
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCopper-catalyzed room-temperature cross-dehydrogenative coupling of secondary amides with terminal alkynes: a chemoselective synthesis of ynamides†
Shuang-Yan
Zhuo
 a,
Jian-Liang
Ye
*b and
Xiao
Zheng
*a
a,
Jian-Liang
Ye
*b and
Xiao
Zheng
*a
aXiamen Key Laboratory of Chiral Drugs, School of Pharmaceutical Sciences, Xiamen University, Xiamen, Fujian 361102, China. E-mail: zxiao@xmu.edu.cn
bFujian Provincial Key Laboratory of Chemical Biology, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen, Fujian 361005, China. E-mail: yejl@xmu.edu.cn
First published on 15th January 2024
Abstract
A copper-catalyzed aerobic oxidative cross-dehydrogenative coupling reaction between secondary amides and terminal alkynes has been developed. With the aid of ligands and 3 Å molecular sieves, ynamides can be efficiently synthesized at room temperature and conveniently scaled up. A legitimate mechanism involving nitrogen-centred radicals and copper trivalent intermediates has been proposed.
Introduction
Ynamides are highly versatile synthetic building blocks. Despite that their first preparation was accomplished by Viehe and coworkers half a century ago,1 the extensive utility of ynamides in organic synthesis has been realized only recently and the field continues to stimulate high research interest. Ynamides can serve as precursors of keteniminium salts2 and α-imino-metal carbenes,3 and pave the foundation for a number of novel ynamide-mediated addition reactions,4 annulation (cycloaddition and cycloisomerization) reactions,5 metal-catalyzed cross-coupling reactions,6 and free radical reactions.7 In addition, useful synthetic reagents can be designed based on the unique reactivity of ynamides, as demonstrated recently by Zhao's group with their efficient and mild racemization-free coupling reagents for the synthesis of amides and peptides.8 Structurally diverse ynamides serve as a crucial foundation for the advancement of ynamide chemistry. The synthesis of such compounds has attracted significant attention, leading to the development of a series of methods9 that can be classified into three main categories: the elimination reaction of halogenated enamides (Scheme 1-1), copper-catalyzed (mediated) coupling reaction of halogenated olefins with amides (Scheme 1-2), and copper-catalyzed (mediated) coupling reaction of alkynes or alkyne derivatives with amides (Scheme 1-3). However, these reactions either require harsh conditions, inconvenient starting materials, or a large excess of coupling partners. The development of greener, safer, more economical and efficient synthesis methods for ynamides remains a challenging topic. | ||
| Scheme 1 Synthesis of ynamides. | ||
Cross-dehydrogenative coupling (CDC) represents a class of atom- and step-economical methods.10 The copper-catalyzed or -mediated CDC reaction of terminal alkynes and amides under an oxygen atmosphere is a highly effective strategy for the synthesis of ynamides,11 owing to the direct use of readily available terminal alkynes and amides as substrates. However, the low reactivity of these substrates necessitates heating for these reactions to occur, leading to undesired alkyne homo-coupling dimers. On the other hand, the mechanism of this reaction remains elusive. Therefore, there is an urgent need for improving the chemoselectivity of this reaction toward ynamides and gaining further insight into its mechanism. Here, we report a facile synthesis of ynamides through a copper-catalyzed CDC reaction of terminal alkynes and amides under an oxygen atmosphere (Scheme 1-4). Promoted by ligands and 3 Å molecular sieves, the reaction can proceed smoothly at room temperature, potentially involving nitrogen-centred radicals and Cu(III) complexes as key intermediates.
Results and discussion
Copper-catalyzed CDC reaction of terminal alkynes and amides
Our investigation into the CDC protocol commenced with the optimization of the model reaction between N-methyl-p-toluenesulfonamide 1a and phenylacetylene 2a. It was observed that many copper(II) salts exhibit catalytic activity in the presence of an oxygen atmosphere. After examining several types of ligands, 1-methylbenzimidazole was found to provide the desired ynamide 3aa with an excellent yield at room temperature. Interestingly, we discovered that molecular sieves are an indispensable additive to promote this reaction, and 3 Å molecular sieves gave the optimal results. Other optimization studies involving bases and solvents were performed (see ESI Tables S1–4†). Finally, the CDC reaction of N-methyl-p-toluenesulfonamide 1a (3.0 equiv.) and phenylacetylene 2a (1.0 equiv.) was conducted using Cu(OTf)2 (0.2 equiv.) as the catalyst and 1-methylbenzimidazole (0.4 equiv.) as the ligand in the presence of 3 Å molecular sieves (360 mg for 1.0 mmol of alkyne) in toluene (0.25 M), under an oxygen atmosphere at room temperature for 20 hours, resulting in the desired ynamide 3aa with 93% yield (Table 1, entry 1). In the absence of the ligand or molecular sieve additive, the reaction failed to provide any detectable product (Table 1, entries 2–4). Other solvents such as dichloromethane led to diminished yields (Table 1, entry 5).
