
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrganocatalytic enantioselective Mannich and retro-Mannich reactions and combinations of these reactions to afford tetrasubstituted α-amino acid derivatives†
Kuan-Lin
Chen
and
Fujie
Tanaka
 *
*
Chemistry and Chemical Bioengineering Unit, Okinawa Institute of Science and Technology Graduate University, 1919-1 Tancha, Onna, Okinawa 904-0495, Japan. E-mail: ftanaka@oist.jp
First published on 15th December 2023
Abstract
Organocatalytic asymmetric Mannich reactions and kinetic resolutions of the products via retro-Mannich reactions that afford enantiomerically enriched tetrasubstituted α-amino acid derivatives (α,α-disubstituted-α-amino acid derivatives) were developed. Furthermore, the combination of the Mannich reaction and the retro-Mannich reaction allowed access to products with almost perfect enantiopurities.
α,α-Disubstituted-α-amino acid derivatives or tetrasubstituted α-amino acid derivatives are found in pharmaceuticals and their building blocks1–4 (Fig. 1). Enantioselective synthesis of these molecules is of interest.1–4 For example, tetrasubstituted α-amino acid derivatives have been synthesized by Mannich reactions with α-ketimino esters,1 addition reactions to α-ketimino esters,2 and α-alkylations and arylations of α-amino acid derivatives.3 However, there remains a need for methods that provide highly enantiomerically enriched, functionalized tetrasubstituted α-amino acid derivatives.1–3 For example, Mannich reactions of aldehydes or ketones with ketimines catalyzed by enamine-forming catalysts (or under aminocatalysis) have been reported,1a,e,5,6 but the ketimines used in these reactions were relatively reactive (Fig. 2) and are not for the syntheses of α-methyl-substituted tetrasubstituted α-amino acid derivatives. Here, we report proline-catalyzed enantioselective Mannich reactions of 1,4-benzoxazinone-derived cyclic ketimino esters 1 and ketones 2 to afford tetrasubstituted α-amino acid derivatives 3 (Scheme 1). We also report kinetic resolutions of (±)-3via retro-Mannich reactions (Scheme 1). Furthermore, we report a strategy that combines the Mannich reactions with the retro-Mannich reaction to afford highly enantiomerically enriched tetrasubstituted α-amino acid derivatives 3 with er >99
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, which are difficult to obtain from either reaction alone (Scheme 1).
:1, which are difficult to obtain from either reaction alone (Scheme 1).
 | ||
| Fig. 1 Examples of bioactive tetrasubstituted-α-amino acid derivatives. | ||
 | ||
| Fig. 2 Ketimines used previously in Mannich reactions under aminocatalysis. | ||
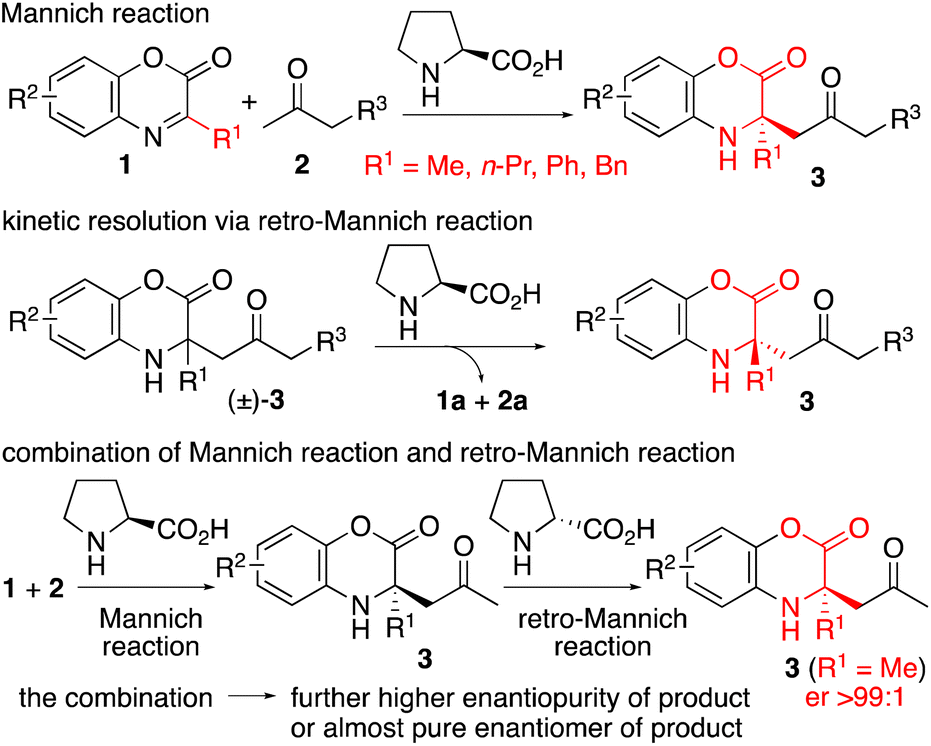 | ||
| Scheme 1 Catalytic asymmetric Mannich reaction, retro-Mannich reaction, and the combination of these two reactions to afford highly enantiomerically enriched tetrasubstituted α-amino acid derivatives. | ||
