
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Alcohol synthesis based on the SN2 reactions of alkyl halides with the squarate dianion†
Kazuto
Sato‡
a,
Tomoyuki
Fujita‡
a,
Takashi
Takeuchi
a,
Takahiro
Suzuki
 b,
Kazutada
Ikeuchi§
b and
Keiji
Tanino
*b
b,
Kazutada
Ikeuchi§
b and
Keiji
Tanino
*b
aGraduate School of Chemical Sciences and Engineering, Hokkaido University, Sapporo 060-0810, Japan
bDepartment of Chemistry, Faculty of Science, Hokkaido University, Sapporo 060-0810, Japan. E-mail: ktanino@sci.hokudai.ac.jp
First published on 11th January 2024
Abstract
A convenient method has been developed for transforming alkyl halides into the corresponding alcohols via an SN2 reaction. Treatment of an alkyl halide with the squarate dianion at high temperature produces mono-alkyl squarate, and a one-pot basic hydrolysis of the intermediate affords the alcohol in good yield.
Synthesis of alcohols from their corresponding alkyl halides is one of the most important and fundamental transformations in organic chemistry.1 Spontaneous hydrolysis in wet polar solvents via an SN1 mechanism is applicable only to highly reactive halides, such as tertiary alkyl halides. Hence, the conversion of primary and secondary alkyl halides into alcohols requires some reactions with an oxygen source.2 The SN2 reaction of an alkyl halide with the hydroxide ion may provide a straightforward method for this purpose, but the “hard” property of the hydroxide ion tends to induce undesirable β-elimination reactions yielding alkenes.3 A dialkyl ether, derived from the desired alcohol and remaining alkyl halide under basic conditions, can also be detected as a side product. Therefore, the use of oxygen nucleophiles with a “soft” property is essential for achieving the SN2 reaction of an alkyl halide in high yields (Scheme 1).
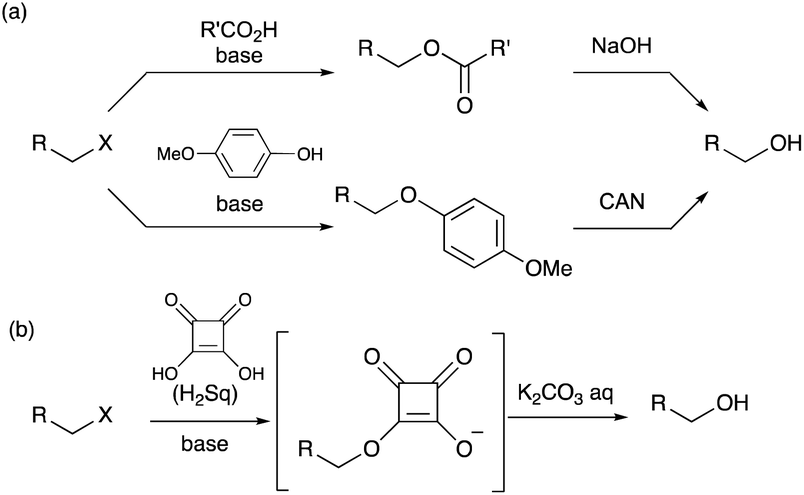 | ||
| Scheme 1 (a) Typical conversions of alkyl halides into the corresponding alcohols through SN2 reactions with oxygen nucleophiles. (b) Conversion mechanism proposed in this work. | ||
Superoxide ions4 are considered as representative nucleophiles for converting alkyl halides into alcohols,5 but the use of hazardous reagents is a serious drawback, especially in large-scale experiments. In contrast, carboxylate anions undergo SN2 reactions with alkyl halides to afford the corresponding esters, which can easily be converted into alcohols by saponification.6 The delocalization of the anionic charge in a carboxylate anion makes it a “soft” nucleophile. However, the carboxylate ions barely react with the sterically hindered alkyl halides. Similarly, phenolate ions function as soft oxygen nucleophiles in the SN2 reactions with alkyl halides. p-Methoxyphenolate ions are widely used as reagents because alkyl p-methoxyphenyl ethers can be converted into the corresponding alcohols upon treatment with cerium(VI) ammonium nitrate (CAN) and water.7 However, this transformation requires a two-step protocol to obtain alcohols from alkyl halides. Herein, we report that squarate dianions conveniently behave as oxygen nucleophiles that can transform primary and secondary alkyl halides. The dianion species exhibited high reactivity in SN2 reactions, and the resulting squarate monoesters readily underwent hydrolysis in one pot (Scheme 1b).
Squaric acid (H2Sq) is a strong acid (pKa1 = 0.52 and pKa2 = 3.48), comparable to trifluoroacetic acid, because of the high stability of the corresponding squarate anion.8,9 The anionic charge of the squarate is completely delocalized across the four-membered ring, and the aromatic character of the conjugated π-system contributes to the exceptionally high stability of the anion.
