
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Increasing the versatility of the biphenyl-fused-dioxacyclodecyne class of strained alkynes†
Sam
Forshaw
 a,
Jeremy S.
Parker
b,
William T.
Scott
ac,
Richard C.
Knighton
ad,
Neelam
Tiwari
a,
Samson M.
Oladeji
a,
Andrew C.
Stevens
a,
Yean Ming
Chew
ac,
Jami
Reber
a,
Guy J.
Clarkson
a,
Mohan K.
Balasubramanian
c and
Martin
Wills
*a
a,
Jeremy S.
Parker
b,
William T.
Scott
ac,
Richard C.
Knighton
ad,
Neelam
Tiwari
a,
Samson M.
Oladeji
a,
Andrew C.
Stevens
a,
Yean Ming
Chew
ac,
Jami
Reber
a,
Guy J.
Clarkson
a,
Mohan K.
Balasubramanian
c and
Martin
Wills
*a
aDepartment of Chemistry, The University of Warwick, Coventry, CV4 7AL, UK. E-mail: M.wills@warwick.ac.uk
bEarly Chemical Development, Pharmaceutical Sciences, IMED Biotech Unit, AstraZeneca, Macclesfield, SK10 2NA, UK
cWarwick Medical School, The University of Warwick, Coventry, CV4 7AL, UK
dSchool of Chemistry, University of Southampton, SO17 1BJ, UK
First published on 13th December 2023
Abstract
Biphenyl-fused-dioxacyclodecynes are a promising class of strained alkyne for use in Cu-free ‘click’ reactions. In this paper, a series of functionalised derivatives of this class of reagent, containing fluorescent groups, are described. Studies aimed at understanding and increasing the reactivity of the alkynes are also presented, together with an investigation of the bioconjugation of the reagents with an azide-labelled protein.
Introduction
Bioconjugations1 using Strain-Promoted Alkyne–Azide Cycloadditions (SPAAC)2 are important reactions, due to their high reaction rates, the lack of a requirement for a catalyst,3 and their bioorthogonal reactivity. A number of strained alkynes have been widely adopted for synthetic and biolabelling applications.4 Early examples such as OCT 1,4a and fluorinated derivatives such as DIFO 24c were followed by highly reactive, strained alkynes such as DIBO 3,4d,e BCN 4,4f DIBAC 54g and BARAC 64h (Fig. 1), which have been employed in numerous biolabelling applications. The second order rate constant for each alkyne with benzylazide provides a convenient means for comparison of their reactivity (Fig. 1). The high reactivity of the strained alkynes is reflected by the distorted sp bond angles. Derivatives of strained alkynes, loaded with a fluorescent group, also undergo cyclisations with azide-containing molecules both in vitro and in vivo without the need for a catalyst. | ||
| Fig. 1 Strained alkynes and their second order rate constants for addition to benzyl azide in MeCN, MeOH or MeCN/H2O at rt.2d R = functional group. | ||
We,5–7 and others,8 recently reported the synthesis of strained alkynes of general structure 7, where X/Y = O, NH, NTs,9 as reagents for copper-free cycloaddition reactions with azides (Fig. 2). Specific examples of this class of alkyne are 8–14 and, although not as reactive as some of the well-established strained alkynes shown in Fig. 1, they benefit from the straightforward introduction of the alkyne through the reaction of a 2,2′-biphenol reagent with 1,4-ditosylbut-2-yne, and readily react with azides at concentrations above ca. 0.1 M. Alabugin et al.8 described how the ‘twisted’ structure of this class of dioxacyclodecyne is alleviated upon approach of the azide. This effect generates improved reactivity when the heteroatom (X, Y) in the structure is an oxygen or a nitrogen atom.
 | ||
| Fig. 2 Biphenyl-fused dioxacyclodecynes and second order rate constants for addition with benzylazide under the conditions shown. Bond angles were established by X-ray crystal structure analyses or DFT calculations. | ||
Altering the heteroatoms X/Y in 7 influences their reactivity; Alabugin8 studied biphenyl-fused-diazacyclodecyne derivative 13 and observed a similar rate constant to that of the unsubstituted biphenyl-dioxacyclodecyne 8, when reacted with benzyl azide in CDCl3 at rt. The p-toluenesulfonamide derivative 14 exhibited a lower rate of reaction, corresponding to its less distorted sp bond angle of 169° (Fig. 2).8 This is less distorted than in the more reactive unsubstituted alkyne 8 which has an sp angle of ca. 166° and significantly less than for highly reactive alkynes such as DIBAC and BARAC (Fig. 1). In this paper, we describe our studies aimed at expanding the range of biphenyl-fused-dioxacyclodecyne reagents, and at increasing their reactivity in Cu-free click reactions.
Results and discussion
Fluorescent enone-containing derivatives
Enones can exhibit fluorescent properties. A report by Xing et al.10 indicated that an effective pairing was a compound containing a methoxy group and a dimethylamino group para- to the ketone and aldehyde enone precursors respectively. Alkyne 156 was reacted under basic conditions with 4-dimethylaminobenzaldehyde to give 16 in good yield (Fig. 3).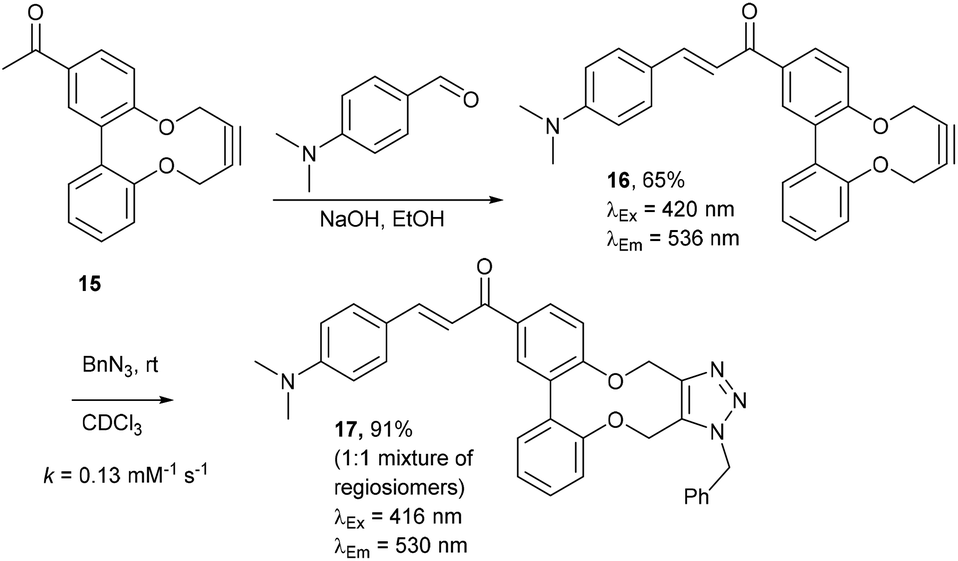 | ||
| Fig. 3 Synthesis of enone alkyne, 16, and subsequent cycloaddition of benzyl azide to form triazole 17. | ||
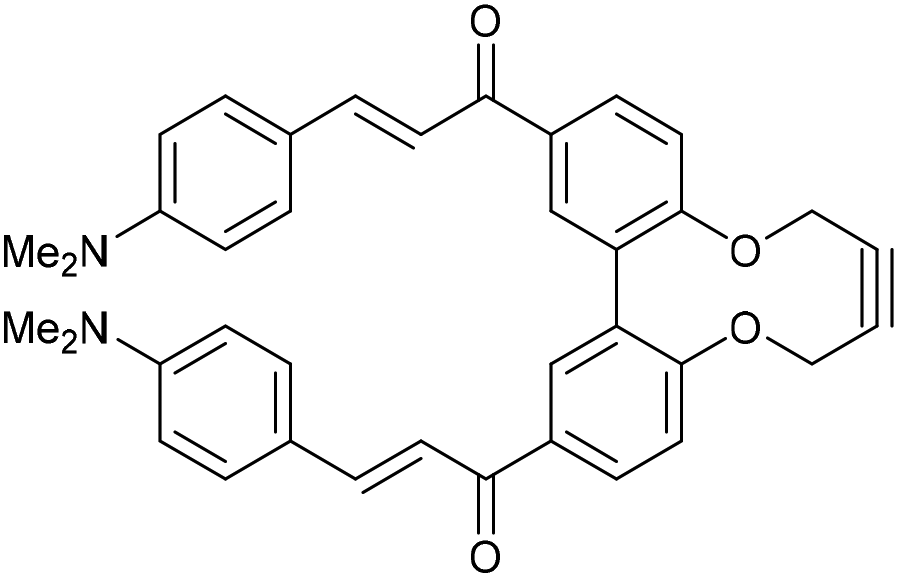
Compound 16 exhibited strong fluorescence excitation and emission maxima at 420 nm and 536 nm respectively. The fluorescent data for the benzyl azide addition product 17 (formed as an inseparable 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of regioisomers) exhibited excitation and emission wavelengths essentially unchanged from alkyne 16. The rate constant for the cycloaddition was 0.13 mM−1 s−1, similar to that of biphenyl-fused-dioxacyclodecyne 8. The reaction of 2,2′-biphenol with an excess of AlCl3 and acetyl chloride gave 18 in moderate yield (47%). The ester groups were then hydrolysed using lithium hydroxide to give the diacetyl biphenol 19, and its cyclisation with ditosylate 20 gave the strained diacetyl alkyne 21 (Fig. 4). Dienone 22 was then formed using the same conditions as for the synthesis of compound 17, using two equivalents of 4-dimethylaminobenzaldehyde. Unexpectedly, the reaction rate for the reaction between dienone 22 and benzyl azide (k = 0.25 mM−1 s−1) to give 23 was double that for enone 16, possibly due to steric effects between the two large enone groups on the opposite side to the alkyne.
:1 mixture of regioisomers) exhibited excitation and emission wavelengths essentially unchanged from alkyne 16. The rate constant for the cycloaddition was 0.13 mM−1 s−1, similar to that of biphenyl-fused-dioxacyclodecyne 8. The reaction of 2,2′-biphenol with an excess of AlCl3 and acetyl chloride gave 18 in moderate yield (47%). The ester groups were then hydrolysed using lithium hydroxide to give the diacetyl biphenol 19, and its cyclisation with ditosylate 20 gave the strained diacetyl alkyne 21 (Fig. 4). Dienone 22 was then formed using the same conditions as for the synthesis of compound 17, using two equivalents of 4-dimethylaminobenzaldehyde. Unexpectedly, the reaction rate for the reaction between dienone 22 and benzyl azide (k = 0.25 mM−1 s−1) to give 23 was double that for enone 16, possibly due to steric effects between the two large enone groups on the opposite side to the alkyne.
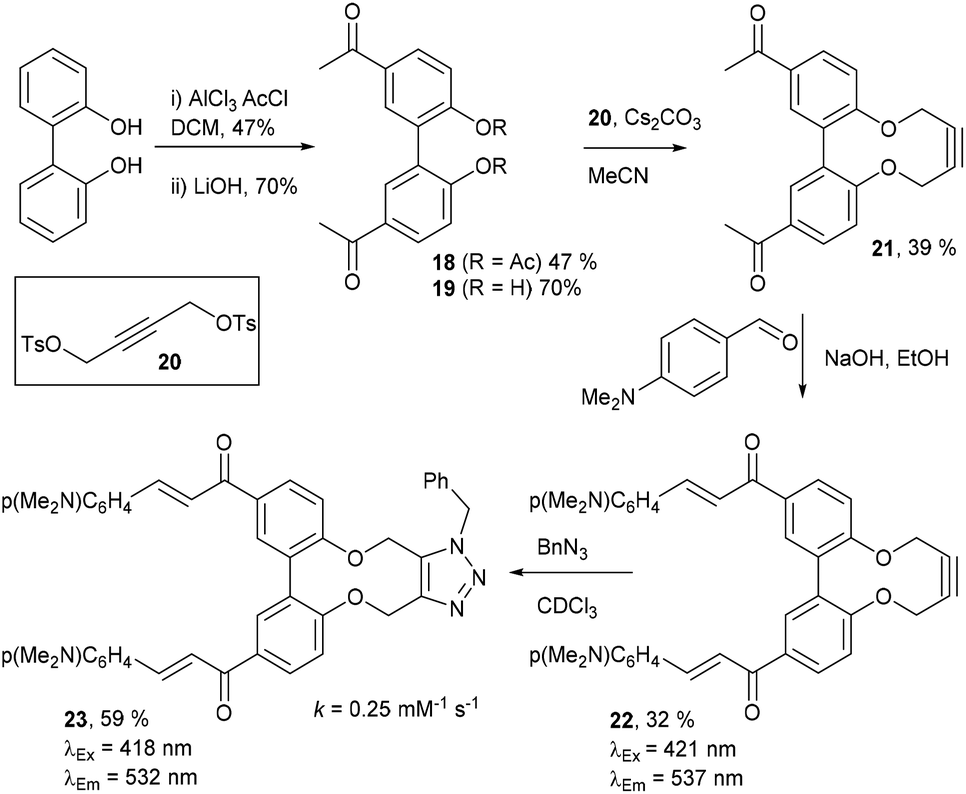 | ||
| Fig. 4 Synthesis of dienone alkyne, 22, and subsequent cycloaddition with benzyl azide to give triazole, 23. | ||
Two further derivatives, 24 and 25, containing fluorescein and rhodamine groups respectively, were prepared through DCC couplings with known fluorescent precursors 26 and 27, and the strained alkyne 28 (Fig. 5).11,12 These were available for subsequent testing with an azide-functionalised protein, which is described in a later section.