The 1,4-benzoxazinone ring structure is often present in bioactive molecules.7,8 Thus, Mannich reaction products bearing the benzoxazinone moiety could be useful for drug discovery.7 In addition, benzoxazinone-derived cyclic amino esters can be transformed into other derivatives by reactions on the ester group and the deprotection of the phenol group attached to the amine group.1a,7a,e,8 The use of cyclic ketimino esters avoids potential hydrolysis of the imine during Mannich reactions. Whereas catalytic enantioselective Mannich reactions involving 1,4-benzoxazinone-derived aldimino esters,5c,8 other cyclic aldimino esters,9 or acyclic aldimino esters10 to afford α-amino acid derivatives have been reported,5c,8–10 only a small number of examples of Mannich reactions of the corresponding ketimino esters to afford tetrasubstituted α-amino acid derivatives have been reported.1,5,11 Highly enantioselective Mannich reactions of ketimino esters in which a simple alkyl group (such as a methyl group) is substituted on the imine carbon of the ketimino ester (i.e., the substituent is not an electron-withdrawing substituent) have rarely been reported.1c We explored the Mannich reactions of ketones with cyclic ketimino esters bearing alkyl and aryl substituents on the imine carbon.
First, the catalysts and conditions of the Mannich reaction between ketimino ester 1a and acetone (2a) to afford Mannich product 3a were evaluated (Table 1).
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst (equiv.) | Solvent | Yieldb (%) | erc |
| a Conditions: 1a (1.0 mmol, 1.0 equiv.), 2a (5.0 mmol, 5.0 equiv.), and catalyst in solvent (1.0 mL) at rt (25 °C) for 48 h. b Determined by 1H NMR analysis based on the ratio between 1a and 3a before purification. c Determined by HPLC analysis of purified 3a. d Reaction for 7 days. | ||||
| 1 | (S)-Proline (0.1) | DMSO | 76 | 96:4 |
| 2 | (R)-β-Proline (0.1) | DMSO | 15 | 65:35 |
| 3 | (S)-Serine (0.1) | DMSO | 0 | — |
| 4 | (S)-Valine (0.1) | DMSO | 0 | — |
| 5 | O-tBu-(2S,3R)-threonine (0.1) | DMSO | 1 | 94:6 |
| 6 | (S)-Proline (0.1) | 2-PrOH | 12 | 97:3 |
| 7 | (S)-Proline (0.1) | DMF | 14 | 99:1 |
| 8 | (S)-Proline (0.1) | NMP | 12 | 98:2 |
| 9 | (S)-Proline (0.1) | CH3CN | 2 | 98:2 |
| 10 | (S)-Proline (0.1) | Toluene | 0 | — |
| 11 | (S)-Proline (0.1) | CHCl3 | 1 | 94:6 |
| 12 | (S)-Proline (0.05) | DMSO | 76 | 96:4 |
| 13 | (S)-Proline (0.3) | DMSO | 75 | 96:4 |
| 14 | (S)-Proline (0.025) | DMSO | 64 | 98:2 |
| 15d | (S)-Proline (0.05) | DMSO | 88 | 97:3 |

The reaction in the presence of (S)-proline (5 to 10 mol%) in DMSO resulted in the formation of 3a in high yields (>75%) with high enantioselectivities (er 96:4 to 97:3) in 48 h (Table 1, entries 1 and 12). The reaction in the presence of (S)-proline (2.5 mol%) also generated 3a with high enantioselectivity but was slower (Table 1, entry 14). For the formation of 3a by the Mannich reaction, the use of (S)-proline (5 mol%) as the catalyst in DMSO (Table 1, entries 12 and 15) was optimal for the catalysts and conditions tested. Based on previously reported information,5c,9a the stereochemistry of the major enantiomer of Mannich reaction product 3a obtained by (S)-proline catalysis was suggested to be R.