To elucidate the utility of the soft and non-basic oxygen nucleophile, we explored the SN2 reactions of the squarate dianion (Sq2−) with primary alkyl bromide 1a under various conditions. The screening of suitable bases and solvents for the reaction revealed that the combination of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) and DMF was optimal for the in situ generation of Sq2− from H2Sq (see ESI-1 for details†). Bromide 1a smoothly underwent the substitution reaction upon heating with H2Sq (1.5 equiv.) and DBU (3.0 equiv.) at 60 °C in DMF, and the reaction mixture containing monoalkyl squarate 2 was treated with a K2CO3 aqueous solution to obtain the desired alcohol 3a in 89% yield (Table 1, entry 1). When i-Pr2NEt was used as the base, 3a was obtained but with a dramatically reduced yield of 27% (entry 2). Investigation of the solvent effects showed that the use of MeCN resulted in good yields of 3a (entry 3), but THF led to a decreased reaction conversion to 3a (43% yield) and a recovery of 1a in 48% yield (entry 4). The absence of H2Sq prevented the formation of 3, leading to a preference for the elimination of 1a (entry 5), thereby demonstrating the role of 2 as the reaction intermediate in our system. Further examination involved changing the amount of H2Sq from 1.5 equivalents to 0.5 equivalents because H2Sq can theoretically react with two equivalents of 1a. However, this reaction resulted in the recovery of 1a in 13% yield and generation of 3a in 56% yield (entry 6), suggesting that the reactivity of 2 was lower than that of Sq2−. The decrease in the amounts of DBU to 2.2 equivalents also caused the low conversion of 1a (entry 7). Although the reaction at rt required a longer time than that at 60 °C, the yield of 3a remained high (entry 8). The use of a salt prepared using H2Sq and 2.0 equivalents of DBU led to a dramatic decrease in the yield of 3a to 30% NMR yield (entry 9). The bis-tetra-n-propylammonium salt of H2Sq10 improved the yield of 3a (entry 10), but the yield was lower than that in entry 1.11
|
|
|||
|---|---|---|---|
| Entry | Variation from the standard conditions | Yield of 3ab (%) | Recovery of 1ab (%) |
| a 0.2 mmol scale. b Isolated yield. c 3.6 equivalents of DBU were used. d The reaction time was 1 h. e The reaction temperature ranged from 60 to 150 °C, and the reaction time was 6 h. f NMR yield. | |||
| 1 | None | 89 | — |
| 2 | i-Pr2NEt | 27 | 20 |
| 3c | MeCN | 82 | — |
| 4c | THF | 43 | 48 |
| 5 | Absence of H2Sq | ND | Trace |
| 6 | 0.5 equiv. of H2Sq | 56 | 13 |
| 7 | 2.2 equiv. of DBU | 60 | 24 |
| 8 | rt, 6 h | 87 | — |
| 9d |
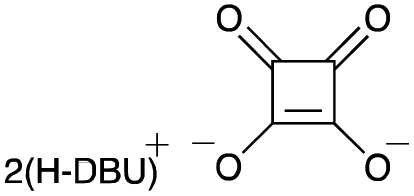
|
30f | — |
| 10e |
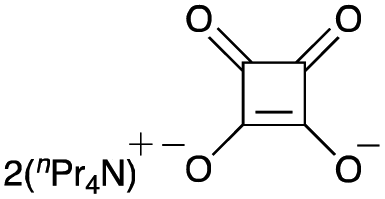
|
39 | — |
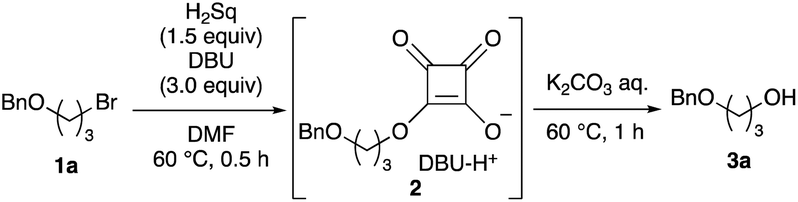
We attempted to isolate mono-alkyl squarate 2; however, its isolation was difficult owing to its high polarity. In contrast, quenching the reaction with an excess amount of methyl iodide produced methyl alkyl ester 4, which was isolated in 73% yield (Scheme 2). Hydrolysis of 4 also occurred when using an aqueous K2CO3 solution, but the yield of 3a was lower than that for the conversion of 1a into 3a (entry 1 in Table 1). This result suggests that the reactivity of the dialkyl squaric ester under basic hydrolysis conditions is low, and the oxy-anion of 2 might accelerate the opening of the squarate moiety under the reaction conditions.