With these optimized conditions in hand, we next investigated the scope of terminal alkynes. As demonstrated in Table 2, CDC reactions of aryl, alkyl and silyl monosubstituted acetylenes (2a–2o) with N-methyl-p-toluenesulfonamide 1a gave the desired ynamides (3aa–3ao) in moderate to good yields (57–93%). For para-substituted aromatic terminal alkynes, both electron-donating and electron-withdrawing groups gave the corresponding ynamides in high yields of 80–93% (3aa–3ag). Sterically more demanding ortho- and meta-substituted aromatic terminal alkynes provided diminished yields of 57–69% (3ah–3ak). Additionally, the reaction conditions are also suitable for aliphatic and heterocyclic terminal alkynes, affording the desired ynamides in 73–75% yields (3al–3ao). Notably, for cyclopropyl and silyl-substituted terminal alkynes, higher yields (3am and 3an: 71% and 75%) were obtained when dichloromethane was used as solvent instead of toluene (3am and 3an: 45% and 55%), and no discernible dimerization byproducts of alkyne were observed. Notably, this mild protocol enables the synthesis of thermolabile ynamides 3ap and 3aq. To our satisfaction, a gram-scale synthesis of 3aa was achieved in 83% yield from 5.0 mmol of alkyne 2a by following the standard procedure.
a General method: amide (1.5 mmol), 1-methylbenzimidazole (0.2 mmol), Cu(OTf)2 (0.1 mmol), Na2CO3 (1.5 mmol), and 3 Å molecular sieves (180 mg) were dissolved in dry solvent (2.0 mL) and the terminal alkyne (0.5 mmol) was successively added. The mixture was stirred at room temperature for 20 h under an oxygen atmosphere.
b Isolated yield.
c Solvent: DCM.
d Solvent: toluene![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DCM = 1:1. :DCM = 1:1.
|
|---|

|
We also explored the scope of amides bearing various electron-withdrawing protecting groups on the nitrogen. In general, the CDC reactions between most secondary amides and phenylacetylene 2a can provide the corresponding ynamides smoothly (Table 3), even for very low reactive γ-lactams (3ha–3ja).11d However, the