
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of the full-length hepatitis B virus core protein and its capsid formation†
Keisuke
Aoki
ab,
Shugo
Tsuda
 c,
Naoko
Ogata
b,
Michiyo
Kataoka
d,
Jumpei
Sasaki
a,
Shinsuke
Inuki
a,
Hiroaki
Ohno
a,
Koichi
Watashi
e,
Taku
Yoshiya
cf and
Shinya
Oishi
*ab
c,
Naoko
Ogata
b,
Michiyo
Kataoka
d,
Jumpei
Sasaki
a,
Shinsuke
Inuki
a,
Hiroaki
Ohno
a,
Koichi
Watashi
e,
Taku
Yoshiya
cf and
Shinya
Oishi
*ab
aGraduate School of Pharmaceutical Sciences, Kyoto University, Sakyo-ku, Kyoto 606-8501, Japan
bLaboratory of Medicinal Chemistry, Kyoto Pharmaceutical University, Yamashina-ku, Kyoto 607-8412, Japan. E-mail: soishi@mb.kyoto-phu.ac.jp; Tel: +81-75-595-4635
cPeptide Institute, Inc. Ibaraki, Osaka 567-0085, Japan
dDepartment of Pathology, National Institute of Infectious Disease, Shinjuku-ku, Tokyo 162-8640, Japan
eResearch Center for Drug and Vaccine Development, National Institute of Infectious Diseases, Shinjuku-ku, Tokyo 162-8640, Japan
fInstitute for Protein Research, Osaka University, Suita, Osaka 565-0871, Japan
First published on 15th February 2024
Abstract
Chronic infection with hepatitis B virus (HBV) is a major cause of cirrhosis and liver cancer. Capsid assembly modulators can induce error-prone assembly of HBV core proteins to prevent the formation of infectious virions, representing promising candidates for treating chronic HBV infections. To explore novel capsid assembly modulators from unexplored mirror-image libraries of natural products, we have investigated the synthetic process of the HBV core protein for preparing the mirror-image target protein. In this report, the chemical synthesis of full-length HBV core protein (Cp183) containing an arginine-rich nucleic acid-binding domain at the C-terminus is presented. Sequential ligations using four peptide segments enabled the synthesis of Cp183 via convergent and C-to-N direction approaches. After refolding under appropriate conditions, followed by the addition of nucleic acid, the synthetic Cp183 assembled into capsid-like particles.
Introduction
Chronic infection with hepatitis B virus (HBV) is a leading cause of cirrhosis and hepatocellular carcinoma.1 Although an effective vaccine is available, approximately 290 million people worldwide have chronic hepatitis B, and more than 800![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 people die annually from HBV-related diseases, as estimated by the World Health Organization.2 Several drugs have been approved for treating chronic HBV infections, including nucleoside and nucleotide analogs targeting the viral reverse transcriptase and interferon-α to stimulate host immunity.1 These agents are effective in inhibiting HBV replication and suppressing hepatitis; however, the treatment is not curative because of their inability to eliminate covalently closed circular DNA (cccDNA), which is the reservoir for persistent HBV infection.3 Therefore, developing novel therapeutic agents via an alternative mechanism(s) is needed.4
000 people die annually from HBV-related diseases, as estimated by the World Health Organization.2 Several drugs have been approved for treating chronic HBV infections, including nucleoside and nucleotide analogs targeting the viral reverse transcriptase and interferon-α to stimulate host immunity.1 These agents are effective in inhibiting HBV replication and suppressing hepatitis; however, the treatment is not curative because of their inability to eliminate covalently closed circular DNA (cccDNA), which is the reservoir for persistent HBV infection.3 Therefore, developing novel therapeutic agents via an alternative mechanism(s) is needed.4
The HBV core protein, a building block of nucleocapsids, plays essential roles during multiple stages of the viral replication cycle, including nucleocapsid assembly, encapsidation of the pregenomic RNA (pgRNA), reverse transcription, and DNA synthesis.5,6 Many compounds targeting the HBV core protein have been identified.7–10 These compounds are classified into two categories based on the post-treatment capsid states. Class I compounds (e.g., heteroaryldihydropyrimidines) induce misfolding of the core protein structure,11 whereas class II compounds (e.g., phenylpropenamides and sulfamoylbenzamides) accelerate the formation of empty capsids without pgRNA.12 Both of these capsid assembly modulators (CAMs) promote error-prone assembly of core proteins, leading to the prevention of infectious virion formation. Thus, the HBV core protein is a promising target for developing anti-HBV agents.13 Some CAMs targeting the HBV core protein are currently in phase 1 and 2 clinical trials for treating HBV infection.4
We have developed a mirror-image screening system to discover drug candidates from unexplored mirror-image structures of natural products.14 Mirror-image libraries of natural products are valuable resources for therapeutic and diagnostic agents because they should have drug-like properties identical to those of natural products, including unique, complex, and sp3-carbon-rich scaffolds.15 In this approach, the bioactivity of the existing library of chiral natural products is evaluated using a synthetic mirror-image target protein (D-target). The enantiomer of the resulting hit molecule(s) should be active toward the native target protein. We previously identified a novel inhibitor against mouse double minute 2 homolog (MDM2)-p53 from a mirror-image library of natural products.14 This technology has also been applied to other therapeutic targets, including Grb2 SH2, Src SH2 and XIAP BIR3 domain-containing proteins.16–18 To identify novel drug candidates to treat chronic HBV infection by mirror-image screening, herein, we focused on the HBV core protein as the next target (ESI Fig. S1†). Developing synthetic and folding protocols of the core protein to obtain functional proteins is essential for mirror-image screening because mirror-image proteins cannot be prepared by recombinant technology.
