
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stimuli-responsive anion transport utilising caged hydrazone-based anionophores†
Manzoor
Ahmad
,
Martin
Flerin
,
Hui Min
Tay
 ,
Amber L.
Thompson
,
Fernanda
Duarte
* and
Matthew J.
Langton
*
,
Amber L.
Thompson
,
Fernanda
Duarte
* and
Matthew J.
Langton
*
Chemistry Research Laboratory, Mansfield Road, Oxford, OX1 3TA, UK. E-mail: matthew.langton@chem.ox.ac.uk
First published on 28th October 2024
Abstract
Ion transport across biological membranes, facilitated by naturally occurring ion channels and pumps, plays a crucial role in biological processes. Gating is an important aspect of these systems, whereby transport is regulated by a range of external stimuli such as light, ligands and membrane potential. While synthetic ion transport systems, especially those with gating mechanisms, are rare, they have garnered significant attention due to their potential applications in targeted therapeutics as anticancer agents or to treat channelopathies. In this work, we report stimuli-responsive anion transporters based on dynamic hydrogen bonding interactions of hydroxyl-functionalised hydrazone anionophores. Caging of the hydroxyl groups with moities that are responsive to light and H2S locks the hydrazone protons through intramolecular hydrogen bonding, rendering them unavailable for anion binding and transport. Upon decaging with light or H2S, the hydrogen bonding pattern is reversed, rendering the hydrazone protons available for anion binding, and leading to efficient switch-on of ion transport across the lipid bilayer membrane.
I. Introduction
Gating of biological ion transport proteins regulates their activity, and is triggered by a wide range of stimuli, including light, membrane potential, and small molecule ligand binding.1–3 Malfunctioning ion transport proteins cause a variety of diseases, collectively called ‘channelopathies’.4–6 Synthetic transport systems have emerged as an important class of compounds that have potential as chemotherapeutics to treat channelopathies, particularly in the context of chloride anionophores for cystic fibrosis, or via exploiting ion transport-triggered apoptosis for cancer therapy.7–10 Stimuli-responsive systems in particular are of significant interest for spatiotemporally controlled activation in targeted therapeutics. To achieve this, various stimuli including pH, light, enzymes, redox and voltage have been employed.11–21 Reversibly gated systems that commonly exploit photoisomerization of azobenzene22–26 and stilbene27 often exhibit background transport activity because of incomplete photoconversion or suffer from fast thermal relaxation which limits their applicability in biological contexts. In contrast, irreversibly controlled transporters that are triggered by decaging reactions enable very effective off-on control over transport, and overcome the challenges of incomplete activation or deactivation in photo-switchable systems. Examples include the use of photo-responsive cages to block the anion binding sites in anion carriers,28,29 modulate the self-assembly pattern of channel forming monomers,30 or control the mobility of anionophores.31Recently, a new approach has been developed in which dynamic hydrogen bonding interactions were employed to generate responsive ion transport systems. Ren and coworkers reported non-covalently stapled self-assembled H+/Cl− channels utilizing photo-decaging of alkyl-functionalized dihydroxy isophthalamide derivatives.32 We have demonstrated that a similar concept could be utilized for accessing multiple stimuli-responsive moieties, in which light, redox or enzymatic decaging of pro-carriers leads to a intramolecular hydrogen bonding switch, and activation of ion transport, including with dual-caged AND logic activation.33
Here, we demonstrate that efficient off-on activation of transport, with negligible background activity in the off state and high transport activities in the on state, can be achieved using stimuli-triggered decaging of hydrazone based anionophores that operate via an intramolecular hydrogen bond switch. We show that this system can be readily modified, to access transporters efficiently activated using light or H2S as a biologically relevant redox stimulus.
II. Results and discussion
Approach
We envisaged that replacing the amide motif in our previously reported system with an acyl hydrazone anion binding motif would enhance the anion binding as it provides a binding core with multiple hydrazone N–H and imine–N–C–H hydrogen bonding interactions, and enhanced ion transport activity. Accordingly, we designed 4,6-dihydroxyisophthalohydrazone-derived transporters to develop dynamic hydrogen bonding-based responsive anionophores that are activated by either light or H2S (Fig. 1). In this design, dual-caging of the hydroxy groups with ortho-nitrobenzyl (ONB) and azido cages locks the anion binding hydrazone protons through intra-molecular hydrogen bonding, inhibiting anion binding and hence anion transport. Decaging of both hydroxy groups, using light or H2S respectively, reverses the hydrogen bonding pattern such that the hydrazone NH protons are available for anion binding and transport (Fig. 1C). A library of four different transporters 1–4 containing variable aromatic moieties were designed and synthesized, varying the nature of the aromatic groups to tune lipophilicity, and optimise the permeability and transport affinity of these anionophores (Fig. 1B). | ||
| Fig. 1 (A) Schematic representation of stimuli-responsive anion transport. (B) Chemical structure of active transporters 1–4 and (C) caged protransporters. (D) Activation mechanisms with light and H2S. | ||
Synthesis and binding of active transporters 1–4
The synthesis of anion transporters 1–4 was achieved by reacting compound 5 with hydrazine hydrate to form compound 6, which upon coupling with different aromatic aldehydes 7a–7d yielded acylhydrazones 1–4 in excellent yield (Fig. 2A). Full experimental details and characterisation are available in the ESI.† Subsequently, chloride anion binding studies were initially carried out by 1H NMR titration experiments. Titration of 1–4 with tetrabutylammonium chloride (TBACl) in DMSO-d6 led to changes in H1, H2, and H3 protons, revealing their role in chloride binding through hydrazone-N–H1⋯Cl−, imine–C–H2⋯Cl−, and ArC–H3⋯Cl− hydrogen bonding interactions (Fig. 2B, S34, S36, S38, and S40†). The data could be fitted to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding isotherm using Bindfit, to afford association constants values (Ka(1:1)/Cl−) of 168 M−1 ± 4% for 1, 165 M−1 ± 4% for 2, 150 M−1 ± 4% for 3, and 167 M−1 ± 3% for 4, respectively (Fig. 2C, S35, S37, S39, and S41†). Moreover, we also performed the chloride binding analysis of the previously reported amide-based transporter 5′ in DMSO-d6 for means of comparison.33 Importantly, compound 5′ displayed very weak binding in the solution phase compared to the hydrazone derivatives ((Ka(1:1)/Cl−) of 64 M−1 ± 1%) demonstrating that replacing amide with hydrazone motifs significantly enhances the anion binding, which in turn should improve the ion transport activity (Fig. 2E, S42 and S43†).