reaction does not occur with primary amides like p-toluenesulfonamide. For secondary amides or alkynes that exhibit poor solubility in toluene, switching the solvent to dichloromethane (3ba and 3ma), a 1:1 mixture of toluene and dichloromethane (3ca, 3da, 3ea, 3la, 3na, 3oa, 3pa and 3qa), or tetrahydrofuran (3ka) can result in improved yields. Additionally, base is a significant factor in some cases. For instance, Na2HPO4 as a base resulted in a higher yield of ynamide 3fa compared to Na2CO3 (65% vs. 45%), which suggests that the deprotonation of amides may be crucial. Notably, N-Cbz and Ts glycine methyl esters can also be used to obtain the corresponding ynamides 3ra and 3sa.
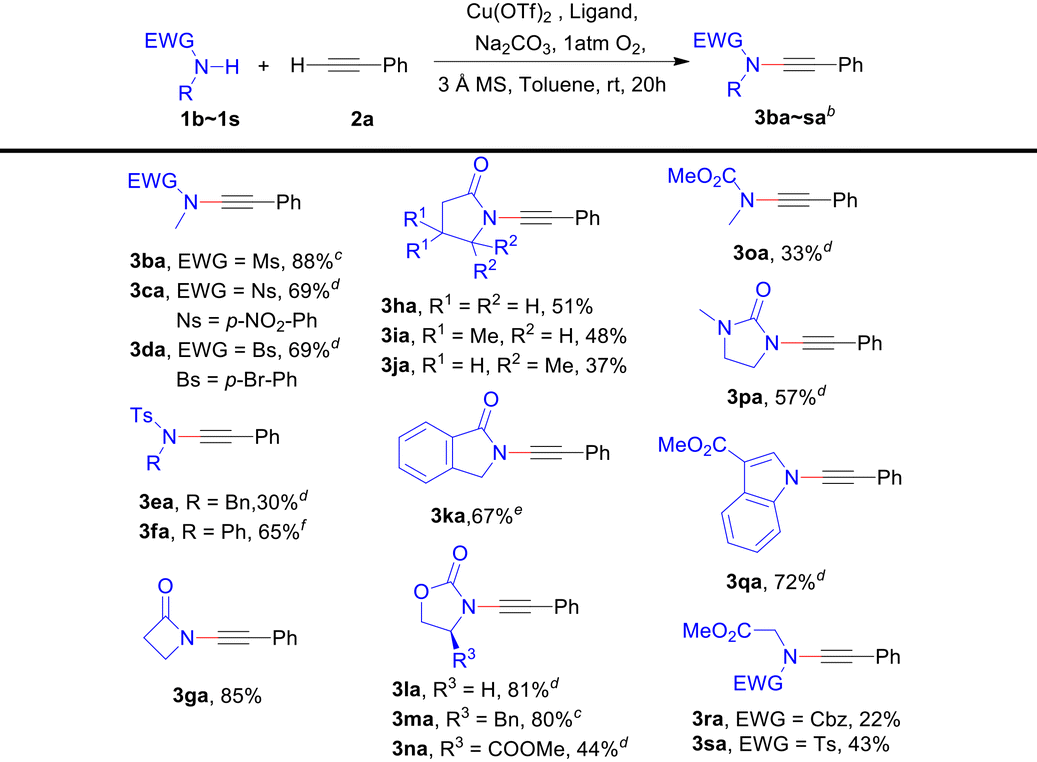
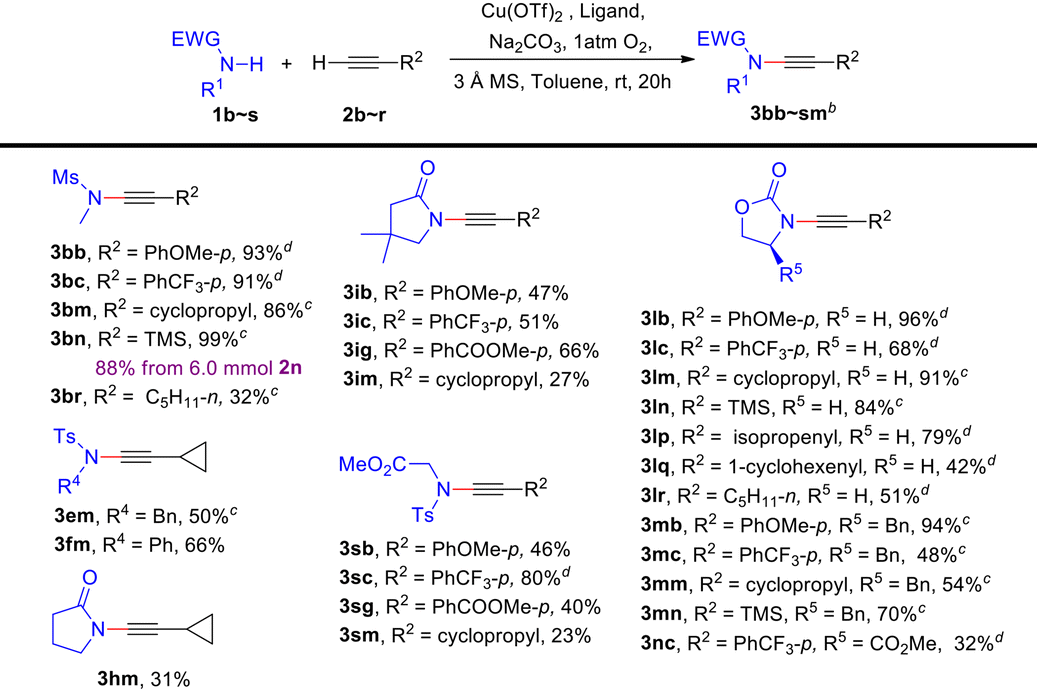
Moreover, we conducted a more comprehensive investigation into the CDC reactions of various amides and terminal alkynes under the general conditions. Table 4 demonstrates the favourable applicability of this protocol towards a wide range of ynamides in moderate to high yields (23%–99%). When using N-methyl methylsulfonamide 1b as a secondary amide substrate, either electron-rich or electron-deficient aromatic terminal alkynes, as well as alkyl-/silyl-substituted terminal alkynes can undergo CDC reactions with satisfactory yields, the same as for the secondary amide substrate 2-oxazolidone 1l (3lb–3lr). Notably, thermolabile ynamides 3lp and 3lq can also be prepared in moderate yields (79% and 42%). Compared with 1l, more sterically hindered (S)-4-benzyl-2-oxazolidinone 1m or (R)-4-methylester-2-oxazolidinone 1n provided diminished reaction yields (3lb–rvs.3mb–3nc). With poorly active γ-lactams 1h and 1i and glycine derivative 1s coupling with terminal alkynes can also give the desired products (3hm, 3ib–3im and 3sb–3sm). Interestingly, electron-deficient aromatic alkyne 2c can provide higher yields than the electron-rich aromatic alkyne 2b (3ibvs.3ic and 3sbvs.3sc). Satisfactorily, a gram-scale synthesis of 3bn was performed with an 88% yield from 6.0 mmol of 2n.
