoms as isotropic.Crystallographic data for 4g
Asymmetric unit contains two independent molecules. This is probably due to a slightly different orientation of the two condensed rings in molecule A compared to B. Molecular formula 2 × (C30H28N3O2Br), M = 2 × 542.46, triclinic, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 9.6503(2), b = 12.2687(2), c = 22.1064(4) Å, α = 81.658(1) β = 84.899(1) γ = 77.132(1) V = 2520.2(1) Å3, Z = 2 Dc = 1.430, μ = 2.483 mm−1, F(000) = 1120. 43937 reflections were collected with a 2.023 < θ < 68.556 range with a completeness to theta 98.7%; 9157 were independent, the parameters were 658 and the final R index was 0.365 for reflections having I > 2σI. Hydrogen atoms were assigned in calculated positions except for H on N2 (A + B) that were found in FD map. No relevant hydrogen bonds are detected.
, a = 9.6503(2), b = 12.2687(2), c = 22.1064(4) Å, α = 81.658(1) β = 84.899(1) γ = 77.132(1) V = 2520.2(1) Å3, Z = 2 Dc = 1.430, μ = 2.483 mm−1, F(000) = 1120. 43937 reflections were collected with a 2.023 < θ < 68.556 range with a completeness to theta 98.7%; 9157 were independent, the parameters were 658 and the final R index was 0.365 for reflections having I > 2σI. Hydrogen atoms were assigned in calculated positions except for H on N2 (A + B) that were found in FD map. No relevant hydrogen bonds are detected.
Enzymatic BACE-1 assay
Primary BACE-1 enzymatic activity was assessed using a fluorometric assay kit (SensoLyte® 520 β-Secretase Assay Kit, Anaspec, USA) employing the fluorescent substrate HiLyte Fluor™ 488-Glu-Val-Asn-Leu-Asp-Ala-Glu-Phe-Lys (QXL™ 520)-OH. All the measurements were performed in 96-well plates with a BMG labtech OptimaStar microplate reader. The inhibitors (10 μM) were pre-incubated with the BACE-1 enzyme (1 μg mL−1) for 10 minutes at 25 °C before starting the reaction by substrate addition. The fluorescence was monitored over 30 minutes (λex = 490 nm, λem = 520 nm) at 25 °C. The percentages of inhibition for the test compounds were determined through the eqn (1 − Vs/Vo) × 100, where Vs is the initial velocity in the presence of the inhibitor and Vo is the initial velocity of the unhibited reaction. The IC50 value of 4g and 5b were obtained by dose–response measurements using 0.01–100 μM as the range of inhibitor concentrations. All the experiments were performed in triplicate and the collected data were analyzed using Graphpad 4.0 software package (Graphpad Prism, San Diego, CA).Docking calculations
Automated docking studies were carried out using the Lamarckian Genetic Algorithm (LGA) as a search engine implemented in the Autodock 4.0.1 program.44 The AutoDockTools 1.4.5 (ADT) graphical interface45 was used to prepare BACE1 and inhibitor PDBQT files. Coordinates of 4g were generated using Spartan (version 5.147), and then energy-minimized through the AM1 semi-empirical method to calculate the equilibrium geometry. The coordinates of BACE1 protease were retrieved from the Protein Data Bank (PDB code: 5CLM), and enzyme-inhibitor complex was unmerged for achieving the free enzyme structure. Water molecules were removed. Hydrogen atoms were added to the enzyme and the ligand, Gasteiger charges were added and non-polar hydrogens were merged. Three-dimensional energy scoring grids of 0.375 Å resolution and 40 Å × 40 Å × 40 Å dimensions were computed. The center of the grid was set using the coordinates of one of the oxygen atoms of D228. A total of 50 runs with a maximum of 2500000 energy evaluations were carried out for 4g, using the default parameters for LGA. Low energy ligand–protein complexes were subjected to AMMP energy minimization using VegaZZ,46 then Cluster analysis was performed on docked results using a root-mean-square (rms) tolerance of 1.5 Å. The analysis of the binding mode of the docked conformations was carried out using the Autodock plugin within PyMol software v0.99.47
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support provided by “Fondo di Beneficenza di Intesa Sanpaolo”, project B/2022/0209, and the MUR - Dipartimenti di Eccellenza 2023-2027 (DICUS 2.0) to the Department of Chemistry “Ugo Schiff” of the University of Florence is acknowledged.References
- C. W. Murray and D. C. Rees, Nat. Chem., 2009, 1, 187–192 CrossRef CAS PubMed.
- J. D. St Denis, R. J. Hall, C. W. Murray, T. D. Heightman and D. C. Rees, RSC Med. Chem., 2020, 12, 321–329 RSC.