 | ||
| Fig. 5 Synthesis of fluorescein and rhodamine-containing strained alkynes. | ||
Further derivatives and attempts to increase the reactivity of the alkynes
To understand the significance of the heteroatoms on the reactivity, compound 29, containing a combination of an oxygen and toluenesulfonamide heteroatoms, was prepared. 2-Iodoaniline was converted to 30 which was coupled under Suzuki conditions with 2-hydroxyphenylboronic acid to give the biphenyl 31, and subsequently converted to 29 in moderate yield (Fig. 6a).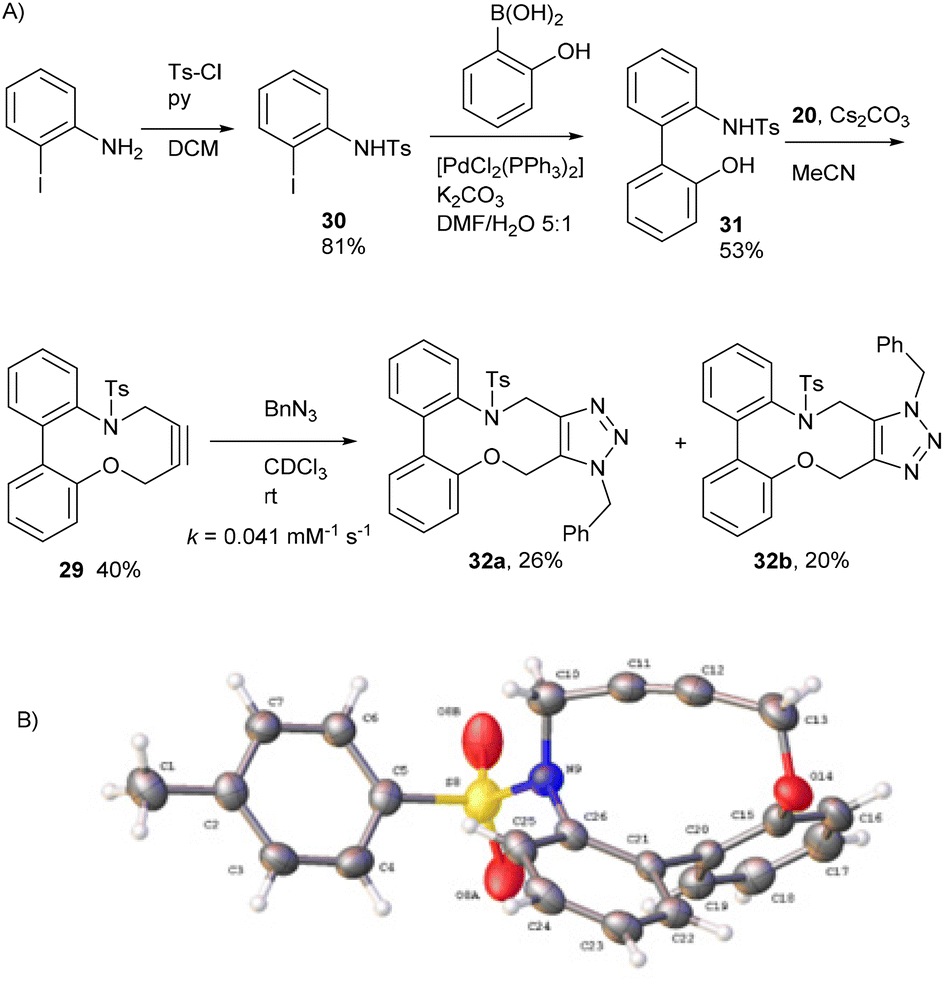 | ||
| Fig. 6 (A) The synthetic route to the heterocyclic alkyne 29 and subsequent reaction with BnN3. The regiochemistry of each isomer of 32 has not been confirmed. (B) X-ray crystal structure of 29. | ||
Alkyne 29 underwent cycloaddition with benzyl azide in CDCl3 with a second order rate constant of just 0.041 mM−1 s−1, to form 32, as a mixture of isomers. This rate is lower than the same reaction of biphenyl-fused-dioxacyclodecyne, 8, but higher than the reaction of biphenyl-NTs-alkyne, 14. The X-ray crystal structure of 29 (Fig. 6b) revealed sp bond angles of ca. 169.5 and 167.4° respectively. The synthesis of other cyclic alkynes was considered, including the use of sulfur as a heteroatom. However, the introduction of sulfur atoms generally diminishes the reactivity of the alkyne due to the larger bond length of the sulfur–carbon bonds.13,14 In an earlier result published by Wills et al.,7 bisalkyne 33 (Fig. 7) was used in ‘protein stapling’ reactions. Analysis of the reaction by NMR, which featured direct formation of 34 without the monoadduct 35, suggested that the first cycloaddition was rate limiting and that the second cycloaddition occurred much more rapidly. Molecular modelling confirmed that the transition state for the second cycloaddition had a lower energy barrier than the first. This increase in reactivity is likely caused by an increased distortion of the remaining alkyne bond.
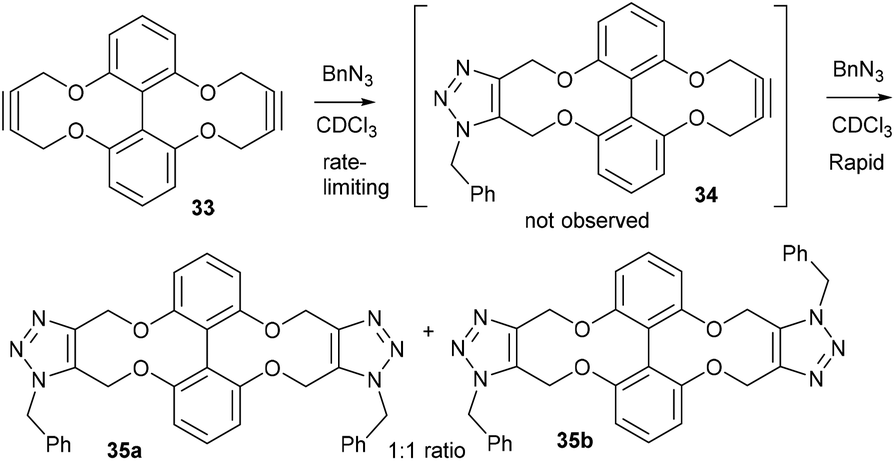 | ||
| Fig. 7 The reaction between bisalkyne, 33, and benzyl azide. | ||
It was speculated that harnessing this effect into a strained alkyne could be beneficial. To achieve this, we studied the effects that functional groups at the 6 and 6′ positions have on the rates of cycloaddition. The known biphenol 36,15 was converted to 6,6′-dimethoxybiphenyl-dioxacyclodecyne 37 in low yield (Fig. 8) but sufficient material was isolated to test the addition reaction. The rate constant for the reaction of 37 with BnN3, forming adduct 38, was 0.20 mM−1 s−1, indicating that methoxy groups at these positions have little effect on the rate of reaction or the structure of the alkyne and the distortion of the alkyne bond.
 | ||
| Fig. 8 Synthetic route to 6,6′-dimethoxybiphenyl-dioxacyclodecyne 37 and subsequent click reaction with benzyl azide in CDCl3. | ||
A route to asymmetric biphenols with a bridge between the 6 and 6′ positions has been reported using a removable chiral bridging group.16,17 Using this approach, dimethylsulfonate, 39 was reacted with the known tetrol 407 under the conditions reported by Harada et al.,16 producing the ethyl bridged biphenol 41,18 in moderate yield. Cyclisation with 1,4-dibromobutane formed the bicyclic compound 42. Lithium di-tert-butylbiphenyl (LiDBB) cleaved the more strained ethyl bridge in 42 selectively to produce 43 in good yield and this was then cyclised with alkyne 20 to give the 4C bridged strained alkyne 44 (Fig. 9).8
 | ||
| Fig. 9 Synthetic route to 44 and subsequent cycloaddition reaction with benzyl azide to produce triazole 45. | ||
The 4C-oxo-bridged biphenyl-fused-dioxabiphenylcyclodecyne 44, reacted with benzyl azide to give adduct 45, with a rate constant of 2.1 mM−1 s−1, representing an increase compared to analogous biphenyl-fused-dioxacyclodecyne 37 (k = 0.17 mM−1 s−1). The increase in reactivity is likely caused by the 6,6′-4C bridge forcing the phenyl rings to lie in a more planar structure and providing more distortion to the alkyne bond angles. To increase this effect further, the synthesis of a three-carbon bridged derivative was attempted, however this was not successful. Given the promising result with 44, an N-containing C4-bridged reagent was prepared. Ullmann homo-coupling of 4619 with activated copper was carried out to give protected biphenol 47 in high yield. An attempt at the Ullmann coupling of the unprotected analogue of 46 was unsuccessful. Deprotection of 47 gave biphenol 48 in high yield, which was then cyclised to the strained alkyne 49 (Fig. 10).
 | ||
| Fig. 10 Synthetic route to the 6,6′-dinitrobiphenyl-dioxacyclodecyne 49 and derivatives, with rate constraints for subsequent cycloadditions with benzyl azide in CDCl3(unless otherwise indicated) Where sp bond angles are given, these were determined by X-ray crystallography (Fig. 11). | ||
It was found that the use of iron powder and ammonium chloride selectively reduced the nitro groups to amines to give 50, leaving the alkyne intact (Fig. 10). 6,6′-Diamino-dioxacyclodecyne 50 was then reacted with toluenesulfonyl chloride, dansyl chloride and mesyl chloride under basic conditions to give 51 (Ts), 52 (Dns) and 53 (Ms) respectively. To create the anticipated more reactive derivatives, each bisulfonamide was reacted with 1,4-dibromobutane under basic conditions using a syringe pump to maintain a pseudo-dilute solution.8 These studies afforded the bridged product, 54, in moderate yield from the ditosylate precursor. Unfortunately, the bis-dansyl precursor 52 gave no corresponding product 55, likely due to increased steric hindrance. Tests on the cyclisation of dimesylate 53 with varying equivalents of 1,4-dibromobutane revealed that the use of three equivalents of the dibromide gave an improved yield of C4-cyclised product 56 over the use of one equivalent. This was surprising as we were concerned that an excess of the dihalide would result in dialkylation of the dimesylate prior to intramolecular cyclisation. However, the improved yield indicates that the intramolecular step must outpace the second N-alkylation. The cycloadditions of the new alkynes with benzyl azide in CDCl3 (at rt) were tested (Fig. 10). For 49, the rate constant calculated for this reaction was 0.64 mM−1 s−1; an improvement of about a factor of four compared to 37, suggesting that the electron-withdrawing nitro group increases the reactivity. The X-ray crystal structure of 49 (Fig. 11a) indicates that the alkyne bond angles average 165.6°, more distorted than the biphenyl-fused-dioxacyclodecyne, 8. For diamine 50, the rate constant was 0.40 mM−1 s−1; slightly lower than that for 49, but slightly higher than that observed for the reaction with biphenyl-fused-dioxacyclodecyne 37. Bisulfonylated compounds 51 and 53 produced the triazole products 57 and 59 respectively upon reaction with benzyl azide, however due to the low solubility of alkyne 52, a rate constant could not be accurately determined and the anticipated product 58 was not isolated. The reaction between 51 (Ts) and 53 (Ms) and benzyl azide gave rate constants of 0.13 mM−1 s−1 in each case, similar to that of biphenyl-fused-dioxacyclodecyne 8. However, the corresponding cycloaddition reactions of 54 and 56 proceeded with significantly higher rate constants of 62.1 and 5.0 mM−1 s−1 respectively, in CDCl3 to give products 60 and 61 respectively. The reaction of 54 in an NMR tube was substantially complete within 5 hours ([alkyne] = 0.04 mM), representing a step change in reactivity for this class of strained alkynes. X-ray structures of 54 and 46 (Fig. 11b and c), revealing the alkyne sp bond angles to be 163.7° and 162.8° in 54 and 164.0°/165.1° in 56. The difference in average alkyne bond angle between compound 54 and compound 49 is only 2.3°, which shows even small changes to the bond angle can have a great influence on the reaction rate. For 56 (diMs) the angles were intermediate, and this was reflected in its reactivity.
 | ||
| Fig. 11 (A) Two views of the X-ray crystal structure of 6,6′-dintrobiphenyl-dioxacyclodecyne 49. (B) Two views of the X-ray crystal structure of bridged alkyne 54. (C) Two views of the X-ray crystal structure of bridged alkyne 56. | ||
When comparing the rate of the reaction between alkyne 54 and benzyl azide with previously published strained alkynes it displays similar reactivity to the difluorinated cyclooctynes, which display rate constants between 42–76 mM−1 s−1.2d The comparative rate constants of the novel compounds in this study give an insight into how electronic and structural effects can combine to produce more reactive alkynes. Although using electron withdrawing groups at the 6 and 6′ position in 49 did improve the reactivity, the largest increases in reactivity came with the addition of the 6,6′-4C bridge, i.e. in 44, 54 and 56. The reason for the difference in reactivity between 54 and 56 may stem from an increase strain created by the bulky tosyl groups creating increased distortion in the alkyne bond.
To establish whether the new alkynes may be compatible with biomolecules, attempts were made to react the novel alkynes with glutathione S-transferase (GST) containing an azidophenylalanine at position 52. In an initial series of tests, an earlier-reported BoDIPY-containing strained alkyne, fluorescein 24 and rhodamine derivative 25 were reacted and a gel indicated that conjugation had occurred in most cases. However, MS analyses of the adducts indicated that this was only the case for the fluorescein derivative 24, hence there may be non-covalent, non-specific interactions between protein and dye in the other cases (see the ESI†). In a second round of tests of non-fluorescent compounds, the dimesylated compound 56 gave an addition product when analysed by mass spectrometry, although the more reactive ditosylated 54 did not. Examination of the second order rate constant for the reaction with BnN3 in DMSO-d6 (i.e. reflecting more closely the conditions used in the enzyme reactions where DMSO/H2O was used) gave k values of 18.3 and 12.5 mM−1 s−1 for 54 and 56 respectively. Hence the rates of each compound were closer in DMSO-d6 than in CHCl3. Coupled to a potential lower solubility of the larger molecule in the water/DMSO mixture used with the enzyme may account for differences in the observed results. Compound 44, bearing a 4C aliphatic linking group, added to the protein, but at a low level. See the ESI† for full details of these tests.
Conclusion
The development of novel strained alkynes for use in bioconjugation is still a focus of international research. Biphenyl-fused-dioxacyclodecynes react with azides without the need for a catalyst. The current investigation into the reactivity of this class of strained alkyne has led to the synthesis of a variety of 6,6′-functionalised biphenyl-fused-dioxacyclodecyne derivatives with rate constants in the region of 0.13–0.64 mM−1 s−1. This inspired the synthesis of a four-carbon bridged class of biphenyl-fused-dioxacyclodecynes, which are more reactive towards azides, with rate constants between 2.13–62.1 mM−1 s−1. There is potential for functionalisation of the sulfonamide groups of the C4-bridged alkynes, e.g. with fluorescent groups, which could provide a reactive, fluorescent strained biphenyl alkyne for use in bioimaging. Further studies of the value of the alkynes, of which 56 represents a promising candidate for further applications, are ongoing.Experimental section
Solvents and reagents were degassed before use and all reactions were carried out under a nitrogen atmosphere using vacuum line apparatus. Reactions were monitored by TLC using aluminium backed silica gel 60 (F254) plates, visualized using UV 254 nm and phosphomolybdic acid or potassium permanganate as appropriate. Flash column chromatography was carried out routinely on silica gel. Reagents were used as received from commercial sources unless otherwise stated. Dry solvents were purchased and used as received. 1H NMR spectra were recorded on a Bruker DPX (300, 400 or 500 MHz) spectrometer. Chemical shifts are reported in δ units, parts per million relative to the singlet at 7.26 ppm for chloroform and 0.00 ppm for TMS. Coupling constants (J) are measured in hertz. Mass spectra for analysis of synthetic products were recorded on a Bruker Esquire 2000 or a Bruker MicroTOF mass spectrometer. IR spectra were recorded on a PerkinElmer Spectrum One FT-IR Golden Gate. Melting points were recorded on a Stuart Scientific SMP 1 instrument and are uncorrected. X-ray crystallography was carried out on a Rigaku Oxford Diffraction SuperNova diffractometer with a duel source (Cu at zero) equipped with an AtlasS2 CCD area detector or an Xcalibur Gemini diffractometer with a Ruby CCD area detector. The procedure and full details of the kinetic 1H NMR runs are given in the ESI.†Safety and hazards
All synthetic organic chemistry has potential hazards, however azides are known to be highly reactive and required full risk assessment and care in handling throughout their preparation, use and disposal. In this study, small amounts of benzylazide (typically <10 mg) were used in NMR-scale tests of reactivity with the strained alkynes.The following compounds were prepared following published methods; ditosyl-1,4-dihydroxybut-2-yne 20,21N-tosyl-2-iodoaniline 30,20 dimethoxydiphenol 36,15 tetrahydroxybiphenyl 40,7 ethanedioldimesylate 39,22 ethylbridged tetrahydroxybiphenyl 4118 and the MOM derivative of 2-iodo-3-nitrophenol 46.19
5-(3-(4-Dimethylaminophenyl)-1-oxo-prop-2-ene)-2,2′-biphenyldioxacyclodecyne (16)
This compound is novel. A solution of 15 (100 mg, 0.360 mmol, 1.0 eq.), 4-dimethylaminobenzaldehyde (58 mg, 0.39 mmol, 1.1 eq.) and NaOH (43 mg, 1.1 mmol, 3.0 eq.) in EtOH (2 mL) was stirred at room temperature for 24 h. H2O (20 mL) was added and the product was extracted with EtOAc (3 × 20 mL). The combined organic extracts were dried over MgSO4 and concentrated. Purification by column chromatography (eluted with 50% EtOAc/hexane) gave the pure product as a yellow solid (96 mg, 0.23 mmol, 65%). Rf = 0.38 (1
 :1 EtOAc/hexane); (found (ESI)) 432.1563 C27H23NNaO3 requires 432.1570; vmax 2916, 2865, 1641, 1565, 1526, 1180, 1170, 966, 802 cm−1; δH (500 MHz, CDCl3) 8.08 (1 H, dd, J = 8.4, 2.1 Hz, ArH) 7.89 (1 H, d, J = 2.1 Hz, ArH) 7.80 (1 H, d, J = 15.4 Hz, COCHCHPh) 7.53 (2 H, d, J = 8.9 Hz, ArH) 7.42 (1 H, ddd, J = 8.0, 6.5, 2.7 Hz, ArH) 7.32 (1 H, d, J = 15.4 Hz, COCHCHPh) 7.28 (1 H, d, J = 8.4 Hz, ArH) 7.19–7.25 (3 H, m, ArH) 6.67 (2 H, d, J = 8.9 Hz, ArH) 4.52–4.62 (2 H, m, OCHaHb) 4.32–4.44 (2 H, m, OCHaHb) 3.03 (6 H, s, NCH3); δC (125 MHz, CDCl3) 189.5, 158.0, 154.5, 152.0, 145.7, 135.8, 135.3, 135.1, 132.5, 131.9, 130.5, 129.5, 129.4, 124.3, 122.9, 122.7, 122.7, 116.7, 111.8, 87.2, 86.3, 63.7, 63.5, 40.1 ppm; m/z (ESI) 410.2 [M + H]+, 432.2 [M + Na]+; Fluorescence (MeCN; λex = 420 nm); λem = 536 nm; UV-Vis (MeCN) λmax (ε/M−1 cm−1): 413 (486976) nm.