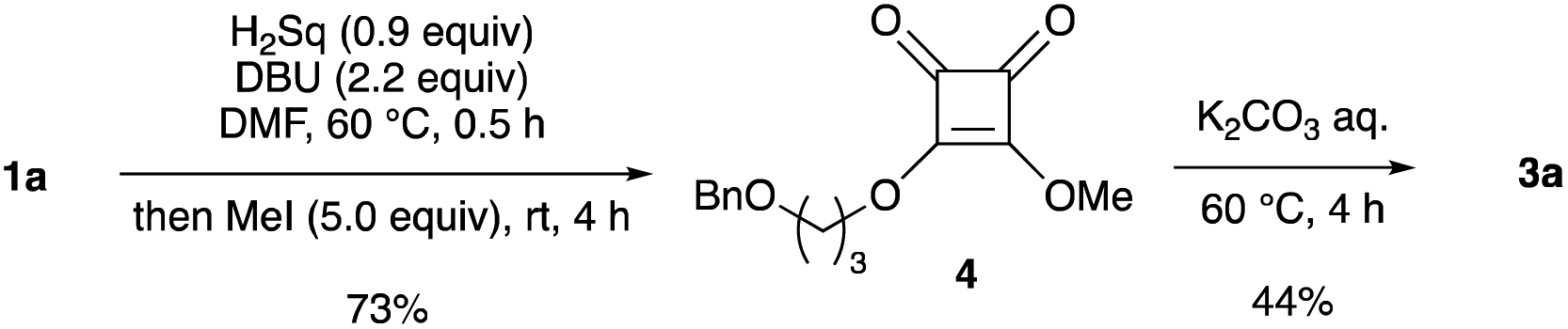 | ||
| Scheme 2 Trapping of monoalkyl squarate 2 with iodomethane and trial hydrolysis of dialkyl squarate 4. | ||
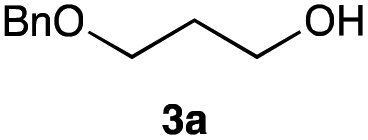
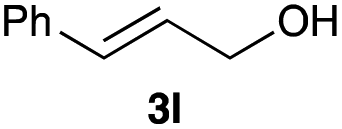
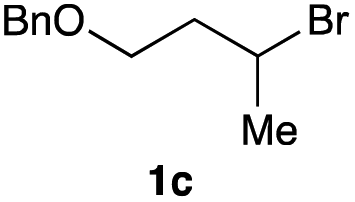
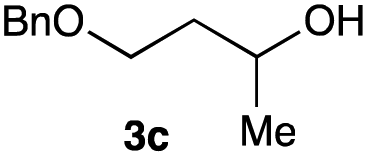
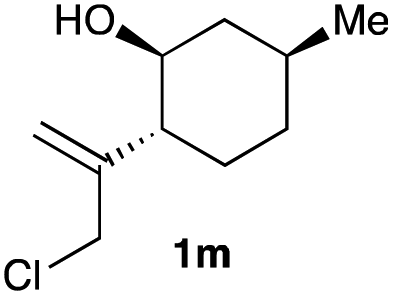
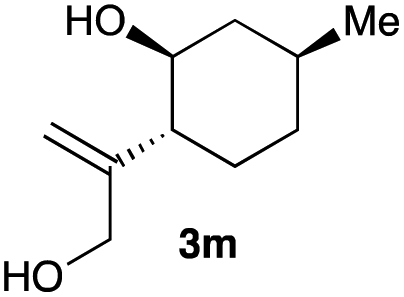
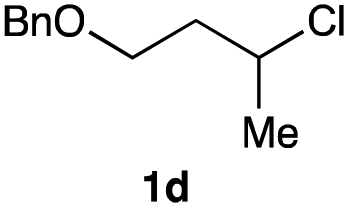
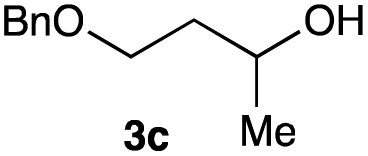
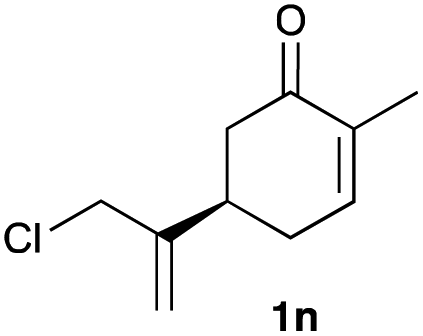
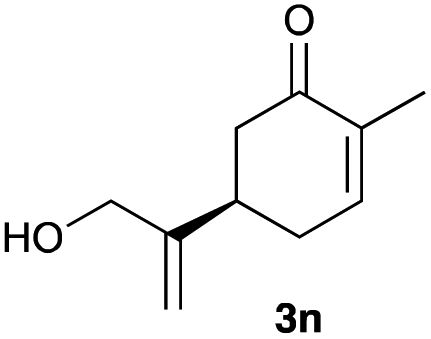
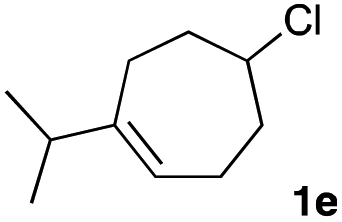
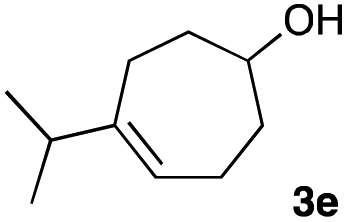
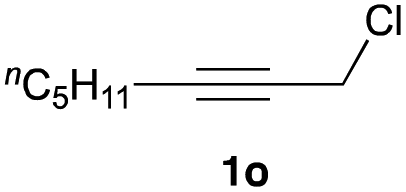
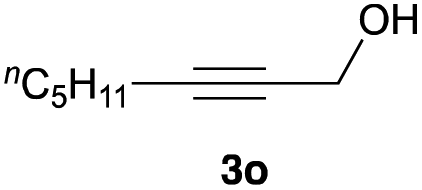
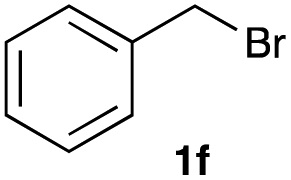
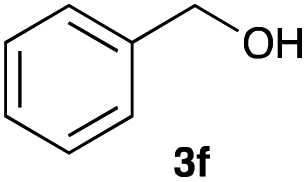
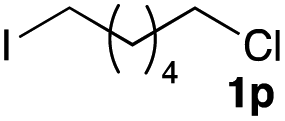
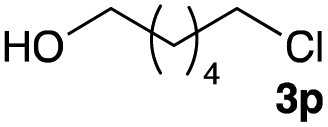
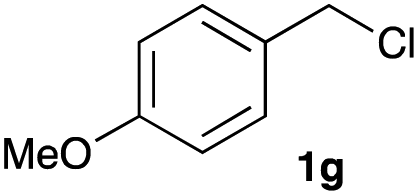
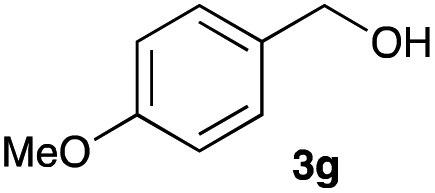
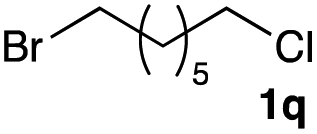
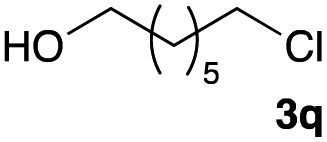
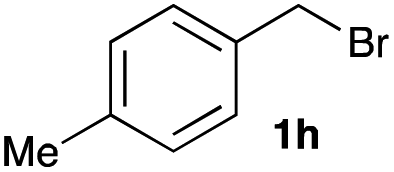
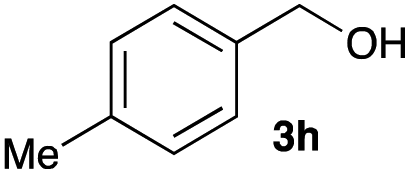
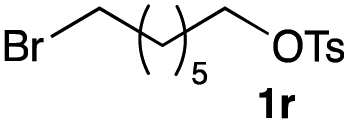
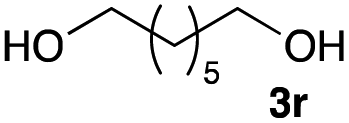
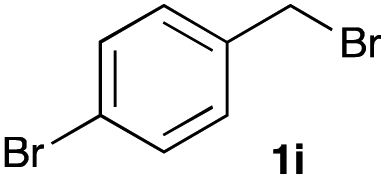
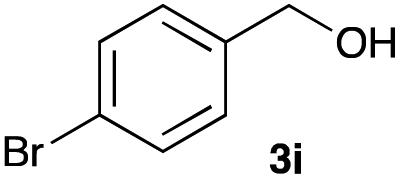
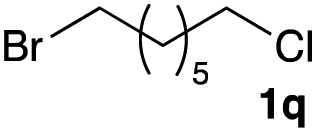
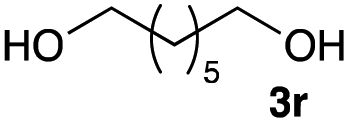
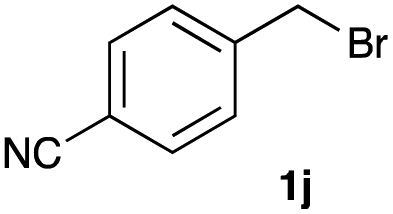
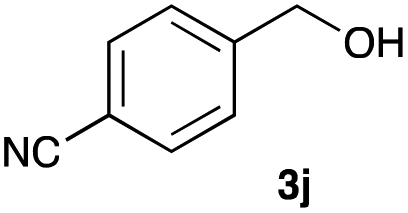
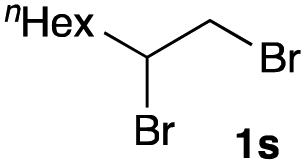
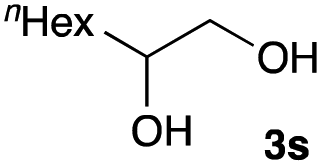
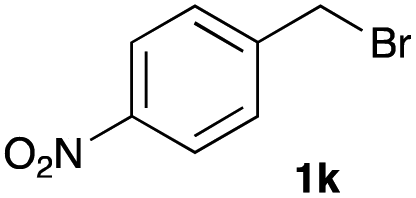
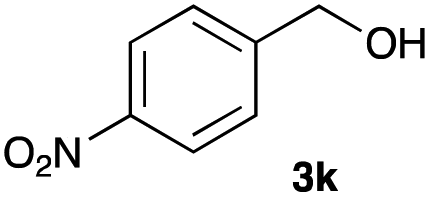
The optimized reaction conditions were applied to the transformation of several alkyl halides into their corresponding alcohols (Table 2). While a prolonged reaction time at a higher temperature was required to achieve the complete consumption of primary chloride 1b, the desired alcohol 3a was obtained in 91% yield after a one-pot hydrolysis (entry 1). Secondary halides 1c and 1d were transformed in a similar manner, giving rise to alcohol 3c in good yields (entries 2 and 3). Note that the reaction of 1c with potassium acetate in DMF at 90 °C did not yield 3c and 1c was recovered in 84% NMR yield. Although the Nozaki group reported that the direct conversion of seven-membered chloride 1e into alcohol 3e failed under various conditions,12 our method facilitated this conversion when the