
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlkene carboamination/oxidative denitrogenation of 3-allyl-3-hydrazinylindolin-2-ones: one-pot entry to spirocyclopropyloxindoles†
Marco
Manenti
 *,
Tommaso
Villa
,
Giovanni
Macetti
and
Alessandra
Silvani
*,
Tommaso
Villa
,
Giovanni
Macetti
and
Alessandra
Silvani
Department of Chemistry, University of Milan, via C. Golgi 19, Milano, 20133, Italy. E-mail: marco.manenti@unimi.it; tommaso.villa7@studenti.unimi.it
First published on 14th February 2024
Abstract
A one-pot protocol, consisting of a Pd-catalysed carboamination reaction, followed by N-deprotection and oxidative denitrogenation, has been developed for the synthesis of diversely substituted spirocyclopropyloxindoles, in yields up to 73% and with diastereoselectivity close to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Readily accessible starting materials, mild reaction conditions, an easy to operate one-pot procedure and good functional group tolerance make this transformation a versatile tool for the synthesis of substituted spirocyclopropyloxindoles. This protocol successfully works on the gram-scale and allows access to both diastereoisomers separately. A plausible mechanism was proposed, and a series of post-transformations were performed on the obtained products, showing their remarkable synthetic versatility.
:1. Readily accessible starting materials, mild reaction conditions, an easy to operate one-pot procedure and good functional group tolerance make this transformation a versatile tool for the synthesis of substituted spirocyclopropyloxindoles. This protocol successfully works on the gram-scale and allows access to both diastereoisomers separately. A plausible mechanism was proposed, and a series of post-transformations were performed on the obtained products, showing their remarkable synthetic versatility.
Introduction
Spirocyclopropyloxindoles are an important class of heterocyclic derivatives found in both natural and synthetic compounds and are endowed with distinctive properties that are associated with their spirocyclic three-dimensional feature.1 They combine the cyclopropyl ring, whose rigidity is suitable for a special range of biological activities, including protein inhibition,2 DNA alkylation3 and interaction with unique relevant targets,4 with the privileged oxindole scaffold, which is widely distributed in a number of natural products and pharmaceuticals, displaying, among others, anticancer,5 antiviral,6 antitubercular7 and antimalarial activities.8It is therefore not surprising that spirocyclopropyloxindoles are becoming increasingly relevant in drug discovery, and several compounds have already been established as reliable sources of new drugs (Fig. 1).9 Among these, CFI-400945 is a potent PLK4 inhibitor, which has entered phase I clinical trials for the treatment of breast cancer,10 while Sisunatovir is an orally bioavailable inhibitor of the respiratory syncytial virus.11 This class of compounds includes even anti-HIV, anti-obesity and different anticancer compounds.12
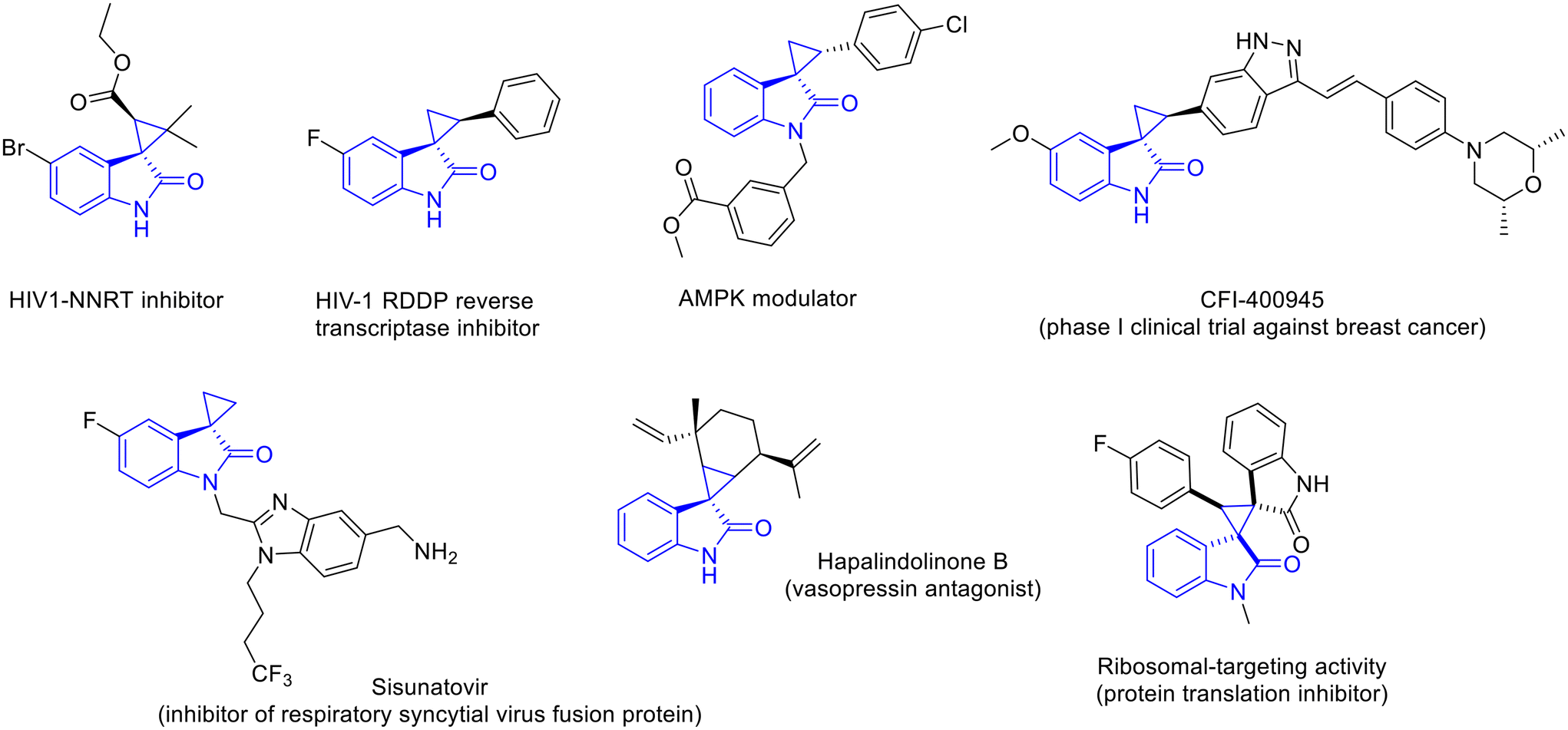 | ||
| Fig. 1 Examples of natural and synthetic spirocyclopropyloxindoles with relevant biological activities. | ||
Significant work has been done over the past few decades to discover viable synthesis methods of spirocyclopropyloxindoles, and the development of efficient and short routes to generate chemical diversity around this distinctive framework remains an active research field today. Most of the reported methodologies rely on the use of sulfur ylides, such as the recent protocol developed by Hajra and co-workers, in which isatin undergoes a domino Corey–Chaykovsky reaction to give unsubstituted spirocyclopropyloxindoles,13 and the protocols described by Feng and co-workers and by Yuan and co-workers, in which activated 3-alkenyl oxindoles undergo a formal [2 + 1] annulation with sulfur and sulfoxonium ylides, both by metal catalysis and under metal-free conditions.14,15 Besides sulfur ylides, phenyliodonium ylides can be employed as C-1 synthons in [2 + 1] annulations, as recently demonstrated by Feng and co-workers,16 as well as Morita–Baylis–Hillman carbonates, as reported by Warghude and co-workers.17 A domino reaction of M–B–H carbonates of isatins and sulfur ylides was exploited by Zheng and co-workers to synthesize complex oxospiro[bicyclo[3.1.0]hexane-6,3′-indoline] scaffolds.18 All these approaches require particularly activated 3-alkenyl oxindoles, such as conjugated ones, in order to give the target spiro derivatives, severely limiting the scope of these types of reactions.