Mechanistic studies of the copper-catalyzed CDC reaction
After establishing the broad substrate scope of this transformation, we next turned our attention to mechanistic investigations, and a plausible reaction mechanism was established with the aid of density functional theory (DFT) calculations. As listed in Fig. 1, Cu(II) complex I is taken as the starting point of this reaction, which is formed from Cu(OTf)2 and two 1-methylbenzimidazoles. Complex I then reacts with terminal alkyne 2a in the presence of a base to generate the copper alkyne intermediate Int1, which further forms Cu(II) complex Int2 (−60.6 kcal mol−1) upon interaction with an amide anion [the formation of the homo-coupling precursor Int3 from Int1 is less favorable (−53.0 kcal mol−1)]. However, the reductive elimination of Int2 towards ynamide 3aa requires a relatively high activation barrier (viaTS1, ΔG‡ = 23.0 kcal mol−1), which is not in agreement with the facile product formation at room temperature. | ||
| Fig. 1 DFT calculations on the assumed Cu(II)-mediated CDC process (the values shown are relative free energies in kcal mol−1). | ||
Alternatively, we assumed that Int2 undergoes single-electron oxidation with a nitrogen-centred radical12 generated by the synergistic oxidative system of copper complex, 3 Å molecular sieves and oxygen prior to reductive elimination. Indeed, DFT calculations indicated that after the nitrogen-centred radical III oxidizes the Cu(II) intermediate Int2 to the thermodynamically stable Cu(III) intermediate Int4, the reductive elimination towards ynamide 3aa and Cu(I) complex IV has a much lower barrier of 12.6 kcal mol−1, and thus occurs at room temperature (Fig. 2, transition state TS3, for details see ESI Fig. S2–4†). However, the calculation results indicate that the reductive elimination of Int5 towards the homo-coupling dimer of alkyne 4a is a spontaneous process due to a very low activation barrier (transition state TS5 cannot be located by DFT calculations, see ESI Fig. S6†). This seems to imply that the generation of ynamide 3aa is not a feasible process. Considering the experimental results, the generation of nitrogen-centred radical III rather than the reductive elimination of Cu(III) intermediates Int4 and Int5 may be the rate-determining step of the reaction.
 | ||
| Fig. 2 DFT calculations on the assumed Cu(III)-mediated CDC process (the values shown are relative free energies in kcal mol−1). | ||
In order to verify the reaction mechanism described by DFT calculations and obtain a more comprehensive understanding of the impact of reaction substrates and molecular sieves on the CDC reaction, kinetic studies were conducted. As shown in Fig. 3a, the plots of [amide], [alkyne] and [ynamide] versus time gave three straight lines which indicates that the CDC reaction can be considered zero-order for secondary amide and terminal alkyne in the presence of excess of amides, and thus can be deemed a typical surface-catalyzed reaction (also see ESI Table S5 and Fig. S8†). Interestingly, when the amount of 3 Å MS was changed in the general conditions, a plot of kobsversus [weight of 3 Å MS] also exhibits a linear relationship (Fig. 3b, also see ESI Table S6 and Fig. S9†). Additionally, since molecular sieves cannot be replaced with dry silica gel or anhydrous magnesium sulfate, and the promotional effect of 3 Å MS on the reaction surpasses that of 4 Å MS, the essential role of molecular sieves in the CDC reaction should be a promoter rather than a desiccant. According to the aforementioned, it can be reasonably inferred that the oxidation process of secondary amides to nitrogen-centred radicals occurs on the surface of molecular sieves and represents the rate-determining step. Therefore, the formation of the homo-coupling dimer 4a from Int5, which is generated by nitrogen-centred radical oxidation of Int3, can be facilitated by the presence of small amounts of secondary amide 1a (∼6% 4a obtained in the absence of 1a). However, Cu(II) intermediate Int2 will be overwhelmingly favored over Int3 in the presence of excess amides, leading to the nitrogen-centred radical-mediated generation of Cu(III) intermediate Int4 and ultimately resulting in ynamide 3aa formation (Fig. 4, also see ESI Table S4 and Fig. S1, S5–7†).
 | ||
| Fig. 3 Kinetic studies of the copper-catalyzed CDC reactions of acetylenes and amides under an oxygen atmosphere. | ||
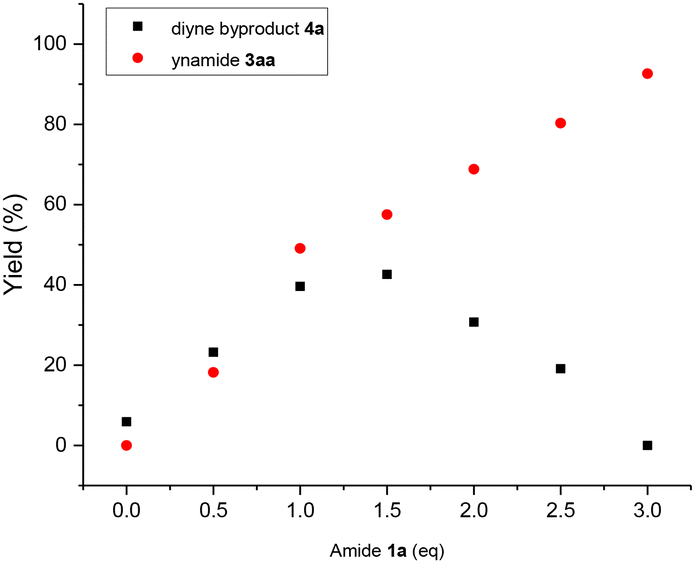 | ||
| Fig. 4 The effect of amide loadings on the CDC reaction between amide 1a and alkyne 2a to yield ynamide 3aa under general conditions. | ||
On the basis of DFT calculations and kinetic studies, the reaction mechanism was postulated as follows (Fig. 5): Cu(II) complex I is initially formed from Cu(OTf)2 and two 1-methylbenzimidazoles. This complex then reacts with terminal alkyne 2 in the presence of a base to generate copper alkyne intermediate Int1, which subsequently forms Cu(II) complex Int2 upon interaction with amide 1 in the presence of a base. Furthermore, Int2 undergoes single-electron oxidation with nitrogen-centred radical III generated by the [Cu(II)Ln-MS-O2] system to form Cu(III) intermediate Int4. The reductive elimination of Int4 produces the desired ynamide 3 and Cu(I) complex IVvia the transition state TS3. Finally, Cu(II) complex Int2 is regenerated via aerobic oxidation of Cu(I) complex IV followed by ligand and alkyne coordination.
