
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reactive deep eutectic solvents for EDC-mediated amide synthesis†
Debora
Procopio
 ,
Carlo
Siciliano
and
Maria Luisa
Di Gioia
*
,
Carlo
Siciliano
and
Maria Luisa
Di Gioia
*
Department of Pharmacy, Health and Nutritional Sciences, University of Calabria, 87036, Arcavacata of Rende, Italy. E-mail: ml.digioia@unical.it
First published on 21st December 2023
Abstract
The sustainability of amide bond formation is an ever-present topic in the pharmaceutical industry, as it represents the common motif in many clinically approved drugs. Despite many procedures for accomplishing eco-friendly amide synthesis having been developed, this transformation still remains a contemporary challenge. Herein, we report a greener approach for amide synthesis by using Reactive Deep Eutectic Solvents (RDESs) acting as both the reaction medium and reactants. The procedure not only avoids the use of hazardous solvents but also provides operationally simple product recovery with high purity and efficiency, without chromatographic purification. This approach was efficiently applied to the synthesis of a key intermediate in the production of an active pharmaceutical ingredient like atenolol. The green metrics of the gram-scale procedure were compared to the conventional industrial strategy showing an advancement in the greening of amide synthesis.
Introduction
Over the last few decades, green chemistry has emerged as a pivotal scientific approach that addresses the intrinsic challenges of conventional chemical processes while prioritizing environmental sustainability,1,2 minimizing waste, toxicity, and energy consumption, and maximizing efficiency, safety, and sustainability.3,4 The syntheses of active pharmaceutical ingredients (APIs) have been associated with resource-intensive procedures, generating substantial waste and environmental impact,5,6 and often involving the use of hazardous chemicals.7 In this context, green chemistry offers an alternative resource, where sustainability is integrated into all stages of drug discovery, development, and production.8 Using safer and more environmentally friendly reagents, it is possible to reduce pollution and minimize health risks during synthetic production.9 In addition, reducing waste generation also leads to cost saving, a fundamental concern in pharmaceutical economics.10,11 Thus, by integrating the principles of sustainability throughout drug discovery, development, and manufacturing, the pharmaceutical industry can mitigate its ecological footprint while advancing therapeutic solutions. The recent literature underscores the sector's increasing awareness of sustainability issues and the imperative to adopt greener alternatives.12 In synthetic medicinal chemistry, the synthesis of amides stands as a crucial cornerstone, often dictating the efficacy and safety of drug candidates (Fig. 1).13 The literature demonstrated that approximately 54% of biologically tested compounds contain at least one amide bond and more than 25% of known drugs consist of amide functionalities as per the Comprehensive Medicinal Chemistry database analysis.14 Moreover, amide synthesis was identified in the top 10 key research areas for the pharmaceutical industry by ACS Green Chemistry Institute Pharmaceutical Roundtable (ACS GCIPR). Notably, the existing processes for the amide coupling reaction in manufacturing of APIs have a gap in reaction process efficiency and sustainability.15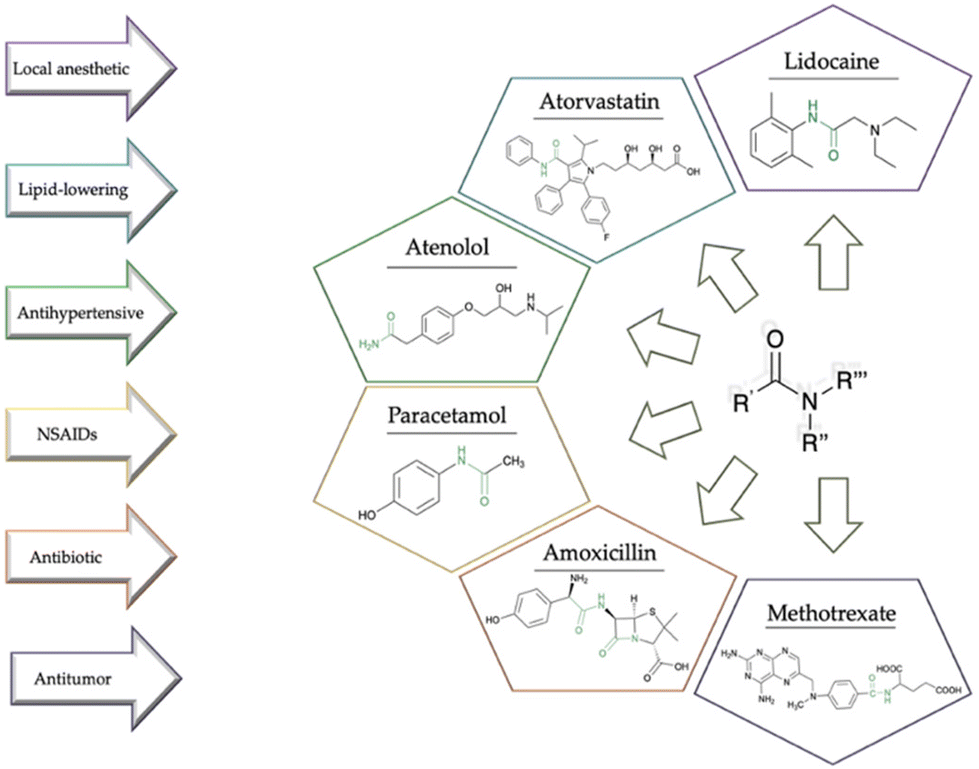 | ||
| Fig. 1 Examples of some amide-based drugs. | ||
The traditional methods for amide formation frequently rely on stoichiometric reagents and harsh reaction conditions, leading to challenges in atom efficiency and undesirable by-products.16 Moreover, the use of conventional solvents, characterized by toxicity and environmental concerns, adds further complexity to the synthetic process.17 Thus, the green synthesis of amides assumes critical importance not only due to its intrinsic relevance but also as a transformative approach to address the limitations of conventional methods.7 In this framework, the search for eco-friendly alternatives to the conventional solvents and reagents employed in amide synthesis becomes overriding. In recent years, the landscape of amide synthesis has witnessed a profound transformation, driven by innovative green strategies that leverage novel reagents, catalytic systems, and reaction media.18 Water, an abundant and benign solvent, has emerged as an attractive medium for amide bond formation under milder conditions, promoting atom efficiency and obviating the need for toxic additives.19 Ionic liquids, with their unique solvent properties and potential for recycling, also offer a sustainable alternative to conventional solvents.20 A particularly promising opportunity involves the utilization of renewable and bio-derived solvents.21,22
The emergence of Deep Eutectic Solvents (DESs), characterized by their tunability and efficiency as both solvents/catalysts and reagents, presents an exciting frontier for a greener synthesis of amides.23 DESs, renowned for their low cost, biodegradability, and low toxicity, have already proven their worth as alternatives to traditional organic solvents.24 They are prepared by combining hydrogen bond acceptors and donors, resulting in a eutectic mixture with a significantly lowered freezing point compared to its individual components.25 Their unique properties make them attractive candidates for various applications, ranging from extraction and separation processes to catalysis and electrochemistry.26
Over the last few years, Reactive Deep Eutectic Solvents (RDESs) have emerged as a new promising class of media that combine the advantageous properties of DESs with the ability to actively participate in chemical reactions.27 The innovation of RDESs builds upon these features, adding an extra dimension of functionality. RDESs can be fine-tuned to exhibit different desired properties, such as acidity, basicity, or redox potential. This dual role as both a solvent and a reactant opens up opportunities for more sustainable, streamlined, and efficient synthetic routes, minimizing the use of external reagents and reducing waste generation. Several research endeavours have emerged, delving into the multiple applications of DESs in amide bond formation. Notably, Azizi and colleagues harnessed the synergistic capabilities of DESs, particularly choline chloride![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :stannous chloride (ChCl:SnCl2), which serve both as a reaction medium and a catalyst (Fig. 2A).28 Nevertheless, the approach was restricted to the formylation of aryl amines. Then, Salomone et al. approached a distinct route, by employing two different DESs (Fig. 2B),29 exploiting a Pd-catalysed aminocarbonylation of (hetero)-aryl iodides. However, the procedure needed chromatographic purification of the obtained product. Parallel to these advances, Saberi's group highlighted the intrinsic potential of the uncomplicated DES ChCl:urea (Fig. 2C) in the direct condensation of amines and carboxylic acids30 using 2,4,6-trichloro-1,3,5-triazine (TCT) as a coupling reagent. However, the protocol involved the use of a base catalyst which enhanced the final amount of waste produced. Recently, we have made a significant contribution by introducing a pioneering approach for synthesizing amides from thioacids, by using visible light irradiation under mild conditions in DESs.31 Hence, as a part of our ongoing interest in the preparation of DESs and investigation of their application in organic synthesis,32 we investigated the possibility to use RDESs for the N-amidation of amines with carboxylic acids. RDESs have been designed in order to contribute actively to the synthetic process, using choline chloride as the hydrogen-bond acceptor and various carboxylic acids as hydrogen-bond donors.