The full-length HBV core protein (Cp183) consists of an assembly domain (Cp149: Met1–Val149) for capsid formation and a C-terminal arginine-rich nucleic acid-binding domain (Arg150–Cys183) (Fig. 1).19,20 Cp149 was reported to be expressed in Escherichia coli and spontaneously assemble to form capsid particles.21 The Cp149 capsids disassemble into core protein dimers in the presence of guanidine or urea, whereas the reassembly of empty Cp149 capsids was triggered by increasing the ionic strength.22,23 Previously, we reported the chemical synthesis of the C-terminal truncated HBV core protein (Cp149).24 However, unlike recombinantly expressed Cp149, the preparation of capsid particles from the synthetic Cp149 failed.25 We postulated that nucleic acid association with the C-terminal nucleic acid-binding domain promotes in vitro assembly to form capsid particles from synthetic proteins.26 Based on these previous findings, we selected the full-length core protein (Cp183) as an alternative synthetic target. In this study, we describe the synthetic process and in vitro capsid assembly of the full-length HBV core protein.
 | ||
| Fig. 1 Domain structure and sequence of the HBV core protein (Cp183). (A) Schematic representation of the HBV core protein domain architecture, showing the assembly domain (amino acids 1–149) and the nucleic acid-binding domain (amino acids 150–183). (B) Sequence of the HBV core protein (subtype adyw).19 Cysteine residues are highlighted in yellow. The underlined sequence is the nucleic acid-binding domain. | ||
Results and discussion
Design of the synthetic process for Cp183
Cp183 contains four cysteine residues (Cys48, Cys61, Cys107, and Cys183) in the 183-residue sequence (Fig. 1). Among these, N-proximal cysteines (Cys48, Cys61, and Cys107) in the assembly domain form intermolecular disulfide bonds to yield Cp183 dimers,27,28 whereas Cys183 in the nucleic acid-binding domain is not required for capsid assembly.29 Thus, we substituted Cys183 with Ala to avoid the potential side reaction(s), including epimerization30 and β-piperidinyl alanine formation31 at this Cys residue during peptide synthesis. Additionally, in our previous synthetic study of Cp149, we had to attach a trityl-based solubilizing tag (Trt-K10) to a Cys residue because the poor solubility of the peptide segments hampered the ligation and purification processes (ESI Fig. S2†).24 We hypothesized that the C-terminal arginine-rich domain in the Cp183 sequence should enhance the solubility of the peptide segments equally as well as the previously developed solubilizing tags containing multiple Arg residues.32,33Native chemical ligation (NCL) is a gold standard approach to chemically synthesize large proteins, in which two unprotected peptide segments are efficiently ligated under mild conditions.34,35 Metal-free desulfurization (MFD) is another essential technique for chemical protein synthesis, which converts the temporarily mutated Cys to Ala in aqueous buffer.36 Several strategies have also been developed to extend the scope of NCL to various ligation junctions beyond Cys or Ala; however, ligation auxiliaries should be prepared in advance for practical applications.37,38 We planned to use the Ag-free thioester method39 because the C-terminal sequence (Arg150–Ala183) of Cp183(C183A) has no Cys or Ala residues in appropriate positions for NCL. This method allows direct amide bond formation between a thioester and amine in the presence of protecting groups for other amino and thiol groups. However, we posited that the ligation reaction proceeds selectively between unprotected peptide segments in this case because the C-terminal sequence of Cp183(C183A) contains no Lys and Cys residues. We selected Gly153–Arg154 as the ligation site to prevent possible epimerization during ligation.
Synthesis of Cp183(C183A) by the convergent route