 | ||
| Fig. 5 Plausible mechanism for the CDC reaction of amides and terminal alkynes. | ||
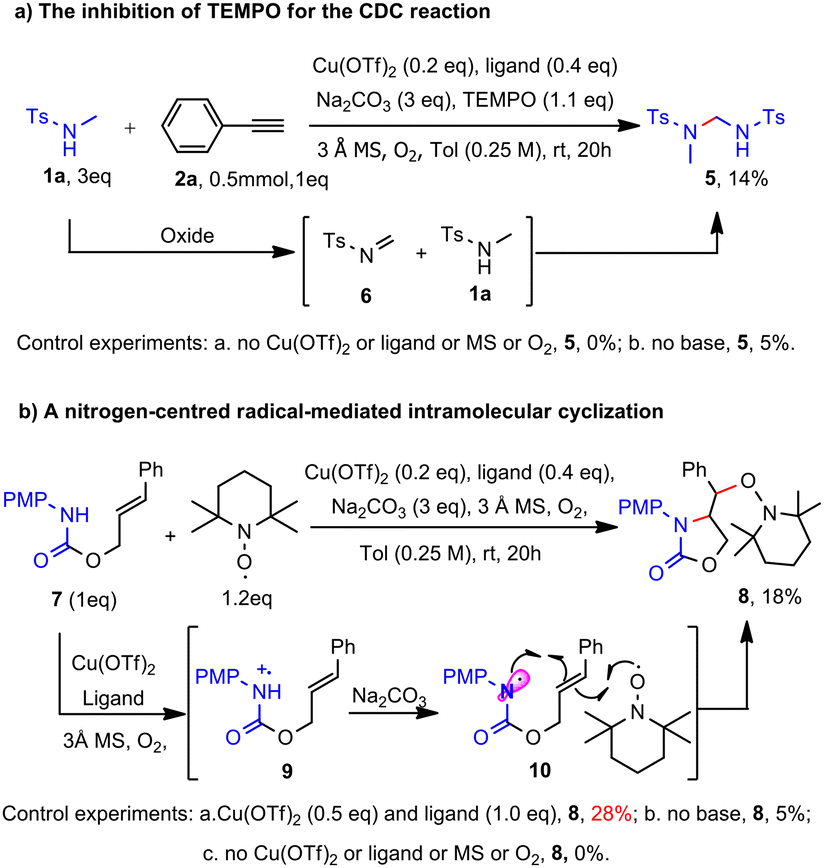
Notably, the involvement of nitrogen-centred radical was also supported by several controlled experiments. Upon the addition of a stoichiometric amount of TEMPO to the CDC reaction, the formation of ynamide and the homo-coupling dimer of alkyne was completely suppressed; alternatively, an unexpected gem-diamido compound 5 was obtained with a yield of 14%. We speculated that the compound could be formed through the dimerization of amide 1a, wherein one molecule of amide is oxidized to imine 6 followed by the nucleophilic addition of another molecule of 1a (Scheme 2a).
 | ||
| Scheme 2 Control experiments for nitrogen radicals. | ||
The presence of imine 6 suggests an oxidative-elimination mechanism from amide 1a. Under aerobic oxidative conditions, it is rational that amide 1a may undergo oxidation by the [Cu(II)Ln-MS-O2] system to generate the nitrogen radical cation, followed by deprotonation to form the proposed nitrogen-centred radical, leading to imine 6 by TEMPO oxidation. Indeed, we synthesized carbamate 7 and subjected it to oxidation by [Cu(II)Ln-MS-O2] system under CDC general conditions. The corresponding nitrogen radical cation 9 was first generated by the [Cu(II)Ln-MS-O2] system, which then underwent deprotonation to form nitrogen-centred radical 10. Subsequent 5-exo-trig radical cyclization of 10 followed by capture with TEMPO13 gave the desired product 8 in 18% yield and the yield dropped to 5% in the absence of base; furthermore, no product 8 was afforded in the absence of any one of Cu/ligand/MS/O2 (Scheme 2b).