:stannous chloride (ChCl:SnCl2), which serve both as a reaction medium and a catalyst (Fig. 2A).28 Nevertheless, the approach was restricted to the formylation of aryl amines. Then, Salomone et al. approached a distinct route, by employing two different DESs (Fig. 2B),29 exploiting a Pd-catalysed aminocarbonylation of (hetero)-aryl iodides. However, the procedure needed chromatographic purification of the obtained product. Parallel to these advances, Saberi's group highlighted the intrinsic potential of the uncomplicated DES ChCl:urea (Fig. 2C) in the direct condensation of amines and carboxylic acids30 using 2,4,6-trichloro-1,3,5-triazine (TCT) as a coupling reagent. However, the protocol involved the use of a base catalyst which enhanced the final amount of waste produced. Recently, we have made a significant contribution by introducing a pioneering approach for synthesizing amides from thioacids, by using visible light irradiation under mild conditions in DESs.31 Hence, as a part of our ongoing interest in the preparation of DESs and investigation of their application in organic synthesis,32 we investigated the possibility to use RDESs for the N-amidation of amines with carboxylic acids. RDESs have been designed in order to contribute actively to the synthetic process, using choline chloride as the hydrogen-bond acceptor and various carboxylic acids as hydrogen-bond donors.
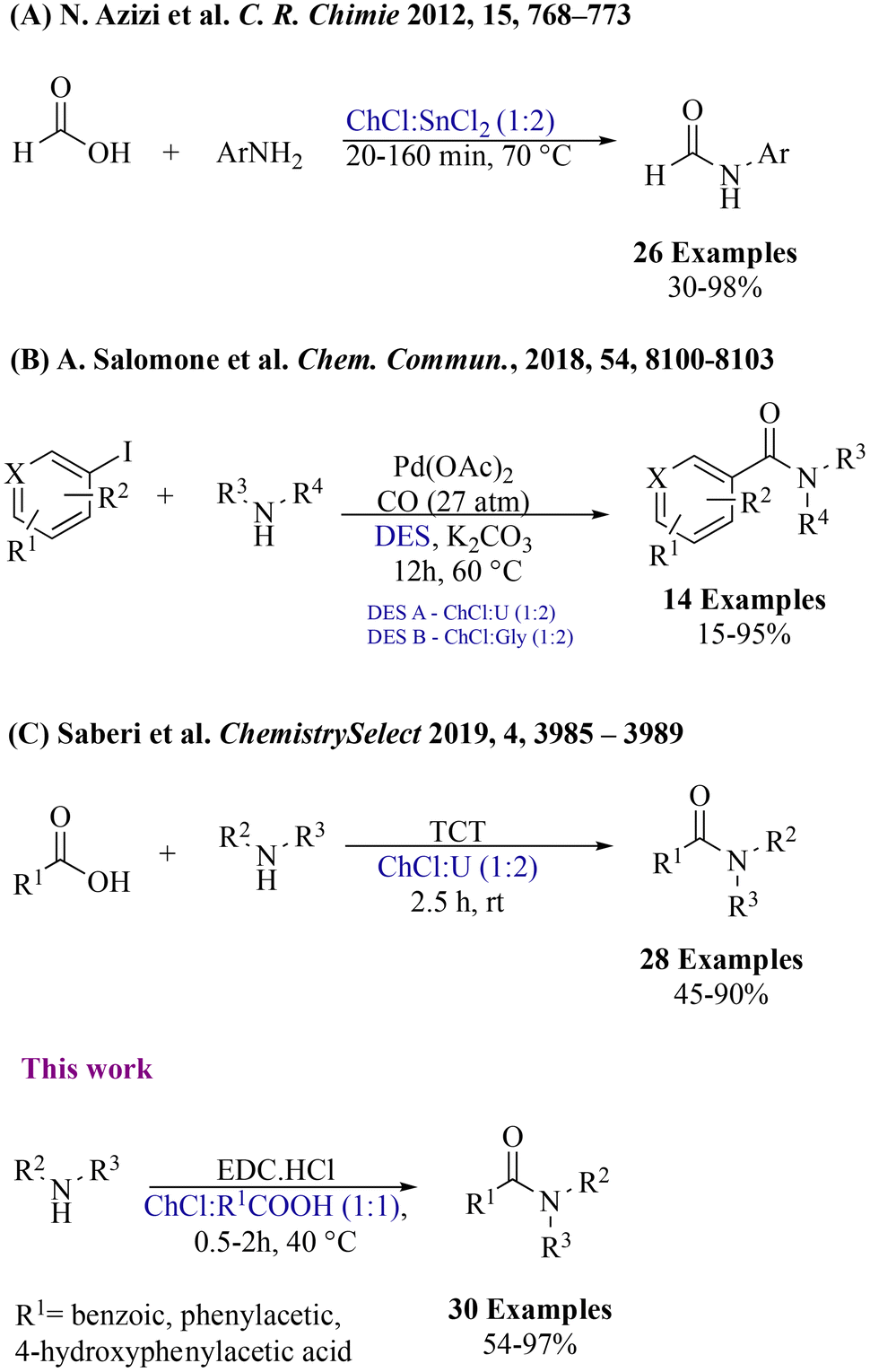 | ||
| Fig. 2 Synthesis of amides using deep eutectic solvents. | ||
Results and discussion
Preparation of RDES media
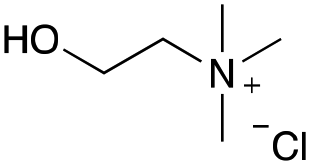
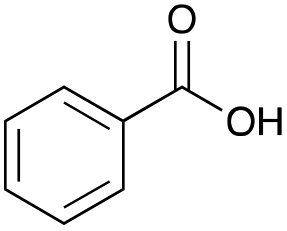
Our investigation started with the preparation of three different RDESs made up of mixtures of HBA/HBD in a 1:1 molar ratio. In particular, the quaternary ammonium salt choline chloride (ChCl) was used as the HBA, whereas benzoic acid (BA), phenylacetic acid (PA) and 4-hydroxyphenylacetic acid (HPA) were chosen as HBDs and reactants (Table 1). The selected HBA, ChCl, exhibits clear biocompatibility and benefits from its natural origin, which represents one of its significant advantages.32
| Hydrogen-bond acceptor (HBA) | Hydrogen-bond donors (HBD) | Molar ratio (HBA/HBD) | ||
|---|---|---|---|---|
| ChCl |
|
BA |
|
1:1 |
| ChCl |
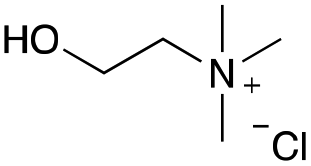
|
PA |
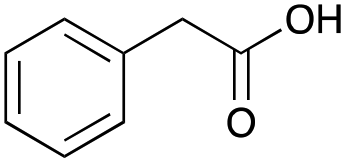
|
1:1 |
| ChCl |
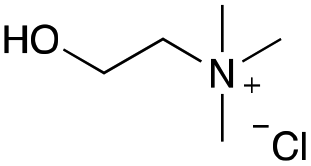
|
HPA |
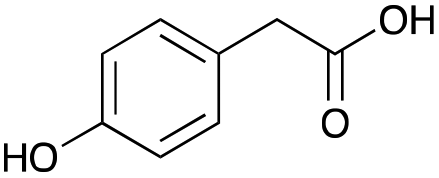
|
1:1 |
All RDESs were prepared by using a well-established protocol, by mixing the components in a defined molar ratio and heating them at 60–90 °C, under constant stirring in a round bottom flask, until a homogeneous colorless liquid phase was formed.33 A differential scanning calorimetry (DSC) analysis of the obtained mixtures as well as of the individual components was performed to prove the successful formation of RDESs (Table 2).