Initially, we planned to synthesize Cp183(C183A) by a convergent ligation strategy from four peptide segments using the intermediate segments for Cp149 synthesis.24 We designed four peptide segments with appropriate accessory groups, which were split at Cys61, Cys107, and Arg154 of Cp183 (Fig. 2): the N1 segment 1 (Cp1831–60) with an alkyl thioester at the C-terminus; the N2 segment 2a (Cp18361–106) with an activable first-generation Dawson linker (Dbz)40 at the C-terminus; the C1 segment 3 ([Thz107]-Cp183107–153) with a thiazolidine carboxylic acid (Thz) at the N-terminus for temporary protection of Cys and an aryl thioester at the C-terminus; and the C2 segment 4 ([Ala183]-Cp183154–183). The N1 segment 1 and N2 segment 2a were synthesized by Boc-SPPS and Fmoc-SPPS, respectively, according to our previous report on the synthesis of Cp149.24 For the preparation of C1 segment 3, the protected peptide resin was synthesized with an o-amino(methyl)aniline (MeDbz)41 moiety, which was converted to the N-acyl-N′-methylacylurea (MeNbz) form by treatment with 4-nitrophenyl chloroformate and iPr2NEt. Global deprotection and cleavage from the resin followed by reaction with 4-mercaptophenylacetic acid (MPAA) provided peptide thioester 3. Similarly, the C2 segment 4 was obtained using the Fmoc-Ala-Wang resin in a satisfactory yield. | ||
| Fig. 2 Synthesis of Cp183(C183A) via the convergent approach. Reagents and conditions: (a) MPAA, TCEP, 6 M guanidine·HCl, and 200 mM phosphate buffer (pH 7.0); (b) NaNO2, 6 M guanidine·HCl, 200 mM phosphate buffer (pH 3.0), then MESNa and TCEP; (c) iPr2NEt, HOOBt, and DMSO; (d) methoxyamine, 6 M guanidine·HCl, and 200 mM phosphate buffer (pH 4.0). Abbreviations: Dbz, 3,4-diaminobenzoic acid; HOOBt, 3,4-dihydro-3-hydroxy-4-oxo-1,2,3-benzotriazine; MES, 2-mercaptethanesulfonate; MPA, 3-mercaptopropionic acid; MPAA, 4-mercaptophenylacetic acid; TCEP, tris(2-carboxyethyl)phosphine; Thz, thiazolidine carboxylic acid. | ||
With the four peptide segments in hand, we investigated the sequential ligations. According to our previous report,24 the N1 thioester segment 1 and N2 segment 2a were ligated in the presence of MPAA to give N-terminal half intermediate 5 (Cp1831–106). NaNO2-mediated activation32 of the Dbz linker in 5 followed by treatment with sodium 2-mercaptethanesulfonate (MESNa) provided thioester 6.42 Next, we performed the synthesis of the C-terminal half of Cp183(C183A). The MPAA thioester 3 and peptide 4 were dissolved in DMSO containing 3,4-dihydro-3-hydroxy-4-oxo-1,2,3-benzotriazine (HOOBt) and iPr2NEt (Fig. 3A).39 The coupling proceeded efficiently to provide thiazolidine-protected 7. Subsequent methoxyamine-mediated deprotection of thiazolidine43 afforded the desired C-terminal half intermediate 8. The final ligation between thioester 6 and peptide 8 proceeded smoothly to afford the desired full-length Cp183(C183A) 9 in 21% yield (Fig. 3B and C). Note that this NCL reaction proceeded under standard peptide concentration for NCL (∼1.0 mM) in aqueous buffer without organic solvents to give the expected Cp183(C183A), whereas NCL at the same position for the synthesis of Cp149 required low peptide concentrations (∼0.3 mM) in aqueous buffer containing 20% NMP to suppress the aggregation of peptides.24 These results suggest that the C-terminal arginine-rich domain contributed significantly to the high solubility of full-length protein and intermediate peptides. In this convergent route, we successfully constructed the full-length sequence of Cp183(C183A) (3.8% overall yield from peptide 1); however, the poor solubility of the hydrophobic intermediates (5 and 6) prompted us to seek an alternative synthetic strategy.
 | ||
| Fig. 3 HPLC trace of each synthetic step via the convergent approach and ESI-MS characterization of Cp183(C183A). (A) HPLC trace of the ligation of peptides 3 and 4 followed by Thz deprotection. HPLC conditions: Cosmosil 5C18-AR300 column (Nacalai Tesque, 4.6 × 250 mm), linear gradient of 10–70% CH3CN containing 0.1% TFA at a flow rate of 1 mL min−1 over 30 min at room temperature. (B) HPLC trace of the NCL between thioester 6 and peptide 8. HPLC conditions: DAISOPAK SP-120-5-ODS-BIO (Osaka Soda, 4.6 × 150 mm), linear gradient of 40–60% CH3CN containing 0.1% TFA at a flow rate of 1 mL min−1 over 20 min at 40 °C. (C) ESI-MS spectrum of Cp183(C183A). See ESI† for details. | ||
Synthesis of Cp183(C183A) by the C-to-N route
Next, we investigated the synthesis of Cp183(C183A) by a C-to-N ligation strategy to avoid using hydrophobic intermediates (Fig. 4). We hypothesized that the C-terminal arginine-rich domain would enhance the solubility of all intermediate peptide segments. Because the N1 segment 1 and C-terminal half 8 can be used for sequential ligations via the C-to-N direction, only the N2 segment 2b ([Thz61]-Cp18361–106) was newly designed and synthesized, which contains a Thz for N-terminal temporary protection and a MeNbz linker for C-terminal activation. The resulting peptides 2b and 8 were subjected to the MPAA-mediated NCL condition (Fig. 5A). The formation of Thz-protected intermediate 10 was observed when the starting material 2b was mostly consumed after a 3-hour incubation. The subsequent methoxyamine-mediated deprotection provided the Cys-free intermediate 11 in 24% overall yield (two steps from the peptide segment 2b). The final ligation between thioester 1 and peptide 11 proceeded smoothly to afford the desired full-length Cp183(C183A) 9 in 32% yield (Fig. 5B). In this C-to-N route, all intermediate peptides (i.e., 8 and 11) contained the arginine-rich sequence. Thus, these peptides were more hydrophilic than peptides 5 and 6 used for the convergent route in HPLC analysis. | ||