A different strategy toward substituted spirocyclopropyloxindoles involves the use of highly reactive diazo compounds, again in combination with 3-alkenyl oxindoles, to perform a [3 + 2] cyclization followed by nitrogen elimination, as demonstrated by Maurya and co-workers19 and by Xiao and co-workers20 (Scheme 1, previous studies). However, diazo compounds have to be synthesized in situ due to their carcinogenicity and highly explosive nature, making the scale-up of the process troublesome and dangerous. Moreover, apart from the safety issues related to diazo compounds, spirocyclopropyloxindoles prepared in this way are always obtained as single diastereoisomers, according to the frontier molecular orbital theory and without the possibility of accessing alternative diastereoisomers.21 By reversing the substrates’ functionalities, Cao and co-workers demonstrated the application of 3-diazo oxindoles in a series of Hg(II)- or Au(I)-catalysed reactions with both electron-rich or electron-neutral olefins, achieving the desired spirocyclopropyloxindoles in good yields and with a broad range of different substituents,22,23 while Iwasa and co-workers performed the same transformation using a chiral Ru(II) catalyst.24 In a solvent and catalyst-free version, developed by Muthusamy and co-workers, 3-diazo oxindoles were heated directly in the presence of electron-deficient olefins,25 while a photochemical variation was described by Chen and co-workers.26 Since 3-diazo oxindoles are also carcinogenic and explosive,27 they must be handled with extreme caution and a blast shield must be used in all their reactions.
 | ||
| Scheme 1 Target spirocyclopropyloxindoles: previous studies using [3 + 2] cyclization/ring contraction approaches and the new strategy herein reported. | ||
For various reasons, it is therefore worthwhile to develop new synthetic protocols that are mild, safe, broadly tolerant and capable of providing access to both diastereoisomers of the desired spirocyclopropyloxindoles.
In our previous work, we described an easy entry to spiropyrazolidinyloxindoles, exploiting a Pd(0)-catalysed carboamination reaction, which gives access to both separable diastereoisomers of the desired products in good yields and with high functional group tolerance.28 Now, we envisioned that such a transformation could be coupled with N-deprotection, mild N–N bond oxidation, nitrogen elimination and ring contraction, leading to the target spirocyclopropyloxindoles in a one-pot process starting from the readily available 3-allyl-3-hydrazinylindolin-2-ones (Scheme 1, this work). To the best of our knowledge, no similar one-pot strategy, involving such a mild oxidative denitrogenation step, has been described so far in the literature for the synthesis of related compounds.
Results and discussion
We began our investigation with the allylation reaction of the known isatin-derived N-Boc-1a and N-Cbz-hydrazones 2a, which afforded the corresponding 3-allyl-3-hydrazinylindolin-2-ones 3a and 4a.While the allylation of 1a was already optimized in our previous work (Table 1, entry 1), the same conditions did not work for the related N-Cbz derivative 2a (entry 2), so alternative procedures were sought.
|
|
||||
|---|---|---|---|---|
| Entry | R | X | Solvent and conditions | Yieldb (%) |
| a Reactions were carried out with 1a (0.15 mmol) and allyl-X (0.30 mmol) in a solvent (1.5 mL) and 2a (0.15 mmol) and allyl-X (0.33 mmol) in a solvent (0.5 mL). b Isolated yields. | ||||
| 1 | Boc | Br | In powder, MeOH, 40 °C, 3 h | 93% |
| 2 | Cbz | Br | In powder, MeOH, 40 °C, 3 h | Traces |
| 3 | Cbz | Bpin | Ti(OiPr)4, THF, 40 °C, 3 h | Traces |
| 4 | Cbz | MgBr | THF, −78 °C to rt, 3 h | 48% |
| 5 | Cbz | Br | Zn powder, THF, 0 °C, 5 min | 92% |

The Brown reaction with allylboronic acid pinacol ester was not effective, even after the addition of the strong Lewis acid Ti(OiPr)4 (entry 3). Using the Grignard reagent allylmagnesium bromide, the target compound 4a could be obtained in 48% yield (entry 4). We were pleased to observe that a Barbier-type reaction, relying on activated zinc powder to form allylzinc bromide in situ, gave the desired product in 92% yield in only five minutes (entry 5).
Following the conditions reported in entries 1 and 5, respectively, a small library of different 3-allyl-3-hydrazinylindolin-2-ones 3 and 4 was prepared (Scheme 2). Apart from the sterically hindered 4-chloro derivatives, all the selected hydrazone substrates gave the corresponding products in good to excellent yields.