| HBA | HBDs | T f (°C) | T m HBD (°C) | Δ (°C) | Aspect |
|---|---|---|---|---|---|
| ChCl | BA | 95 | 122.3 | 27.3 | Viscous transparent liquid |
| ChCl | PA | 25 | 76–77 | 51–52 | Clear and transparent liquid |
| ChCl | HPA | 19 | 147.6 | 129.6 | Yellow viscous liquid |
All the RDESs prepared were used without the need for further purification.
Optimization of the reaction conditions
In order to investigate DESs for their dual roles as solvents and reactants in the synthesis of amides, we performed the N-amidation of aniline with phenylacetic acid, employed as a component of the RDES. To date, the preferred method in the pharmaceutical industry for large-scale amide synthesis is the activation of carboxylic acids using carbodiimides in combination with additives.34 At first, the model reaction was performed in the presence of N,N-dicyclohexylcarbodiimide (DCC)/1-hydroxybenzotriazole (HOBt) as a coupling reagent combination. After 60 minutes of magnetic stirring at room temperature, the desired amide was obtained in 61% yield (entry 1, Table 3), indicating that the RDES could promote the amidation reaction. Encouraged by this promising outcome, we proceeded to further optimize the coupling reaction within the context of the RDES medium. To this end, we systematically assessed various combinations of coupling reagents across different temperature ranges, but in the absence of a basic catalyst. The progress of all reactions was monitored through the utilization of thin layer chromatography (TLC) and gas chromatography/mass spectrometry (GC/MS) analysis. As shown in Table 2, the RDES-mediated synthesis method was applicable to all coupling combinations considered. The greatest efficiency in terms of coupling performance and product purity was achieved by using 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) as the coupling reagent (entry 6, Table 3). This was justified by the fact that EDC and the corresponding urea byproduct that is produced are soluble in water, thus allowing an easier work up procedure at the end of the reaction. As a matter of fact, since the amide is insoluble in water, it precipitates and can be easily afforded by washing it with water, without any further purification. Also, from a green chemistry point of view, EDC is greener than other carbodiimides according to the GlaxoSmithKline reaction-selection guide, since the urea byproduct generated does not pose health and environmental hazard.35,36 Finally, it is important to highlight that raising the temperature in EDC-mediated reactions did not affect the final product yield. On the other hand, when the reaction was performed in a conventional organic solvent like dichloromethane, the desired amide was afforded in a good yield (entry 8, Table 3). Nevertheless, the use of an organic base like DIPEA was required and the product was recovered after chromatographic purification, thus revealing the strength of the RDES mediated procedure.|
|
|||||
|---|---|---|---|---|---|
| Entry | Coupling reagent | Additive | T (°C) | Time (min) | Yieldd (%) |
| a Reaction conditions: HOBt (1 equiv.) and the coupling reagent (1 equiv.) were sequentially added in the RDES (1 equiv.) and stirred for 10 min at the temperatures reported above. Successively, aniline (1 equiv.) was added and stirred for the time reported above. b Reaction conditions: the coupling reagent (1 equiv.) was added in the RDES (1 equiv.) and stirred for 10 min at different temperatures. Successively, aniline (1 equiv.) was added and stirred for the time reported above. c The reaction was performed in dichloromethane in the presence of DIPEA as the base. The amide product was purified by column chromatography. d % conversion calculated by GC/MS. | |||||
| 1a | DCC | HOBt | rt | 60 | 61 |
| 2a | DCC | HOBt | 40 °C | 60 | 76 |
| 3a | DIC | HOBt | 40 °C | 60 | 68 |
| 4b | DIC | — | 40 °C | 30 | 79 |
| 5a | EDC | HOBt | 40 °C | 30 | 89 |
| 6b | EDC | — | 40 °C | 60 | 93 |
| 7b | EDC | — | 70 °C | 30 | 91 |
| 8c | EDC | — | rt | 240 | 60 |
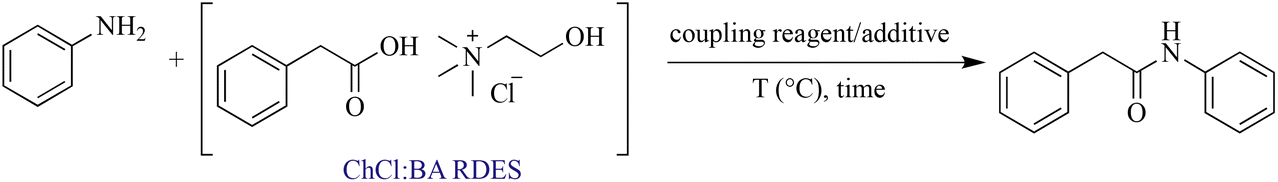
Scope of amines
In order to investigate the scope and limitations of the optimized methodology, we examined the coupling reaction for a series of amines, by employing the same RDES system. First, a phenylacetic-based RDES was subjected to the reaction with different aliphatic primary amines (entries 3c, 3g–3i, Table 4) affording the corresponding N-alkyl amides with good to excellent yields. The reaction of the same RDES with the secondary amine N-ethylbenzylamine led to the corresponding compound in 78% yield, while the one with aniline afforded 3b with 93% yield. When p-toluidine or p-anisidine was used as an amine source, a slight reduction in the yield was observed (entries 3d and 3e, Table 4), probably due to the reduced reactivity of the aromatic amine. Moreover, the presence of an additional functional group in the amine framework did not interfere in the yield of the desired amide (entries 3g and 3i, Table 4). Besides the benzylamine, the reaction was also carried out with other diversely substituted amines, confirming that the amine scope is wide. We were also pleased to find that halogen substituents such as chloride are tolerated in the reaction (3m, Table 3). The reaction proceeded well with anilines bearing strong electron-withdrawing substituents like para-nitro or trifluoromethyl (3n, 3l, Table 3). Tertiary amides could also be prepared, including examples derived from both cyclic and acyclic (3f, 3o, and 3p, Table 3) secondary amines.| a Determined by chiral HPLC (see the ESI† for details). |
|---|
|
The additive HOBt is regularly employed in combination with EDC or other coupling reagents in order to reduce the risk of racemization in amide synthesis.37,38 In particular, it acts as an activating agent for the carboxylic acid group, making it more reactive towards the amine. This activation reduces the time that the intermediates are exposed to potentially racemizing conditions, thus minimizing the chances of racemization occurring during the reaction. However, the inherent risks associated with HOBt have led to its regulation under the REACH (Registration, Evaluation, Authorization, and Restriction of Chemicals) framework.39 This is due to its potential hazards to individuals for its capacity in causing skin and ocular irritation, as well as inhalation-related risks when not handled in accordance with the established safety protocols. Furthermore, its release into the environment can cause adverse ecological effects. In this context, it is worth noting that no racemisation was observed when using enantiopure amines such as (R)- and (S)-1-phenylethan-1-amine (entries 3j and 3k, Table 4). Consequently, the rapidity of the conversion achieved through the developed protocol enabled us to avoid the necessity of employing HOBt, thus enhancing the overall sustainability of the process.