Next, using the optimized conditions identified, the scope of the (S)-proline-catalyzed Mannich reactions was examined using various 1,4-benzoxazinone-derived ketimino esters 1 and ketones 2 (Scheme 2). The imines bearing substituents at the p-position of the ester oxygen of the benzoxazinone moiety reacted with acetone to afford Mannich products 3b, 3c, and 3h in high yields with high enantioselectivities (er 97:3 to 96:4). The reactions of the imines bearing the substituents at the p-position of the nitrogen of the benzoxazinone moiety were relatively slow (formations of 3d, 3e, 3f, and 3g). For these reactions, a longer reaction time did not further increase the yield of the product; it seems that the formation of the Mannich product was in equilibrium with the decomposition of the Mannich product via the retro-Mannich reaction after the Mannich reaction product accumulated to a certain level. Reactions of the imines bearing substituents larger than the methyl groups also afforded the Mannich products 3i, 3j, and 3k. The reaction of 2-butanone with imine 1a also afforded product 3l, although the reaction rate was slow. With the exception of 3f, which has a nitro group substituent, all Mannich products 3 were obtained with high enantioselectivities (er 99:1 to 91:9).
 | ||
| Scheme 2 Scope of the Mannich reactions of 1 and 2 to afford 3. Conditions: 1a (1.0 mmol), 2a (5.0 mmol), and (S)-proline (0.05 mmol) in DMSO (1.0 mL) at rt (25 °C). | ||
To investigate the Mannich reactions in more detail and test the possibility of kinetic resolutions of (±)-3, the Mannich reaction to afford 3a and the decomposition of 3a in the presence of (S)-proline were analyzed at various time points (Scheme 3). In the Mannich reaction that forms 3a, the er ((R)-isomer to (S)-isomer) of 3a was 99:1 at 7% conversion and was gradually decreased to 97:3 at 58% conversion at 24 h, and this er was almost unchanged at 87% conversion at 120 h (Scheme 3a).
 | ||
| Scheme 3 Time-dependent analyses of the Mannich reaction to form 3a and of the retro-Mannich reaction of 3a. | ||
When (±)-3a was treated with (S)-proline, (S)-isomer-enriched 3a was obtained with the formation of imine 1a and acetone, indicating that the decomposition of (R)-3a was more favorable than the decomposition of (S)-3a in the presence of (S)-proline (Scheme 3b). For the kinetic resolution of (±)-3a, the use of 0.3 equiv. of proline led to better results than the use of 0.05 equiv. The preferential decomposition of the (R)-isomer of 3a by (S)-proline was further confirmed by comparison of the reactions of (R)-enriched 3a and (S)-enriched 3a in the presence of (S)-proline (Scheme 3c). The rate of the decomposition of (R)-3a in the presence of (S)-proline was more than 10 times faster than that of (S)-3a based on the imine formation after 24 h (Scheme 3c). Notably, even when the concentration of (R)-3a was low or when the ratio of (R)-3a/(S)-3a was less than 2:98, (R)-3a was efficiently decomposed in the presence of (S)-proline, resulting in (S)-3a with er >99:1 (Scheme 3c, reaction of (S)-3a (R:S = 2:98)).