:1 EtOAc/hexane); (found (ESI)) 432.1563 C27H23NNaO3 requires 432.1570; vmax 2916, 2865, 1641, 1565, 1526, 1180, 1170, 966, 802 cm−1; δH (500 MHz, CDCl3) 8.08 (1 H, dd, J = 8.4, 2.1 Hz, ArH) 7.89 (1 H, d, J = 2.1 Hz, ArH) 7.80 (1 H, d, J = 15.4 Hz, COCHCHPh) 7.53 (2 H, d, J = 8.9 Hz, ArH) 7.42 (1 H, ddd, J = 8.0, 6.5, 2.7 Hz, ArH) 7.32 (1 H, d, J = 15.4 Hz, COCHCHPh) 7.28 (1 H, d, J = 8.4 Hz, ArH) 7.19–7.25 (3 H, m, ArH) 6.67 (2 H, d, J = 8.9 Hz, ArH) 4.52–4.62 (2 H, m, OCHaHb) 4.32–4.44 (2 H, m, OCHaHb) 3.03 (6 H, s, NCH3); δC (125 MHz, CDCl3) 189.5, 158.0, 154.5, 152.0, 145.7, 135.8, 135.3, 135.1, 132.5, 131.9, 130.5, 129.5, 129.4, 124.3, 122.9, 122.7, 122.7, 116.7, 111.8, 87.2, 86.3, 63.7, 63.5, 40.1 ppm; m/z (ESI) 410.2 [M + H]+, 432.2 [M + Na]+; Fluorescence (MeCN; λex = 420 nm); λem = 536 nm; UV-Vis (MeCN) λmax (ε/M−1 cm−1): 413 (486976) nm.
(E)-1-(1-Benzyl-4,15-dihydro-1H-dibenzo[7,8:9,10][1,6]dioxecino[3,4-d][1,2,3]triazol-8-yl)-3-(4-(dimethylamino)phenyl)prop-2-en-1-one (17)
This compound is novel. A solution of 16 (10 mg, 0.024 mmol, 1 eq.) and benzyl azide (3.2 mg, 0.024 mmol, 1 eq.) in CDCl3 (0.5 mL) was monitored by NMR until completion of the reaction. The solvent was removed under vacuum and the crude material was purified by column chromatography (eluted with 50% EtOAc/hexane) to give the pure product as two inseparable isomers in a 1
 :1 ratio as an orange solid (11.8 mg, 0.022 mmol, 91%).
:1 ratio as an orange solid (11.8 mg, 0.022 mmol, 91%).
R
f = 0.16 (1:1 EtOAc/hexane); (found (ESI)) 543.2379 C34H31N4O3 requires 543.2391; vmax 2923, 1570, 1521, 1495, 1443, 1433, 1332, 1261, 1180, 1167, 1107, 810, 750 cm−1; δH (500 MHz, CDCl3) 8.00 (0.5 H, dd, J = 8.5, 2.1 Hz, PhCHCHCO) 7.92 (1 H, dd, J = 6.6, 2.1 Hz, ArH) 7.82 (0.5 H, dd, J = 8.5, 2.1 Hz, PhCHCHCO) 7.78 (1 H, d, J = 15.4 Hz, PhCHCHCO) 7.53 (2 H, d, J = 8.8 Hz, ArH), 7.38–7.42 (2 H, m, ArH), 7.32–7.37 (3 H, m, ArH), 7.26–7.05 (7 H, m, ArH), 6.78 (0.5 H, d, J = 7.9 Hz, PhCHCHCO) 6.68 (2 H, d, J = 8.8 Hz, ArH) 6.59 (0.5 H, d, J = 8.5 Hz, PhCHCHCO) 5.81 (0.5 H, d, J = 16.0 Hz, CHeHf) 5.77 (0.5 H, d, J = 16.0 Hz, CHeHf) 5.63 (0.5 H, d, J = 13.7 Hz, CHcHd) 5.35–5.45 (2.5 H, m, CHeHf + CHcHd + CHcHd) 5.22 (1.5 H, m, 2 × OCHaHb + OCHaHb) 5.05 (0.5 H, d, J = 13.0 Hz, OCHaHb) 3.03 (6 H, s, NCH3); (125 MHz, CDCl3) 188.8, 188.8, 159.4, 158.8, 156.9, 156.0, 152.0, 151.9, 145.5, 145.2, 144.8, 144.5, 134.6, 134.4, 134.1, 133.1, 132.3, 132.1, 131.2, 131.2, 130.7, 130.5, 130.4, 130.3, 130.1, 129.7, 129.6, 129.6, 129.4, 129.3, 129.2, 128.9, 128.9, 128.7, 127.2, 127.1, 123.6, 122.8, 122.7, 122.4, 116.5, 116.4 116.0, 114.7, 114.4, 113.5, 111.8, 63.6, 62.9, 61.0, 60.4, 52.6, 52.4, 40.1 ppm; m/z (ESI) 543.2 [M + H]+, 565.2 [M + Na]+; fluorescence (MeCN; λex = 416 nm); λem = 530 nm; UV-Vis (MeCN) λmax (ε/M−1 cm−1): 410 (70000) nm.
5,5′-Diacetyl-[1,1′-biphenyl]-2,2′-diyl diacetate (18)
This compound is novel. A solution of AlCl3 (6.00 g, 45 mmol, 8.3 eq.) in DCM (4 mL) was cooled to 0 °C. Acetyl chloride (4.40 g, 56.0 mmol, 10.4 eq.) was added to the solution and the reaction mixture was stirred for 30 min. A solution of 2,2′-biphenol (1.00 g, 5.4 mmol, 1.0 eq.) in DCM (10 mL) was added to the reaction mixture at 0 °C and the mixture was stirred for a further 30 min. The reaction was then refluxed until completion, at which point H2O (30 mL) was added dropwise to quench. The product was extracted with EtOAc (3 × 30 mL) and the combined organic extracts were dried over MgSO4, which was removed by filtration, and concentrated under vacuum to give the crude product. Purification by column chromatography gave the pure product as a white solid (892 mg, 2.5 mmol, 47%).

R
f = 0.60 (1:1 EtOAc/Pet Ether); mp = 205–209 °C; (found (ESI)) 377.0982 C20H18NaO6 requires 377.0996; vmax 1740, 1683, 1600, 1355, 1191, 910, 619 cm−1; δH (500 MHz, CDCl3) 8.07 (2 H, dd, J = 8.5, 2.1 Hz, ArH) 7.97 (2 H, d, J = 2.1 Hz, ArH) 7.33 (2 H, d, J = 8.5 Hz, ArH) 2.65 (6 H, s, COCH3) 2.09 (6 H, s, OCOCH3) ppm; δC (125 MHz, CDCl3) 196.5, 168.6, 151.7, 135.0, 131.6, 130.0, 129.6, 123.0, 26.7, 20.7 ppm; m/z (ESI) 377.1 [M + Na]+.
1,1′-(6,6′-Dihydroxy-[1,1′-biphenyl]-3,3′-diyl)bis(ethan-1-one) (19)
This compound is novel. A solution of compound 18 (848 mg, 2.25 mmol, 1 eq.) and LiOH (302 mg, 12.6 mmol, 5.6 eq.) in MeOH/H2O 1
 :1 (10 mL) was refluxed for 2 h. The mixture was then cooled to room temperature before 2 M HCl (20 mL) was added. The product was then extracted with EtOAc (3 × 20 mL), the combined organic extracts were dried over MgSO4 and concentrated and recrystalised in MeOH to give the pure product as a white solid (472 mg, 1.76 mmol, 70%).
:1 (10 mL) was refluxed for 2 h. The mixture was then cooled to room temperature before 2 M HCl (20 mL) was added. The product was then extracted with EtOAc (3 × 20 mL), the combined organic extracts were dried over MgSO4 and concentrated and recrystalised in MeOH to give the pure product as a white solid (472 mg, 1.76 mmol, 70%).
R
f = 0.2 (1:1 EtOAc/Pet Ether); mp = 177–181 °C; (found (ESI)) 293.0782 C16H14NaO4 requires 293.0784; vmax 3222, 1651, 1579, 1383, 1354, 1255, 818, 583 cm−1; δH (500 MHz, DMSO-d6) 10.30 (2 H, s, OH) 7.84 (2 H, dd, J = 8.5, 2.3 Hz, ArH) 7.78 (2 H, d, J = 2.3 Hz, ArH) 7.00 (2 H, d, J = 8.5 Hz, ArH) 2.50 (6 H, s, OCH3); δC (125 MHz, d6-DMSO) 196.2, 159.6, 132.3, 129.6, 128.3, 124.9, 115.4, 26.3 ppm; m/z (ESI) 293.1 [M + Na]+.
5,5′-Diacetyl-2,2′-biphenyldioxacyclodecyne (21)
This compound is novel. To a solution of Cs2CO3 (1.33 g, 4.1 mmol, 2.2 eq.) in MeCN (43 mL), at 60 °C, was added a solution of ditosylate 20 (733 mg, 1.86 mmol, 1 eq.) and compound 19 (500 mg, 1.86 mmol, 1 eq.) in MeCN (8.7 mL) over 6 h. The mixture was stirred for a further 12 h before being cooled to room temperature and the solvent removed under vacuum. The residue was dissolved in H2O (30 mL) and the product was extracted with EtOAc (3 × 30 mL). The combined organic extracts were dried over MgSO4 and concentrated to give the crude product, which was purified by column chromatography (eluted with 50% EtOAc/hexane) to give the crude product as a white solid (233 mg, 0.73 mmol, 39%).

R
f = 0.42 (1:1 EtOAc/Pet. Ether); mp = 188–189 °C; (found (ESI)) 343.0935 C20H16NaO4 requires 343.0941; vmax 3060, 2919, 1673, 1594, 1477, 1238, 1191, 956, 676 cm−1; δH (500 MHz, CDCl3) 8.05 (2 H, dd, J = 8.5, 2.3 Hz, ArH), 7.82 (2 H, d, J = 2.3 Hz, ArH) 7.28 (2 H, d, J = 8.5 Hz, ArH) 4.55–4.64 (2 H, m, OCHaHb) 4.36–4.44 (2 H, m, OCHaHb) 2.60 (6 H, s, COCH3); δC (125 MHz, CDCl3) 197.1, 158.7, 135.2, 133.4, 132.6, 129.7, 123.1, 86.7, 63.7, 26.7 ppm; m/z (ESI) 343.1 [M + Na]+.
5,5′-Bis(3-(4-dimethylaminophenyl)-1-oxo-prop-2-ene)-2,2′-biphenyldioxacyclodecyne (22)
This compound is novel. A solution of compound 21 (99.2 mg, 0.310 mmol, 1.0 eq.), 4-dimethylamino benzaldehyde (101 mg, 0.680 mmol, 2.2 eq.) and NaOH (74 mg, 1.9 mmol, 6 eq.) in EtOH (2 mL) was stirred at room temperature for 12 h. H2O (20 mL) was added, and the product was extracted with EtOAc (3 × 20 mL). The combined organic extracts were then dried over MgSO4 and concentrated. Purification by column chromatography (eluted with 50% EtOAc/hexane) gave the pure product as an orange solid (59 mg, 0.10 mmol, 32%).
R
f = 0.26 (1:1 EtOAc/hexane); (found (ESI)) 605.2396 C38H34N2NaO4 requires 605.2411; vmax 2906, 2854, 1647, 1569, 1518, 1331, 1163, 1109, 810, 747 cm−1; δH (500 MHz, CDCl3) 8.12 (2 H, dd, J = 8.4, 2.1 Hz, ArH), 7.91 (2 H, d, J = 2.1 Hz, ArH), 7.81 (2 H, d, J = 15.4 Hz, COCHCHPh), 7.55 (4 H, d, J = 8.9 Hz, ArH), 7.34 (2 H, d, J = 15.4 Hz, COCHCHPh), 7.29–7.34 (2 H, m, ArH), 6.69 (4 H, d, J = 8.9 Hz, ArH), 4.52–4.68 (2 H, m, OCHaHb), 4.33–4.49 (2 H, m, OCHaHb), 3.09 (12 H, s, NCH3) ppm; δC (125 MHz, CDCl3) 189.4, 158.0, 152.0, 145.8, 135.2, 132.4, 130.5, 129.8, 123.0, 122.7, 116.7, 111.8, 86.8, 63.7, 40.1 ppm; m/z (ESI) 583.3 [M + Na]+, 605.2 [M + Na]+; fluorescence (MeCN; λex = 420 nm); λem = 536 nm; UV-Vis (MeCN) λmax (ε/M−1 cm−1): 416 (148000) nm.
(2E,2′E)-1,1′-(1-Benzyl-4,15-dihydro-1H-dibenzo[7,8:9,10][1,6]dioxecino[3,4-d][1,2,3]triazole-8,11-diyl)bis(3-(4-(dimethylamino)phenyl)prop-2-en-1-one) (23)
This compound is novel. A solution of compound 22 (10 mg, 0.017 mmol, 1 eq.) and benzyl azide (2.3 mg, 0.017 mg, 1 eq.) in CDCl3 (0.5 mL) was monitored by NMR until completion. The solvent was removed under vacuum and the crude material was purified by column chromatography (eluted with EtOAc) to give the pure product as a red solid (7.0 mg, 0.010 mmol, 58%).