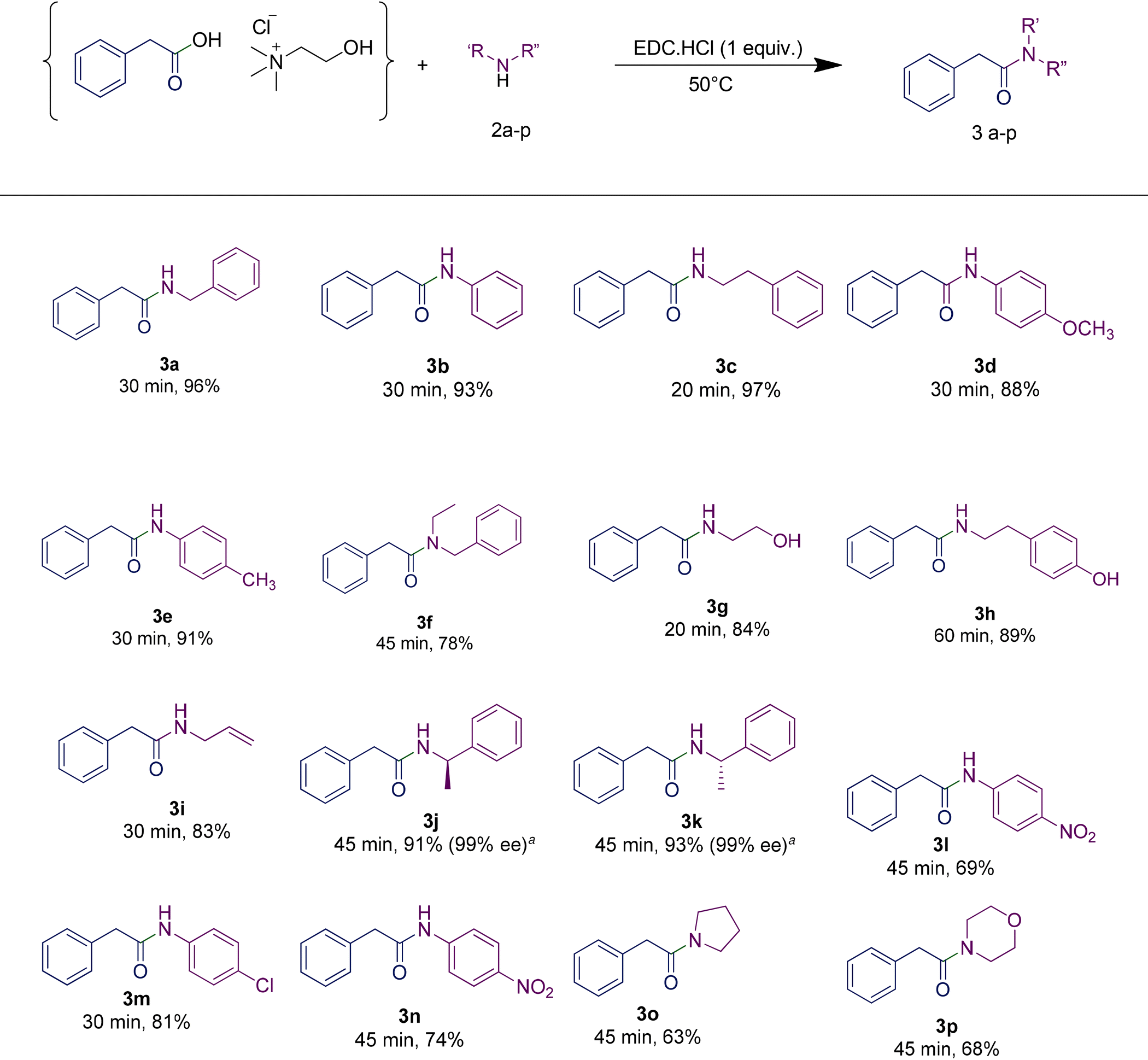
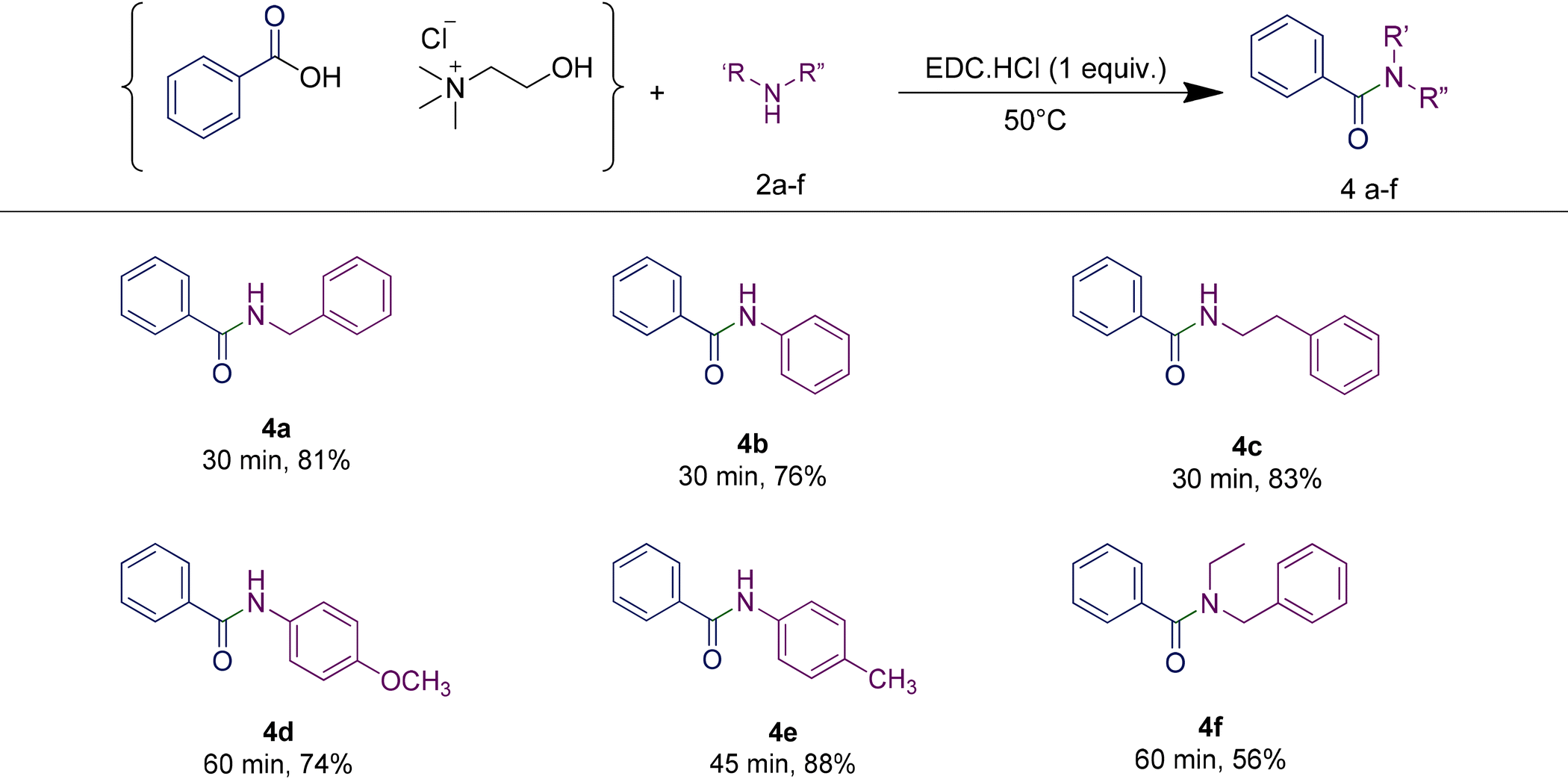
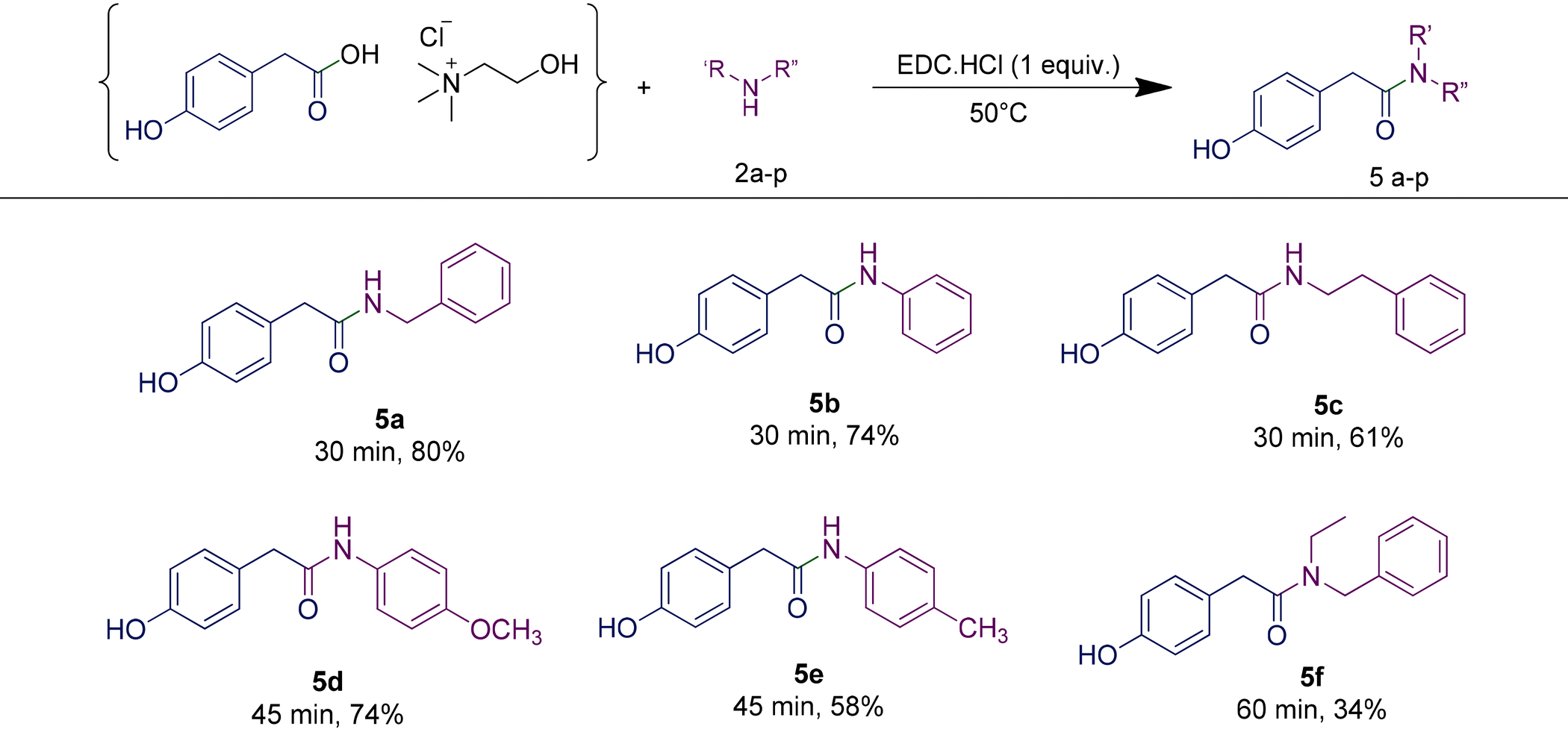
In order to highlight the generality of the process, some of the same amine moieties were used to perform coupling reactions with the other two RDESs prepared. The results showed that even upon changing the acidic component in the RDES, the performances of the systems were comparable with the first one, confirming and widening the scope of the protocol developed. The benzoic acid-based RDES reacted with aromatic and primary and secondary amines with acceptable yields (Table 5). A p-hydroxyphenylacetic acid-based RDES was also evaluated as a substrate. A slight drop in the yield was observed and the corresponding amides were isolated in fair to good yields (Table 6).
| a Reaction conditions: the coupling reagent (1 equiv.) was added in a benzoic acid-based RDES (1 equiv.) and stirred for 10 min at different temperatures. Successively, the amine (1 equiv.) was added and stirred for the time reported above. |
|---|
|
|
In order to investigate the utility of the developed methodology in the pharmaceutical industry, the optimized reaction conditions were applied to synthesize 4-hydroxyphenylacetamide, a key synthetic intermediate in the industrial production of atenolol. Atenolol represents one of the top-five bestselling drugs in the world, today used to treat angina and hypertension and to reduce the risk of death after a heart attack.40 In this regard, the synthesis of 4-hydroxyphenylacetamide represents the first step in the industrial production of atenolol. From the reaction between ammonium chloride and a 4-hydroxyphenylacetic acid-based RDES, the desired amide was recovered in 94% yield, using EDC·HCl as the coupling reagent (Scheme 1).