reaction temperature was elevated to 120 °C, affording 3e in 64% yield (entry 4). These results demonstrate the usefulness of squarate dianions as oxygen-incorporating nucleophiles in SN2 reactions. The use of benzyl halides (1g–k) enabled the formation of the corresponding alcohols (3g–k) without the influence of the electron-donating or electron-withdrawing groups (entries 5–10). Cinnamyl bromide 1l exhibited good reactivity in the SN2 reaction of Sq2−, and cinnamyl alcohol (3l) was obtained in 57% yield (entry 11). Additionally, allylic chlorides 1m and 1n, derived from isopulegol13 and carvone,14 respectively, also efficiently induced the desired reactions, producing diol 3m and ketoalcohol 3n in 79% and 71% yields, respectively (entries 12 and 13). Propargyl chloride 1o also afforded the corresponding alcohol 3o in 86% yield (entry 14). Next, Sq2−-mediated chemoselective reactions were examined using acyclic compounds 1p–r bearing two different halides/pseudohalides. The reaction of 6-iodo-1-chlorohexane (1p) with Sq2− at rt occurred at the alkyl iodide moiety chemoselectively, which afforded 3p in 87% yield (entry 15). The subjection of 7-bromo-1-chloroheptane 1q to similar reaction conditions also induced the chemoselective substitution with the Br group, providing 3q in high yield (entry 16). In contrast, the adoption of 7-bromo-1-heptyl tosylate (1r) to the chemoselective reaction was difficult because the eliminated bromo ion easily underwent substitution with the tosyloxy group. On the other hand, the coincident conversions of the bromo and chloro/tosyloxy groups in 1r/1q proceeded smoothly using 3.0 equivalents of Sq2− at 60–80 °C affording the same product, diol 3r, in 83% yield from 1r and in 85% yield from 1q, respectively (entries 17 and 18). Furthermore, the simultaneous conversion of the 1,2-dibromide moiety of 1s15 was achieved via the exposure of 2.4 equivalents of H2Sq and DBU at 120 °C, which afforded 3s in 59% yield after a one-pot hydrolysis (entry 19). The E2 reaction of 1s competed when the proportion of DBU exceeded that of H2Sq; thus, we used the squarate monoanion, generated from the same equivalents of H2Sq and DBU, as a nucleophile.
| Entry | Substrate | Temp. (°C) | Product | Yieldb (%) | Entry | Substrate | Temp. (°C) | Product | Yieldb (%) |
|---|---|---|---|---|---|---|---|---|---|
| time (h) | time (h) | ||||||||