R
f = 0.20 (1:1 EtOAc/hexane); (found (ESI)) 716.3214 C45H42N5O4 requires 716.3231; vmax 1575, 1521, 1334, 1167, 1117, 1026, 979, 809 cm−1; δH (500 MHz, CDCl3) 8.06 (1 H, dd, J = 8.5, 2.1 Hz, ArH), 8.02 (1 H, d, J = 2.1 Hz, ArH) 8.00 (1 H, d, J = 2.1 Hz, ArH), 7.94–7.99 (1 H, m, ArH) 7.90–7.95 (1 H, m, ArH) 7.78–7.84 (2 H, m, PhCHCHCO) 7.55 (4 H, m, J = 8.7, 3.5 Hz, ArH) 7.40 (2 H, m, PhCHCHCO) 7.33–7.38 (3 H, m, ArH) 7.20 (3 H, m, J = 6.1, 2.9 Hz, ArH) 6.68 (4 H, m, J = 8.9 Hz, ArH) 5.82 (1 H, d, J = 15.7 Hz, CHeHf) 5.58 (1 H, d, J = 13.4 Hz, OCHcHd) 5.39–5.47 (2 H, m, OCHcHd + CHeHf) 5.29 (1 H, d, J = 13.4 Hz, OCHaHb) 5.15 (1 H, d, J = 13.4 Hz, OCHaHb) 3.03 (12 H, s, NCH3); δC (125 MHz, CDCl3) 188.7, 159.7, 159.1, 152.0, 152.0, 145.7, 145.4, 144.5, 134.4, 134.4, 133.4, 132.0, 131.1, 131.1, 130.5, 130.4, 130.1, 130.0, 129.8, 129.4, 129.0, 128.9, 127.2, 122.7, 122.7, 116.4, 116.3, 115.6, 114.0, 111.8, 63.5, 60.8, 52.7, 40.1 ppm; m/z (ESI) 716.3 [M + H]+, 738.3 [M + Na]+; fluorescence (MeCN; λex = 418 nm); λem = 532 nm; UV-Vis (MeCN) λmax (ε/M−1 cm−1): 409 (199900) nm.
Fluorescein amide 2,2′-biphenyldioxacyclodecyne (24)
This compound is novel. 6-Hydroxy-9-(2-(piperazine-1-carbonyl)phenyl)-3H-xanthen-3-one 26, synthesised as previously described,11 (78.0 mg, 0.195 mmol) and acid alkyne 28 (54.6 mg, 0.195 mmol) were dissolved in anhydrous DMF (2 mL). DMAP (59.5 mg, 0.287 mmol) and EDCI (74.9 mg, 0.390 mmol) were added and the reaction stirred under N2 at room temperature for 18 hours. H2O (10 mL) was added, extracted with CH2Cl2/IPA (4
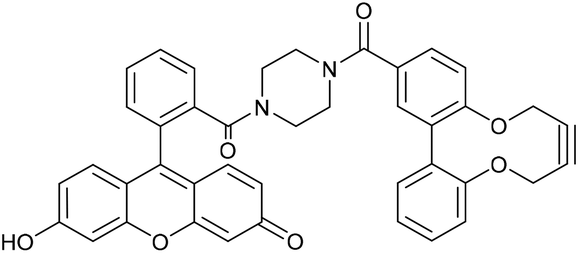 :1) (3 × 10 mL) and the combined organic layers dried over MgSO4. The crude mixture was purified by column chromatography (SiO2; CH2Cl2/MeOH; 100:0 → 90:10) to afford the compound as an orange solid (74 mg, 0.111 mmol, 57%). Rf = 0.60 (4:1 DCM/MeOH); mp 187–198 (dec) °C; (found (ESI) [M + H]+, 663.2125. C41H31N2O7 requires [M + H]+, 663.2126); νmax 1591, 1417, 1379, 1195, 1001, 964 and 847 cm−1; δH (500 MHz, CD3OD) 7.90 (2 H, s, ArH), 7.80–7.61 (3 H, m, ArH), 7.53–7.46 (1 H, m, ArH), 7.45–7.35 (2 H, m, ArH), 7.29 (1 H, d, J = 8.3, ArH), 7.20–7.12 (4 H, m, ArH), 6.76–6.68 (3 H, m, ArH), 4.57–4.43 (2 H, m, OCHaHb), 4.43–4.30 (2 H, m, OCHaHb), 3.45 (8 H, br. s, NCH2); δC (126 MHz, CD3OD) 172.2, 169.8, 157.9, 156.0, 153.7, 137.7, 136.5, 136.4, 132.9, 132.8, 132.6, 132.2, 131.8, 131.6, 131.2, 131.1, 130.6, 129.4, 128.9, 125.1, 124.5, 123.8, 104.4, 88.0, 87.3, 64.4, 64.3, 64.3, 64.1; m/z (ESI) 663 (M+ + H, 30%) and 685 (M+ + Na, 30); UV-Vis (MeCN) lmax (ε/M−1 cm−1): 487 (13200), 457 (18800), 430 (16700), 353 (9200), 227 (59000) nm; fluorescence (MeCN; λex = 531 nm); λem 545 nm.
:1) (3 × 10 mL) and the combined organic layers dried over MgSO4. The crude mixture was purified by column chromatography (SiO2; CH2Cl2/MeOH; 100:0 → 90:10) to afford the compound as an orange solid (74 mg, 0.111 mmol, 57%). Rf = 0.60 (4:1 DCM/MeOH); mp 187–198 (dec) °C; (found (ESI) [M + H]+, 663.2125. C41H31N2O7 requires [M + H]+, 663.2126); νmax 1591, 1417, 1379, 1195, 1001, 964 and 847 cm−1; δH (500 MHz, CD3OD) 7.90 (2 H, s, ArH), 7.80–7.61 (3 H, m, ArH), 7.53–7.46 (1 H, m, ArH), 7.45–7.35 (2 H, m, ArH), 7.29 (1 H, d, J = 8.3, ArH), 7.20–7.12 (4 H, m, ArH), 6.76–6.68 (3 H, m, ArH), 4.57–4.43 (2 H, m, OCHaHb), 4.43–4.30 (2 H, m, OCHaHb), 3.45 (8 H, br. s, NCH2); δC (126 MHz, CD3OD) 172.2, 169.8, 157.9, 156.0, 153.7, 137.7, 136.5, 136.4, 132.9, 132.8, 132.6, 132.2, 131.8, 131.6, 131.2, 131.1, 130.6, 129.4, 128.9, 125.1, 124.5, 123.8, 104.4, 88.0, 87.3, 64.4, 64.3, 64.3, 64.1; m/z (ESI) 663 (M+ + H, 30%) and 685 (M+ + Na, 30); UV-Vis (MeCN) lmax (ε/M−1 cm−1): 487 (13200), 457 (18800), 430 (16700), 353 (9200), 227 (59000) nm; fluorescence (MeCN; λex = 531 nm); λem 545 nm.
Rhodamine amide 2,2′-biphenyldioxacyclodecyne (25)
This compound is novel. N-(6-(Diethylamino)-9-(2-(piperazine-1-carbonyl)phenyl)-3H-xanthen-3-ylidene)-N-ethylethanaminium 27, synthesised as previously described,12 (100 mg, 0.195 mmol) and acid alkyne 28 (54.6 mg, 0.195 mmol) were dissolved in anhydrous DMF (2 mL). DMAP (59.5 mg, 0.287 mmol) and EDCI (74.9 mg, 0.390 mmol) were added and the reaction stirred under N2 at room temperature for 18 hours. H2O (10 mL) was added, extracted with CH2Cl2/IPA (4
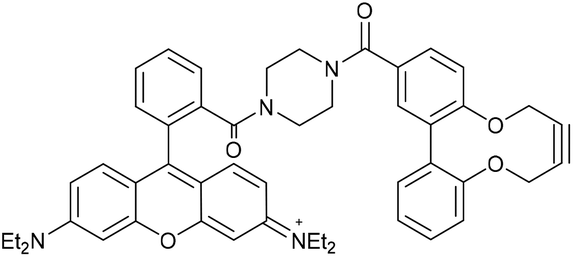 :1) (3 × 10 mL) and the combined organic layers dried over MgSO4. The crude mixture was purified by column chromatography (SiO2; CH2Cl2/MeOH; 100:0 → 90:10) to afford the compound as a dark purple solid (36 mg, 0.049 mmol, 24%).
:1) (3 × 10 mL) and the combined organic layers dried over MgSO4. The crude mixture was purified by column chromatography (SiO2; CH2Cl2/MeOH; 100:0 → 90:10) to afford the compound as a dark purple solid (36 mg, 0.049 mmol, 24%).
R
f = 0.70 (4:1 DCM/MeOH); mp 169–170 (dec) °C; (found (ESI) [M + H]+, 773.3692. C49H49N4O5 requires [M + H]+, 773.3697); νmax 1586, 1334, 1244, 1178, 1122, 1070, 1002 and 759 cm−1; δH (500 MHz, CD3OD) 7.90 (2 H, s, ArH), 7.81–7.74 (2 H, m, ArH), 7.70 (1 H, d, J = 6.8, ArH), 7.54–7.49 (1 H, m, ArH), 7.46–7.39 (2 H, m, ArH), 7.32–7.25 (3 H, m, ArH), 7.23–7.12 (5 H, m, ArH), 7.05 (2 H, d, J = 9.7, ArH), 6.95 (2 H, t, J = 3.1, ArH), 4.55–4.43 (2 H, m, OCHaHb), 4.40–4.33 (2 H, m, OCHaHb), 3.66 (8 H, app. pent., J = 7.3, NCH2CH3), 3.58–3.37 (8 H, m, NCH2), 1.29 (12 H, app. q, J = 7.1, NCH2CH3); δC (126 MHz, CD3OD) 172.2, 169.6, 159.3, 157.9, 157.2, 157.1, 156.0, 137.7, 136.5, 136.4, 133.2, 132.8, 132.4, 132.2, 131.8, 131.6, 131.2, 130.7, 129.4, 128.9, 125.2, 124.6, 123.8, 114.9, 97.4, 88.0, 87.4, 64.4, 64.3, 46.9, 12.8. m/z (ESI) 773 (M+ + H, 100%); UV-Vis (MeCN) lmax (ε/M−1 cm−1): 558 (57400), 522 (35100), 352 (24700), 522 (36200), 250 (59800) nm; fluorescence (MeCN; λex = 566 nm); λem 578 nm.
N-(2′-Hydroxy-[1,1′-biphenyl]-2-yl)-4-methylbenzenesulfonamide (31)
This compound is novel. A solution of 2-hydroxyphenyl boronic acid (175 mg, 1.27 mmol, 1.3 eq.), compound 30 (365 mg, 0.981 mmol, 1.0 eq.), K2CO3 (270 mg, 1.96 mmol, 2.0 eq.) and [PdCl2(PPh3)2] (68 mg, 0.098 mmol, 0.1 eq.) in 5
 :1 DMF-H2O (9 mL) was stirred at 80 °C for 12 h. The reaction was cooled to room temperature and then diluted with H2O (20 mL). The product was then extracted with EtOAc (3 × 20 mL) and the combined organic extracts were dried over MgSO4 before being concentrated. The crude product was then subjected to column chromatography (graduated eluent: 9:1 Hex/EtOAc–7:3 Hex/EtOAc) to give the pure product as a white solid (177 mg, 0.523 mmol, 53%).
:1 DMF-H2O (9 mL) was stirred at 80 °C for 12 h. The reaction was cooled to room temperature and then diluted with H2O (20 mL). The product was then extracted with EtOAc (3 × 20 mL) and the combined organic extracts were dried over MgSO4 before being concentrated. The crude product was then subjected to column chromatography (graduated eluent: 9:1 Hex/EtOAc–7:3 Hex/EtOAc) to give the pure product as a white solid (177 mg, 0.523 mmol, 53%).
R
f = 0.63 (2:3 EtOAc/DCM); mp = 142–146 °C; (found (ESI) [M + Na]+ 362.0821 C16H17NNaO3S requires 362.0821); vmax 3422, 3321, 3231, 1596, 1484, 1163, 700 and 527 cm−1; δH (CDCl3, 500 MHz) 7.74 (1 H, d, J = 8.0 Hz, ArH), 7.42 (1 H, t, J = 8.1 Hz, ArH), 7.36 (2 H, d, J = 8.1 Hz, 2 × ArH), 7.22–7.28 (1 H, m, ArH), 7.15 (1 H, d, J = 7.6 Hz, ArH), 7.09 (2 H, d, J = 8.1 Hz, 2 × ArH), 6.94 (2 H, d, J = 8.0 Hz, ArH), 6.85 (1 H, t, J = 7.5 Hz, ArH) 6.57 (1 H, d, J = 7.5 Hz, ArH) 5.07–5.12 (1 H, br. s, NH) 2.39 (3 H, s, ArCH3) ppm; δC (CDCl3, 125 MHz) 151.8, 143.6, 135.9, 134.6, 131.0, 130.0, 130.0, 129.5, 129.3, 126.9, 126.0, 123.8, 121.3, 116.0, 21.5 ppm; m/z (ESI) 362.2 [M + Na]+.
Compound 32
This compound is novel. A solution of 20 (116 mg, 0.294 mmol, 1.0 eq.), 30 (99.3 mg, 0.294 mmol, 1.0 eq.) and Cs2CO3 (383 mg, 1.18 mmol, 4.0 eq.) in CH3CN (15 mL) was stirred at room temperature for 2 weeks. The solvent was then removed under vacuum and the residue taken up in water (20 mL) the product was extracted with EtOAc (3 × 20 mL). The combined organic extracts were dried over MgSO4 and concentrated to give the crude product. This was then subjected to column chromatography (eluent: 1
 :1 Hex/EtOAc) to give the pure product as a white solid (46.6 mg, 0.12 mmol, 40%).
:1 Hex/EtOAc) to give the pure product as a white solid (46.6 mg, 0.12 mmol, 40%).
R
f = 0.82 (1:1 EtOAc/hexane); mp = 122–128 °C; (found (ESI) [M + Na]+ 412.0981 C23H19NNaO3S requires 412.0978); vmax 3059, 2922, 1596, 1501, 1452, 1106, 1056, 966, 688 and 575 cm−1; δH (CDCl3, 500 MHz) 7.72 (1 H, d, J = 7.5 Hz, ArH), 7.65 (2 H, d, J = 8.2 Hz, 2 × ArH), 7.28–7.37 (2 H, m, 2 × ArH), 7.23–7.28 (3 H, m, 3 × ArH), 7.17–7.23 (2 H, m, 2 × ArH), 7.08 (1 H, d, J = 8.1 Hz, ArH), 6.88 (1 H, d, J = 8.0 Hz, ArH), 4.35 (1 H, d, J = 15.0 Hz, CHaHb) 4.18–4.25 (2 H, m, CH2) 3.50 (1H, d, J = 15.0 Hz, CHaHb), 2.39 (3 H, s, Ar–CH3) ppm; δC (CDCl3, 125 MHz) 154.3, 144.1, 142.1, 136.9, 135.8, 135.6, 133.1, 132.4, 129.8, 129.5, 128.4, 128.2, 128.2, 127.6, 124.3, 122.3, 84.9, 84.3, 63.0, 43.5, 21.6 ppm; m/z (ESI) 362.2 [M + Na]+.
(3-Benzyl-14-tosyl-3,4,14,15-tetrahydrodibenzo[b,d][1,2,3]triazolo[4,5-h][1,6]oxazecine) and 32B (1-benzyl-14-tosyl-1,4,14,15-tetrahydrodibenzo-[b,d][1,2,3]triazolo[4,5-h][1,6]oxazecine) (32A)
These compounds are novel. To a solution of compound 31 (20 mg, 0.051 mmol, 1.0 eq.) in CDCl3 (0.5 mL) was added benzyl azide (6.8 mg, 6.4 μL, 0.051 mmol, 1 eq.). The mixture was left undisturbed at room temperature and monitored by proton NMR until full conversion was observed. The chloroform was removed by evaporation and the residue purified by column chromatography (eluted with DCM) to provide the product as two isolable isomers A (white solid) (6.8 mg, 0.013 mmol, 26%) and B (white solid) (5.4 mg, 0.010 mmol, 20%). The stereochemical assignments are arbitrary.