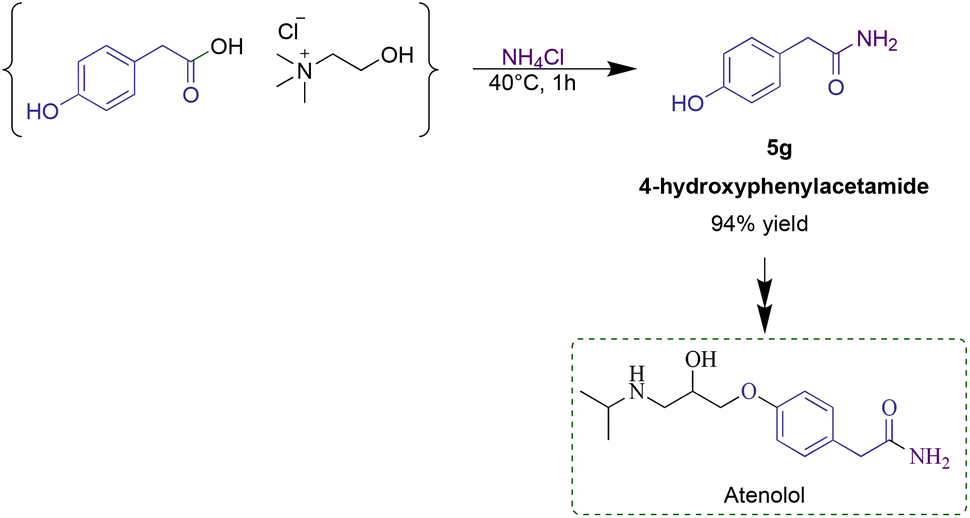 | ||
| Scheme 1 Amidation step with an RDES during the synthesis of atenolol. | ||
It is important to emphasize that one of the advantages of the developed method is that for all these amidations, the devised workup procedure gave the pure amide product without the requirement for chromatography.
By considering the possibility of adopting the developed synthetic route for the industrial synthesis of amides, the gram-scale synthesis of N-phenethyl-2-phenylacetamide was carried out. By adding water to the reaction mixture at the end of the coupling reaction for quenching, the precipitated product was successfully recovered in high yields and purity by simple filtration, without the need for chromatographic procedures.
In order to demonstrate the environmental benefits of our approach, we calculated the green chemistry metrics for the synthesis of the pilot reaction (for calculations, see the ESI†).41 The method was highly efficient to produce the desired amide in 97% yield (on a gram scale) with PMI (Process Mass Intensity) and E-factor values superior when compared to the conventional industrial method (Fig. 3).42
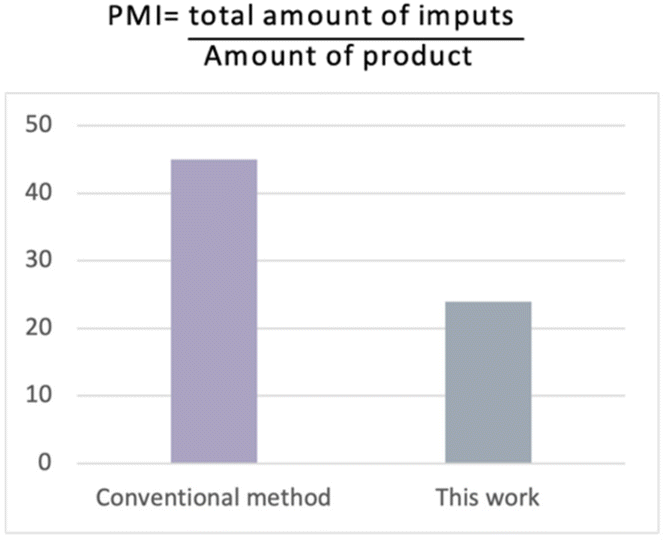 | ||
| Fig. 3 Comparison of the PMI data with the conventional industrial method. | ||
Proposed mechanism
On the basis of previous literature results and our experiments, a plausible mechanism for the amidation reaction in the presence of an RDES is shown in Scheme 2. In the first reaction step, the proton transfer from the carboxylic acid to the weakly basic nitrogen atom on EDC was facilitated by hydrogen bond interactions between the RDES and the coupling reagent causing a nucleophilic attack of the resulting carboxylate anion to lead to the formation of the O-acylisourea intermediate (1). Then, the same hydrogen bond interactions allowed the aminolysis step (2). Finally, anchimeric assistance guided the formation of the amide product (3), along with the production of urea as a byproduct.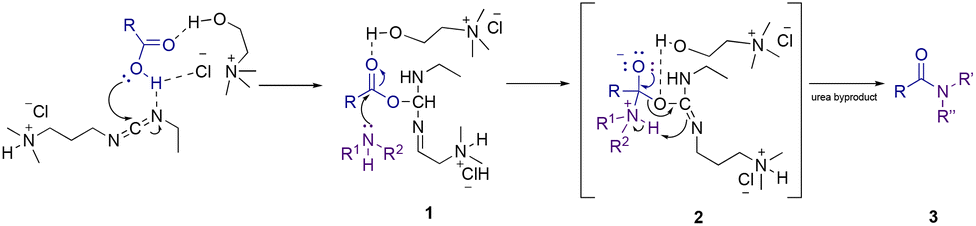 | ||
| Scheme 2 Proposed N-amidation mechanism in an RDES. | ||
Experimental
General information
All starting materials were purchased from Aldrich (Merck) and used without further purification unless stated otherwise. Thin layer chromatography (TLC) was carried out on Schleicher & Schuell F1400/LS 254 plates coated with a 0.2 mm layer of silica gel; detection by UV254 light. Mass spectra (EI) were obtained at 70 eV on a Shimazdu QP-5000 spectrometer, giving fragment ions in m/z with relative intensities (%) in parentheses. 1H NMR (300 MHz) spectra were recorded on a Bruker AC-300 NMR spectrometer respectively in proton coupled mode. 13C NMR (75.5 MHz) spectra were recorded on a Bruker AC-300 NMR spectrometer respectively in proton decoupled mode at 20 °C. Chemical shifts are given in δ (parts per million) and coupling constants (J) in hertz. HPLC analysis was performed to determine the ee % value (DIACEL Chiralcel OD-H, n-hexane:2-propanol = 90:10 (1 mL min−1), tr = 13.4 min (R) and tr = 18.1 min (S), 99% ee, S-enantiomer).
General procedure for RDES preparation
The different RDESs were prepared by mixing choline chloride and the carboxylic acid (benzoic, phenylacetic or 4-hydroxyphenylacetic acid) in a round-bottom flask under an inert atmosphere in a 1:1 molar ratio, respectively. The resulting mixture was magnetically stirred at 60–80 °C, until a clear liquid was observed. The obtained DESs were used without further purification.
General procedure for the synthesis of amides in RDESs
In a typical procedure, 1 mmol of the coupling reagent was added to a ChCl:carboxylic acid (1:1)-based RDES (1 mL). After stirring at 50 °C for 10–15 minutes, the amine was added in an equimolar ratio and the resulting mixture was vigorously stirred at the given temperature for an additional time (30–60 min). The progress of the reaction was monitored by TLC and GC/MS analysis. Upon completion of the reaction, 2 mL of H2O were added. The resulting aqueous suspension was then extracted with ethyl acetate (3 × 2 mL). The organic phases were dried over sodium sulphate, followed by evaporation under reduced pressure to give the corresponding amides. The reaction conversions were determined by GC/MS analysis. The spectral data were in accordance with the literature (see the ESI†).
Conclusions
In conclusion, we present a sustainable method for amide synthesis using Reactive Deep Eutectic Solvents as both solvents and reagents. The exploitation of RDESs overcomes the use of hazardous solvents and additives, significantly reducing the environmental footprint of amide synthesis. RDES-mediated amide synthesis demonstrates excellent efficiency, allowing for the formation of amide bonds with high purity and yield. The method is versatile and tuneable to a wide range of substrates, both aliphatic and aromatic, including chiral compounds, without any observed racemization. This innovative approach is not limited to laboratory-scale applications but can be readily scaled up for industrial manufacturing. This sustainable approach aligns with the principles of green chemistry, contributing to a cleaner and more responsible pharmaceutical industry.Author contributions
Debora Procopio: Investigation, writing – original draft, writing – review and editing. Carlo Siciliano: Investigation, writing – review and editing; Maria Luisa Di Gioia: conceptualization, resource, methodology, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.References
- M. K. M. Lane, H. E. Rudel, J. A. Wilson, H. C. Erythropel, A. Backhaus, E. B. Gilcher, M. Ishii, C. F. Jean, F. Lin, T. D. Muellers, T. Wang, G. Torres, D. E. Taylor, P. T. Anastas and J. B. Zimmerman, Nat. Sustain., 2023, 3, 502–512 CrossRef.