| Fig. 4 Synthesis of Cp183(C183A) via C-to-N native chemical ligations. Reagents and conditions: (a) MPAA, TCEP, 6 M guanidine·HCl, and 200 mM phosphate buffer (pH 7.0); (b) methoxyamine, 6 M guanidine·HCl, and 200 mM phosphate buffer (pH 4.0). Abbreviations: MeNbz, N-acyl-N′-methylacylurea. | ||
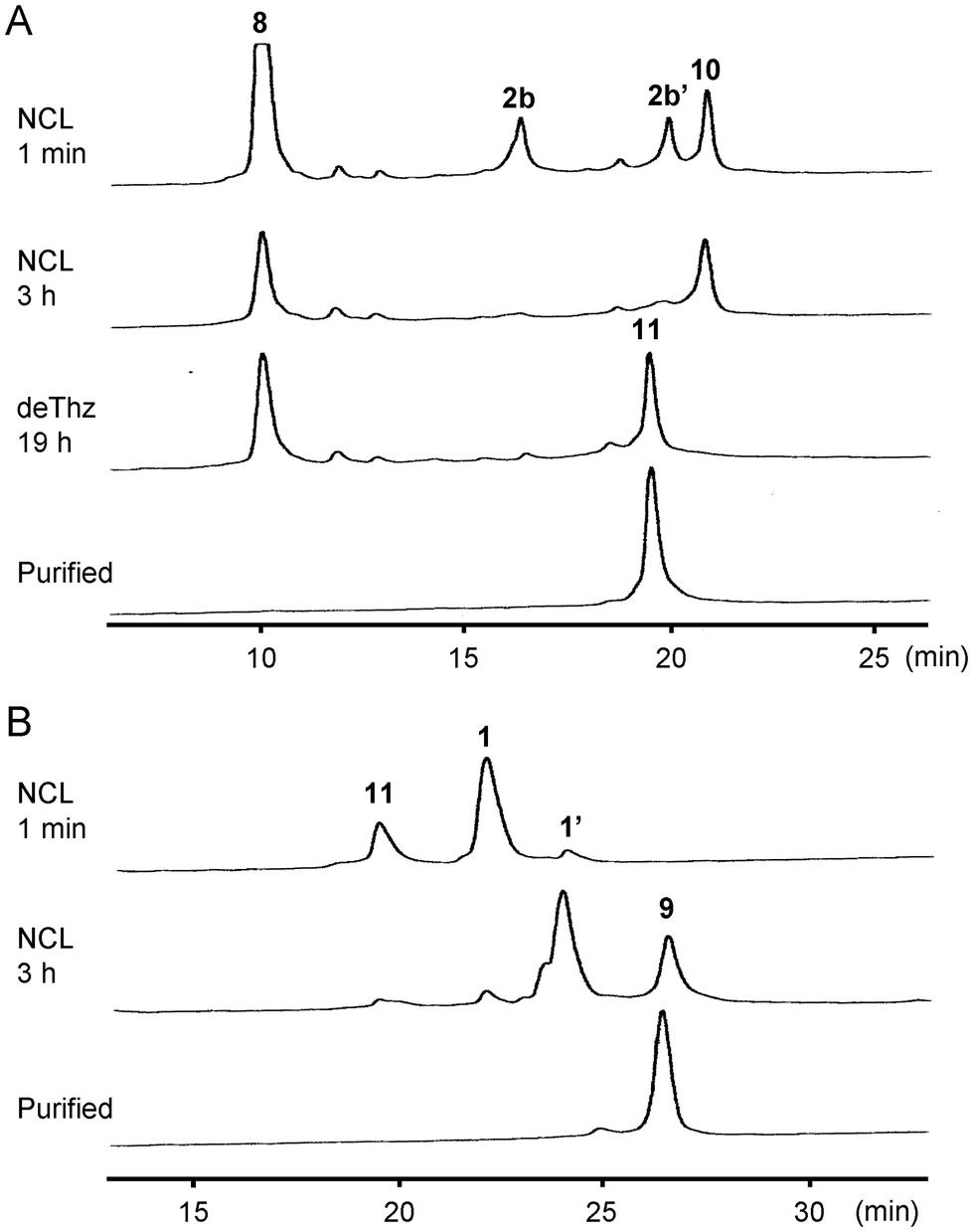 | ||
| Fig. 5 HPLC trace of each synthetic step for Cp183(C183A) via C-to-N native chemical ligations. (A) HPLC trace of the NCL between MeNbz-peptide 2b and 8 followed by Thz deprotection. Peptide 2b′: MPAA form of 2b. HPLC conditions: Cosmosil 5C4-AR300 column (Nacalai Tesque, 4.6 × 150 mm), linear gradient of 30–60% CH3CN containing 0.05% TFA at a flow rate of 1 mL min−1 over 30 min at 40 °C. (B) HPLC trace of the NCL between thioester 1 and peptide 11. Peptide 1′: MPAA form of 1. HPLC conditions: Cosmosil 5C4-AR300 column (Nacalai Tesque, 4.6 × 150 mm), linear gradient of 30–60% CH3CN containing 0.05% TFA at a flow rate of 1 mL min−1 over 30 min at 40 °C. | ||
Folding and capsid assembly of synthetic Cp183(C183A)
With full-length Cp183(C183A) in hand, we investigated the folding conditions to obtain the synthetic protein for capsid assembly. The predominant HBV capsid consists of 120 copies of the core protein dimer arranged in a T = 4 icosahedral symmetry.44 Zlotnick et al. reported that Cp183 expressed in E. coli assembles to form empty capsids or RNA-filled capsids in vitro under appropriate conditions, including ionic strength, Cp183 dimer concentration and the presence of nucleic acids.26 In contrast to the high stability of Cp149 even under low ionic strength conditions, the Cp183 dimer precipitated when the guanidine concentration was less than 0.5 M. According to the reported procedure for capsid assembly from recombinant Cp183,26 we initially investigated the assembly conditions from synthetic Cp183 for empty capsids. After lyophilized synthetic Cp183(C183A) was denatured in 6 M guanidine, the concentration of guanidine was gradually reduced using dialysis steps to 1.5 M.26 Circular dichroism (CD) analysis of Cp183(C183A) in 1.5 M guanidine exhibited the negative band at 222 nm indicative of the existence of an α-helix structure, which is consistent with the canonical secondary structure of the Cp183 monomer (ESI Fig. S3A†).45,46 The negative minimum at 222 nm disappeared when the concentration of guanidine was reduced further from 1.5 M. The negative band at 222 nm was not observed in 1.5 M NaCl (ESI Fig. S3B†). These results revealed that 1.5 M guanidine used in a previous report is optimal for stabilizing the Cp183 protein.26 We investigated several other conditions to prepare empty capsids in low-ionic-strength buffer, including the concentration of Cp183(C183A), the type of denaturing agents and the use of additives or molecular chaperons;46,47 however, synthetic Cp183(C183A) was highly prone to aggregation.We next investigated the in vitro assembly of nucleic acid-filled capsids, which contain single-strand DNA (ssDNA) in the particles (Fig. 6A). After preparing the folded Cp183(C183A) in 1.5 M guanidine using the abovementioned protocol, ssDNA was added to this protein solution. Subsequently, the mixture was diluted 3-fold (to 0.5 M guanidine) according to the assembly protocol for recombinant Cp183.26 Particle formation was observed by negative-stain electron microscopy (Fig. 6B). The size of the observed particles was 30–40 nm, which is consistent with the reported T = 3 (∼32 nm) or T = 4 (∼36 nm) HBV capsids.26 Notably, a high concentration of synthetic Cp183 for the assembly experiment resulted in aggregation without forming the expected particles.48 Although further optimization of assembly conditions in the presence of ssDNA is required, these results demonstrated that the synthetic full-length HBV core protein may be used for preparing homogenous samples for mirror-image screening. To our knowledge, this is the first report describing the preparation of HBV capsids from synthetic proteins.