These results indicate that (R)-3a is kinetically formed in the (S)-proline-catalyzed Mannich reaction and that (R)-3a, the major enantiomer formed in the Mannich reaction, is kinetically decomposed by the (S)-proline-catalyzed retro-Mannich reaction. This is similar to what was observed in previously reported amine-catalyzed aldol reactions that construct chiral tertiary alcohol centers; in these reactions, stereoselective retro-aldol reactions catalyzed by the catalyst erode the enantiopurity of the aldol product during the aldol reactions.12
Based on the results of the decomposition of 3a catalyzed by proline, kinetic resolutions of various (±)-3 in the presence of (S)-proline were performed (Scheme 4). Through the kinetic resolutions catalyzed by (S)-proline, the opposite enantiomers of 3a relative to the (S)-proline-catalyzed Mannich reactions were obtained.
 | ||
| Scheme 4 Kinetic resolutions of (±)-3. Conditions: (±)-3a (0.5 mmol) and (S)-proline (0.15 mmol) in DMSO (5.0 mL) at 40 °C. a(R)-Proline was used instead of (S)-proline. | ||
Both the Mannich reactions and the kinetic resolutions via retro-Mannich reactions afforded enantiomerically enriched 3, but the enantiopurities of 3 were not perfect. To obtain 3 with er >99:1, the (S)-proline-catalyzed Mannich reaction and the (R)-proline-catalyzed retro-Mannich reaction were combined (Scheme 5). When Mannich products 3 formed by the Mannich reaction in the presence of (S)-proline were treated with (R)-proline, the minor enantiomers were decomposed, and products 3 with er >99:1 were obtained.
 | ||
| Scheme 5 Combination of the (S)-proline-catalyzed Mannich reaction and the (R)-proline-catalyzed retro-Mannich reaction. | ||
In summary, we have developed catalytic enantioselective Mannich reactions of cyclic ketimino esters with ketones that afford highly enantiomerically enriched α-amino acid derivatives bearing tetrasubstituted carbon centers at the α-positions. We have also developed kinetic resolutions via retro-Mannich reactions to yield the α-amino acid derivatives. Importantly, we have demonstrated that a combination of catalytic bond formation in the presence of a homochiral catalyst and the decomposition of the minor enantiomer product leads to the formation of highly enantiomerically enriched products (er >99:1). The combination of the (S)-proline-catalyzed Mannich reaction and the (R)-proline-catalyzed retro-Mannich reaction provided tetrasubstituted α-amino acid derivatives with very high enantiopurities.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Michael Chandro Roy, Instrumental Analysis Section, Okinawa Institute of Science and Technology Graduate University for mass analyses. This study was supported by the Okinawa Institute of Science and Technology Graduate University.References
- (a) T. Kano, S. Song, Y. Kubota and K. Maruoka, Angew. Chem., Int. Ed., 2012, 51, 1191 CrossRef CAS PubMed; (b) K.-Q. Hou, F. Zhou, X.-P. Chen, Y. Ge, A. S. C. Chan and X.-F. Xiong, J. Org. Chem., 2020, 85, 9661 CrossRef CAS PubMed; (c) Y. Deng, X. Shi, G. Shi, X. Lu, J. Luo and L. Deng, JACS Au, 2022, 2, 2678 CrossRef CAS PubMed; (d) S. Saaby, K. Nakama, M. A. Lie, R. G. Hazell and K. A. Jørgensen, Chem. – Eur. J., 2003, 9, 6145 CrossRef CAS PubMed; (e) W. Zhuang, S. Saaby and K. A. Jørgensen, Angew. Chem., Int. Ed., 2004, 43, 4476 CrossRef PubMed.