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, 1998 Search PubMed.
- V. K. Tiwari, A. Kumar, S. Rajkhowa, G. Tripathi and A. K. Singh, Green Chemistry: Introduction to the Basic Principles, in Green Chemistry, Springer, Singapore, 2022 Search PubMed.
- R. A. Sheldon and D. Brady, ChemSusChem, 2022, 15, e202102628–e202102648 CrossRef CAS PubMed.
- J. Becker, C. Manske and S. Randl, Curr. Opin. Green Sustainable Chem., 2022, 33, 100562 CrossRef CAS.
- S. Kar, H. Sanderson, K. Roy, E. Benfenati and J. Leszczynski, Chem. Rev., 2022, 3, 3637–3710 CrossRef PubMed.
- (a) M. C. Bryan, P. J. Dunn, D. Entwistle, F. Gallou, S. G. Koenig, J. D. Hayler, M. R. Hickey, S. Hughes, M. E. Kopach, G. Moine, P. Richardson, F. Roschangar, A. Steven and F. J. Weiberth, Green Chem., 2018, 20, 5082–5103 RSC; (b) L. Maiuolo, P. Merino, V. Algieri, M. Nardi, M. L. Di Gioia, B. Russo, I. Delso, M. A. Tallarida and A. De Nino, RSC Adv., 2017, 7(77), 48980–48988 RSC; (c) R. De Marco, M. L. Di Gioia, A. Leggio, A. Liguori, F. Perri, C. Siciliano and M. C. A. Viscomi, Amino Acids, 2010, 38(3), 691–700 CrossRef PubMed.
- (a) R. A. Sheldon, Green Chem., 2016, 18, 3180–3183 RSC; (b) M. Nardi, M. L. Di Gioia, P. Costanzo, A. De Nino, L. Maiuolo, M. Oliverio, F. Olivito and A. Procopio, Catalysts, 2017, 7(9), 269 CrossRef.
- A. Kumar, E. K. Zavadskas, S. K. Mangla, V. Agrawal, K. Sharma and D. Gupta, Int. J. Prod. Res., 2019, 57, 3554–3576 CrossRef.
- A. P. Abbott, D. Boothby, G. Capper, D. L. Davies and R. K. Rasheed, J. Am. Chem. Soc., 2004, 126, 9142–9147 CrossRef CAS PubMed.
- K. Zobaidul, S. Z. Emon and S. Karim, Microbiology for Cleaner Production and Environmental Sustainability, CRC Press, Boca Raton, 2023 Search PubMed.
- M. Mishra, M. Sharma, R. Dubey, P. Kumari, V. Ranjan and J. Pandey, Curr. Res. Green Sustainable Chem., 2021, 4, 100174 CrossRef CAS.
- (a) V. Pattabiraman and J. Bode, Nature, 2011, 480, 471–479 CrossRef CAS PubMed; (b) J. S. Carey, D. Laffan, C. Thomson and M. T. Williams, Org. Biomol. Chem., 2006, 4, 2337–2347 RSC; (c) S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451–3479 CrossRef CAS PubMed.
- S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451–3479 CrossRef CAS PubMed.
- F. Albericio and A. El-Faham, Org. Process Res. Dev., 2018, 22, 760–772 CrossRef CAS.
- (a) E. Massolo, M. Pirola and M. Benaglia, Eur. J. Org. Chem., 2020, 4641–4651 CrossRef CAS; (b) L. J. Gooßen, D. M. Ohlman and P. Lange, Rev. Syn., 2009, 1, 160–164 Search PubMed; (c) R. Cassano, A. Trapani, M. L. Di Gioia, D. Mandracchia, R. Pellitteri, G. Tripodo, S. Trombino, S. Di Gioia and M. Conese, Int. J. Pharm., 2020, 589, 119829 CrossRef CAS PubMed; (d) H. Charville, D. A. Jackson, G. Hodges, A. Whiting and M. R. Wilson, J. Org. Chem., 2011, 2011, 5981–5990 CAS; R. M. De Figueiredo, J. S. Suppo and J. M. Campagne, Chem. Rev., 2016, 116, 12029–12122 Search PubMed; (e) J. R. Dunetz, J. Magano and G. A. Weisenburger, Org. Process Res. Dev., 2016, 20, 140–177 CrossRef CAS.
- (a) P. A. Fail, J. D. George, T. B. Grizzle and J. J. Heindel, Reprod. Toxicol., 1998, 12, 3 Search PubMed; (b) J. Sherwood, Angew. Chem., Int. Ed., 2018, 57, 14286–14290 CrossRef CAS PubMed; (c) D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehad and P. J. Dunn, Green Chem., 2016, 18, 288–296 RSC; (d) A. Leggio, E. L. Belsito, G. De Luca, M. L. Di Gioia, V. Leotta, E. Romio, C. Siciliano and A. Liguori, RSC Adv., 2016, 6(41), 34468–34475 RSC; (e) D. S. MacMillan, J. Murray, H. F. Sneddon, C. Jamieson and A. J. B. Watson, Green Chem., 2012, 14, 3016–3019 RSC.
- D. Procopio, C. Siciliano, S. Trombino, D. E. Dumitrescu, F. Suciu and M. L. Di Gioia, Org. Biomol. Chem., 2022, 20, 1137–1149 RSC.
- (a) F. Gallou, P. Guo, M. Parmentier and J. Zhou, Org. Process Res. Dev., 2016, 20, 1388–1391 CrossRef CAS; (b) M. Cortes-Clerget, J. Y. Berthon, I. Krolikiewicz-Renimel, L. Chaisemartin and B. H. Lipshutz, Green Chem., 2017, 19, 4263–4267 RSC; (c) M. Cortes-Clerget, N. R. Lee and B. H. Lipshutz, Nat. Protoc., 2019, 14, 1108–1129 CrossRef CAS PubMed; (d) M. Shi, N. Ye, W. Chen, H. Wang, C. Cheung, M. Parmentier, F. Gallou and B. Wu, Org. Process Res. Dev., 2020, 24, 1543–1154 CrossRef CAS; (e) S. Sharma, N. W. Buchbinder, W. M. Braje and S. Handa, Org. Lett., 2020, 22, 5737–5740 CrossRef CAS PubMed; (f) M. Konwar, A. A. Ali and D. Sarma, Tetrahedron Lett., 2016, 57, 2283–2285 CrossRef CAS; (g) M. Badland, R. Crook, B. Delayre, S. J. Fussell, I. Gladwell, M. Hawksworth, R. M. Howard, R. Walton and G. A. Weisenburger, Tetrahedron Lett., 2017, 58, 4391–4394 CrossRef CAS; N. Fattahi, M. Ayubi and A. Ramazani, Tetrahedron, 2018, 74, 4351–4356 Search PubMed.