- D. A. Erlanson, S. W. Fesik, R. E. Hubbard, W. Jahnke and H. Jhoti, Nat. Rev. Drug Discovery, 2016, 15, 605–619 CrossRef CAS PubMed.
- I. J. P. de Esch, D. A. Erlanson, W. Jahnke, C. N. Johnson and L. Walsh, J. Med. Chem., 2022, 65, 84–99 CrossRef CAS PubMed.
- G. Chessari, R. Grainger, R. S. Holvey, R. F. Ludlow, P. N. Mortenson and D. C. Rees, Chem. Sci., 2021, 12, 11976–11985 RSC.
- L. Guillemard, N. Kaplaneris, L. Ackermann and M. J. Johansson, Nat. Rev. Chem., 2021, 5, 522–545 CrossRef CAS PubMed.
- R. Jana, H. M. Begam and E. Dinda, Chem. Commun., 2021, 57, 10842–10866 RSC.
- L. Zhang and T. Ritter, J. Am. Chem. Soc., 2022, 144, 2399–2414 CrossRef CAS PubMed.
- C. W. Murray, O. Callaghan, G. Chessari, A. Cleasby, M. Congreve, M. Frederickson, M. J. Hartshorn, R. McMenamin, S. Patel and N. Wallis, J. Med. Chem., 2007, 50, 1116–1123 CrossRef CAS PubMed.
- Y. Cheng, T. C. Judd, M. D. Bartberger, J. Brown, K. Chen, R. T. Fremeau, D. Hickman, S. A. Hitchcock, B. Jordan, V. Li, P. Lopez, S. W. Louie, Y. Luo, K. Michelsen, T. Nixey, T. S. Powers, C. Rattan, E. A. Sickmier, D. J. St Jean, R. C. Wahl, P. H. Wen and S. Wood, J. Med. Chem., 2011, 54, 5836–5857 CrossRef CAS PubMed.
- M. Congreve, D. Aharony, J. Albert, O. Callaghan, J. Campbell, R. A. Carr, G. Chessari, S. Cowan, P. D. Edwards, M. Frederickson, R. McMenamin, C. W. Murray, S. Patel and N. Wallis, J. Med. Chem., 2007, 50, 1124–1132 CrossRef CAS PubMed.
- E. Lenci, L. Calugi and A. Trabocchi, ACS Chem. Neurosci., 2021, 12(3), 378–390 CrossRef CAS PubMed.
- S. Rej, Y. Ano and N. Chatani, Chem. Rev., 2020, 120, 1788–1887 CrossRef CAS PubMed.
- G. Meng, N. Y. S. Lam, E. L. Lucas, T. G. Saint-Denis, P. Verma, N. Chekshin and J.-Q. Yu, J. Am. Chem. Soc., 2020, 142, 10571–10591 CrossRef CAS PubMed.
- O. Verho, M. Maetani, B. Melillo, J. Zoller and S. L. Schreiber, Org. Lett., 2017, 19, 4424–4427 CrossRef CAS PubMed.
- M. Maetani, J. Zoller, B. Melillo, O. Verho, N. Kato, J. Pu, E. Comer and S. L. Schreiber, J. Am. Chem. Soc., 2017, 139, 11300–11306 CrossRef CAS PubMed.
- V. G. Zaitsev, D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2005, 127, 13154–13155 CrossRef CAS PubMed.
- T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS PubMed.
- G. Li Petri, M. V. Raimondi, V. Spanò, R. Holl, P. Barraja and A. Montalbano, Top. Curr. Chem., 2021, 379, 34 CrossRef CAS PubMed.
- S. Poyraz, H. A. Döndaş, N. Y. Döndaş and J. M. Sansano, Front. Pharmacol., 2023, 14, 1239658 CrossRef CAS PubMed.
- J. Palmer and S. Love, Pharmacol. Res., 2011, 63, 525–531 CrossRef CAS PubMed.
- J. R. M. Coimbra, P. I. Moreira, A. E. Santos and J. A. R. Salvador, Drug Discovery Today, 2023, 28, 103644 CrossRef CAS PubMed.
- J. Moreira, M. Machado, M. Dias-Teixeira, R. Ferraz, C. Delerue-Matos and C. Grosso, Acta Pharm. Sin. B, 2023, 13, 3208–3237 CrossRef PubMed.
- Á. Cores, S. Abril, P. Michalska, P. Duarte, A. I. Olives, M. A. Martín, M. Villacampa, R. León and J. C. Menéndez, Antioxidants, 2021, 10, 941 CrossRef PubMed.
- C. Wei, X. L. Cheng, Y. J. Chen, C. Liang and D. L. Mo, Adv. Synth. Catal., 2023, 365, 2976–2981 CrossRef CAS.
- Y. Zhang, H. Ma, X. Liu, X. Cui, S. Wang, Z. Zhan, J. Pu and G. Huang, Org. Biomol. Chem., 2018, 16, 4439–4442 RSC.