| a The reaction was generally conducted on a 0.2 mmol scale. b Isolated yield. c 0.1 mmol scale. d 3.0 equivalents of H2Sq and 6.0 equivalents of DBU were used and one-pot hydrolysis was conducted at 100 °C for 2.5 h. e 2.4 equivalents of H2Sq and 2.4 equivalents of DBU were used and one-pot hydrolysis was conducted at 100 °C for 1 h. | |||||||||
| 1 |
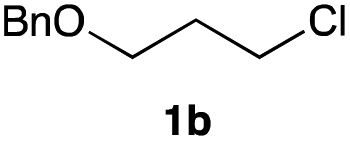
|
80 |
|
91 | 11 |
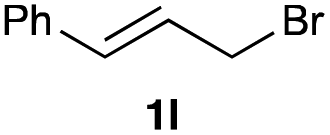
|
60 |
|
57 |
| 2 | 0.7 | ||||||||
| 2 |
|
80 |
|
76 | 12 |
|
80 |
|
79 |
| 2 | 2 | ||||||||
| 3 |
|
90 |
|
70 | 13 |
|
80 |
|
71 |
| 3 | 2 | ||||||||
| 4c |
|
120 |
|
64 | 14 |
|
80 |
|
86 |
| 6 | 1 | ||||||||
| 5 |
|
60 |
|
71 | 15 |
|
rt |
|
87 |
| 0.5 | 2 | ||||||||
| 6 |
|
60 |
|
96 | 16 |
|
rt |
|
73 |
| 0.5 | 6 | ||||||||
| 7 |
|
60 |
|
69 | 17d |
|
60 |
|
83 |
| 0.5 | 1 | ||||||||
| 8 |
|
60 |
|
74 | 18d |
|
80 |
|
85 |
| 0.5 | 2 | ||||||||
| 9 |
|
60 |
|
87 | 19e |
|
120 |
|
59 |
| 0.5 | 2 | ||||||||
| 10 |
|
600.5 |
|
66 | |||||

|
|||||||||
To demonstrate another application of squarates, we explored the synthesis of highly functionalized alcohol, namely glycerol (Scheme 3). In the presence of a primary alcohol, triisopropylsilyl ether 516 was reacted with N-iodosuccinimide (NIS) to afford iodoether 6 in good yields. The use of methanol, benzyl alcohol (BnOH), and propargyl alcohol resulted in the formation of ethers 6a, 6b, and 6c, respectively. Upon heating with Sq2− generated in situ in DMF at 120 °C for 4 h, followed by a one-pot hydrolysis, alcohols 7a, 7b, and 7c were obtained in high yields (Scheme 3a). On the other hand, the bromoetherification of allyl silyl ether 817 using 1,3-dibromo-5,5-dimethylhydantoin and BnOH provided a 1.8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 inseparable mixture of primary bromide 9 and secondary bromide 10. This mixture was subjected to 1.5 equivalents of Sq2− at 100 °C, leading to the desired SN2 reactions in both compounds. Subsequent hydrolysis resulted in the formation of primary alcohol 11 and secondary alcohol 12 in 47% and 15% yields, respectively. A decrease in the reaction temperature to 60 °C induced a selective reaction of 9, which afforded 11 in 48% yield and 10 was recovered in 30% yield (Scheme 3b). The alcohols 7, 11 and 12 are regarded as analogues of glycerol possessing two different protective groups; therefore, they are expected to be versatile building blocks for the synthesis of various complex lipids.