Conclusions
In summary, we have developed a copper-catalyzed aerobic oxidative CDC reaction between secondary amides and terminal alkynes. Through the synergistic promotion of appropriate ligands and 3 Å molecular sieves, ynamides can be efficiently prepared at room temperature and conveniently scaled up. Moreover, a mechanism involving the generation of nitrogen-centred radicals and reductive elimination of copper trivalent intermediates has been proposed legitimately. This also provides an explanation for substrate proportions, as well as the roles of ligands, molecular sieves and bases. Taking advantage of this catalytic mechanism, homo-coupling reactions of alkynes were predominantly suppressed. Furthermore, a preliminary mechanistic application was conducted for the cyclization reaction mediated by a nitrogen-centred radical. These deep insights revealed the potential copper-catalyzed aerobic oxidative process occurring on the surface of molecular sieves, which can inspire further research and application of related reactions.Experimental
General information
Melting points (mp) were determined on SGW® X-4A micromelting point apparatus and were uncorrected. Infrared (IR) spectra were measured with a Bruker Alpha spectrometer using film KBr pellet techniques. 1H and 13C NMR spectra were recorded on a Bruker Avance III 600 MHz spectrometer. The 1H and 13C NMR chemical shifts are expressed in parts per million (δ) referenced to either the internal standard Me4Si or solvent signals (Me4Si at 0 ppm for 1H NMR and CDCl3 at 77.0 ppm for 13C NMR). The NMR data are presented as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, dd = doublet of doublet, dt = doublet of triplet, dq = doublet of quartet, td = triplet of doublet, qd = quartet of doublet, ddd = doublet of doublet of doublet, m = multiplet, br s = broad singlet), coupling constant (Hz) and integration. High-resolution mass spectra (HRMS) were recorded on a Thermo Scientific Q Exactive LC-MS mass spectrometer.Unless otherwise noted, materials were purchased from commercial suppliers and used without further purification. Toluene was treated with CaCl2. DCM was purchased from Energy Chemical (extra dry, with molecular sieves, water ≤50 ppm, in a resealable bottle) and THF was purchased from Energy Chemical (extra dry, with molecular sieves, water ≤30 ppm, in a resealable bottle). Flash column chromatography was performed using 200–300 mesh silica gel. All reactions were carried out in flame-dried glassware under a dry oxygen atmosphere. Reactions were monitored by TLC and visualized using a dual short wave/long wave UV lamp.
General procedure for the copper-catalyzed CDC reaction of amides with terminal alkynes
In a 25 mL round bottom flask, the amide (1.5 mmol, 3.0 equiv.), 1-methylbenzimidazole (0.2 mmol, 0.4 equiv.), Cu(OTf)2 (0.1 mmol, 0.2 equiv.), Na2CO3 (1.5 mmol, 3.0 equiv.), and 3 Å molecular sieves (180 mg) were dissolved in dry solvent (2 mL) and the terminal alkyne (0.5 mmol, 1.0 equiv.) was successively added. The mixture was degassed three times by applying vacuum and backfilling with oxygen while stirring vigorously. The mixture was stirred at room temperature for 20 h, filtered using diatomaceous earth over a plug of silica gel (washed with EtOAc), and the solvent was removed under reduced pressure. The crude residue was purified by flash chromatography over silica gel.Discovery of the nitrogen-centered radical in the copper-catalyzed CDC reaction
Author contributions
X. Zheng conceived this project. X. Zheng and J.-L. Ye supervised the investigation. S.-Y. Zhuo performed the research. J.-L. Ye conducted the DFT calculations. All authors wrote and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for the financial support from the NSF of China (91856110). We also thank Dr An-An Wu, Dr Gang Fu, Dr Feng-Ru Fan of XMU and Dr Chen-Xi Ye for kind and helpful discussions on this paper.References
- For the first synthesis of ynamides, see: (a) Z. Janousek, J. Collard and H. G. Viehe, Angew. Chem., Int. Ed. Engl., 1972, 11, 917–918 CrossRef CAS. For some selected reviews, see: (b) D. Campeau, D. F. L. Rayo, A. Mansour, K. Muratov and F. Gagosz, Chem. Rev., 2021, 121, 8756–8867 CrossRef CAS PubMed; (c) J. Luo, G.-S. Chen, S.-J. Chen, J.-S. Yu, Z.-D. Li and Y.-L. Liu, ACS Catal., 2020, 10, 13978–13992 CrossRef CAS; (d) C. C. Lynch, A. Sripada and C. Wolf, Chem. Soc. Rev., 2020, 49, 8543–8583 RSC; (e) K. A. DeKorver, H. Li, A. G. Lohse, R. Hayashi, Z. Lu, Y. Zhang and R. P. Hsung, Chem. Rev., 2010, 110, 5064–5106 CrossRef CAS PubMed; (f) G. Evano, A. Coste and K. Jouvin, Angew. Chem., Int. Ed., 2010, 49, 2840–2859 CrossRef CAS PubMed.