A; Rf = 0.28 (DCM); (found (ESI) [M + Na]+ 545.1615 C30H26N4NaO3S requires 545.1618); vmax 3028, 2925, 2854, 1596, 1478, 1439, 1353, 1331, 1159 and 738 cm−1; δH (CDCl3, 500 MHz) 7.28–7.44 (7 H, m, ArH), 7.13–7.20 (3 H, m, ArH), 7.03 (2 H, d, J = 8.1 Hz, ArH), 6.98–7.02 (1 H, m, ArH), 6.91 (2 H, d, J = 8.1 Hz, ArH), 6.88–6.90 (2 H, m, ArH), 5.76 (1 H, d, J = 15.7 Hz, CHeHf), 5.64 (1 H, d, J = 15.7 Hz, CHeHf), 5.54 (1 H, d, J = 14.5 Hz, CHcCHd), 5.14 (1H, d, J = 14.5 Hz, CHcHd), 4.91 (1 H, d, J = 15.7 Hz CHaHb), 4.47 (1 H, d, J = 15.7 Hz, CHaHb), 2.39 (3 H, s, Ar–CH3) ppm; δC (CDCl3, 125 MHz) 154.4, 144.6, 143.9, 140.2, 140.0, 135.5, 134.8, 131.3, 131.2, 131.0, 130.1, 129.3, 129.2, 129.1, 128.7, 128.6, 128.5, 128.4, 127.8, 127.1, 121.8, 112.7, 62.0, 52.4, 44.4, 21.5 ppm; m/z (ESI) 523.3 [M + H]+ 545.2 [M + Na]+.
B; Rf = 0.1 (DCM); (found (ESI) [M + Na]+ 545.1615 C30H26N4NaO3S requires 545.1618); vmax 3028, 2925, 2854, 1596, 1478, 1439, 1353, 1331, 1159 and 738 cm−1; δH (CDCl3, 500 MHz) 7.33–7.40 (5 H, m, ArH), 7.28–7.31 (2 H, m, ArH),7.19–7.21 (2H, m, ArH), 7.13–7.18 (4 H, m, ArH), 7.03–7.08 (3 H, m, ArH), 6.45 (1 H, m, ArH), 5.65 (1 H, d, J = 15.7 Hz, CHcHd), 5.31 (1 H, d, J = 15.7 Hz, CHcHd) 5.05 (1 H, d, J = 13.7 Hz, CHeHf) 5.04 (1H, d, J = 14.8 Hz, CHaHb), 4.96 (1 H, d, J = 13.7 Hz CHeHf) 4.59 (1 H, d, J = 14.8 Hz, CHaHb) 2.43 (3 H, s, Ar–CH3) ppm; δC (CDCl3, 125 MHz) 155.4, 143.7, 142.4, 140.6, 139.4, 134.4, 134.0, 133.0, 132.0, 131.7, 131.1, 129.2, 129.2, 128.8, 128.7, 128.6, 128.4, 128.3, 127.2, 126.7, 123.0, 114.8, 60.0, 52.4, 47.2, 21.6 ppm; m/z (ESI) 523.3 [M + H]+ 545.2 [M + Na]+.
6,6-Dimethoxy-2,2′-biphenyldioxacyclodecyne (37)
This compound is novel. To a stirring solution of compound 20 (145 mg, 0.368 mmol, 1 eq.) and compound 3615 (90 mg, 0.37 mmol, 1 eq.) in MeCN (20 mL) was added Cs2CO3 (475 mg, 1.46 mmol, 4 eq.). The mixture was stirred for 10 days at 50 °C and monitored by TLC. Solvents were removed under vacuum and residue was dissolved in H2O (30 mL). The aqueous layer was then extracted with EtOAc (3 × 30 mL) and the combined organic extracts were dried over MgSO4, which was removed by filtration. The solvents were removed under vacuum to give the crude product. The crude product was then purified by column chromatography (eluted with 0–25% EtOAc/heptane) to give the pure product as a white solid (10 mg, 0.034 mmol, 10%).

Mp = 134–136 °C; (found (ESI)) 297.1180 C18H17O4 requires 297.1127; δH (500 MHz, CDCl3) 7.37 (2 H, t, J = 8.2 Hz, ArH) 6.78–6.87 (4 H, m, ArH) 4.48–4.58 (2 H, m, CHaHb) 4.35–4.44 (2 H, m, CHaCHb) 3.76 (6 H, s, OCH3); δC (125 MHz, CDCl3) 158.1, 156.6, 129.3, 120.4, 114.0, 107.7, 87.2, 63.2, 56.3 ppm; m/z (ESI) 297.2 [M + Na]+.
1-Benzyl-9,10-dimethoxy-4,15-dihydro-1H-dibenzo[7,8:9,10][1,6]dioxecino[3,4-d][1,2,3]triazole (38)
This compound is novel. To a solution of compound 37 (7.0 mg, 0.023 mmol, 1.0 eq.) in CDCl3 (0.5 mL) was added benzyl azide (3.1 mg, 0.023 mmol, 1.0 eq.). The reaction was monitored by proton NMR. Once the reaction was complete the solvent was evaporated to give the crude product which was purified by column chromatography (eluted 0–25% EtOAc
 :heptane) to give the pure product as a white solid (7.5 mg, 0.017 mmol, 85%).
:heptane) to give the pure product as a white solid (7.5 mg, 0.017 mmol, 85%).
(Found (ESI)) 452.1580 C25H23N3NaO4 requires 452.1581; vmax 2902, 2839, 1588, 1577, 1466, 1435, 1075 and 726 cm−1; δH (500 MHz, CDCl3) 7.33–7.36 (3 H, m, ArH) 7.23 (1 H, t, J = 8.3 Hz, ArH) 7.18 (1 H, t, J = 8.4 Hz, ArH) 7.12–7.16 (2 H, m, ArH) 6.83 (1 H, d, J = 8.3 Hz, ArH) 6.71 (1 H, d, J = 8.3 Hz, ArH) 6.63 (1 H, d, J = 8.4 Hz, ArH) 6.46 (1 H, d, J = 8.3 Hz, ArH) 5.73 (1 H, d, J = 15.9 Hz, CHeHf) 5.48 (1 H, d, J = 13.7 Hz, CHcHd) 5.41 (1 H, d, J = 15.9 Hz, CHeCHf) 5.29 (1 H, d, J = 13.7 Hz, CHcHd) 5.15 (1 H, d, J = 13.2 Hz, CHaHb) 4.96 (1 H, d, J = 13.2 Hz, CHaHb) 3.75 (3 H, s, OCH3) 3.72 (3 H, s, OCH3); δC (125 MHz, CDCl3) 158.5, 158.3, 158.0, 156.2, 144.7, 134.8, 132.1, 129.0, 128.9, 128.5, 127.0, 115.4, 114.0, 113.8, 109.1, 107.7, 106.4, 105.2, 63.2, 61.3, 56.0, 55.9, 52.3 ppm; m/z (ESI) 452.2 [M + Na]+.
6,7,8,9-Tetrahydro-1,14-(epoxyethanooxy)dibenzo[b,d][1,6]dioxecine (42)
This compound is novel. A solution of compound 41 (357 mg, 1.47 mol, 1 eq.) and 1,4-dibromobutane (317 mg, 1.47 mmol, 1 eq.) in MeCN (15 mL) was added to a solution of C2CO3 (1.2 g, 3.7 mmol, 2.5 eq.) in MeCN (50 mL) at 60 °C over 5 h. The resulting solution was stirred for a further 12 h before the solvent was removed under vacuum. The residue was dissolved in H2O (30 mL) and the product was extracted with EtOAc (3 × 30 mL). The combined organic extracts were dried over MgSO4, which was removed by filtration, and then concentrated to give the crude product. Purification by column chromatography (eluted 25–100% EtOAc/hexane) gave the pure product as a white solid (352 mg, 1.18 mmol, 81%).

R
f = 0.53 (1:1 EtOAc/hexane); mp = 157–160 °C; (found (ESI)) 321.1094 C18H18NaO4 requires 321.1097; vmax 2928, 1590, 1563, 1257, 1221, 1065, 1023, 785 cm−1; δH (500 MHz CDCl3) 7.31 (2 H, t, J = 8.2 Hz, ArH), 6.89 (2 H, d, J = 8.2 Hz, ArH), 6.86 (2 H, d, J = 7.9 Hz, ArH), 4.40 (2 H, d, J = 8.5 Hz, OCH2), 4.38–4.45 (2 H, m, OCH2), 4.25 (2 H, m, OCH2), 4.12 (2 H, d, J = 8.5 Hz, OCH2), 1.91–2.01 (2 H, m, CH2), 1.79–1.88 (2 H, m, CH2); δC (125 MHz, CDCl3) 160.3, 157.9, 129.3, 118.9, 115.6, 111.6, 74.0, 70.7, 26.7; m/z (ESI) 321.0 [M + Na]+.
6,7,8,9-Tetrahydrodibenzo[b,d][1,6]dioxecine-1,14-diol (43)
This compound is known and fully characterised.23 A solution of di-tert-butyl biphenyl (574 mg, 2.16 mmol, 8 eq.) in THF (11 mL) was cooled to 0 °C. To the solution, Li (13 mg, 1.89 mmol, 7 eq.) was added and the solution was stirred until dark blue in colour. This solution was then added to compound 42 (80 mg, 0.27 mmol, 1 eq.) and the mixture was stirred for 1 h. The mixture was then quenched with 1 M HCl (7 mL) and the product was extracted with EtOAc (3 × 10 mL). The combined organic extracts were then dried over MgSO4, which was removed by filtration, and the solution concentrated to give the crude product. Purification by column chromatography (eluted with 25–50% EtOAc
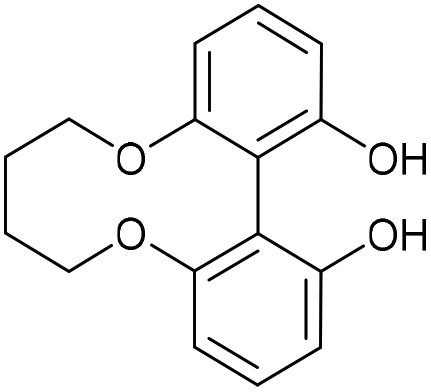 :hexane) gave the pure product as a white solid (48.4 mg, 0.17 mmol, 66%).
:hexane) gave the pure product as a white solid (48.4 mg, 0.17 mmol, 66%).
R
f = 0.46 (1:1 EtOAc/hexane); vmax 3240, 2929, 1601, 1572, 1441, 1227, 1040, 775 cm−1; δH (500 MHz, CDCl3) 7.28 (2 H, t, J = 8.3 Hz, ArH), 6.73 (2 H, d, J = 8.2 Hz, ArH), 6.72 (2 H, d, J = 8.2 Hz, ArH), 5.15 (2 H, s, OH), 4.27–4.36 (2 H, m, OCH2), 4.18–4.27 (2 H, m, OCH2), 1.83–1.96 (2 H, m, CH2), 1.70–1.83 (2 H, m, CH2); δC (125 MHz, CDCl3) 154.1, 130.1, 109.7, 108.6, 70.5, 26.0; m/z (ESI) 271.1 [M − H]−, 295.1 [M + H]+.
6,6′-Oxybutyloxy-linked-2,2′-biphenyldioxacyclodecyne (44)
This compound is novel. A solution of compound 43 (127 mg, 0.467 mmol, 1.0 eq.) and compound 20 (184 mg, 0.467, 1.0 eq.) in MeCN (2.2 mL) was added to a solution of Cs2CO3 (336 mg, 1.03 mmol, 2.2 eq.) in MeCN (11 mL) at 60 °C over 5 h. The mixture was stirred for a further 12 h before the solvent was removed. The remaining residue was dissolved in H2O (20 mL) and extracted with EtOAc (3 × 20 mL). The combined organic extracts were dried over MgSO4, which was removed by filtration, and concentrated. The crude product was then purified by column chromatography (eluted with 25% EtOAc/hexane) to give the pure product as a white solid (56 mg, 0.17 mmol, 36%).
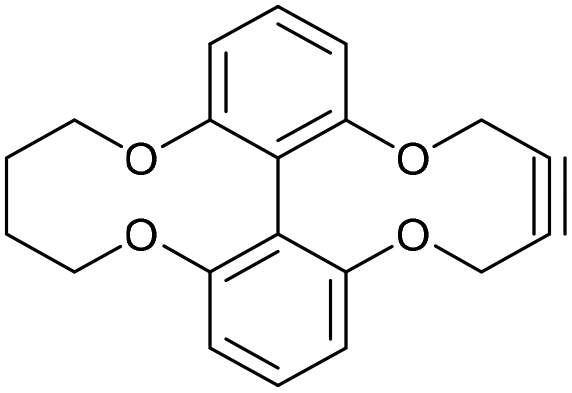
R
f = 0.20 (1:3 EtOAc/hexane); mp = 166–168 °C; (found (ESI)) 345.1088 C20H18NaO4 requires 345.1097; vmax 2953, 2922, 2852, 1569, 1457, 1251, 1218, 1031, 739 cm−1; δH (500 MHz, CDCl3) 7.37 (2 H, t, J = 8.2 Hz, ArH), 7.00 (2 H, d, J = 8.2 Hz, ArH), 6.94 (2 H, d, J = 8.2 Hz, ArH), 4.54 (4 H, s, 2 × OCH2), 4.27 (2 H, d, J = 11.9 Hz, OCHaHb), 3.80 (2 H, t, J = 11.9 Hz, CHaHb), 1.34–1.53 (4 H, m, 2 × CH2); δC (125 MHz, CDCl3) 156.7, 156.4, 129.2, 125.8, 115.8, 115.2, 88.4, 73.0, 62.4, 23.9; m/z (ESI) 345.1 [M + Na]+.
15-Benzyl-5,6,7,8,15,18-hexahydro-14H-4,9,13,19-tetraoxa-15,16,17-triazadibenzo[hi,qr]cyclopenta[d]decalene (45)
This compound is novel. To a solution of compound 44 (10 mg, 0.031 mmol, 1 eq.) in CDCl3 (0.6 mL) was added benzyl azide (4.1 mg, 0.031 mmol, 1 eq.). The reaction was monitored by NMR until completion after which the solvent was removed and the residue was purified by column chromatography (eluted with 10% EtOAc/hexane) to give the pure product as a white solid (13 mg, 0.028 mmol, 95%).
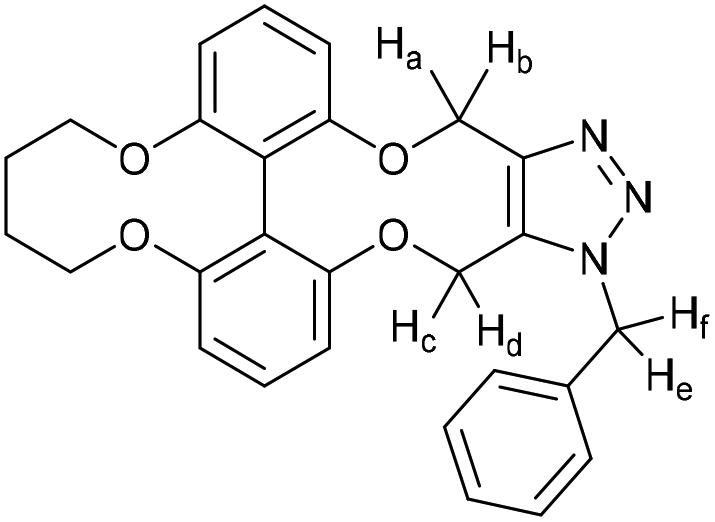
R
f = 0.34; (1:1 EtOAc/hexane); (found (ESI)) 478.1735 C27H25N3NaO4 requires 478.1737; vmax 2939, 1589, 1573, 1448, 1222, 1058, 716 cm−1; δH (500 MHz, CDCl3) 7.33–7.39 (3 H, m, ArH), 7.21 (1 H, t, J = 8.2 Hz, ArH), 7.15–7.19 (2 H, m, ArH), 7.12 (1 H, t, J = 8.2 Hz, ArH), 6.82 (1 H, d, J = 8.2 Hz, ArH), 6.78 (1 H, d, J = 8.2 Hz, ArH), 6.72 (1 H, d, J = 8.2 Hz, ArH), 6.39 (1 H, d, J = 8.2 Hz, ArH), 5.73 (1 H, d, J = 15.7 Hz, CHfHe), 5.47 (1 H, d, J = 13.6 Hz, CHcHd), 5.38 (1 H, d, J = 15.7 Hz, CHfHe), 5.30 (1 H, d, J = 13.6 Hz, CHcHd), 5.17 (1 H, d, J = 13.3 Hz, CHaHb), 5.03 (1 H, d, J = 13.3 Hz, CHaHb), 4.16–4.35 (4 H, m, 2 × OCH2), 1.70–1.94 (4 H, m, 2 × CH2); δC (125 MHz, CDCl3) 158.0, 157.8, 156.6, 144.9, 134.7, 132.3, 129.1, 128.7, 128.6, 128.5, 127.1, 116.9, 115.4, 110.9, 109.9, 108.9, 107.7, 70.8, 70.7, 62.8, 60.8, 52.3, 26.5; m/z (ESI) 478.2 [M + Na]+.