:1 inseparable mixture of primary bromide 9 and secondary bromide 10. This mixture was subjected to 1.5 equivalents of Sq2− at 100 °C, leading to the desired SN2 reactions in both compounds. Subsequent hydrolysis resulted in the formation of primary alcohol 11 and secondary alcohol 12 in 47% and 15% yields, respectively. A decrease in the reaction temperature to 60 °C induced a selective reaction of 9, which afforded 11 in 48% yield and 10 was recovered in 30% yield (Scheme 3b). The alcohols 7, 11 and 12 are regarded as analogues of glycerol possessing two different protective groups; therefore, they are expected to be versatile building blocks for the synthesis of various complex lipids.
 | ||
| Scheme 3 (a) Synthesis of 2-methylglycerol derivatives. (b) Synthesis of doubly protected glycerols. (c) Preparation of an azidoalcohol. | ||
We further investigated the synthesis of azidoalcohol 13, a precursor for the synthesis of sphingosine (Scheme 3c).18 The primary bromo group in dibromide 14, prepared from 8via bromination, underwent a chemoselective reaction upon treatment with 1.5 equivalents of H2Sq and 2.9 equivalents of DBU. Although this reaction proceeded smoothly to produce 15,19 the subsequent one-pot hydrolysis of 15 using aqueous K2CO3 solution generated bromohydrin 16 and epoxide 17 in a 1:1 ratio. In contrast, the treatment of the reaction mixture containing 15 with excess hydrazine monohydrate suppressed the formation of 17, affording 16 in 59% yield. The typical conditions for the replacement of the bromo group with an azido group allowed the conversion of 16 into 13 in 87% yield. The conventional protocol for synthesizing azidoalcohol 13 from olefin 8 involves 5 steps: dihydroxylation, selective protection of the primary alcohol, sulfonylation of the secondary alcohol, azidization, and removal of the protecting group. Our synthetic method of 13 from 8 can occur in 3 steps, indicating the utility of Sq2− as an oxygen nucleophile.
Finally, we demonstrated the Walden inversion of secondary alkyl halides/pseudohalides with a stereogenic center (Scheme 4). The reaction of tosylate 18, which was derived from epiandrosterone, with Sq2− proceeded at 100 °C to provide α-alcohol 19 in 70% yield. Although the application of bicyclic compound 2020 required an excess amount of Sq2− and a higher reaction temperature than that required for 18, β-alcohol 21 was obtained in 50% yield.