 | ||
| Scheme 2 Substrate scope of N-Boc allyl-oxindoles 3 and N-Cbz allyl-oxindoles 4. Reactions were performed on a 1.50 mmol scale. Conditions A: allyl bromide (2 eq.), indium powder (2 eq.) in MeOH (0.1 M), 40 °C, 3 h. Conditions B: allyl bromide (2.2 eq.), activated zinc powder (5 eq.) in THF (0.3 M), 0 °C, 5 min. Isolated yields. | ||
Next, we focused our attention on the one-pot conversion of allyl derivatives 3 and 4 to the desired spirocyclopropyloxindoles, envisioning nitrogen deprotection and subsequent N–N bond oxidation as the key steps after the Pd-catalysed alkene carboamination reaction (Table 2). In the model reaction, starting from compound 3a and 4-bromo-1,10-biphenyl as the reference aryl halide, the intermediate spiropyrazolidinyloxindole was achieved by reaction with Pd2(dba)3 as the catalyst, paired with X-phos, according to an optimised protocol that is able to drive the reaction towards the key intramolecular carboamination, rather than towards the competing intermolecular Heck reaction (see below, Scheme 7).28
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Conditions 1 (N-Boc deprotection) | T (°C) | Conv.e (%) | Entry | Conditions 2 (oxidation)d | Conv.e (%) |
| a Reactions were carried out with 3a (0.10 mmol), Pd2(dba)3 (0.004 mmol), XPhos (0.016 mmol), tBuONa (0.13 mmol), and Br-biphenyl (0.13 mmol) in toluene (2.0 mL). b The volume of the added acid solution was equal to the starting volume of toluene. c TMSI (0.50 mmol) in MeOH (5.0 mL). d Oxidants (0.60 mmol) were used as a solid or added as an aqueous solution. e Determined by 1H NMR of the crude reaction mixture. f Not detected. | ||||||
| 1 | TFAb | 30 | 74 (8 h) | 5 | Pb(OAc)4 | Quant. |
| 2 | 4 N HCl aq.b | 30 | Traces (8 h) | 6 | KMnO4 (aq.) | Quant. |
| 3 | 4 N HCl in dioxaneb | 30 | 77 (3 h) | 7 | CuSO 4 (aq.) | Quant. |
| 4 | TMSI/MeOHc | 30 | n.d.f | 8 | AgOTf | 40 |

Instead of isolating, it was subjected to N-Boc deprotection in situ, evaluating a series of different protocols (conditions 1). Trifluoroacetic acid afforded the desired NH–NH pyrazolidine intermediate in 74% conversion, as detected by 1H NMR, but with a long reaction time (entry 1). Aqueous 4 N HCl was ineffective, probably due to the decreased solubility of the substrate in the biphasic reaction medium (entry 2), as confirmed by the good result conversely obtained using 4 N HCl in dioxane, which actually afforded the desired deprotected intermediate with a conversion of 77% in three hours (entry 3). However, the N-Boc deprotection was not achieved using trimethylsilyl iodide solution in methanol (entry 4). Next, a series of different oxidants were investigated for the following step. Besides Pb(OAc)4 and KMnO4 (entries 5 and 6), which proved to be effective, but are strong environmental pollutants, an aqueous solution of CuSO4 performed well, undergoing quantitative mild oxidation and ring contraction to afford the desired target 5a in a satisfactory 73% overall yield from precursor 3a (entry 7). Finally, AgOTf afforded the desired product with lower conversion (40%), probably due to the low solubility of the silver salt in the solvent (entry 8). It is noteworthy that, under the described conditions, the nitrogen elimination seems to be almost instantaneous after oxidation, preventing the observation of the intermediate spiropyrazolinyloxindole by TLC.
On the other hand, starting from the N-Cbz allyl derivative 4a, the alkene carboamination reaction was followed by N-Cbz deprotection by NH4(HCO2) in methanol and 10% Pd/C, and finally by CuSO4 oxidation, to afford the expected spirocyclopropyloxindole 5a, again by a one-pot procedure, in a comparable 67% overall yield (Scheme 3).
 | ||
| Scheme 3 One-pot protocol for the synthesis of the target compound 5a starting from the N-Cbz protected compound 4a. Reaction was carried out with 4a (0.25 mmol), NH4(HCO2) (25 mmol), 10% Pd/C (121 mg), and methanol (15 mL). Copper sulphate pentahydrate (1.50 mmol) in water (3.0 mL). Yield reported is the isolated yield over three steps. | ||
The availability of two classes of differently N-protected starting substrates (compounds 3 and 4) allows to select the best one-pot reaction conditions, on the basis of the different functional groups and substituents that are present on the entire molecule. This greatly increases the versatility of the protocol, as widely demonstrated by the substrate scope study that was carried out.
Hence, relying on the optimised conditions and starting from compounds 3a and 4a and various aryl and heteroaryl bromides, a library of different spirocyclopropyloxindoles was synthesized. In all cases, two diastereoisomers were formed in a ratio close to 1:1. They can be easily separated by flash chromatography and were individually fully characterised by NMR spectroscopy and high-resolution mass spectrometry (HRMS) (Scheme 4, for the relative configuration assignment see later).
 | ||
| Scheme 4 Substrate scope of different aryl or heteroaryl bromides R2–Br for the synthesis of substituted spirocyclopropyloxindoles 5a–5l (as pairs of diastereoisomers a and b). Reactions were performed on a 0.25 mmol scale. In brackets: conditions A (for substrate 3a: 4 N HCl in dioxane) or conditions B (for substrate 4a: NH4(HCO2), 10% Pd/C, MeOH). Yields reported are the isolated yields over three steps. aReaction performed on a 1 gram scale. | ||
The protocol proved to be very suitable, tolerating both electron-rich (4-methoxy-phenyl) and electron-poor (4-nitro-phenyl) aryl bromides as reaction partners, although the presence of the nitro group slightly lowers the yield, due to non-investigated side reactions (5b and 5c). Sterically hindered naphthyl and mesityl bromides show similar reactivity in the one-pot process, affording the desired spirocyclic derivatives 5d and 5e in comparable yields. Pleasingly, the 2-bromo-6-phenylpyridine reacted smoothly to give compound 5f in good yield, demonstrating the applicability of the protocol to the synthesis of spirocyclopropyloxindoles containing pharmacologically relevant heterocycles. With R2 = vinyl, the reaction was effective, but the terminal double bond, inserted with the carboamination step, was reduced under the subsequent reaction conditions, so the final spirocyclopropyloxindole 5g, bearing a saturated alkyl chain, was recovered. A carbonyl group on the aryl bromide reagent was also found to be compatible with the reaction conditions (5h). 3-Bromo-chlorobenzene can be used as a reaction component, displaying both good reactivity and complete selectivity for bromide in the step of oxidative addition to Pd(0) (5i). 4-Bromo-N,N-dimethylaniline and 5-bromo-1-methyl-1H-indole do not react completely in the carboamination step, affording the corresponding cyclopropyloxindoles 5j and 5k only in moderate yields. Finally, 3-Br-thiophene was used and the corresponding product 5l was obtained in acceptable yield, but after a slightly longer reaction time.
Next, after the screening of aryl, heteroaryl and alkenyl bromides suitable for the reaction, we moved our attention to differently substituted oxindole precursors (Scheme 5).
 | ||
| Scheme 5 Substrate scope of different substituted allyloxindoles 3 and 4 for the synthesis of substituted spirocyclopropyloxindoles 5m–5r (as pairs of diastereoisomers a and b). Reactions were performed on a 0.25 mmol scale. Conditions A (for substrates 3): 4 N HCl in dioxane. Conditions B (for substrates 4): NH4(HCO2), 10% Pd/C, MeOH. Yields reported are isolated yields over three steps. aReaction performed on a 1 gram scale. | ||
With regard to R3, the reaction proceeded well with the bulky trityl (5m), while with R3 = Me, the overall yield decreased slightly (5n). Moving to R2, oxindoles bearing halogen substituents, both at positions C-5 (5o) and C-6 (5q), proved to be suitable substrates. The electron-rich 5-methoxy-oxindole substrate was observed to undergo minimal side reactions during the deprotection step, affording the corresponding product 5p in a slightly lower yield. On the other hand, the electron-withdrawing trifluoromethyl substituent at C7 was well tolerated and compound 5r was obtained smoothly. Finally, the 4-chloro-oxindole substrate afforded the undesired Buchwald coupling product 6 after the Pd-catalysed step, probably due to the steric hindrance near the carboamination reaction site.
To demonstrate the scalability and reliability of the process, a gram-scale synthesis of compounds 5a and 5m was performed. It afforded the desired products in comparable yields and diastereomeric ratios, despite requiring slightly longer reaction times for the deprotection step. It is worth underlining that the reported final yields, even if seemingly moderate, are actually highly satisfactory, representing the overall yields over three different reactions carried out as a one-pot process.