 | ||
| Fig. 6 Assembly of capsids from synthetic Cp183(C183A). (A) Process for folding and assembly of capsids. (B) Negative-stain electron microscope image of viral capsid-like particles from synthetic Cp183(C183A). Conditions: 1.0 μM peptide 9 [synthetic Cp183(C183A)], 13 μM ssDNA, 0.5 M guanidine·HCl, 17 mM HEPES, 0.67 mM DTT, pH 7.5. | ||
Conclusions
In this report, we established a synthetic process for preparing the full-length HBV core protein (Cp183) using NCL and thioester methods. Full-length Cp183(C183A) was constructed by sequential ligations from four peptide segments by a convergent or C-to-N route. For synthesis using the C-to-N direction, the C-terminal arginine-rich domain improved the solubility of all intermediate peptides. The resulting synthetic Cp183(C183A) was appropriately refolded to form HBV capsid particles. The application of synthetic Cp183 to mirror-image screening for identifying novel capsid assembly modulators is ongoing.Experimental section
General procedure of peptide synthesis
All reagents and solvents were purchased from Watanabe Chemical Industries, Ltd (Hiroshima, Japan), Kokusan Chemical Industries, Ltd (Kanagawa, Japan), Sigma-Aldrich Japan (Tokyo, Japan), Wako Pure Chemical Industries, Ltd (Osaka, Japan), Tokyo Chemical Industry Co., Ltd (Tokyo, Japan) or Nacalai Tesque, Inc. (Kyoto, Japan). For analytical high-performance liquid chromatography (HPLC), a Cosmosil 5C18-AR300 column (4.6 × 250 mm, Nacalai Tesque, Inc.), Cosmosil 5C4-AR300 column (4.6 × 150 mm or 4.6 × 250 mm, Nacalai Tesque, Inc.), or DAISOPAC SP-120-5-ODS-BIO (Osaka Soda, 4.6 × 150 mm) was employed with a linear gradient of CH3CN containing 0.05% or 0.1% (v/v) TFA at a flow rate of 1 mL min−1 (25 °C or 40 °C). The products were detected by UV absorbance at 220 nm. For preparative HPLC, a Cosmosil 5C18-AR300 column (20 × 250 mm, Nacalai Tesque, Inc.) or a Cosmosil 5C4-AR300 column (20 × 150 mm, Nacalai Tesque, Inc.) was employed with a linear gradient of CH3CN containing 0.05% or 0.1% TFA at a flow rate of 8 mL min−1 (room temperature). All peptides were characterized by ESI-MS (micromass ZQ, Waters). Peptides 1, 2a, 2b, 5, and 6 were synthesized according to the previously reported procedures.24Fmoc-based solid-phase peptide synthesis
Standard Fmoc-based solid-phase peptide synthesis (Fmoc-SPPS) was performed using an automatic peptide synthesizer (PSSM-8, Shimadzu, Japan). The following side chain protected amino acids were employed: Arg(Pbf), Asn(Trt), Asp(OtBu), Cys(Trt), Gln(Trt), Glu(OtBu), His(Trt), Lys(Boc), Ser(tBu), Thr(tBu), and Tyr(tBu). Fmoc protected amino acids (5 equiv.) were coupled using HBTU (5 equiv.), HOBt·H2O (5 equiv.) and iPr2NEt (10 equiv.) in DMF for 60 min twice. Fmoc protection group was deprotected by 20% piperidine in DMF for 4 min twice unless otherwise stated.[Thz107]-Cp183107–153-MPAA (3)
By the standard procedure of automated Fmoc-SPPS, the peptide sequence was constructed from Fmoc-Gly-MeDbz-[Arg(Pbf)]3-Rink amide resin (0.16 mmol). For the coupling of an N-terminal cysteine in peptide 3, Boc-Thz-OH was employed with DIC/Oxyma pure in DMF for 2 h. Then, the resin was treated with 50 mM 4-nitrophenyl chloroformate in CH2Cl2 (16 mL) for 1 h followed by 0.5 M iPr2NEt in DMF (16 mL) for 15 min. Global deprotection and cleavage from resin was performed by TFA/H2O/m-cresol/thioanisole/EDT (80:5:5:5:5) for 2 h. After the removal of the resin by filtration, the crude products were precipitated and washed using ice-cold dry Et2O. The resulting precipitate was dissolved in minimum amount of MPAA buffer (100 mM MPAA, 50 mM TCEP, 6 M guanidine·HCl, 100 mM phosphate buffer, pH 7.2) and incubated for 1 h. The crude products were purified by preparative HPLC to afford desired peptide 3 (41 mg, 4.7% yield). MS (ESI): calcd for C253H393N67O66S2: 5493.45; observed: [M + 6H]6+m/z = 916.85, [M + 5H]5+m/z = 1099.99, [M + 4H]4+m/z = 1374.55, [M + 3H]3+m/z = 1831.68.