- (a) H. Miyabe, Y. Yamaoka and Y. Takemoto, J. Org. Chem., 2006, 71, 2099 CrossRef CAS PubMed; (b) K. Yeung, F. J. T. Talbot, G. P. Howell, A. P. Pulis and D. J. Procter, ACS Catal., 2019, 9, 1655 CrossRef CAS; (c) U. Bhakta, P. V. Kattamuri, J. H. Siitonen, L. B. Alemany and L. Kürti, Org. Lett., 2019, 21, 9208 CrossRef CAS PubMed; (d) X.-H. Deng, J.-X. Jiang, Q. Jiang, T. Yang, B. Chen, L. He, W.-D. Chu, C.-Y. He and Q.-Z. Liu, Org. Lett., 2022, 24, 4586 CrossRef CAS PubMed.
- (a) T. Ooi, M. Takeuchi, M. Kameda and K. Maruoka, J. Am. Chem. Soc., 2000, 122, 5228 CrossRef CAS; (b) J. Kikuchi, N. Momiyama and M. Terada, Org. Lett., 2016, 18, 2521 CrossRef CAS PubMed; (c) M. Wang, M. Zhou, L. Zhang, Z. Zhang and W. Zhang, Chem. Sci., 2020, 11, 4801 RSC; (d) L. Chen, M.-J. Luo, F. Zhu, W. Wen and Q.-X. Guo, J. Am. Chem. Soc., 2019, 141, 5159 CrossRef CAS PubMed; (e) X. Huo, R. He, J. Fu, J. Zhang, G. Yang and W. Zhang, J. Am. Chem. Soc., 2017, 139, 9819 CrossRef CAS PubMed; (f) L. Wei, Q. Zhu, S.-M. Xu, X. Chang and C.-J. Wang, J. Am. Chem. Soc., 2018, 140, 1508 CrossRef CAS PubMed; (g) Y. Matsumoto, J. Sawamura, Y. Murata, T. Nishikata, R. Yazaki and T. Ohshima, J. Am. Chem. Soc., 2020, 142, 8498 CrossRef CAS PubMed; (h) K. Tomohara, T. Yoshimura, R. Hyakutake, P. Yang and T. Kawabata, J. Am. Chem. Soc., 2013, 135, 13294 CrossRef CAS PubMed; (i) Z.-C. Liu, Z.-Q. Wang, X. Zhang and L. Yin, Nat. Commun., 2023, 14, 2187 CrossRef CAS PubMed; (j) D. J. Leonard, J. W. Ward and J. Clayden, Nature, 2018, 562, 105 CrossRef CAS PubMed; (k) C. Cativiela, M. Ordóñez and J. L. Viveros-Ceballos, Tetrahedron, 2020, 76, 130875 CrossRef CAS.
- (a) C. Najera and J. M. Sansano, Chem. Rev., 2007, 107, 4584 CrossRef CAS PubMed; (b) N. Yasukawa and S. Nakamura, Chem. Commun., 2023, 59, 8343 RSC; (c) Y. Kondo, Y. Hirazawa, T. Kadota, K. Yamada, K. Morisaki, H. Morimoto and T. Ohshima, Org. Lett., 2022, 24, 6594 CrossRef CAS PubMed; (d) N. S. Chowdari and C. F. Barbas, Org. Lett., 2005, 7, 867 CrossRef CAS PubMed.
- (a) S. Zhang, L. Li, Y. Hu, Z. Zha, Z. Wang and T.-P. Loh, Org. Lett., 2015, 17, 1050 CrossRef CAS PubMed; (b) G. Li, M. Liu, S. Zou, X. Feng and L. Lin, Org. Lett., 2020, 22, 8708 CrossRef CAS PubMed; (c) M. Viji, J. Sim, S. Li, H. Lee, K. Oh and J.-K. Jung, Adv. Synth. Catal., 2018, 360, 4464 CrossRef CAS; (d) S. Mondal, R. D. Aher, V. Bethi, Y.-J. Lin, T. Taniguchi, K. Monde and F. Tanaka, Org. Lett., 2022, 24, 1853 CrossRef CAS PubMed; (e) V. Bethi and F. Tanaka, Org. Lett., 2022, 24, 6711 CrossRef CAS PubMed; (f) Y. Garg, J. Osborne, S. Vasylevskyi, N. Velmurugan and F. Tanaka, J. Org. Chem., 2023, 88, 11096 CrossRef CAS PubMed.