Since oxindoles bearing N–H are often more suitable for biological activity, with respect to the N-protected analogues, easy deprotection of compounds 5ma and 5mb was also demonstrated, allowing the achievement of compounds 7a and 7b in good yields. Compound 7b was also crystallized through slow evaporation from chloroform and subjected to single crystal X-ray diffraction analysis, which proved the relative configuration of the two stereogenic centers (Scheme 6).
 | ||
| Scheme 6 Removal of the trityl group (left) and the asymmetric unit of compound 7b at 100 K (right). Non-H atom numbering scheme. Thermal ellipsoids of non-H atoms were drawn at the 50% probability level. The usual colour code was employed for atoms (grey: C; white: H; blue: N; red: O). For crystallographic details see the ESI.† | ||
With regard to the one-pot reaction mechanism, we have to consider the Pd-catalyzed carboamination and the subsequent ring contraction (Scheme 7). The mechanism of the first transformation is already described,29 and in our case, it relies on the known ability of the Pd2(dba)3 complex to reduce the rate of β-hydrogen elimination in the intermediate Pd(II)–σ-alkyl complex,30 driving the reaction towards intramolecular cyclization rather than the β-hydrogen elimination involved in the Heck reaction. To shed light on the ring contraction mechanism, the synthesis of the model compound 5a was carried out in the presence of TEMPO as the radical scavenger. Since the obtained yield does not differ from that in the absence of the radical scavenger, it is plausible to assume that the ring contraction involves a stabilized zwitterionic intermediate, rather than a di-radical species, as sometimes reported in the literature for similar denitrogenation processes31 (Scheme 7).
 | ||
| Scheme 7 Pd-catalyzed carboamination reaction and ring contraction: plausible zwitterionic mechanism, in comparison with the di-radical alternative. | ||
To investigate the synthetic versatility of the obtained spirocyclopropyloxindoles, some post-transformations were evaluated. We considered first the amazing conversion of cyclopropyl rings into different heteroatom-containing five-membered rings via MgI2-catalysed [3 + 2] cycloaddition with carbonyl compounds and imines, described by Carreira and co-workers some years ago,32 and applied it to the synthesis of biologically relevant compounds, such as the alkaloids (±)-horsfiline,33 (±)-strychnofoline34 and (−)-rhynchophylline.35 However, this reaction, as well as the more recently described related methodologies,36 works only with activated donor–acceptor cyclopropanes, namely compounds endowed with a specific substitution pattern at the cyclopropane ring, where the combined effects of vicinal electron-donating and electron-accepting moieties allow for particularly mild, efficient, and selective transformations.
In our case, relying on molecules that lack suitable substitution for activating [3 + 2] cycloaddition pathways, a new rearrangement reaction was explored, taking advantage of the unique presence of a reactive benzylic methylene on the cyclopropyl ring. Starting from spirocyclopropyloxindoles 5 or 7, via reaction with MgI2 in combination with triethylamine hydrochloride, acting both as a magnesium-enolate quencher and an E2BC promoter, the ring opening was achieved, leading to the formation of unprecedented 3-substituted oxindoles (Scheme 8).
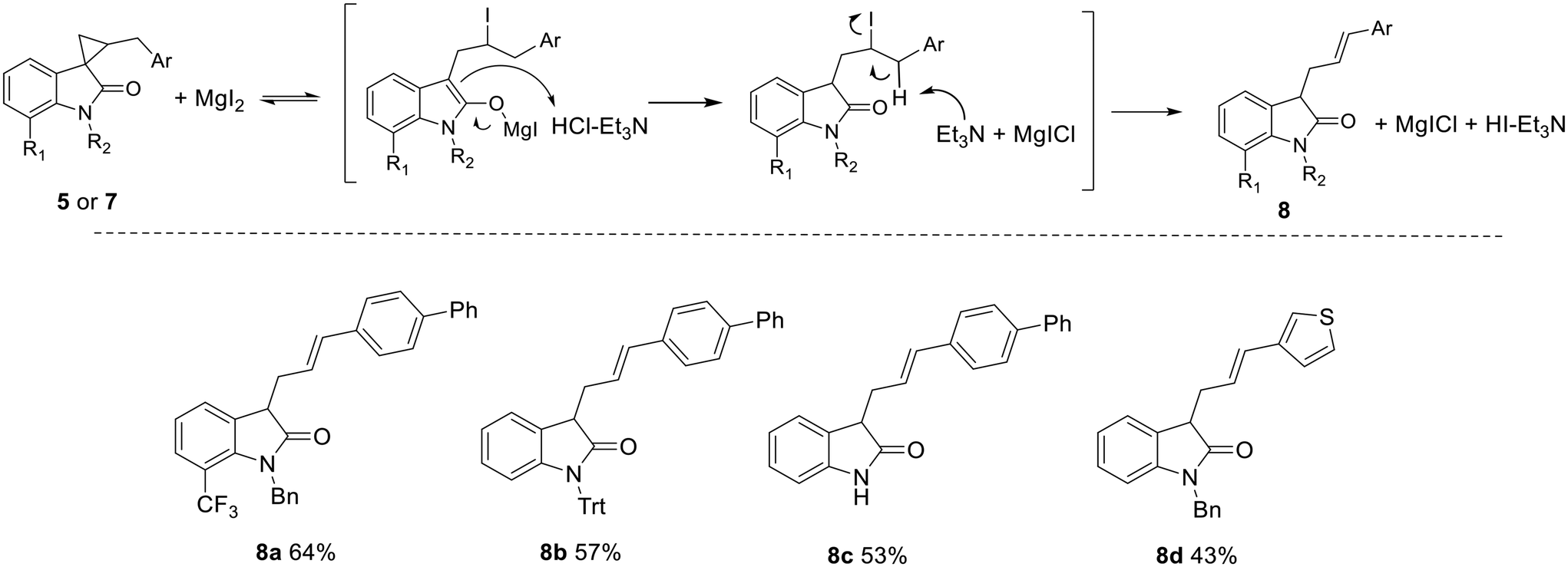 | ||
| Scheme 8 Rearrangement of spirocyclopropyloxindoles 5 and 7 into 3-substituted oxindoles 8. Reactions were performed on a 0.20 mmol scale. Magnesium iodide (1.2 eq.), triethylammonium hydrochloride (6.0 eq.), THF:DMF 3:1 (0.4 M), 130 °C, 48 h. Isolated yields. | ||
Indeed, triethylamine hydrochloride, as a proton source, drives the reaction towards the intermolecular quenching of the intermediate magnesium enolate and the subsequent elimination of HI, discouraging in this way the intramolecular iodide displacement, which would regenerate the starting three-membered ring and free MgI2. No reaction was observed when the temperature was decreased to 90 °C or in the absence of one of the two reagents. Under such conditions, four different spirocyclopropyloxindoles were converted into the corresponding 3-substituted oxindoles 8, without any significant difference in reactivity and with comparable yields.