[Ala183]-Cp183154–183 (4)
By the standard procedure of automated Fmoc-SPPS, the peptide sequence was constructed from Fmoc-Ala-Wang resin (0.025 mmol). Global deprotection and cleavage from resin was performed by TFA/H2O/m-cresol/thioanisole/EDT (80:5:5:5:5) for 2 h. After the removal of the resin by filtration, the crude products were precipitated and washed using ice-cold dry Et2O. The crude products were purified by preparative HPLC to afford desired peptide 4 (41 mg, 38% yield). MS (ESI): calcd for C146H264N72O44: 3732.18; observed: [M + 6H]6+m/z = 622.99, [M + 5H]5+m/z = 747.13, [M + 4H]4+m/z = 933.93, [M + 3H]3+m/z = 1245.15, [M + 2H]2+m/z = 1866.85.
[Ala183]-Cp183107–183 (8)
MPAA thioester 3 (10 mg, 1.8 μmol) and peptide 4 (7.7 mg, 2.0 μmol) were reacted with HOOBt (8.9 mg, 55 μmol) and iPr2NEt (6.2 μL, 36 μmol) in DMSO (600 μL) for 15 h at room temperature. Then, deThz buffer (1 M methoxyamine·HCl, 6 M guanidine·HCl, 200 mM phosphate buffer, pH 4.0; 3.4 mL) was added and the reaction mixture was incubated for 3 h at 37 °C. The reaction was monitored by LC-MS. The crude products were purified by preparative HPLC to afford the desired peptide 8 (6.5 mg, 40% yield). MS (ESI): calcd for C390H649N139O108S: 9045.41; observed: [M + 8H]8+m/z = 1131.63, [M + 7H]7+m/z = 1293.24, [M + 6H]6+m/z = 1508.22, [M + 5H]5+m/z = 1809.56.[Ala183]-Cp1831–183 (9)
010.06; observed: [M + 15H]15+m/z = 1402.08, [M + 14H]14+m/z = 1502.13, [M + 13H]13+m/z = 1617.73, [M + 12H]12+m/z = 1752.00, [M + 11H]11+m/z = 1912.00.
Cp18361–183 (11)
MeNbz-peptide 2b (1.2 mg, 0.17 μmol) and cysteine-peptide 8 (1.6 mg, 0.14 μmol) were reacted in ligation buffer (100 mM MPAA, 50 mM TCEP, 6 M guanidine·HCl, 200 mM phosphate buffer, pH 7.0; 0.28 mL) for 3 h at 37 °C. Then, the same amount of deThz buffer (1 M methoxyamine·HCl, 6 M guanidine·HCl, 200 mM phosphate buffer, pH 4.0; 0.28 mL) was added and the reaction mixture was incubated for 19 h at 37 °C. The reaction was monitored by LC-MS. The crude products were purified by preparative HPLC to afford the desired peptide 11 (0.57 mg, 24% yield). MS (ESI): calcd for C631H1013N201O174S3: 14295.46; observed: [M + 11H]11+m/z = 1300.95, [M + 10H]10+m/z = 1431.64, [M + 9H]9+m/z = 1589.93.
CD analysis of synthetic Cp183(C183A)
Lyophilized Cp183(C183A) peptide 9 was dissolved in denaturing buffer (6 M guanidine·HCl, 50 mM HEPES, 2 mM DTT, pH 7.5) at a concentration of ∼0.2 mg mL−1, and the mixture was incubated overnight at 4 °C. The denatured protein solution was dialyzed against 200 volumes of dialysis buffer (1.5 M guanidine·HCl, 50 mM HEPES, 2 mM DTT, pH 7.5) overnight at 4 °C. The protein solution was then further dialyzed against 200 volumes of various buffer conditions (1.0 M guanidine·HCl [or 0.5 M guanidine·HCl or 1.5 M NaCl], 50 mM HEPES, 2 mM DTT, pH 7.5) overnight at 4 °C. CD spectra of proteins were recorded using a JASCO J-720 circular dichroism spectrometer (JASCO, Tokyo, Japan) at 20 °C.Folding and assembly of capsids from synthetic Cp183(C183A)
Lyophilized Cp183(C183A) peptide 9 was dissolved in denaturing buffer (6 M guanidine·HCl, 50 mM HEPES, 2 mM DTT, pH 7.5) at a concentration of ∼0.2 mg mL−1, and the mixture was incubated overnight at 4 °C. The denatured protein solution was dialyzed against 200 volumes of dialysis buffer (1.5 M guanidine·HCl, 50 mM HEPES, 2 mM DTT, pH 7.5) overnight at 4 °C. UV absorption spectra were obtained using a V-630 BIO spectrophotometer (JASCO, Tokyo, Japan). The concentration of Cp183(C183A) was calculated using the extinction coefficient of 58900 M−1 cm−1 (280 nm) per Cp183(C183A) dimer.49 Then, 3 μL of 10 μM ssDNA (5′-ATGAATAACCAACGAAAAAAGGCGAGAAATACGCCTTTCAATATGCTGAA-3′; Thermo Fisher Scientific, Tokyo, Japan) was added to 7.5 μL of 3.1 μM Cp183(C183A). After incubating overnight at 4 °C, 12 μL H2O was added to the protein solution. The final capsid solution (1.0 μM Cp183(C183A), 13 μM ssDNA, 0.5 M guanidine·HCl, 17 mM HEPES, 0.67 mM DTT, pH 7.5) was analyzed by transmission electron microscopy.