- (a) Y.-B. Chen, P.-C. Qian and L.-W. Ye, Chem. Soc. Rev., 2020, 49, 8897–8909 RSC; (b) G. Evano, M. Lecomte, P. Thilmany and C. Theunissen, Synthesis, 2017, 49, 3183–3214 CrossRef CAS; (c) X.-J. Li, Y. Sun, L. Zhang and B. Peng, Chin. J. Org. Chem., 2016, 36, 2530–2544 CrossRef CAS.
- (a) P. W. Davies, Chem. Rec., 2021, 21, 3964–3977 CrossRef CAS PubMed; (b) G.-X. Ru, T.-T. Zhang, M. Zhang, X.-L. Jiang, Z.-K. Wan, X.-H. Zhu, W.-B. Shen and G.-Q. Gao, Org. Biomol. Chem., 2021, 19, 5274–5283 RSC; (c) D. V. Patil and S. Shin, Asian J. Org. Chem., 2019, 8, 63–73 CrossRef CAS; (d) Y. Liao, L. Zhu, Y.-H. Yu, G. Chen and X.-L. Huang, Chin. J. Org. Chem., 2017, 37, 2785–2799 CrossRef CAS.
- For reviews on addition reactions of ynamides, see: (a) B. Zhou, T.-D. Tan, X.-Q. Zhu, M. Shang and L.-W. Ye, ACS Catal., 2019, 9, 6393–6406 CrossRef CAS; (b) X. Li, Y. Sun, L. Zhang and B. Peng, Chin. J. Org. Chem., 2016, 36, 2530–2544 CrossRef CAS.
- For reviews on annulation reactions of ynamides, see: (a) Y.-C. Hu, Y. Zhao, B. Wan and Q.-A. Chen, Chem. Soc. Rev., 2021, 50, 2582–2625 RSC; (b) L. Li, W.-F. Luo and L.-W. Ye, Synlett, 2021, 32, 1303–1308 CrossRef CAS; (c) X. Zhou, Z. Liang and X.-N. Wang, Chin. J. Org. Chem., 2021, 41, 1288–1318 CrossRef CAS; (d) F.-L. Hong and L.-W. Ye, Acc. Chem. Res., 2020, 53, 2003–2019 CrossRef CAS PubMed; (e) G. Duret, V. L. Fouler, P. Bisseret and V. Bizet, Eur. J. Org. Chem., 2017, 6816–6830 CrossRef CAS; (f) B. Prabagar, N. Ghosh and A. K. Sahoo, Synlett, 2017, 28, 2539–2555 CrossRef CAS; (g) F. Pan, C. Shu and L.-W. Ye, Org. Biomol. Chem., 2016, 14, 9456–9465 RSC; (h) X.-N. Wang, H.-S. Yeom, L.-C. Fang, S. He, Z.-X. Ma, B. L. Kedrowski and R. P. Hsung, Acc. Chem. Res., 2014, 47, 560–578 CrossRef CAS PubMed.
- For reviews on transition metal-catalyzed cross-coupling reactions of ynamides, see: (a) F.-L. Hong and L.-W. Ye, Acc. Chem. Res., 2020, 53, 2003–2019 CrossRef CAS PubMed; (b) L. Li, T.-D. Tan, Y.-Q. Zhang, X. Liu and L.-W. Ye, Org. Biomol. Chem., 2017, 15, 8483–8492 RSC.
- For reviews on radical reactions of ynamides, see: (a) T.-D. Tan, Z.-S. Wang, P.-C. Qian and L.-W. Ye, Small Methods, 2021, 5, 2000673 CrossRef CAS PubMed; (b) C. Mahe and K. Cariou, Adv. Synth. Catal., 2020, 362, 4820–4832 CrossRef CAS.