- (a) N. Galy, M. R. Mazières and J. C. Plaquevent, Tetrahedron Lett., 2013, 54, 2703–2705 CrossRef CAS; (b) M. Konwar, N. D. Khupse, P. J. Saikia and D. Sarma, J. Chem. Sci., 2018, 130, 6016–6019 CrossRef; (c) C. Li, M. Wang, X. Lu, L. Zhang, J. Jiang and L. Zhang, ACS Sustainable Chem. Eng., 2020, 8, 4353–4361 CrossRef CAS.
- (a) Y. E. Jad, G. A. Acosta, S. N. Khattab, B. G. de la Torre, T. Govender, H. G. Kruger, A. El-Faham and F. Albericio, Amino Acids, 2016, 48, 419–426 CrossRef CAS PubMed; (b) Y. E. Jad, G. A. Acosta, S. N. Khattab, B. G. de la Torre, T. Govender, H. G. Kruger, A. El-Faham and F. Albericio, Org. Biomol. Chem, 2015, 13, 2393–2398 RSC; (c) Y. E. Jad, G. A. Acosta, T. Govender, H. G. Kruger, A. El-Faham, B. G. de la Torre and F. Albericio, ACS Sustainable Chem. Eng., 2016, 4, 6809–6814 CrossRef CAS; (d) K. L. Wilson, J. Murray, C. Jamieson and A. J. B. Watson, Org. Biomol. Chem., 2018, 16, 2851–2854 RSC; (e) T. W. Bousfield, K. P. R. Pearce, S. B. Nyamini, A. Angelis-Dimakis and J. E. Camp, Green Chem., 2019, 21, 3675–3681 RSC.
- (a) A. Kumar, Y. E. Jad, J. M. Collins, J. M. F. Albericio and B. G. de la Torre, Chem. Eng., 2018, 6, 8034–8039 CAS; (b) A. Kumar, A. Thompson-Adewunmi, K. P. Nandhini, J. M. Collins, F. Albericio and B. G. de la Torre, Org. Process Res. Dev., 2019, 23, 1096–1100 CrossRef CAS; (c) T. H. M. Petchey, J. W. Comerford, T. J. Farmer, D. J. Macquarie, J. Sherwood and J. H. Clark, ACS Sustainable Chem. Eng., 2018, 6, 1550–1554 CrossRef CAS; (d) G. Paggiola, N. Darrien, J. D. Moseley, A. Green, S. L. Flitch, J. H. Clark, C. R. McElroy and A. J. Hunt, Pure Appl. Chem., 2020, 92, 579–586 CrossRef CAS.
- (a) E. L. Smith, A. P. Abbott and K. S. Ryder, Chem. Rev., 2014, 114, 11060–11082 CrossRef CAS PubMed; (b) A. P. Abbott, G. Capper, D. L. Davies, R. K. Rasheed and V. Tambyrajah, Chem. Commun., 2003, 7, 70–71 RSC.
- D. A. Alonso, A. Baeza, R. Chinchilla, G. Guillena, I. M. Pastor and D. J. Ramón, Eur. J. Org. Chem., 2016, 612–632 CrossRef CAS.
- M. L. Di Gioia, M. Nardi, P. Costanzo, A. De Nino, L. Maiuolo, M. Oliverio and A. Procopio, Molecules, 2018, 23, 1891 CrossRef PubMed.
- (a) F. Liu, Z. Xue, X. Zhao, H. Mou, J. He and T. Mu, Chem. Commun., 2018, 54, 6140–6143 RSC; (b) A. E. Ünlü, A. Arıkaya and S. Takaç, Green Process. Synth., 2019, 8, 355–372 Search PubMed; (c) M. L. Di Gioia, R. Cassano, P. Costanzo, N. Herrera Cano, L. Maiuolo, M. Nardi, F. P. Nicoletta, M. Oliverio and A. Procopio, Molecules, 2019, 24, 2885 CrossRef PubMed; (d) S. M. Taklimi, A. Divsalar, B. Ghalandari, X. Ding, M. L. Di Gioia, K. A. Omar and A. A. Saboury, J. Mol. Liq., 2023, 377, 121562 CrossRef CAS.
- C. Allegretti, F. G. Gatti, S. Marzorati, L. A. M. Rossato, S. Serra, A. Strini and P. D'Arrigo, Catalysts, 2021, 11, 655 CrossRef CAS.
- N. Azizi, E. Gholibeglo, M. Babapour, H. Ghafuri and S. M. Bolourtchian, C. R. Chim., 2012, 15, 768–773 CrossRef CAS.
- F. Messa, S. Perrone, M. Capua, F. Tolomeo, L. Troisi, V. Capriati and A. Salomone, Chem. Commun., 2018, 54, 8100–8103 RSC.
- K. Salimiyan and D. Saberi, ChemistrySelect, 2019, 4, 3985–3989 CrossRef CAS.
- D. Procopio, X. Marset, G. Guillena, M. L. Di Gioia and D. J. Ramón, Adv. Synth. Catal., 2023, 365, 1–8 CrossRef.
- (a) S. Trombino, C. Siciliano, D. Procopio, F. Curcio, A. S. Laganà, M. L. Di Gioia and R. Cassano, Pharmaceutics, 2022, 14, 333 CrossRef CAS PubMed; (b) D. Procopio, C. Siciliano, R. De Rose, S. Trombino, R. Cassano and M. L. Di Gioia, Catalysts, 2022, 12, 1480 CrossRef CAS; (c) M. L. Di Gioia, A. R. C. Duarte and M. B. Gawande, Front. Chem., 2023, 11, 1258718 CrossRef PubMed.
- (a) C. Florindo, F. S. Olivera, L. P. N. Rebelo, A. M. Fernandes and I. M. Marrucho, ACS Sustainable Chem. Eng., 2014, 2(10), 2416–2425 CrossRef CAS; (b) M. Zhang, T. Tian, H. Han, K. Wu, B. Wang, Y. Liu, Y. Zhu and H. Lu, J. Clean. Prod., 2022, 342, 131028 CrossRef.
- C. Jimenez-Gonzalez, C. S. Ponder, Q. B. Broxterman and J. B. Manleyres, Org. Process Res. Dev., 2011, 15, 912–917 CrossRef CAS.
- (a) J. R. Dunetz, J. Magano and G. A. Weisenburger, Org. Process Res. Dev., 2016, 20, 140–177 CrossRef CAS; (b) E. Valeur and M. Badley, Chem. Rev., 2011, 111, 6557–6602 CrossRef PubMed.
- S. Sharma, G. Kaur and S. Handa, Org. Process Res. Dev., 2021, 25, 1960–1965 CrossRef CAS.
- A. I. Llobet, M. N. Kenworthy, S. Mukherjee, M. E. Kopach, K. Wegner, F. Gallou, A. G. Smith and F. Roschangar, J. Org. Chem., 2019, 84(8), 4615–4628 CrossRef PubMed.
- T. I. Al-Warhi, H. M. A. Al-Hazimi and A. El-Faham, J. Saudi Chem. Soc., 2012, 16, 97–116 CrossRef CAS.
- Related web page: https://echa.europa.eu/it/substance-information/-/substanceinfo/100.018.173.
- M. Saquib, M. F. Khan, J. Singh, B. K. Priti, P. Kumar and M. K. Hussain, Sustainable Chem. Pharm., 2022, 30, 100860 CrossRef CAS.
- R. A. Sheldon, ACS Sustainable Chem. Eng., 2018, 6, 32–48 CrossRef CAS.
- M. T. Sabatini, L. T. Boulton, H. F. Sneddon and T. D. Sheppard, Nat. Catal., 2019, 2, 10–17 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ob01673k |
| This journal is © The Royal Society of Chemistry 2024 |