2,2′-Bis(methoxymethoxy)-6,6′-dinitro-1,1′-biphenyl (47)
This compound is novel. Precursor 46 was prepared by the published method.19 It was purified by chromatography on silica gel using DCM as eluant. A solution of compound 46 (3.4 g, 11 mmol, 1 eq.)19 in dry DMF (100 mL) was added to Cu powder (2.8 g, 44 mmol, 4 eq.) and the mixture heated to 100 °C overnight. The mixture was cooled to room temperature and filtered to remove solid residues. The solids in the filter paper were washed through and EtOAc (2 × 50 mL). To the combined filtrates, water (100 mL) was added and the product was extracted with EtOAc (3 × 100 mL). The combined organic extracts were washed with H2O (3 × 100 mL) and brine (100 mL) before being dried over MgSO4, which was removed by filtration, and concentrated. The crude product was then purified by column chromatography (eluted with 0–25% EtOAc/pet. Ether 40
 :60) to give the pure product as a yellow solid (1.56 g, 4.34 mmol, 79%).
:60) to give the pure product as a yellow solid (1.56 g, 4.34 mmol, 79%).
R
f = 0.54 (1:1 EtOAc/hexane); mp = 104–106 °C; (found (ESI)) 387.0797 C16H16N2NaO8 requires 387.0799; vmax 3075, 2959, 2831, 1580, 1531, 1456, 1253, 1205, 1083, 1001, 733 cm−1; δH (500 MHz, CDCl3) 7.79–7.85 (2 H, m, ArH), 7.45–7.52 (4 H, m, ArH), 5.01–5.06 (4 H, m, OCH2), 3.30 (6 H, s, OCH3); δC (125 MHz, CDCl3) 154.6, 149.1, 129.4, 119.5, 119.2, 117.7, 94.9, 56.1; m/z (ESI) 387.1 [M + Na]+. In another run a product was obtained in 92% yield without the need for purification by column chromatography.
6,6′-Dinitro-[1,1′-biphenyl]-2,2′-diol (48)
This compound is novel. To a solution of compound 47 (1.22 g, 3.35 mmol, 1 eq.) in MeOH (10 mL) was added conc. HCl (10 mL) dropwise. The mixture was stirred for 24 h and then H2O (30 mL) was added. The product was extracted with EtOAc (3 × 30 mL) and the combined organic extracts were dried over MgSO4, which was removed by filtration, and concentrated to give the crude product. Purification by column chromatography (eluted 50% EtOAc/pet.ether 40
 :60) gave the pure product (856 mg, 2.86 mmol, 81%).
:60) gave the pure product (856 mg, 2.86 mmol, 81%).
R
f = 0.61 (1:1 EtOAc/Pet. Ether); mp = >200 °C (decomposition); (found (ESI)) 299.0269 C12H8N2NaO6 requires 299.0275; vmaz 3311, 1510, 1331, 1288, 1160, 1002, 733 cm−1; δH (500 MHz, CD3CN) 7.64 (2 H, dd, J = 8.2, 0.8 Hz, ArH), 7.62 (2 H, br. s, OH), 7.45 (2 H, t, J = 8.2 Hz, ArH), 7.23 (2 H, dd, J = 8.2, 0.8 Hz, ArH); δC (125 MHz, CD3CN) 156.3, 151.1, 131.1, 121.8, 117.6, 117.3; m/z (ESI) 299.0 [M + Na]+ In an alternative workup the final product was purified by recrystallisation from MeOH in 88% yield.
6,6′-Dinitro-2,2′-biphenyldioxacyclodecyne (49)
This compound is novel. A solution of compound 48 (856 mg, 3.10 mmol, 1.0 eq.) and compound 20 (1.22 g, 3.10 mmol, 1.0 eq.) in DMF (14 mL) was added to a solution of Cs2CO3 (2.50 g, 7.75 mmol, 2.5 eq.) in DMF (70 mL) at 60 °C over 5 h (using a syringe pump) and the resulting mixture was stirred for a further 12 h. The reaction mixture was then cooled to room temperature. Water (100 mL) was added and the product was extracted with EtOAc (3 × 100 mL). The combined organic extracts were then washed with H2O (2 × 50 mL) and brine (50 mL) and dried over MgSO4, which was removed by filtration, and concentrated. The water washings are required to remove DMF. The crude product was then purified by column chromatography (eluted with 50% EtOAc/pet. Ether 40
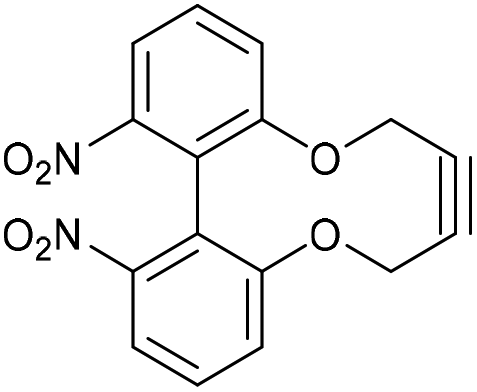 :60) to give the pure product as a yellow solid (530 mg, 1.63 mmol, 53%).
:60) to give the pure product as a yellow solid (530 mg, 1.63 mmol, 53%).
R
f = 0.55 (1:1 EtOAc/hexane); mp 220–230 °C (dec); (found (ESI)) 349.0427 C16H10N2NaO6 requires 349.0431; vmax 3088, 2926, 2852, 1519, 1349, 1337, 990, 1238, 1175, 990, 735 cm−1; δH (500 MHz, CDCl3) 8.01 (2 H, d, J = 8.2 Hz, ArH), 7.60 (2 H, t, J = 8.2 Hz, ArH), 7.43 (2 H, d, J = 8.2 Hz, ArH), 4.47–4.55 (2 H, m, OCHaHb), 4.37–4.47 (2 H, m, OCHaHb), ppm; δC (125 MHz, CDCl3) 155.2, 149.2, 129.9, 126.9, 126.5, 121.2, 87.4, 63.6 ppm; m/z (ESI) 349 [M + Na]+. In another procedure, chromatography on silica gel using DCM as eluant instead, and this gave a product in 39% yield.
Benzyl azide cycloadduct of compound 49. 1-Benzyl-9,10-dinitro-4,15-dihydro-1H-dibenzo[7,8:9,10][1,6]dioxecino[3,4-d][1,2,3]triazole
This compound is novel. To a solution of compound 49 (9.0 mg, 0.027 mmol, 1 eq.) in CDCl3 (0.5 mL) was added benzyl azide (3.6 mg, 0.027 mmol, 1 eq.). The reaction was monitored by proton NMR until completion and the solvent was removed under vacuum. The residue was purified by column chromatography (eluted with 50% EtOAc/hexane) to give the pure product as an oil (5.0 mg, 0.011 mmol, 40%).

R
f = 0.10 (1:1 EtOAc/hexane); (found (ESI)) 482.1067 C23H17N5NaO6 requires 482.1071; vmax 2923, 2853, 1521, 1346, 1266, 1182, 1076, 902, 722 cm−1; δH (500 MHz, CDCl3) 7.85–7.92 (1 H, m, ArH), 7.76 (1 H, dd, J = 7.7, 1.6 Hz, ArH), 7.39–7.46 (3 H, m, ArH), 7.29–7.35 (3 H, m, ArH), 7.09 (1 H, d, J = 8.1 Hz, ArH), 7.01–7.07 (2 H, m, ArH), 5.72 (1 H, d, J = 15.9 Hz, CHaHb), 5.58 (1 H, d, J = 13.9 Hz, OCHcHd), 5.44 (1 H, d, J = 15.9 Hz, CHaHb), 5.32 (1 H, d, J = 13.9 Hz, OCHcHd), 5.30 (1 H, d, J = 13.2 Hz, OCHeHf), 4.89 (1 H, d, J = 13.2 Hz, OCHeHf), ppm; (125 MHz, CDCl3) 157.9, 155.2, 148.8, 148.6, 143.5, 134.3, 130.8, 129.8, 129.4, 129.2, 128.8, 126.8, 122.2, 122.1, 120.7, 120.5, 119.6, 118.0, 63.9, 62.9, 52.5 ppm; m/z (ESI) 482.1 [M + Na]+.
6,6′-Diamino-2,2′-biphenyldioxacyclodecyne (50)
This compound is novel. To a solution of compound 49 (238 mg, 0.730 mmol, 1 eq.) in 6
 :1 EtOH/H2O (3.0 mL) was added NH4Cl (39 mg, 0.73 mmol, 1 eq.) and Fe powder (204 mg, 3.65 mmol, 5 eq.). The mixture was heated to 70 °C and stirred for 1 h. The solution was allowed to cool to rt then the crude reaction was filtered through filter paper using MeOH (4 × 30 mL). Solvent was removed, then the residue was filtered through cotton wool using DCM (4 × 30 mL). Removal of the solvent gave 50 as an amorphous solid (179 mg, 0.673 mmol, 92%) without the need for column chromatography.
:1 EtOH/H2O (3.0 mL) was added NH4Cl (39 mg, 0.73 mmol, 1 eq.) and Fe powder (204 mg, 3.65 mmol, 5 eq.). The mixture was heated to 70 °C and stirred for 1 h. The solution was allowed to cool to rt then the crude reaction was filtered through filter paper using MeOH (4 × 30 mL). Solvent was removed, then the residue was filtered through cotton wool using DCM (4 × 30 mL). Removal of the solvent gave 50 as an amorphous solid (179 mg, 0.673 mmol, 92%) without the need for column chromatography.
R
f = 0.38 (1:1 EtOAc/hexane); (found (ESI)) 289.0946 C16H14N2NaO2 requires 289.0947; vmax 3465, 3360, 2960, 2914, 2864, 1611, 1565, 1461, 1302, 1248, 1116, 1020, 920, 729 cm−1; δH (500 MHz, CDCl3) 7.21 (2 H, t, J = 8.1 Hz, ArH), 6.65 (2 H, d, J = 8.1 Hz, ArH), 6.61 (2 H, d, J = 8.1 Hz, ArH), 4.54–4.61 (2 H, m, CHaHb), 4.40–4.47 (2 H, m, CHaHb), 3.63 (4 H, br. s., NH2) ppm; δC (125 MHz, CDCl3) 156.2, 146.2, 129.9, 116.5, 112.3, 111.8, 87.2, 63.3 ppm; m/z (ESI) 267.1 [M + H]+, 289.1 [M + Na]+.
Benzyl azide cycloadduct of compound 50. 1-Benzyl-4,15-dihydro-1H-dibenzo[7,8:9,10][1,6]dioxecino[3,4-d][1,2,3]triazole-9,10-diamine
This compound is novel. To a solution of compound 50 (3.0 mg, 0.011 mmol, 1 eq.) in CDCl3 (0.5 mL) was added benzyl azide (1.5 mg, 0.011 mmol, 1 eq.). The reaction was monitored by proton NMR until completion, at which point the solvent was removed under vacuum. The crude material was purified by column chromatography (eluted with 25%–100% EtOAc/pet. Ether 40
 :60) to give the pure product as a white waxy solid (4 mg, 0.01 mmol, 91%).
:60) to give the pure product as a white waxy solid (4 mg, 0.01 mmol, 91%).
R
f = 0.30 (1:1 EtOAc/hexane); (found (ESI)) 422.1590 C23H21N5NaO2 requires 422.1587; vmax 3450, 3356, 2926, 2854, 1615, 1574, 1456, 1231, 1072, 909, 724 cm−1; δH (500 MHz, CDCl3) 7.32–7.38 (3 H, m. ArH), 7.11–7.16 (2 H, m, ArH), 7.08 (1 H, t, J = 8.0 Hz, ArH), 7.01 (1 H, t, J = 8.0 Hz, ArH), 6.60 (1 H, d, J = 8.1 Hz, ArH), 6.50 (1 H, d, J = 8.0 Hz, ArH), 6.42 (1 H, d, J = 8.1 Hz, ArH), 6.22 (1 H, d, J = 8.0 Hz, ArH), 5.73 (1 H, d, J = 15.8 Hz, CHcHd), 5.43 (1 H, d, J = 13.6 Hz, CHaHb), 5.42 (1 H, d, J = 15.8 Hz, CHcHd), 5.28 (1 H, d, J = 13.6 Hz, CHaHb), 5.12 (1 H, d, J = 13.3 Hz, CHeHf), 4.94 (1 H, d, J = 13.3 Hz, CHeHf), 3.74 (4 H, br. s., NH2); δC (125 MHz, CDCl3) 158.7, 157.0, 145.6, 145.4, 144.8, 134.8, 132.1, 129.5, 129.1, 128.5, 127.0, 111.7, 110.6, 110.3, 109.6, 106.5, 105.4, 63.3, 61.2, 52.3 ppm; m/z (ESI) 398.1 [M − H]−.
6,6′-Bis(tosylamido)-2,2′-biphenyldioxacyclodecyne (51)
This compound is novel. To a solution of compound 50 (61 mg, 0.23 mmol, 1 eq.) and toluenesulfonyl chloride (109 mg, 0.58 mmol, 2.5 eq.) in DCM (1.7 mL) was added pyridine (45 mg, 0.58 mmol, 2.5 eq.). The reaction was stirred for 4 h at room temperature. H2O was added (10 mL) and the product was extracted with EtOAc (3 × 10 mL) and the combined organic extracts were dried over MgSO4, which was removed by filtration, and concentrated. The product was a white solid (96 mg, 0.16 mmol, 73%) which was taken forward without further purification.

R
f = 0.51 (1:1 EtOAc/Pet. Ether); (found (ESI)) 597.1114 C30H26N2NaO6S2 requires 597.1124; vmax 3365, 2917, 1596, 1576, 1454, 1378, 1321, 1212, 1157, 1029, 1004, 935, 658, 536 cm−1; δH (500 MHz, CDCl3) 7.51 (4 H, d, J = 8.1 Hz, ArH), 7.51 (2 H, d, J = 7.9 Hz, ArH), 7.38 (2 H, t, J = 8.1 Hz, ArH), 7.19 (4 H, d, J = 8.1 Hz, ArH), 6.90 (2 H, d, J = 7.9 Hz, ArH), 5.95 (2 H, br. s, NH), 4.18–4.26 (2 H, mCHaHb), 4.08–4.18 (2 H, m, CHaHb); δC (125 MHz, CDCl3) 154.4, 144.3, 136.4, 135.8, 131.0, 129.6, 127.3, 120.7, 118.6, 117.6, 86.7, 63.3, 21.6 ppm; m/z (ESI) 597.1 [M + Na]+.