The synthetic utility of compounds 8 was illustrated through two post-transformation reactions. By electrophilic fluorination, compound 8a was converted into the highly fluorinated derivative 9, whose chemical structure resembles that of the potassium channel modulator Maxipost. Compound 8b was indeed alkylated at C-3 to afford derivative 10, which was subjected to a photoinduced dearomative [2 + 2] cyclization, leading to the densely functionalized sp3-rich spiro tetracyclic compound 11 (Scheme 9).
 | ||
| Scheme 9 Post-transformation reactions to give compounds 9 and 11. Isolated yields. Symmetric unit of 11 at RT, with the non-H atom numbering scheme. Thermal ellipsoids of non-H atoms were drawn at the 50% probability level. Atom color code is grey: C; white: H; blue: N; red: O; gold: Br. | ||
The remarkable selectivity in the light-promoted cyclization step could possibly be related to the favourable π–π interactions between the aromatic rings of the molecule in the transition state. The relative configuration was assigned unambiguously by 2D-NOESY-NMR, and fully confirmed by single-crystal X-ray diffraction analysis (see the ESI†). It is noteworthy that compound 11 can likely be functionalized even further, exploiting both the aryl bromide for Pd-catalysed reactions and the easily deprotectable oxindole N-position.
Conclusion
A safe and scalable one-pot protocol for the synthesis of spirocyclopropyloxindoles was developed, exploiting an alkene carboamination/oxidative denitrogenation strategy, starting from 3-allyloxindole derivatives. Under mild conditions, a broad scope of the desired products, as two separable diastereoisomers, was easily achieved, even when carrying out the reaction on the gram-scale. An unprecedented rearrangement reaction was discovered for the conversion of the obtained spirocyclopropyloxindoles into 3-susbstituted oxindoles in acceptable yields. Finally, the possible conversion into new densely functionalized polycyclic scaffolds with remarkable stereoselectivity, via a photoinduced [2 + 2] dearomative cyclization, was also demonstrated.Efforts to apply this protocol to the synthesis of spirocyclopropyloxindoles and derivatives within programs of drug discovery are currently underway in our laboratory.
Experimental section
General information
Isatin-derived hydrazones 1 and 2 were prepared according to the method reported in the literature.28 Unless noted, all the reactions were performed under an inert atmosphere (nitrogen or argon) and using flame-dried glassware. All employed reagents, including catalysts and aryl-, heteroaryl- and alkenyl-bromides, are commercially available and used without further purification. Solvents were purchased as “anhydrous” and used without further purification. 1H NMR, 13C NMR and 19F NMR spectra were recorded at room temperature using a Bruker AV 400 Ultrashield spectrometer. 1H NMR and 13C NMR chemical shifts are reported in parts per million (ppm) downfield from tetramethylsilane, 19F NMR chemical shifts were determined relative to CFCl3. Coupling constants (J) are reported in hertz (Hz). The residual solvent peak was used as the internal reference: 1H NMR (CDCl3 7.26 ppm), 13C NMR (CDCl3 77.0 ppm). The following abbreviations were used to explain the multiplicities: s = singlet, d = doublet, t = triplet, m = multiplet, br = broad, dd = double doublet, dt = double triplet, and tt = triple triplet. In some spectra, the residual water signal can be seen as a singlet around 1.56 ppm. The melting points of crystalline compounds were determined using a Büchi apparatus in open tubes and are uncorrected. The photochemical reaction was performed using a 15 W Kessel blue lamp (wavelength 456 nm), a fan to keep the reaction temperature below 30 °C and the solvent previously degassed, following the standard literature protocol.37 Zinc powder was “activated” before use by washing it with 2 M HCl aqueous solution (3 times), and then with distilled water, absolute ethanol and diethyl ether, before drying under vacuum. Chromatographic purifications were performed by flash chromatography (FC), using Merck Silica gel 60.General procedure for the synthesis of compounds 3 and 4 (Scheme 2)
Conditions A: under air, in a round-bottom flask, the desired compound 1 (1.50 mmol, 1 eq.) and indium powder (350 mg, 3.00 mmol, 2 eq.) were suspended in methanol (15.0 mL, 0.1 M). Allyl-bromide (0.27 mL, 3.00 mmol, 2 eq.) was added and the reaction mixture was stirred at 40 °C for 3 hours. The reaction was cooled to room temperature and partitioned between saturated aqueous NH4Cl solution and ethyl acetate. The water phase was extracted with ethyl acetate 3 times; then the combined organic phases were dried over anhydrous sodium sulphate and the solvent was removed under reduced pressure. The crude reaction mixture was purified by FC (hexane/ethyl acetate) to afford pure compound 3.Conditions B: in a round-bottom flask, the desired compound 1 or 2 (1.50 mmol, 1 eq.) and activated zinc powder (490 mg, 7.50 mmol, 5 eq.) were suspended in dry THF (4.5 mL, 0.33 M) and cooled to 0 °C. Allyl bromide (0.30 mL, 3.30 mmol, 2.2 eq.) was added and the reaction mixture was stirred at 0 °C for 5 minutes. Saturated aqueous NH4Cl solution (4.5 mL) was added at 0 °C and the reaction mixture was stirred for 30 minutes at room temperature; it was then extracted with ethyl acetate 4 times. The combined organic phases were dried over anhydrous sodium sulphate and the solvent was removed under reduced pressure. The crude reaction mixture was purified by FC (hexane/ethyl acetate) to afford pure compound 3 or 4.
General procedure for the synthesis of compounds 5 (Schemes 4 and 5)
In a round-bottom flask, the desired compound 3 or 4 (0.25 mmol, 1 eq.), sodium tert-butoxide (31 mg, 0.32 mmol, 1.3 eq.), tris(dibenzylideneacetone)dipalladium (0) (9 mg, 0.01 mmol, 0.04 eq.), XPhos (19 mg, 0.04 mmol, 0.16 eq.) and the desired aryl or alkenyl bromide (0.32 mmol, 1.3 eq.) were dissolved in dry toluene (5 mL, 0.05 M) and stirred at 90 °C until the complete consumption of the starting material (generally 3–5 hours, monitored by TLC hexane/ethyl acetate 7:3 or dichloromethane/ethyl acetate 9:1, two new spots were observed). The reaction mixture was then cooled down to 30 °C and then treated under conditions A or B depending on the starting compound (conditions A were used for compounds 3, while conditions B were used for compounds 4).
Conditions A: HCl (4 N in dioxane, 5 mL) was added and the reaction mixture was stirred at 30 °C until the newly observed two spots disappear (monitored by TLC).
Conditions B: anhydrous, finely-ground, ammonium formate (1.58 g, 25.0 mmol, 100 eq.) was added, followed by dry methanol (15 mL) and 10% palladium on carbon (121 mg). The reaction mixture was stirred at 30 °C until the newly observed two spots disappear (monitored by TLC).
After that, copper sulphate pentahydrate (370 mg, 1.50 mmol, 6 eq.) was dissolved in water (3 mL, 0.5 M) and the solution was added to the reaction mixture. The reaction mixture was stirred at 30 °C for 2 hours; then the reaction was diluted with brine (50 mL) and extracted with ethyl acetate (4 × 50 mL). The combined organic phases were dried over anhydrous sodium sulphate and the solvent removed under reduced pressure. The crude product was purified by FC (hexane/ethyl acetate) to afford pure compounds 5, as diastereoisomer a (the second one to elute) and diastereoisomer b (the first one to elute).