Electron microscopy
Copper mesh grids coated with formvar and carbon (Veco grids, Nisshin EM, Tokyo, Japan) were glow-discharged and placed on drops of the specimen for 1 min, rinsed with distilled water, stained with a 2% uranyl acetate solution and examined with a transmission electron microscope (HT7700, Hitachi Ltd, Japan) at 80 kV.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI, Japan (JP18H02555, JP20K21252, JP21J20063; JP22H02747; JP22K19376); Research Program on Hepatitis (JP17fk0310114; JP22fk0310504) from AMED, Japan; The Tokyo Biochemical Research Foundation; Astellas Foundation for Research on Metabolic Disorders; Takeda Science Foundation; TERUMO Life Science Foundation; and JSPS WISE program. K. A. is grateful for “The Graduate Program for Medical Innovation (MIP)” and for Research Fellowships for Young Scientists from JSPS, Japan.References
- M.-F. Yuen, D.-S. Chen, G. M. Dusheiko, H. L. A. Janssen, D. T. Y. Lau, S. A. Locarnini, M. G. Peters and C.-L. Lai, Nat. Rev. Dis. Primers, 2018, 4, 18035 CrossRef PubMed.
- WHO, Hepatitis B. Key facts. July 18, 2023. https://www.who.int/news-room/fact-sheets/detail/hepatitis-b (accessed November 2023).
- M. Nassal, Gut, 2015, 64, 1972–1984 CrossRef CAS PubMed.
- G. Dusheiko, K. Agarwal and M. K. Maini, N. Engl. J. Med., 2023, 388, 55–69 CrossRef CAS PubMed.
- M. Nassal, J. Virol., 1992, 66, 4107–4116 CrossRef CAS PubMed.
- Z. Tan, K. Pionek, N. Unchwaniwala, M. L. Maguire, D. D. Loeb and A. Zlotnick, J. Virol., 2015, 89, 3275–3284 CrossRef CAS PubMed.
- R. W. King, S. K. Ladner, T. J. Miller, K. Zaifert, R. B. Perni, S. C. Conway and M. J. Otto, Antimicrob. Agents Chemother., 1998, 42, 3179–3186 CrossRef CAS PubMed.
- K. Deres, C. H. Schröder, A. Paessens, S. Goldmann, H. J. Hacker, O. Weber, T. Krämer, U. Niewöhner, U. Pleiss, J. Stoltefuss, E. Graef, D. Koletzki, R. N. A. Masantschek, A. Reimann, R. Jaeger, R. Groß, B. Beckermann, K.-H. Schlemmer, D. Haebich and H. Rübsamen-Waigmann, Science, 2003, 299, 893–896 CrossRef CAS PubMed.
- M. R. Campagna, F. Liu, R. Mao, C. Mills, D. Cai, F. Guo, X. Zhao, H. Ye, A. Cuconati, H. Guo, J. Chang, X. Xu, T. M. Block and J.-T. Guo, J. Virol., 2013, 87, 6931–6942 CrossRef CAS PubMed.
- J.-A. Kang, S. Kim, M. Park, H.-J. Park, J.-H. Kim, S. Park, J.-R. Hwang, Y.-C. Kim, Y. J. Kim, Y. Cho, M. S. Jin and S.-G. Park, Nat. Commun., 2019, 10, 2184 CrossRef PubMed.
- S. J. Stray, C. R. Bourne, S. Punna, W. G. Lewis, M. G. Finn and A. Zlotnick, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 8138–8143 CrossRef CAS PubMed.
- J. J. Feld, D. Colledge, V. Sozzi, R. Edwards, M. Littlejohn and S. A. Locarnini, Antiviral Res., 2007, 76, 168–177 CrossRef CAS PubMed.
- U. Viswanathan, N. Mani, Z. Hu, H. Ban, Y. Du, J. Hu, J. Chang and J.-T. Guo, Antiviral Res., 2020, 182, 104917 CrossRef CAS PubMed.
- T. Noguchi, S. Oishi, K. Honda, Y. Kondoh, T. Saito, H. Ohno, H. Osada and N. Fujii, Chem. Commun., 2016, 52, 7653–7656 RSC.
- A. G. Atanasov, S. B. Zotchev, V. M. Dirsch, the International Natural Product Sciences Taskforce and C. T. Supuran, Nat. Rev. Drug Discovery, 2021, 20, 200–216 CrossRef CAS PubMed.
- T. Noguchi, H. Ishiba, K. Honda, Y. Kondoh, H. Osada, H. Ohno, N. Fujii and S. Oishi, Bioconjugate Chem., 2017, 28, 609–619 CrossRef CAS PubMed.
- K. Shu, T. Noguchi, K. Honda, Y. Kondoh, H. Osada, H. Ohno, N. Fujii and S. Oishi, RSC Adv., 2017, 7, 38725 RSC.
- K. Shu, N. Iwamoto, K. Honda, Y. Kondoh, H. Hirano, H. Osada, H. Ohno, N. Fujii and S. Oishi, Bioconjugate Chem., 2019, 30, 1395–1404 CrossRef CAS PubMed.
- C. R. Bourne, M. G. Finn and A. Zlotnick, J. Virol., 2006, 80, 11055–11061 CrossRef CAS PubMed.