6,6′-Bis(dansylamido)-2,2′-biphenyldioxacyclodecyne (52)
This compound is novel. To a solution of compound 50 (50 mg, 0.19 mmol, 1 eq.) and dansyl chloride (128 mg, 0.48 mmol, 2.5 eq.) in DCM (1.4 mL) was added pyridine (37.5 mg, 0.48 mmol, 2.5 eq.). The reaction was stirred for 4 h before H2O (10 mL) was added and the product was extracted with EtOAc (3 × 10 mL). The combined organic extracts were then dried over MgSO4 and concentrated to give the crude product. Purification by column chromatography (eluted with 25–50% EtOAc/pet.ether 40
 :60) gave the pure product as a yellow solid (22.6 mg, 0.0310 mmol, 16%).
:60) gave the pure product as a yellow solid (22.6 mg, 0.0310 mmol, 16%).
R
f = 0.37 (1:1 EtOAc/hexane); (found (ESI)) 733.2153 C40H37N4O6S2 requires 733.2149; vmax 3356, 2939, 1572, 1453, 1321, 1144, 1032, 786, 622, 566 cm−1; δH (400 MHz, CDCl3) 8.47 (2 H, d, J = 8.5 Hz, ArH), 8.03–8.10 (2 H, m, ArH), 7.96 (2 H, d, J = 8.7 Hz, ArH), 7.35–7.43 (4 H, m, ArH), 7.20–7.24 (2 H, m, ArH), 7.16 (4 H, t, J = 7.9 Hz, ArH), 6.64 (2 H, dd, J = 8.0, 1.0 Hz, ArH), 6.20 (2 H, br. s, NH), 3.98–4.09 (4 H, m, OCH2), 2.85–2.92 (12 H, s, NCH3); δC (125 MHz, CDCl3) 154.4, 151.7, 136.5, 135.2, 130.6, 130.4, 129.8, 129.5, 128.9, 128.21, 123.1, 121.7, 118.9, 118.7, 118.7, 115.1, 86.7, 63.1, 45.4 ppm; m/z (ESI) 733.2 [M + H]+, 755.2 [M + Na]+.
6,6-Bis(mesylamido)-2,2′-biphenyldioxacyclodecyne (53)
This compound is novel. To a stirred solution of compound 50 (69 mg, 0.259 mmol, 1.0 eq.) in anhydrous DCM (1.9 mL) was added mesyl chloride (0.1 mL, 148 mg, 1.29 mmol, >5 eq.) and pyridine (0.1 mL, 98 mg, 1.83 mmol, >5 eq.). The solution was degassed, then stirred at room temperature for six hours. Water (10 mL) was added, then the product was extracted with ethyl acetate (3 × 10 mL). The combined organic extracts were dried over MgSO4, filtered and concentrated under reduced pressure. The crude product was purified by column chromatography (eluent: DCM – 5
 :1 DCM/MeOH) to yield the pure product as a white solid (73 mg, 0.173 mmol, 70%). Rf 0.55 (99:1 DCM/MeOH); m.p. > 200 °C; found (ESI-TOF) 445.0494, [M + Na]+ calcd for C18H18N2O6S2Na 445.0498; νmax 3271, 2355, 2330, 1578, 1454, 1356, 1323, 1216, 1154, 1014, 965, 745, 524 cm−1; δH (500 MHz, CDCl3) 7.61 (2H, d, J = 8.0, ArH), 7.52 (2H, t, J = 8.0, ArH), 7.08 (2H, d, J = 8.0, ArH), 6.14 (2H, br. s, –NH–), 4.55–4.42 (4H, m, –OCH2–), 2.89 (6H, s, –SO2CH3); δC (125 MHz, CDCl3) 154.9, 136.8, 131.7, 121.8, 119.5, 118.4, 86.9, 63.8, 39.7; m/z (ESI) 445.0 [M + Na]+.
:1 DCM/MeOH) to yield the pure product as a white solid (73 mg, 0.173 mmol, 70%). Rf 0.55 (99:1 DCM/MeOH); m.p. > 200 °C; found (ESI-TOF) 445.0494, [M + Na]+ calcd for C18H18N2O6S2Na 445.0498; νmax 3271, 2355, 2330, 1578, 1454, 1356, 1323, 1216, 1154, 1014, 965, 745, 524 cm−1; δH (500 MHz, CDCl3) 7.61 (2H, d, J = 8.0, ArH), 7.52 (2H, t, J = 8.0, ArH), 7.08 (2H, d, J = 8.0, ArH), 6.14 (2H, br. s, –NH–), 4.55–4.42 (4H, m, –OCH2–), 2.89 (6H, s, –SO2CH3); δC (125 MHz, CDCl3) 154.9, 136.8, 131.7, 121.8, 119.5, 118.4, 86.9, 63.8, 39.7; m/z (ESI) 445.0 [M + Na]+.
6,6′-Bis(tosylamide)-2,2′-biphenyldioxacyclodecyne butyl bridged derivative (54)
This compound is novel. A solution of 1,4-dibromobutane (34.5 mg, 0.160 mmol, 1 eq.) and compound 51 (96 mg, 0.160 mmol, 1 eq.) in MeCN (1.6 mL) was added to a mixture of Ca2CO3 (130 mg, 0.400 mmol, 2.5 eq.) in MeCN (6 mL) at 75 °C over 5 h. The reaction was stirred for a further 12 h before the solvent was removed under vacuum and H2O (20 mL) was added. The product was extracted with EtOAc (3 × 20 mL) and the combined organic extracts were dried over MgSO4, which was removed by filtration, and concentrated. The crude material was then purified by column chromatography (eluted with 50% EtOAc/pet. ether 40
 :60) to give the pure product as an oil (43 mg, 0.068 mmol, 44%).
:60) to give the pure product as an oil (43 mg, 0.068 mmol, 44%).
R
f = 0.48 (1:1 EtOAc/hexane); (found (ESI)) 651.1586 C34H32N2NaO6S2 requires 651.1594; vmax 3026, 2924, 2872, 1592, 1453, 1371, 1125, 1041, 813, 684, 571, 555 cm−1; δH (500 MHz, CDCl3) 7.62 (4 H, d, J = 8.2 Hz, ArH), 7.49 (2 H, t, J = 7.6 Hz, ArH), 7.31 (2 H, d, J = 7.6 Hz, ArH), 7.21 (2 H, d, J = 7.6 Hz, ArH), 7.16 (4 H, d, J = 8.2 Hz, ArH), 4.58–4.69 (4 H, m, 2 × OCH2), 3.06–3.13 (2 H, m, NCH2), 2.95–3.02 (2 H, m, NCH2), 2.38 (6 H, s, 2 × CH3), 1.00–1.12 (2 H, m, CH2), 0.84–0.98 (2 H, m, CH2); δC (125 MHz, CDCl3) 158.0, 143.5, 141.1, 135.9, 132.8, 123.0, 129.2, 128.5, 124.5, 121.1, 89.8, 62.1, 52.8, 22.1, 21.5 ppm; m/z (ESI) 629.2 [M + H]+, 651.2 [M + Na]+.
6,6′-Bis(mesylamide)-2,2′-biphenyldioxacyclodecyne butyl bridged derivative (56)
This compound is novel. To a solution of caesium carbonate (147 mg, 0.45 mmol, 2.5 eq.) in dry acetonitrile (6.8 mL), stirred and heated at 75 °C was added dropwise a solution of compound 53 (76 mg, 0.18 mmol, 1.0 eq.) and 1,4-dibromobutane (65 μL, 0.54 mmol, 3.0 eq.) in acetonitrile (20 mL) over 24 hours. The solution was then stirred for a further 24 hours, before being cooled to room temperature. Solvents were removed in vacuo. Water (20 mL) was added, and the product was extracted with ethyl acetate (3 × 20 mL). The combined organic layers were dried over MgSO4, filtered and concentrated under reduced pressure. The resulting crude product was purified by column chromatography (eluent: DCM/MeOH gradient) to afford the pure product as a brown solid (53 mg, 0.11 mmol, 62%). Rf: 0.28 (29
 :1 DCM/MeOH); m.p. 131–132 °C (dec.); found 499.0959 (ESI-TOF) m/z: [M + Na]+ calcd for C22H24N2O6S2Na 499.0968; νmax 3034, 2932, 2871, 2331, 1723, 1573, 1449, 1322, 1150, 1047, 969, 935, 894, 748, 664, 529; δH (500 MHz, CDCl3) 7.49 (2H, t, J = 8.0, ArH), 7.29–7.27 (2H, m, ArH) 7.19 (2H, d, J = 8.0, ArH), 4.57–4.45 (4H, m, –OCH2), 3.33–3.26 (4H, m, MsNCH2–), 2.89 (6H, s, –SO2CH3) 1.33–1.09 (4H, m, MsNCH2CH2–); δC (125 MHz, CDCl3) 156.4, 141.0, 133.0, 130.4, 126.9, 121.5, 88.3, 62.5, 52.3, 40.3, 23.7; m/z (ESI) [M + Na]+ 499.0.
:1 DCM/MeOH); m.p. 131–132 °C (dec.); found 499.0959 (ESI-TOF) m/z: [M + Na]+ calcd for C22H24N2O6S2Na 499.0968; νmax 3034, 2932, 2871, 2331, 1723, 1573, 1449, 1322, 1150, 1047, 969, 935, 894, 748, 664, 529; δH (500 MHz, CDCl3) 7.49 (2H, t, J = 8.0, ArH), 7.29–7.27 (2H, m, ArH) 7.19 (2H, d, J = 8.0, ArH), 4.57–4.45 (4H, m, –OCH2), 3.33–3.26 (4H, m, MsNCH2–), 2.89 (6H, s, –SO2CH3) 1.33–1.09 (4H, m, MsNCH2CH2–); δC (125 MHz, CDCl3) 156.4, 141.0, 133.0, 130.4, 126.9, 121.5, 88.3, 62.5, 52.3, 40.3, 23.7; m/z (ESI) [M + Na]+ 499.0.
N,N’-(1-Benzyl-4,15-dihydro-1H-dibenzo[7,8:9,10][1,6]dioxecino[3,4-d][1,2,3]triazole-9,10-diyl)bis(4-methylbenzenesulfonamide) (57)
This compound is novel. To a solution of compound 51 (28.8 mg, 0.050 mmol, 1 eq.) in CDCl3 (0.5 mL) was added benzyl azide (6.7 mg, 0.050 mmol, 1 eq.). The reaction was monitored by proton NMR until completion. The solvent was removed under vacuum and the crude material was purified by column chromatography (eluted with 50–100% EtOAc/hexane) to give the pure product (12.1 mg, 0.017 mmol, 34%).

R
f = 0.27 (1:1 EtOAc/hexane); (found (ESI)) 730.1770 C37H33N5NaO6S2 requires 730.1764; vmax 3337, 3059, 2925, 2854, 1596, 1455, 1288, 1043, 728, 553 cm−1; δH (500 MHz, CDCl3) 7.70 (2 H, d, J = 8.1 Hz, ArH), 7.54 (2 H, d, J = 8.1 Hz, ArH), 7.31–7.35 (3 H, m, ArH), 7.23–7.29 (4 H, m, ArH), 7.14–7.22 (3 H, m, ArH), 7.10 (1 H, d, J = 8.2 Hz, ArH), 7.03 (2 H, m, J = 5.0 Hz, ArH), 6.77 (1 H, d, J = 8.2 Hz, ArH), 6.50 (1 H, d, J = 8.1 Hz, ArH), 6.18 (2 H, br. s, NH), 5.62 (1 H, d, J = 15.8 Hz, CHeHf), 5.41 (1 H, d, J = 15.8 Hz, CHeHf), 5.17 (1 H, d, J = 13.6 Hz, CHaHb), 5.01 (1 H, d, J = 13.6 Hz, CHaHb), 4.93 (1 H, d, J = 13.4 Hz, CHcHd), 4.68 (1 H, d, J = 13.4 Hz, CHcHd); δC (125 MHz, CDCl3) 158.1, 156.7, 144.3, 144.1, 144.0, 136.2, 136.1, 136.1, 136.0, 134.4, 131.3, 130.8, 130.8, 129.9, 129.7, 129.2, 128.7, 127.5, 127.4, 126.9, 115.6, 114.9, 113.7, 113.7, 112.6, 112.1, 63.9, 61.5, 52.3, 21.7, 21.6 ppm; m/z (ESI)706.2 [M − H]−.
N,N’-(1-Benzyl-4,15-dihydro-1H-dibenzo[7,8:9,10][1,6]dioxecino[3,4-d][1,2,3]triazole-9,10-diyl)dimethanesulfonamide (59)
This compound is novel. Compound 53 (10 mg, 23.7 μmol, 1.0 eq.) was dissolved in CDCl3 (0.6 mL) to which was added benzyl azide (3.1 μL, 24.8 μmol, 1.0 eq.). After 9 days, the reaction was deemed complete; solvents were removed in vacuo and the residue was purified by column chromatography (eluent: DCM/MeOH gradient) to yield the pure product as a yellow-brown solid (20 mg, since the yield is >100%, an NMR-silent impurity is likely present). Rf: 0.20 (99
 :1 DCM/MeOH); found 578.1136 (ESI-TOF) m/z: [M + Na]+ calcd for C25H25N5O6S2Na 578.1138; δH (500 MHz, CDCl3) 7.42–7.30 (9H, m, ArH), 7.11–7.09 (2H, m, ArH), 6.15 (1H, s, –NH–), 6.04 (1H, s, –NH–), 5.71 (1H, d, J = 15.5, PhCHeHf–), 5.49 (1H, d, J = 16.0, PhHeHf–), 5.46 (1H, d, J = 14.0, –OCHaHb–), 5.32 (1H, d, J = 14.0, –OCHaHb–), 5.18 (1H, d, J = 13.5, –OCHcHd–), 4.94 (1H, d, J = 13.0, –OCHcHd–); δC (125 MHz, CDCl3); 158.4, 157.0, 144.0, 136.4, 136.1, 134.4, 131.7, 131.4, 129.5, 129.3, 128.8, 119.5, 115.7, 114.7, 114.4, 113.8, 113.7, 112.7, 64.2, 61.8, 52.5, 40.3, 40.2; m/z (ESI) [M + Na]+ 578.1.
:1 DCM/MeOH); found 578.1136 (ESI-TOF) m/z: [M + Na]+ calcd for C25H25N5O6S2Na 578.1138; δH (500 MHz, CDCl3) 7.42–7.30 (9H, m, ArH), 7.11–7.09 (2H, m, ArH), 6.15 (1H, s, –NH–), 6.04 (1H, s, –NH–), 5.71 (1H, d, J = 15.5, PhCHeHf–), 5.49 (1H, d, J = 16.0, PhHeHf–), 5.46 (1H, d, J = 14.0, –OCHaHb–), 5.32 (1H, d, J = 14.0, –OCHaHb–), 5.18 (1H, d, J = 13.5, –OCHcHd–), 4.94 (1H, d, J = 13.0, –OCHcHd–); δC (125 MHz, CDCl3); 158.4, 157.0, 144.0, 136.4, 136.1, 134.4, 131.7, 131.4, 129.5, 129.3, 128.8, 119.5, 115.7, 114.7, 114.4, 113.8, 113.7, 112.7, 64.2, 61.8, 52.5, 40.3, 40.2; m/z (ESI) [M + Na]+ 578.1.