The same procedure was applied for the gram-scale reactions.
Synthesis of compound 6
In a round-bottom flask, compound 3g (88 mg, 0.25 mmol, 1 eq.), sodium tert-butoxide (31 mg, 0.32 mmol, 1.3 eq.), tris(dibenzylideneacetone)dipalladium (0) (9 mg, 0.01 mmol, 0.04 eq.), XPhos (19 mg, 0.04 mmol, 0.16 eq.) and 4-Br-biphenyl (76 mg, 0.32 mmol, 1.3 eq.) were dissolved in dry toluene (5 mL, 0.05 M) and stirred at 90 °C for 3 hours. The reaction was cooled down to room temperature; then the reaction was diluted with brine (50 mL) and extracted with ethyl acetate (4 × 50 mL). The combined organic phases were dried over anhydrous sodium sulphate and the solvent was removed under reduced pressure. The crude product was purified by FC (hexane/ethyl acetate 60:40) to afford pure compound 6 as a pale-yellow sticky gum (77 mg, 61%); 1H NMR (400 MHz, CDCl3) δ 7.54–7.49 (m, 4H), 7.39 (t, J = 7.6 Hz, 2H), 7.32–7.28 (m, 3H), 7.15 (t, J = 8.1 Hz, 1H), 6.82 (d, J = 8.1 Hz, 1H), 6.61 (d, J = 8.1 Hz, 1H), 5.00, 4.75 (br m, 2H), 3.19–3.08 (m, 2H), 3.02 (s, 3H), 2.71–2.66 (m, 1H), 2.26–2.21 (m, 1H), 1.54 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 175.4, 156.3, 146.4, 140.9, 139.5, 136.9 (2C), 131.3, 130.5 (2C), 128.8 (2C), 127.3 (2C), 127.2, 127.0 (2C), 123.4, 121.0, 107.1, 80.6, 67.2, 60.3, 40.1, 38.1, 28.5 (3C), 26.1; HRMS (ESI) calcd for C29H30N3ClO3Na [M + Na]+ 526.1868 (35-Cl) found 526.1873.
Synthesis of compounds 7
In a round-bottom flask, the desired diastereoisomer of compound 5m (85 mg, 0.15 mmol, 1 eq.) was dissolved in dry dichloromethane (7.2 mL, 0.02 M) and anisole (320 μL, 3.0 mmol, 20 eq.); then the reaction mixture was cooled to 0 °C using an ice bath. Trifluoroacetic acid (0.8 mL) was added dropwise, then the ice bath was removed, and the reaction was stirred at room temperature for 2 hours (the reaction colour changes from light-yellow to dark-yellow). The volatiles were removed under reduced pressure and the crude reaction mixture was directly purified by FC (hexane/ethyl acetate 70:30) to afford the desired pure compound 7 as a pale-yellow powder.
General procedure for the synthesis of compounds 8
Under argon, in a sealed tube, the desired compound 5 or 7 (0.20 mmol, 1 eq.) was charged as a mixture of the two diastereoisomers, followed by the addition of anhydrous magnesium iodide (65 mg, 0.24 mmol, 1.2 eq.) and triethylamine hydrochloride (169 mg, 1.20 mmol, 6.0 eq.). The tube was closed, evacuated and back-filled with argon; then a 3:1 mixture of dry THF and dry DMF (0.5 mL, 0.4 M) was added and the reaction mixture was stirred at 130 °C for 48 hours. The reaction was cooled down to room temperature, before being quenched with saturated aqueous NH4Cl solution (5 mL), followed by the addition of few milligrams of Na2S2O3, to destroy any remaining traces of iodine. The reaction mixture was then partitioned between water (10 mL) and ethyl acetate (15 mL), the organic phase was removed, and the aqueous phase extracted again with ethyl acetate (4 × 15 mL). The combined organic phases were washed with water (10 mL) and brine (10 mL) and dried over anhydrous Na2SO4 and the solvents were removed under reduced pressure. The crude product was purified by FC (hexane/ethyl acetate) to afford the desired pure compound 8.
Synthesis of compound 9
In a round-bottom flask, compound 8a (73 mg, 0.15 mmol, 1 eq.) was dissolved in dry DMF (3.0 mL, 0.05 M) and cooled down to 0 °C. Sodium hydride 60% suspension in mineral oil (10 mg, 0.18 mmol, 1.2 eq.) was added and the reaction was stirred at 0 °C for 20 minutes. Selectfluor (70 mg, 0.20 mmol, 1.3 eq.) was added at 0 °C, then the reaction was stirred at room temperature for 10 minutes before being heated up to 60 °C for 1 hour. Upon completion (monitored by TLC), the reaction was cooled down to room temperature, quenched with saturated NH4Cl aq. solution (10 mL) and extracted with ethyl acetate (3 × 20 mL). The combined organic phases were washed with water and brine (2 times) and dried over anhydrous Na2SO4 and the volatiles were removed under reduced pressure. The crude product was purified by FC (hexane/ethyl acetate 90:10) to afford pure compound 9 as a dark-yellow film (62 mg, 83%); 1H NMR (400 MHz, CDCl3) δ 7.71 (br d, J = 7.5 Hz, 1H), 7.67 (br d, J = 7.5 Hz, 1H), 7.59 (d, J = 7.4 Hz, 2H), 7.54 (d, J = 8.4 Hz, 2H), 7.45 (t, J = 7.4 Hz, 2H), 7.35 (t, J = 7.4 Hz, 1H, partially overlapped with adjacent C–H signal), 7.33 (d, J = 8.4 Hz, 2H), 7.24 (t, J = 7.5 Hz, 1H), 7.15–7.03 (m, 5H), 6.54 (d, J = 16.2 Hz, 1H), 5.99–5.91 (m, 1H), 5.25 (d, J = 17.2 Hz, 1H), 5.10 (d, J = 17.2 Hz, 1H), 3.28–3.21 (m, 1H), 3.16–3.06 (m, 1H); 13C NMR (101 MHz, CDCl3) δ 173.8 (d, J2 = 20.3 Hz), 141.7, 140.6 (d, J2 = 22.5 Hz), 136.3 (2C), 135.4, 135.3, 129.3 (q, J3 = 6.0 Hz), 128.8 (2C), 128.7, 128.4 (2C), 127.4, 127.3 (2C), 126.9 (4C), 126.8 (2C), 125.5 (2C), 123.0 (q, J1 = 272.7 Hz), 122.9, 119.1 (d, J3 = 8.9 Hz), 113.6 (q, J2 = 33.2 Hz), 90.1 (d, J1 = 189.2 Hz), 45.7 (q, J = 4.9 Hz), 38.8 (d, J2 = 28.7 Hz); 19F NMR (376 MHz, CDCl3) δ −55.00 (s, 3F), −152.87 (dd, J3 = 10.4 Hz, J3 = 15.7 Hz, 1F); HRMS (ESI) calcd for C31H23NF4ONa [M + Na]+ 524.1608 found 524.1611.