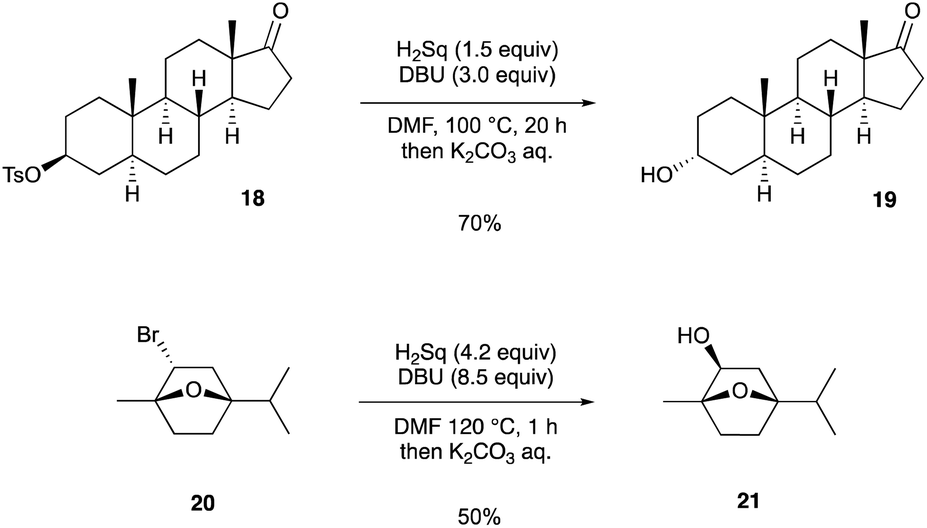 | ||
| Scheme 4 Inversion of the stereochemistry of secondary alkyl bromides and tosylates. | ||
Conclusions
In conclusion, we developed a convenient method for transforming alkyl halides into the corresponding alcohols through an SN2 reaction using Sq2−. The reaction of primary- and secondary-alkyl halides with the dianion species proceeded in DMF at 60–120 °C, and the resulting mono-alkyl squarate was readily hydrolyzed by treatment with an aqueous K2CO3 solution. Examples for the inversion of a stereogenic center were also described. Furthermore, methods for synthesizing versatile building blocks of complex lipids have also been developed. We are further exploring the utility of Sq2−, a potent and safe oxygen nucleophile, in synthetic organic chemistry.Data availability
All experimental procedures and spectral data are available in the ESI.†Author contributions
K. T. conceived the research theme and designed the experiments. K. S., T. F., and T. T. performed the experiments and analyzed the data. K. I. and K. T. commanded this work and wrote the manuscript. T. S. assisted in writing and editing the manuscript. All authors contributed to the discussions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI (grant numbers JP20K05485, JP21H01923, and JP21K14616) and the Photo-Excitonix Project of Hokkaido University.References
- (a) T. A. Hamlin, M. Swart and F. M. Bickelhaupt, ChemPhysChem, 2018, 19, 1315 CrossRef CAS; (b) V. Lankea and I. Marek, Chem. Sci., 2020, 11, 9378 RSC.
- (a) E. Nakamura, T. Inubushi, S. Aoki and D. Machii, J. Am. Chem. Soc., 1991, 113, 8980 CrossRef CAS; (b) M. Sawamura, Y. Kawaguchi, K. Sato and E. Nakamura, Chem. Lett., 1997, 26, 705 CrossRef; (c) A. Abad, C. Agulló, A. C. Cuñat and I. Navarro, Synlett, 2005, 3355 CAS; (d) B. M. Chougala, S. Samundeeswari, M. Holiyachi and L. A. Shastri, ChemistrySelect, 2017, 2, 1290 CrossRef CAS; (e) Y.-M. Cai, Y.-T. Xu, X. Zhang, W.-X. Gao, X.-B. Huang, Y.-B. Zhou, M.-C. Liu and H.-Y. Wu, Org. Lett., 2019, 21, 8479 CrossRef CAS PubMed; (f) H. Liu, J. Liu, X. Cheng, X. Jia, L. Yu and Q. Xu, ChemSusChem, 2019, 12, 2994 CrossRef CAS.
- T. Hansen, J. C. Roozee, F. M. Bickelhaupt and T. A. Hamlin, J. Org. Chem., 2022, 87, 1805 CrossRef CAS PubMed.
- (a) M. Hayyan, M. A. Hashim and I. M. AlNashef, Chem. Rev., 2016, 116, 3029 CrossRef CAS PubMed; (b) D. T. Sawyer and J. S. Valentine, Acc. Chem. Res., 1981, 14, 393 CrossRef CAS.
- (a) R. A. Johnson and E. G. Nidy, J. Org. Chem., 1975, 40, 1680 CrossRef CAS; (b) M. A. Brimble, G. M. Williams, R. Baker and M. James, Tetrahedron Lett., 1990, 31, 3043 CrossRef CAS; (c) A. Matsuzawa, J. Shiraiwa, A. Kasamatsu and K. Sugita, Org. Lett., 2018, 20, 1031 CrossRef CAS PubMed.
- (a) S. Winstein and R. E. Buckles, J. Am. Chem. Soc., 1942, 64, 2780 CrossRef CAS; (b) R. C. Larock, J. Org. Chem., 1974, 39, 3721 CrossRef CAS; (c) N. Ono, T. Yamada, T. Saito, K. Tanaka and A. Kaji, Bull. Chem. Soc. Jpn., 1978, 51, 2401 CrossRef CAS; (d) S. Yamamoto, H. Itani, H. Takahashi, T. Tsuji and W. Nagata, Tetrahedron Lett., 1984, 25, 4545 CrossRef CAS; (e) H. A. Zahalka and Y. Sasson, Synthesis, 1986, 763 CrossRef CAS; (f) J. Alexander, M. L. Renyer and H. Veerapanane, Synth. Commun., 1995, 25, 3875 CrossRef CAS.
- (a) Y. Masaki, K. Yoshizawa and A. Itoh, Tetrahedron Lett., 1996, 37, 9321 CrossRef CAS; (b) L. F. Tietze and J. Görlitzer, Synthesis, 1998, 873 CrossRef CAS; (c) L. He, H.-S. Byun, J. Smit, J. Wilschut and R. Bittman, J. Am. Chem. Soc., 1999, 121, 3897 CrossRef CAS; (d) T. Toma, Y. Kita and T. Fukuyama, J. Am. Chem. Soc., 2010, 132, 10233 CrossRef CAS PubMed; (e) W. Szeja, G. Grynkiewicz, T. Bieg, P. Swierk, A. Byczek, K. Papaj, R. Kitel and A. Rusin, Molecules, 2014, 19, 7072 CrossRef PubMed.