15-Benzyl-4,9-ditosyl-4,5,6,7,8,9,15,18-octahydro-14H-13,19-dioxa-4,9,15,16,17-pentaazadibenzo[hi,qr]cyclopenta-[d]decalene (60)
This compound is novel. To a solution of compound 54 (12.5 mg, 0.020 mmol, 1 eq.) in CDCl3 (0.5 mL) was added benzyl azide (2.7 mg, 0.020 mmol, 1 eq.). The reaction was monitored by proton NMR until completion. The solvents were removed under vacuum and the crude material was purified by column chromatography (eluted with 50% EtOAc/pet. ether 40
 :60) to give the pure product as a colourless oil (9.2 mg, 0.12 mmol, 60%).
:60) to give the pure product as a colourless oil (9.2 mg, 0.12 mmol, 60%).
R
f = 0.15 (1:19 MeOH/DCM); (found (ESI)) 784.2222 C41H39N5NaO6S2 requires 784.2234; vmax 2950, 1452, 1348, 1163, 1068, 728, 694 cm−1; δH (500 MHz, CDCl3) 7.30–7.40 (5 H, m, ArH), 7.22–7.30 (5 H, m, ArH), 7.10–7.20 (6 H, m, ArH), 6.88 (1 H, d, J = 7.9 Hz, ArH), 6.50 (1 H, d, J = 8.1 Hz, ArH), 6.20 (1 H, d, J = 7.8 Hz, ArH), 5.76 (1 H, d, J = 15.4 Hz, CHeHf), 5.65 (1 H, d, J = 13.4 Hz, OCHcHd), 5.52 (1 H, d, J = 15.4 Hz, CHeHf), 5.42 (1 H, d, J = 13.4 Hz, OCHcHd), 5.37 (1 H, d, J = 12.5 Hz, OCHaHb), 5.12 (1 H, d, J = 12.5 Hz, OCHaHb), 3.24–3.56 (4 H, m, 2 × NCH2), 2.41 (6 H, s, 2 × CH3), 1.77–1.97 (2 H, m, CH2), 1.55–1.77 (2 H, m, CH2); δC (125 MHz, CDCl3) 159.7, 157.4, 145.0, 143.5, 143.5, 140.4, 140.1, 134.8, 133.9, 133.3, 132.3, 130.1, 129.3, 129.1, 129.1, 128.7, 128.6, 128.6, 128.5, 128.3, 127.2, 120.5, 118.6, 115.9, 113.6, 62.8, 61.6, 51.1, 51.0, 24.7, 24.3, 21.6 ppm; m/z (ESI) 784.2 [M + Na]+.
15-Benzyl-4,9-bis(methylsulfonyl)-4,5,6,7,8,9,15,18-octahydro-14H-13,19-dioxa-4,9,15,16,17-pentaazadibenzo-[hi,qr]cyclopenta[d]decalene (61)
This compound is novel. Compound 56 (10.4 mg, 21.8 μmol, 1.0 eq.) was dissolved in CDCl3 (0.6 mL), to which was added benzyl azide (2.72 μL, 21.8 μmol, 1.0 eq.). After 28 hours, the reaction was deemed complete, and solvents were removed in vacuo. The reaction was performed again with Compound 11 (5.2 mg, 10.9 μmol, 1.0 eq.) and benzyl azide (1.35 μL, 10.8 μmol, 1.0 eq.). After 26 hours, the reaction was deemed complete, and solvents were removed in vacuo. The residues from each attempt at this reaction were purified together by column chromatography (eluent: DCM/MeOH gradient) to afford the pure product as a yellow-brown solid (8 mg, 13.1 μmol, 40%). Rf: 0.19 (99
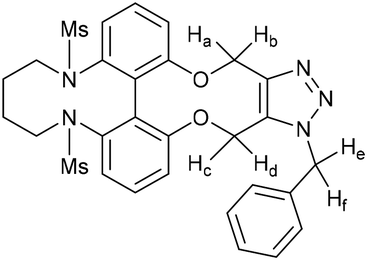 :1 DCM/MeOH); HRMS (ESI-TOF) m/z: found 632.1603 [M + Na]+ calcd for C25H31N5O6S2Na 632.1608; νmax 2928, 2253, 1714, 1573, 1452, 1336, 1149, 1066, 971, 904, 723, 647, 513 cm−1; δH (500 MHz, CDCl3) 7.36–7.32 (4H, m, ArH), 7.23 (1H, t, J = 8.0, ArH), 7.17–7.15 (3H, m, ArH), 7.08 (1H, d, J = 8.0, ArH), 6.99 (1H, d, J = 8.0, ArH), 6.68 (1H, d, J = 7.5, ArH), 5.73 (1H, d, J = 16.0, PhCHeHf–), 5.52 (1H, d, J = 14.0, –OCHaHb–), 5.41 (1H, d, J = 16.0, PhCHeHf–), 5.37 (1H, d, J = 14.0, –OCHaHb–), 5.25 (1H, d, J = 13.0, –OCHcHd–), 5.05 (1H, d, J = 13.0, –OCHcHd–), 4.17–3.64 (4H, m, 2× –NCH2–), 2.63 (3H, s, –SO2CH3), 2.57 (3H, s, –SO2CH3), 2.10–1.69 (4H, m, –NCH2CH2–); δC (125 MHz, CDCl3) 159.0, 157.5, 144.5, 140.6, 140.0, 134.6, 132.0, 129.4, 129.3, 129.0, 128.7, 128.3 128.1, 127.1, 121.1, 118.9, 115.5, 114.2, 63.4, 61.2, 52.4, 51.3, 51.2, 36.5, 35.3, 25.4, 24.1; m/z (ESI) 632.1 [M + Na]+
:1 DCM/MeOH); HRMS (ESI-TOF) m/z: found 632.1603 [M + Na]+ calcd for C25H31N5O6S2Na 632.1608; νmax 2928, 2253, 1714, 1573, 1452, 1336, 1149, 1066, 971, 904, 723, 647, 513 cm−1; δH (500 MHz, CDCl3) 7.36–7.32 (4H, m, ArH), 7.23 (1H, t, J = 8.0, ArH), 7.17–7.15 (3H, m, ArH), 7.08 (1H, d, J = 8.0, ArH), 6.99 (1H, d, J = 8.0, ArH), 6.68 (1H, d, J = 7.5, ArH), 5.73 (1H, d, J = 16.0, PhCHeHf–), 5.52 (1H, d, J = 14.0, –OCHaHb–), 5.41 (1H, d, J = 16.0, PhCHeHf–), 5.37 (1H, d, J = 14.0, –OCHaHb–), 5.25 (1H, d, J = 13.0, –OCHcHd–), 5.05 (1H, d, J = 13.0, –OCHcHd–), 4.17–3.64 (4H, m, 2× –NCH2–), 2.63 (3H, s, –SO2CH3), 2.57 (3H, s, –SO2CH3), 2.10–1.69 (4H, m, –NCH2CH2–); δC (125 MHz, CDCl3) 159.0, 157.5, 144.5, 140.6, 140.0, 134.6, 132.0, 129.4, 129.3, 129.0, 128.7, 128.3 128.1, 127.1, 121.1, 118.9, 115.5, 114.2, 63.4, 61.2, 52.4, 51.3, 51.2, 36.5, 35.3, 25.4, 24.1; m/z (ESI) 632.1 [M + Na]+
Data availability
The research data (and/or materials) supporting this publication can be accessed at https://wrap.warwick.ac.uk/.Author contributions
Sam Forshaw, Richard C. Knighton, Neelam Tiwari, Samson M. Oladeji and Andrew C. Stevens carried out synthetic chemistry, contributed to the design of the project and contributed to the writing up. Jeremy S. Parker contributed to the project design. William T. Scott carried out synthetic chemistry and molecular biology, contributed to the design of the project and contributed to the writing up. Yean Ming Chew and Jami Reber carried out molecular biology, contributed to the design of the project and contributed to the writing up. Guy J. Clarkson completed the X-ray crystal structure analyses and provided data for the project. Mohan K. Balasubramanian and Martin Wills conceived and directed the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the EPSRC and AstraZeneca for support of SF through a National Productivity Investment Fund (NPIF) studentship and the EPSRC for support of RCK through research grant EP/M006670/1. Lijiang Song is thanked for the Mass Spectrometry analyses. The X-ray diffraction instrument was obtained through the Science City Project with support from Advantage West Midlands (AWM) and part funded by the European Regional Development Fund (ERDF). YMC and WS were supported by PhD studentships from the MRC funded Doctoral Training Partnership in Interdisciplinary Biomedical Research (grant numbers MR/R502212/1 and MR/N014294/1, respectively). SMO was supported by a Scholarship from the Commonwealth Scholarships Commission. SMO and JR were students supported by the Warwick University Analytical Sciences and Instrumentation MSc course, MKB thanks the Welcome Trust (grant 101885/C/13/Z). The authors acknowledge Dr Cleidi Zampronio and the Warwick Proteomics Research Technology Platform for Mass Spectrometry analyses. The authors thank Dr Saravanan Palani for assistance and advice with the molecular biology.References
- (a) N. Stephanopoulos and M. B. Francis, Nat. Chem. Biol., 2011, 7, 876–884 CrossRef CAS PubMed; (b) S. M. Ryan, G. Mantovani, X. Wang, D. M. Haddleton and D. J. Brayden, Expert Opin. Drug Delivery, 2008, 5, 371–383 CrossRef CAS PubMed; (c) A. Beck, L. Goetsch, C. Dumontet and N. Corvaia, Nat. Rev. Drug Discovery, 2017, 16, 315–337 CrossRef CAS PubMed; (d) J. A. Prescher and C. R. Bertozzi, Nat. Chem. Biol., 2005, 1, 13–21 CrossRef CAS PubMed.
- (a) G. Wittig and A. Krebs, Chem. Ber., 1961, 94, 3260 CrossRef CAS; (b) T. Harris and I. V. Alabugin, Mendeleev Commun., 2019, 29, 237–248 CrossRef CAS; (c) D. H. Ess, G. O. Jones and K. N. Houk, Org. Lett., 2008, 10, 1633–1636 CrossRef CAS PubMed; (d) J. Dommerholt, F. P. J. T. Rutjes and F. L. van Delft, Top. Curr. Chem., 2016, 374, 16 CrossRef PubMed.
- (a) R. Huisgen, Angew. Chem., Int. Ed. Engl., 1963, 2, 565–598 CrossRef; (b) V. V. Rostovtsev, L. G. Green, V. V. Fokin and B. K. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS; (c) C. W. Tornoe, C. Christensen and M. Meldel, J. Org. Chem., 2002, 67, 3057–3064 CrossRef CAS PubMed.
- (a) N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS PubMed; (b) N. J. Agard, J. M. Baskin, J. A. Prescher, A. Lo and C. R. Bertozzi, ACS Chem. Biol., 2006, 1, 644–648 CrossRef CAS PubMed; (c) J. M. Baskin, J. A. Prescher, S. T. Laughlin, N. J. Agard, P. V. Chang, I. A. Miller, A. LoJ, A. Codelli and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 16793–11697 CrossRef CAS PubMed; (d) X. Ning, J. Guo, M. A. Wolfert and G. J. Boons, Angew. Chem., Int. Ed., 2008, 47, 2253–2255 CrossRef CAS PubMed; (e) N. E. Mbua, J. Guo, M. A. Wolfert, R. Steet and G. J. Boons, Chem. Biochem., 2011, 12, 1912–1921 CrossRef CAS PubMed; (f) J. Dammerholt, S. Schmidt, R. Temming, L. J. A. Hendricks, F. P. J. T. Rutjes, J. C. M. van Hest, D. J. Lefeber, P. Friedl and F. L. van Delft, Angew. Chem., Int. Ed., 2010, 49, 9422–9425 CrossRef PubMed; (g) M. F. Debets, S. S. van Berkel, S. Schoffelen, F. P. J. T. Rutjes, J. C. M. van Hest and F. L. van Delft, Chem. Commun., 2010, 46, 97–99 RSC; (h) J. Jewett, E. Sletten and C. R. Bertozzi, J. Am. Chem. Soc., 2010, 132, 3688–3690 CrossRef CAS PubMed.
- A. Del Grosso, L. D. Galanapoulos, C. K. C. Chiu, G. J. Clarkson, P. B. O'Conner and M. Wills, Org. Biomol. Chem., 2017, 15, 4517–4521 RSC.
- A. Mistry, R. C. Knighton, S. Forshaw, Z. Dualeh, J. S. Parker and M. Wills, Org. Biomol. Chem., 2018, 16, 8965–8975 RSC.
- R. C. Knighton, K. Sharma, N. S. Robertson, D. R. Spring and M. Wills, ACS Omega, 2019, 4, 2160–2167 CrossRef CAS PubMed.
- T. Harris, G. dos Passos Gomes, S. Ayad, R. J. Clark, V. V. Lobodin, M. Tuscan, K. Hanson and I. V. Alabugin, Chem, 2017, 3, 629–640 CAS.
- U. Koch-Pomeranz, H. J. Hansen and H. Schmidt, Helv. Chim. Acta, 1973, 56, 2981–3004 CrossRef CAS.
- B. Zhou, P. Jiang, J. Lu and C. Xing, Arch. Pharma. Chem. Life Sci., 2016, 349, 539–552 CrossRef CAS PubMed.
- C. Huang, Q. Yin, J. Meng, W. Zhu, Y. Yang, X. Qian and Y. Xu, Chem. – Eur. J., 2013, 19, 7739–7747 CrossRef CAS PubMed.
- T. Nguyen and M. B. Francis, Org. Lett., 2003, 5, 3245–3248 CrossRef CAS PubMed.
- G. de Almeida, E. M. Sletten, H. Nakamura, K. K. Palaniappan and C. R. Bertozzi, Angew. Chem., Int. Ed., 2012, 51, 2443–2447 CrossRef CAS PubMed.
- R. Ni, N. Mitsuda, T. Kashiwagi, K. Igawa and K. Tomooka, Angew. Chem., Int. Ed., 2015, 54, 1190–1194 CrossRef CAS PubMed.
- J. M. Yoon, C. Y. Lee, Y. I. Jo and C. H. Cheon, J. Org. Chem., 2016, 81, 8464–8469 CrossRef CAS PubMed.
- T. Harada, S. Ueda, T. Yoshida, A. Inoue, M. Takeuchi, N. Ogawa, A. Oku and M. Shiro, Org. Lett., 2000, 2, 1319–1322 CrossRef CAS PubMed.
- Y. Zhuang, Y. He, Z. Zhou, W. Xia, C. Cheng, M. Wang, B. Chen, Z. Zhou, J. Pang and L. Qiu, J. Org. Chem., 2015, 80, 6968–6975 CrossRef CAS PubMed.
- G. Delogu, D. Fabbri, S. Menichetti and C. Nativi, Tetrahedron, 2003, 59, 2131–2136 CrossRef CAS.
- T. He, L. Peng, S. Li, F. Hu, C. Xie, S. Huang, S. Jia, W. Qin and H. Yan, Org. Lett., 2020, 22, 6966–6971 CrossRef CAS PubMed.
- C. Bressy, D. Alberico and M. Lautens, J. Am. Chem. Soc., 2005, 127, 13148–13149 CrossRef CAS PubMed.
- T. Geiger, A. Haupt, C. Maichle-Mössmer, C. Schrenk, A. Schnepf and H. F. Bettinger, J. Org. Chem., 2019, 84, 10120–10135 CrossRef CAS PubMed.
- C. Tahtaoui, I. Parrot, P. Klotz, F. Guillier, J. L. Galzi, M. Hibert and B. Ilien, J. Med. Chem., 2004, 47, 4300–4315 CrossRef CAS PubMed.
- Y. Liu, W. M. Ren, C. Liu, S. Fu, M. Wang, K. K. He, R. R. Li, R. Zhang and X. B. Lu, Macromolecules, 2014, 47, 7775–7788 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopic data, X-ray crystal structure and bioconjugation results. CCDC 2273606–2273609. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ob01712e |
| This journal is © The Royal Society of Chemistry 2024 |