Synthesis of compound 10
In a round-bottom flask, compound 8b (57 mg, 0.10 mmol, 1 eq.), cesium carbonate (98 mg, 0.30 mmol, 3 eq.) and 5-bromo-2-(chloromethyl)benzofuran (80 mg, 0.30 mmol, 3.0 eq.) were dissolved in dry DMF (2 mL, 0.05 M). The reaction was heated up to 100 °C and stirred for 18 hours (note: compound 8b and compound 10 have almost the same Rf in hexane/ethyl acetate 80:20). After that, the reaction was cooled down to room temperature, diluted with saturated NH4Cl aq. solution (10 mL) and extracted with ethyl acetate (3 × 25 mL). The combined organic phases were washed with water and brine (2 times) and dried over anhydrous Na2SO4 and the volatiles were removed under reduced pressure. The crude product was purified by FC (hexane/ethyl acetate 90:10) to afford pure compound 10 as a light-yellow foam (52 mg, 67%); 1H NMR (400 MHz, CDCl3) δ 7.59 (br d, J = 7.3 Hz, 2H), 7.52–7.49 (m, 3H), 7.45 (t, J = 7.3 Hz, 2H), 7.40–7.35 (m, 2H), 7.29–7.25 (m, 4H), 7.16 (d, J = 7.4 Hz, 6H), 7.06 (t, J = 7.4 Hz, 3H), 7.00 (dt, J3 = 7.7 Hz, J4 = 1.0 Hz, 1H), 6.94 (t, J = 7.4 Hz, 6H), 6.85 (dt, J3 = 7.7 Hz, J4 = 1.2 Hz, 1H), 6.49 (d, J = 15.9 Hz, 1H), 6.18 (d, J3 = 8.2 Hz, 1H), 5.89–5.83 (m, 1H), 5.82 (d, J4 = 0.8 Hz, 1H), 3.44 (d, J = 14.8 Hz, 1H), 3.31 (d, J = 14.8 Hz, 1H), 2.96 (dd, J2 = 13.5 Hz, J3 = 8.7 Hz, 1H), 2.85 (dd, J2 = 13.5 Hz, J3 = 6.6 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 178.7, 155.8, 153.2, 143.6, 142.4, 141.8 (3C), 140.8, 140.3, 136.0, 134.0, 130.6, 129.5, 129.4 (6C), 128.8 (2C), 127.5, 127.3 (6C), 127.2 (2C), 126.9 (2C), 126.83, 126.78 (2C), 126.7 (2C), 126.4, 123.8, 123.3, 122.9, 122.0, 116.1, 115.6, 112.4, 104.3, 74.5, 52.9, 42.4, 36.4; HRMS (ESI) calcd for C51H38NO2BrNa [M + Na]+ 798.1978 (79-Br) found 798.1984.
Synthesis of compound 11
Under argon, in a sealed tube, compound 10 (77 mg, 0.10 mmol, 1 eq.) was charged followed by 4-CzIPN (0.8 mg, 0.001 mmol, 1% mol). The tube was closed, evacuated and back-filled with argon; then freshly degassed acetone (1.0 mL, 0.1 M) was added, the tube closed, and the reaction stirred for 2 days under blue light irradiation (456 nm, 15 W) at a temperature below 30 °C (reaction changes from limpid yellow to turbid light-yellow). Upon completion, the solvent was evaporated under reduced pressure and the crude product was dissolved in a small amount of DCM and subjected to FC (hexane/ethyl acetate 95:5) to afford pure compound 11 as an off-white powder (46 mg, 60%), m.p. 252–254 °C; 1H NMR (400 MHz, CDCl3) δ 7.57 (br d, J = 7.6 Hz, 2H), 7.49–7.46 (m, 7H), 7.45–7.40 (m, 3H), 7.30–7.27 (m, 7H), 7.25–7.18 (m, 5H), 7.02–6.98 (m, 3H), 6.88 (dt, J3 = 7.7 Hz, J4 = 1.6 Hz, 1H), 6.75 (d, J = 8.4 Hz, 1H), 6.52 (d, J4 = 1.7 Hz, 1H), 6.28 (d, J = 8.4 Hz, 1H), 4.35 (d, J = 8.4 Hz, 1H), 3.93–3.89 (m, 1H), 3.53–3.48 (m, 1H), 2.90 (d, J = 14.7 Hz, 1H), 2.53 (d, J = 14.7 Hz, 1H), 2.44–2.42 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 181.3, 159.8, 142.2 (3C), 142.1, 140.9, 139.7, 138.1, 135.0, 131.1, 130.9, 130.4, 129.2 (5C), 128.7 (2C), 128.4 (2C), 127.9 (2C), 127.7 (5C), 127.2, 127.1 (2C), 126.9 (3C), 126.8 (2C), 126.5, 122.7, 122.4, 115.6, 112.4, 111.4, 97.3, 74.2, 58.4, 52.5, 51.0, 47.1, 45.1, 44.9; HRMS (ESI) calcd for C51H38NO2BrNa [M + Na]+ 798.1978 (79-Br) found 798.1973.
Author contributions
M. M. and A. S. conceived and supervised the project. M. M., T. V. and G. M. performed the experiments, the purification of the products and their full characterization. A. S., M. M. and G. M. wrote the manuscript and the ESI.† All the authors have proofread the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.References
- A. P. Sakla, P. Kansal and N. Shankaraiah, Eur. J. Org. Chem., 2021, 757–772 CrossRef CAS.
- J. Salaun, Top. Curr. Chem., 2000, 207, 1–67 CrossRef CAS.
- T. T. Talele, J. Med. Chem., 2016, 59, 8712–8757 CrossRef CAS PubMed.
- K. X. Rodriguez, E. N. Howe, E. P. Bacher, M. Burnette, J. L. Meloche, J. Meisel, P. Schnepp, X. Tan, M. Chang, J. Zartman, S. Zhang and B. L. Ashfeld, ChemMedChem, 2019, 14, 1653–1661 CrossRef CAS PubMed.
- R. Tokala, S. Thatikonda, U. S. Vanteddu, S. Sana, C. Godugu and N. Shankaraiah, ChemMedChem, 2018, 13, 1909–1922 CrossRef CAS PubMed.
- N. Ye, H. Chen, E. A. Wold, P. Y. Shi and J. Zhou, ACS Infect. Dis., 2016, 2, 382–392 CrossRef CAS PubMed.
- V. V. Vintonyak, K. Warburg, H. Kruse, S. Grimme, K. Hubel, D. Rauh and H. Waldmann, Angew. Chem., Int. Ed., 2010, 49, 5902–5905 CrossRef CAS PubMed.
- C. L. Woodard, Z. Li, A. K. Kathcart, J. Terrell, L. Gerena, M. Lopez-Sanchez, D. E. Kyle, A. K. Bhattacharjee, D. A. Nichols, W. Ellis and S. T. Prigge, J. Med. Chem., 2003, 46, 3877–3882 CrossRef CAS.