- R. Innocenti, E. Lenci, G. Menchi, A. Pupi and A. Trabocchi, Bioorg. Med. Chem., 2017, 25, 5077–5083 CrossRef CAS PubMed.
- L. Calugi, E. Lenci, R. Innocenti and A. Trabocchi, Bioorg. Med. Chem. Lett., 2020, 30, 127211 CrossRef PubMed.
- L. Calugi, E. Lenci, F. Bianchini, A. Contini and A. Trabocchi, Bioorg. Med. Chem., 2022, 63, 116746 CrossRef CAS PubMed.
- L. Usmanova, D. Dar'in, M. S. Novikov, M. Gureev and M. Krasavin, J. Org. Chem., 2018, 83, 5859–5868 CrossRef CAS PubMed.
- D. Dar'in, O. Bakulina, M. Chizhova and M. Krasavin, Org. Lett., 2015, 17, 3930–3933 CrossRef PubMed.
- D. Q. Tan, A. Younai, O. Pattawong, J. C. Fettinger, P. H. Y. Cheong and J. T. Shaw, Org. Lett., 2013, 15, 5126–5129 CrossRef CAS PubMed.
- T. T. Wager, X. Hou, P. R. Verhoest and A. Villalobos, ACS Chem. Neurosci., 2010, 1, 435–449 CrossRef CAS PubMed.
- B. De Strooper, R. Vassar and T. Golde, Nat. Rev. Neurol., 2010, 6, 99–107 CrossRef CAS PubMed.
- R. Vassar, B. D. Bennett, S. Babu-Khan, S. Kahn, E. A. Mendiaz, P. Denis, D. B. Teplow, S. Ross, P. Amarante, R. Loeloff, Y. Luo, S. Fisher, J. Fuller, S. Edenson, J. Lile, M. A. Jarosinski, A. L. Biere, E. Curran, T. Burgess, J. C. Louis, F. Collins, J. Treanor, G. Rogers and M. Citron, Science, 1999, 286, 735–741 CrossRef CAS PubMed.
- X. Hu, B. Das, H. Hou, W. He and R. Yan, J. Exp. Med., 2018, 215, 927–940 CrossRef CAS PubMed.
- Z. Zhang, M. Song, X. Liu, S. Su Kang, D. M. Duong, N. T. Seyfried, X. Cao, L. Cheng, Y. E. Sun, S. Ping Yu, J. Jia, A. I. Levey and K. Ye, Nat. Commun., 2015, 6, 8762 CrossRef CAS PubMed.
- K. P. Guzen, A. S. Guarezemini, A. T. G. Órfão, R. Cella, C. M. P. Pereira and H. A. Stefani, Tetrahedron Lett., 2007, 48, 1845–1848 CrossRef CAS.
- S. V. Ryabukhin, D. M. Panov, D. S. Granat, E. N. Ostapchuk, D. V. Kryvoruchko and O. O. Grygorenko, ACS Comb. Sci., 2014, 16, 146–153 CrossRef CAS PubMed.
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 42717 CrossRef PubMed.
- ACD/pKa, version 6.0, Advanced Chemistry Development, Inc., Toronto, ON, Canada, https://www.acdlabs.com Search PubMed.
- J. B. Jordan, D. A. Whittington, M. D. Bartberger, E. A. Sickmier, K. Chen, Y. Cheng and T. Judd, J. Med. Chem., 2016, 59, 3732–3749 CrossRef CAS PubMed.
- A. Stamford and C. Strickland, Curr. Opin. Chem. Biol., 2013, 17, 320–328 CrossRef CAS PubMed.
- G. M. Morris, D. S. Goodsell, R. S. Halliday, R. Huey, W. E. Hart, R. K. Belew and A. J. Olson, J. Comput. Chem., 1998, 19, 1639–1662 CrossRef CAS.
- A. Gillet, M. Sanner, D. Stoffler and A. Olson, Structure, 2005, 13, 483–491 CrossRef CAS PubMed.
- A. Pedretti, L. Villa and G. Vistoli, J. Comput. Aided Mol. Des., 2004, 18, 167–173 CrossRef CAS PubMed.
- W. L. DeLano, The PyMOL Molecular Graphics System. DeLano Scientific LLC, San Carlos, CA, USA. [https://www.pymol.org] Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra for compounds 1–3, 4a–4j, 5a, 5b, crystallographic data for 4g, kinetic NMR studies, inhibition curves of 4g and 5b on BACE-1 enzyme, CNS MPO calculations for compounds 2–3, 4a–4j, 5a, 5b, coordinates of top-ranked pose of 4g. CCDC 2165903. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ob02117c |
| This journal is © The Royal Society of Chemistry 2024 |