- A. Zlotnick, B. Venkatakrishnan, Z. Tan, E. Lewellyn, W. Turner and S. Francis, Antiviral Res., 2015, 121, 82–93 CrossRef CAS PubMed.
- A. Gallina, F. Bonelli, L. Zentilin, G. Rindi, M. Muttini and G. Milanesi, J. Virol., 1989, 63, 4645–4652 CrossRef CAS PubMed.
- P. T. Wingfield, S. J. Stahl, R. W. Williams and A. C. Steven, Biochemistry, 1995, 34, 4919–4932 CrossRef CAS PubMed.
- P. Ceres and A. Zlotnick, Biochemistry, 2002, 41, 11525–11531 CrossRef CAS PubMed.
- S. Tsuda, M. Mochizuki, H. Ishiba, K. Yoshizawa-Kumagaye, H. Nishio, S. Oishi and T. Yoshiya, Angew. Chem., Int. Ed., 2018, 57, 2105–2109 CrossRef CAS PubMed.
- We investigated several conditions for capsid assembly from synthetic Cp149, including sample concentrations, detergents, buffers for refolding; however, we failed to obtain the capsid-like particles.
- J. Z. Porterfield, M. S. Dhason, D. D. Loeb, M. Nassal, S. J. Stray and A. Zlotnick, J. Virol., 2010, 84, 7174–7184 CrossRef CAS PubMed.
- M. Nassal, A. Rieger and O. Steinau, J. Mol. Biol., 1992, 225, 1013–1025 CrossRef CAS PubMed.
- Disulfide bonds in the Cp183 dimer are not essential for HBV capsid particle formation, to see: J. Zheng, F. Schödel and D. L. Peterson, J. Biol. Chem., 1992, 267, 9422–9429 CrossRef CAS PubMed.
- F. Birnbaum and M. Nassal, J. Virol., 1990, 64, 3319–3330 CrossRef CAS PubMed.
- Y. Fujiwara, K. Akaji and Y. Kiso, Chem. Pharm. Bull., 1994, 42, 724–726 CrossRef CAS PubMed.
- J. Lukszo, D. Patterson, F. Albericio and S. A. Kates, Lett. Pept. Sci., 1996, 3, 157–166 CrossRef CAS.
- J.-X. Wang, G.-M. Fang, Y. He, D.-L. Qu, M. Yu, Z.-Y. Hong and L. Liu, Angew. Chem., Int. Ed., 2015, 54, 2194–2198 CrossRef CAS PubMed.
- S. Tsuda, S. Masuda and T. Yoshiya, Org. Biomol. Chem., 2019, 17, 1202–1205 RSC.
- P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. H. Kent, Science, 1994, 266, 776–779 CrossRef CAS PubMed.
- Y. Tan, H. Wu, T. Wei and X. Li, J. Am. Chem. Soc., 2020, 142, 20288–20298 CrossRef CAS PubMed.
- Q. Wan and S. J. Danishefsky, Angew. Chem., Int. Ed., 2007, 46, 9248–9252 CrossRef CAS PubMed.
- V. Agouridas, O. El Mahdi, V. Diemer, M. Cargoët, J.-C. M. Monbaliu and O. Melnyk, Chem. Rev., 2019, 119, 7328–7443 CrossRef CAS PubMed.
- O. Fuchs, S. Trunschke, H. Hanebrink, M. Reimann and O. Seitz, Angew. Chem., Int. Ed., 2021, 60, 19483–19490 CrossRef CAS PubMed.
- H. Hojo, Y. Murasawa, H. Katayama, T. Ohira, Y. Nakahara and Y. Nakahara, Org. Biomol. Chem., 2008, 6, 1808–1813 RSC.
- J. B. Blanco-Canosa and P. E. Dawson, Angew. Chem., Int. Ed., 2008, 47, 6851–6855 CrossRef CAS PubMed.
- J. B. Blanco-Canosa, B. Nardone, F. Albericio and P. E. Dawson, J. Am. Chem. Soc., 2015, 137, 7197–7209 CrossRef CAS PubMed.
- NaNO2-mediated activation of 5 followed by NCL without purification of 6 failed due to the significant production of a hydrolyzed product.
- D. Bang and S. B. H. Kent, Angew. Chem., Int. Ed., 2004, 43, 2534–2538 CrossRef CAS PubMed.
- R. A. Crowther, N. A. Kiselev, B. Böttcher, J. A. Berriman, G. P. Borisova, V. Ose and P. Pumpens, Cell, 1994, 77, 943–950 CrossRef CAS PubMed.
- In the CD analysis, only a low signal was observed due to the formation of insoluble aggregates during the folding process.
- I. F. Bin Mohamed Suffian, M. Garcia-Maya, P. Brown, T. Bui, Y. Nishimura, A. R. Bin Mohammad Johari Palermo, C. Ogino, A. Kondo and K. T. Al-Jamal, Sci. Rep., 2017, 7, 43160 CrossRef PubMed.
- S. Machida, S. Ogawa, S. Xiaohua, T. Takaha, K. Fujii and K. Hayashi, FEBS Lett., 2000, 486, 131–135 CrossRef CAS PubMed.
- Under the similar conditions for capsid assembly from recombinant Cp183 (5–15 μM Cp183),26 synthetic Cp183(C183A) was prone to aggregation. Capsid assembly from synthetic Cp183(C183A) at a lower concentration (1.0 μM protein) provided the capsid-like particles, which was similar to those from recombinant Cp183.26.
- C. N. Pace, F. Vajdos, L. Fee, G. Grimsley and T. Gray, Protein Sci., 1995, 4, 2411–2423 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ob02099a |
| This journal is © The Royal Society of Chemistry 2024 |