- (a) C. V. Galliford and K. A. Scheidt, Angew. Chem., Int. Ed., 2007, 46, 8748–8758 CrossRef CAS PubMed; (b) L. Hong and R. Wang, Adv. Synth. Catal., 2013, 355, 1023–1052 CrossRef CAS; (c) L. Hoffmann, V. Demoulin, D. Klein, D. Daloze and J. C. Braekman, J. Nat. Prod., 1995, 58, 1781–1785 CrossRef; (d) B. Trost and M. Brennan, Synthesis, 2009, 3003–3025 CrossRef CAS.
- P. B. Sampson, Y. Liu, B. Forrest, G. Cumming, S. W. Li, N. K. Patel, L. Edwards, R. Laufer, M. Feher, F. Ban, D. E. Awrey, G. Mao, O. Plotnikova, R. Hodgson, I. Beletskaya, J. M. Mason, X. Luo, V. Nadeem, X. Wei, R. Kiarash, B. Madeira, P. Huang, T. W. Mak, G. Pan and H. W. Pauls, J. Med. Chem., 2015, 58, 147–169 CrossRef CAS PubMed.
- G. S. Cockerill, R. M. Angell, A. Bedernjak, I. Chuckowree, I. Fraser, J. Gascon-Simorte, M. S. A. Gilman, J. A. D. Good, R. Harland, S. M. Johnson, J. H. Ludes-Meyers, E. Littler, J. Lumley, G. Lunn, N. Mathews, J. S. McLellan, M. Paradowski, M. E. Peeples, C. V. Barber, S. E. Ward, D. Watterson, G. Williams, P. Young and K. Powell, J. Med. Chem., 2021, 64, 3658–3676 CrossRef CAS PubMed.
- (a) Z. Zhang, Z. Wang, K. Huang, Y. Liu, C. Wei, J. Zhou, W. Zhang, Q. Wang, H. Liang, A. Zhang, G. Wang, Y. Zhen and L. Han, Cancer Lett., 2019, 443, 91–107 CrossRef CAS; (b) T. Jiang, K. L. Kuhen, K. Wolff, H. Yin, K. Bieza, J. Caldwell, B. Bursulaya, T. Y.-H. Wu and Y. He, Bioorg. Med. Chem. Lett., 2006, 16, 2105–2108 CrossRef CAS PubMed.
- S. Hajra, S. Roy and S. A. Saleh, Org. Lett., 2018, 20, 4540–4544 CrossRef CAS PubMed.
- X. Feng, L. Hu, H. Mei, W. Cao and L. Wang, Adv. Synth. Catal., 2018, 360, 4089–4093 CrossRef.
- W.-C. Yuan, J.-Q. Zhao, Z.-H. Wang, B.-X. Quan and Y. You, Org. Biomol. Chem., 2020, 18, 4560–4565 RSC.
- X. Feng, L. Lin, X. Liu, X. Li, Y. Liu and J. Guo, Chem. Sci., 2016, 74, 2717–2721 Search PubMed.
- P. K. Warghude, P. D. Dharpure and R. G. Bhat, Tetrahedron Lett., 2018, 59, 4076–4079 CrossRef CAS.
- Z. Meng, Q. Wang, D. Lu, T. Yue, P. Ai, H. Liu, W. Yang and J. Zheng, J. Org. Chem., 2020, 85, 15026–15037 CrossRef CAS PubMed.
- J. S. Kapure, C. N. Reddy, P. R. Adiyala, R. Nayak, V. L. Nayak, J. B. Nanubolu, K. K. Singarapu and R. A. Maurya, RSC Adv., 2014, 4, 38425–38432 RSC.
- T.-R. Li, S.-W. Duan, W. Ding, Y.-Y. Liu, J.-R. Chen, L.-Q. Lu and W.-J. Xiao, J. Org. Chem., 2014, 79, 2296–2302 CrossRef CAS PubMed.
- A. Rastelli, R. Gandolfi and M. S. Amadè, J. Org. Chem., 1998, 63, 7425–7436 CrossRef CAS PubMed.
- Z.-Y. Cao, F. Zhou, Y.-H. Yu and J. A. Zhou, Org. Lett., 2013, 15, 42–45 CrossRef CAS PubMed.
- Z.-Y. Cao, X. Wang, C. Tan, X.-L. Zhao, J. Zhou and K. Ding, J. Am. Chem. Soc., 2013, 135, 8197–8200 CrossRef CAS PubMed.
- M. Tone, Y. Nakagawa, S. Chanthamath, I. Fujisawa, N. Nakayama, H. Goto, K. Shibatomi and S. Iwasa, RSC Adv., 2018, 8, 39865–39869 RSC.
- S. Muthusamy and R. Ramkumar, Tetrahedron Lett., 2014, 55, 6389–6393 CrossRef CAS.
- S. Zhao, X.-X. Chen, N. Gao, M. Qian and X. Chen, J. Org. Chem., 2021, 86, 7131–7140 CrossRef CAS PubMed.
- S. P. Green, K. M. Wheelhouse, A. D. Payne, J. P. Hallett, P. W. Miller and J. A. Bull, Org. Process Res. Dev., 2020, 17, 67–84 CrossRef PubMed.
- S. Gazzotti, M. Manenti, L. Lo Presti and A. Silvani, RSC Adv., 2019, 9, 37788–37800 RSC.
- J. P. Wolfe and N. C. Giampietro, J. Am. Chem. Soc., 2008, 130, 12907–12911 CrossRef.
- L. Firmansjah and G. C. Fu, J. Am. Chem. Soc., 2007, 129, 11340–11341 CrossRef CAS PubMed.
- (a) R. J. Crawford and A. Mishra, J. Am. Chem. Soc., 1966, 88(17), 3963–3969 CrossRef CAS; (b) M. Hamaguchi, M. Nakaishi, T. Nagai, T. Nakamura and M. Abe, J. Am. Chem. Soc., 2007, 129(43), 12981–12988 CrossRef CAS PubMed.
- P. B. Alper, C. Meyers, A. Lerchner, D. R. Siegel and E. M. Carreira, Angew. Chem., Int. Ed., 1999, 38(21), 3186–3189 CrossRef CAS PubMed.
- C. Fischer, C. Meyers and E. M. Carreira, Helv. Chim. Acta, 2000, 83, 1175–1181 CrossRef CAS.
- A. Lerchner and E. M. Carreira, J. Am. Chem. Soc., 2002, 124(50), 14826–14827 CrossRef CAS.
- Z. Zhang, W. Zhang, F. Kang, F. C. Ip and N. Y. Ip, J. Org. Chem., 2019, 84(17), 11359–11365 CrossRef CAS PubMed.
- T. F. Schneider, J. Kaschel and D. B. Werz, Angew. Chem., Int. Ed., 2014, 53(22), 5504–5523 CrossRef CAS PubMed.
- G. Zhen, G. Zeng, F. Wang, X. Cao and B. Yin, Adv. Synth. Catal., 2023, 365, 43–52 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H NMR, 13C NMR and 19F NMR spectra of all new compounds, the 1H NMR and NOESY studies conducted on compounds 5, 7 and 11 and CIF files for compounds 7b and 11. CCDC 2307637 and 2307638. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ob02115g |
| This journal is © The Royal Society of Chemistry 2024 |