- (a) S. Zhang, L. Li, Y. Hu, Y. Li, Y. Yang, Z. Zha and Z. Wang, Org. Lett., 2015, 7, 5036 CrossRef PubMed; (b) V. A. Sukach, N. M. Golovach, V. V. Pirozhenko, E. B. Rusanov and M. V. Vovk, Tetrahedron: Asymmetry, 2008, 19, 761 CrossRef CAS.
- (a) H. Miyabe, Y. Yamaoka and Y. Takemoto, Synlett, 2004, 2597 CAS; (b) B. Eftekhari-Sis and M. Zirak, Chem. Rev., 2017, 117, 8326 CrossRef CAS PubMed; (c) M. F. Bhat, A. P. Luján, M. Saifuddin and G. J. Poelarends, ACS Catal., 2022, 12, 11421 CrossRef CAS PubMed; (d) L.-Q. Lu, Y. Li, K. Junge and M. Beller, J. Am. Chem. Soc., 2015, 137, 2763 CrossRef CAS PubMed; (e) X. Zhang, B. Xu and M.-H. Xu, Org. Chem. Front., 2016, 3, 944 RSC; (f) H. Lou, Y. Wang, E. Jin and X. Lin, J. Org. Chem., 2016, 81, 2019 CrossRef CAS PubMed.
- Y.-H. Geng, Y.-Z. Hua, S.-K. Jia and M.-C. Wang, Chem. – Eur. J., 2021, 27, 5130 CrossRef CAS PubMed.
- (a) B. T. Hahn, R. Fröhlich, K. Harms and F. Glorius, Angew. Chem., Int. Ed., 2008, 47, 9985 CrossRef CAS PubMed; (b) S. A. Moteki, J. Han, S. Arimitsu, M. Akakura, K. Nakayama and K. Maruoka, Angew. Chem., Int. Ed., 2012, 51, 1187 CrossRef CAS PubMed.
- (a) K. Juhl, N. Gathergood and K. A. Jørgensen, Angew. Chem., Int. Ed., 2001, 40, 2995 CrossRef CAS; (b) D. Ferraris, B. Young, C. Cox, T. Dudding, W. J. Drury, L. Ryzhkov, A. E. Taggi and T. Lectka, J. Am. Chem. Soc., 2002, 124, 67 CrossRef CAS PubMed; (c) S. Kobayashi, T. Hamada and K. Manabe, J. Am. Chem. Soc., 2002, 124, 5640 CrossRef CAS PubMed; (d) B. M. Trost and L. R. Terrell, J. Am. Chem. Soc., 2003, 125, 338 CrossRef CAS PubMed; (e) T. Kano, Y. Yamaguchi, O. Tokuda and K. Maruoka, J. Am. Chem. Soc., 2005, 127, 16408 CrossRef CAS PubMed; (f) A. Córdova, W. Notz, G. Zhong, J. M. Betancort and C. F. Barbas, J. Am. Chem. Soc., 2002, 124, 1842 CrossRef PubMed; (g) H. Zhang, S. Mitsumori, N. Utsumi, M. Imai, N. Garcia-Delgado, M. Mifsud, K. Albertshofer, P. H.-Y. Cheong, K. N. Houk, F. Tanaka and C. F. Barbas, J. Am. Chem. Soc., 2008, 130, 875 CrossRef CAS PubMed.
- B. Qiao, Y.-J. Huang, J. Nie and J.-A. Ma, Org. Lett., 2015, 17, 4608 CrossRef CAS PubMed.
- J. Wang, Z.-X. Deng, C.-M. Wang, P.-J. Xia, J.-A. Xiao, H.-Y. Xiang, X.-Q. Chen and H. Yang, Org. Lett., 2018, 20, 7535 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ob01855e |
| This journal is © The Royal Society of Chemistry 2024 |