- For reviews on ynamide coupling reagents, see: (a) T. Liu, S.-L. Xu and J.-F. Zhao, Chin. J. Org. Chem., 2021, 41, 873–887 CrossRef CAS; (b) Q. Song, L.-Y. Kong, L.-L. Zhu, R. Hong and S.-H. Huang, Chin. J. Chem., 2021, 39, 1022–1024 CrossRef CAS; (c) L. Hu and J.-F. Zhao, Synlett, 2017, 28, 1663–1670 CrossRef CAS. For some selected reports, see: (d) S.-L. Xu, D.-D. Jiang, Z.-J. Peng, L. Hu, T. Liu, L.-L. Zhao and J.-F. Zhao, Angew. Chem., Int. Ed., 2022, 61, e202212247 CrossRef CAS PubMed; (e) T. Liu, X. Zhang, Z.-J. Peng and J.-F. Zhao, Green Chem., 2021, 23, 9916–9921 RSC; (f) C.-C. Yao, J.-H. Yang, X.-B. Lu, S.-Y. Zhang and J.-F. Zhao, Org. Lett., 2020, 22, 6628–6631 CrossRef CAS PubMed; (g) X.-W. Wang, Y. Yang, Y.-L. Zhao, S. Wang, W.-C. Hu, J.-M. Li, Z. Wang, F.-L. Yang and J.-F. Zhao, J. Org. Chem., 2020, 85, 6188–6194 CrossRef CAS PubMed; (h) M. Yang, X.-W. Wang and J.-F. Zhao, ACS Catal., 2020, 10, 5230–5235 CrossRef CAS; (i) J.-H. Yang, C.-L. Wang, C.-C. Yao, C.-Q. Chen, Y.-F. Hu, G.-F. He and J.-F. Zhao, J. Org. Chem., 2020, 85, 1484–1494 CrossRef CAS PubMed; (j) J.-H. Yang, C.-L. Wang, S.-L. Xu and J.-F. Zhao, Angew. Chem., Int. Ed., 2019, 58, 1382–1386 CrossRef CAS PubMed; (k) L. Hu, S.-L. Xu, Z.-G. Zhao, Y. Yang, Z.-Y. Peng, M. Yang, C.-L. Wang and J.-F. Zhao, J. Am. Chem. Soc., 2016, 138, 13135–13138 CrossRef CAS PubMed.
- (a) Y.-L. Zhao, Y.-L. Tu, M.-Z. Cai and J.-F. Zhao, Chin. J. Org. Chem., 2022, 42, 85–99 CrossRef CAS; (b) W.-X. Peng, E. Vessally, S. Arshad, A. Monfared, A. Hosseinian and L. Edjlali, Top. Curr. Chem., 2019, 377, 20–41 CrossRef PubMed; (c) G. Evano, K. Jouvin and A. Coste, Synthesis, 2013, 45, 17–26 CrossRef CAS. For very recent selected reports, see: (d) K. Kagami, X.-Y. Liang, N. Ishibashi, S. Ohrui, M. Tayu and N. Saito, Chem. Commun., 2023, 59, 8274–8277 RSC; (e) R. Kawakami, S. Usui, N. Tada and A. Itoh, Chem. Commun., 2023, 59, 450–453 RSC.
- For very recent selected reviews, see: (a) J.-B. Li, C.-Y. Huang and C.-J. Li, Trends Chem., 2022, 4, 479–494 CrossRef CAS; (b) A. Bera, L. M. Kabadwal, S. Bera and D. Banerjee, Chem. Commun., 2022, 58, 10–28 RSC; (c) T. Tian, Z.-P. Li and C.-J. Li, Green Chem., 2021, 23, 6789–6862 RSC; (d) C. M. A. Afsina, T. Aneeja, M. Neetha and G. Anilkumar, Eur. J. Org. Chem., 2021, 1776–1808 CrossRef CAS; (e) K. Peng and Z.-B. Dong, Adv. Synth. Catal., 2021, 363, 1185–1201 CrossRef CAS; (f) W.-H. Zhuang, X.-F. Zhang and Q.-F. Huang, Chin. J. Org. Chem., 2021, 41, 529–542 CrossRef CAS.
- (a) For a review, see ref. 9b; ; (b) H. T. N. Le, T. V. Tran, N. T. S. Phan and T. Truong, Catal. Sci. Technol., 2015, 5, 851–859 RSC; (c) X.-J. Jin, K. Yamaguchi and N. Mizuno, Chem. Commun., 2012, 48, 4974–4976 RSC; (d) X.-G. Tong, G.-H. Ni, X. Deng and C.-F. Xia, Synlett, 2012, 23, 2497–2500 CrossRef CAS; (e) T. Hamada, X. Ye and S. S. Stahl, J. Am. Chem. Soc., 2008, 130, 833–835 CrossRef CAS PubMed.
- For reviews on nitrogen-centred radicals, see: (a) H. Song, X.-Y. Liu and Y. Qin, Acta Chim. Sin., 2017, 75, 1137–1149 CrossRef; (b) T. Xiong and T. Zhang, Chem. Soc. Rev., 2016, 45, 3069–3087 RSC; (c) J.-R. Chen, X.-Q. Hu, L.-Q. Lu and W.-J. Xiao, Chem. Soc. Rev., 2016, 45, 2044–2056 RSC; (d) S. Z. Zard, Chem. Soc. Rev., 2008, 37, 1603–1618 RSC.
- F. Xu, L. Zhu, S.-B. Zhu, X.-M. Yan and H.-C. Xu, Chem. – Eur. J., 2014, 20, 12740–12744 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ob02032k |
| This journal is © The Royal Society of Chemistry 2024 |