- (a) L. M. Schwartz and L. O. Howard, J. Phys. Chem., 1970, 74, 4374 CrossRef CAS; (b) H. Schmidt, Synthesis, 1980, 961 CrossRef; (c) G. Seltz and P. Imming, Chem. Rev., 1992, 92, 1227 CrossRef.
- Analogues of squaric acid or squaramide are widely used in various fields. For selected reviews see: (a) M. Ohno, Y. Yamamoto and S. Eguchi, Synlett, 1998, 1167 CrossRef CAS; (b) J. Alemán, A. Parra, H. Jiang and K. A. Jørgensen, Chem. – Eur. J., 2011, 17, 6890 CrossRef PubMed; (c) L. A. Marchetti, L. K. Kumawat, N. Mao, J. C. Stephens and R. B. P. Elmes, Chem, 2019, 5, 1398 CrossRef CAS; (d) K. A. Agnew-Francis and C. M. Williams, Chem. Rev., 2020, 120, 11616 CrossRef CAS PubMed; (e) J. Chasák, V. Šlachtová, M. Urban and L. Brulíková, Eur. J. Med. Chem., 2021, 209, 112872 CrossRef; (f) T. Grus, H. Lahnif, B. Klasen, E.-S. Moon, L. Greifenstein and F. Roesch, Bioconjugate Chem., 2021, 32, 1223 CrossRef CAS.
- M. G. Topuzova, S. V. Kotov and T. M. Kolev, Appl. Catal., A, 2005, 281, 157 CrossRef CAS.
- We also used a di-sodium salt of H2Sq ( Q. Zhao, J. Wang, Y. Lu, Y. Li, G. Liang and J. Chen, Angew. Chem., Int. Ed., 2016, 55, 1252 CrossRef PubMed (8)) as the reagent, but, the reaction did not occur and 1 was recovered in 80% NMR yield.
- A. Itoh, T. Saito, K. Oshima and H. Nozaki, Bull. Chem. Soc. Jpn., 1981, 54, 1456 CrossRef CAS.
- (a) S. G. Hegde, M. K. Vogel, J. Saddler, T. Hrinyo, N. Rockwell, R. Haynes, M. Oliver and J. Wolinsky, Tetrahedron Lett., 1980, 21, 441 CrossRef CAS; (b) S. G. Hegde, D. Beckwith, R. Doti and J. Wolinsky, J. Org. Chem., 1985, 50, 894 CrossRef CAS.
- M. Xuan, I. Paterson and S. M. Dalby, Org. Lett., 2012, 14, 5492 CrossRef CAS PubMed.
- I. Ryu, H. Matsubara, S. Yasuda, H. Nakamura and D. P. Curran, J. Am. Chem. Soc., 2002, 124, 12946 CrossRef CAS PubMed.
- K. Zhao and R. R. Knowles, J. Am. Chem. Soc., 2022, 144, 137 CrossRef CAS.
- N. Saygili, R. J. Brown, P. Day, R. Hoelzl, P. Kathirgamanathan, E. R. Mageean, T. Ozturk, M. Pilkington, M. M. B. Qayyum, S. S. Turner, L. Vorwerg and J. D. Wallis, Tetrahedron, 2001, 57, 5015 CrossRef CAS.
- (a) P. M. Koskinen and A. M. P. Koskinen, Synthesis, 1998, 1075 CrossRef CAS; (b) R. Sridhar, B. Srinivas and K. R. Rao, Tetrahedron, 2009, 65, 10701 CrossRef CAS; (c) Y. Gao, X. He, F. Ding and Y. Zhang, Synthesis, 2016, 4017 CAS.
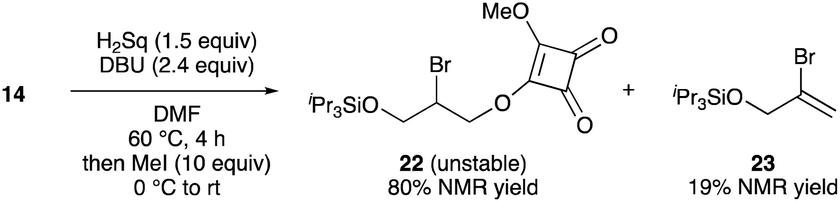
- Reaction of 14 with H2Sq and DBU, followed by the addition of excess amounts of MeI, provided methyl squarate 22 and vinyl bromide 23 in 80% and 19% NMR yields, respectively. Purification of the crude product via silica gel chromatography resulted in the decrease of the isolated yield of 22 (ca. 27% yield) owing to the instability of 22.
. - R. M. Carman and B. N. Venzke, Aust. J. Chem., 1973, 26, 2235 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedure, compound characterization, and copies of NMR data. See DOI: https://doi.org/10.1039/d3ob01507f |
| ‡ These authors contributed equally to this work. |
| § Present address: Graduate School of Pharmaceutical Sciences, Nagoya City University, 467-8603, Japan |
| This journal is © The Royal Society of Chemistry 2024 |