:1 binding isotherm using Bindfit, to afford association constants values (Ka(1:1)/Cl−) of 168 M−1 ± 4% for 1, 165 M−1 ± 4% for 2, 150 M−1 ± 4% for 3, and 167 M−1 ± 3% for 4, respectively (Fig. 2C, S35, S37, S39, and S41†). Moreover, we also performed the chloride binding analysis of the previously reported amide-based transporter 5′ in DMSO-d6 for means of comparison.33 Importantly, compound 5′ displayed very weak binding in the solution phase compared to the hydrazone derivatives ((Ka(1:1)/Cl−) of 64 M−1 ± 1%) demonstrating that replacing amide with hydrazone motifs significantly enhances the anion binding, which in turn should improve the ion transport activity (Fig. 2E, S42 and S43†).
 | ||
| Fig. 2 (A) Chemical synthesis of active transporters 1–4. (B) The plot of chemical shift (δ) of H1 proton vs. concentration of TBACl added, fitted to 1:1 binding model of BindFit v0.5 for the active transporters 1–4. (C) Binding constant values of transporters 1–4. (D) Chemical structure of amide-based transporter 5′. (E) The plot of chemical shift (δ) of H1 proton vs. concentration of TBACl added, fitted to 1:1 binding model of BindFit v0.5 for hydrazone-based transporters 1 and amide-based transporter 5′. | ||
Crystallographic studies of 1 with TBACl
Further evidence for the binding mode of chloride within the isophthalohydrazone binding cavity was obtained by single crystal X-ray diffraction studies. Crystals of 1 with Cl− were obtained by mixing 1 and tetrabutylammonium chloride (TBACl) in dimethyl sulfoxide in 1:5 ratio. Slow evaporation led to the formation of colourless needle-like crystals. The structure reveals the complementarity of the binding cavity for the chloride anion, which interacts with the receptor molecule through convergent amide–N–H1⋯Cl−, imine–C–H2⋯Cl−, and ArC–H3⋯Cl− hydrogen bonding interactions, in agreement with the change of chemical shift value of these protons observed in 1H NMR binding studies (Fig. 3). Furthermore, both NMR and crystallographic studies revealed a 1:1 receptor:Cl− binding stoichiometry.
 | ||
| Fig. 3 Molecular structure of 1 with chloride anion from single crystal X-ray diffraction studies. The tetrabutylammonium (TBA) cation, selected hydrogen atoms and minor components of disorder are omitted for clarity. | ||
Ion transport studies
Having confirmed that compounds 1–4 were effective chloride receptors in the solution phase, ion transport experiments in large unilamellar vesicles (LUVs) encapsulating the chloride sensitive fluorophore lucigenin were performed. LUVs were prepared by entrapping lucigenin (1 mM) and sodium nitrate (200 mM) buffered at pH of 6.5 using the phosphate buffer (10 mM). Ion transport was monitored by creating a 33.3 mM Cl− gradient across the membrane by adding sodium chloride as a pulse (50 μL, 2.0 M) in the extravesicular buffer. The rate of fluorescence quenching for lucigenin was recorded upon the addition of transporters 1–4 and 5′. These experiments revealed an activity sequence for the transport of 1 > 2 > 3 > 5′> 4, determined by monitoring the chloride transport at a loading of 0.055 mol% with respect to lipid (Fig. 4A). Dose–response curves for each transporter revealed the EC50 values of 0.035 ± 0.002 mol% for 1, 0.042 ± 0.002 mol% for 2, and 0.063 ± 0.002 mol% for 3, respectively (Table 1, Fig. S49–S51†). Hill analysis could not be performed for transporter 4 due to precipitation issues at higher concentration (Fig. S52†). The above activity sequences of 1 > 2 > 3 > 4 is most likely determined by variation in the lipophilicities of the transporters, given the similarities of the chloride binding constants across the series, with most active transporter 1 having optimum clogP value. Notably, the hydrazone transporter 1 showed almost three-fold enhancement in the ion transport activity compared to the amide-based transporter 5′ that was used in the previous studies. This result is likely the outcome of the enhanced anion binding affinity. Hill coefficients of ∼2 is indicative of two molecules of the carrier binding to the anion to facilitate transport.
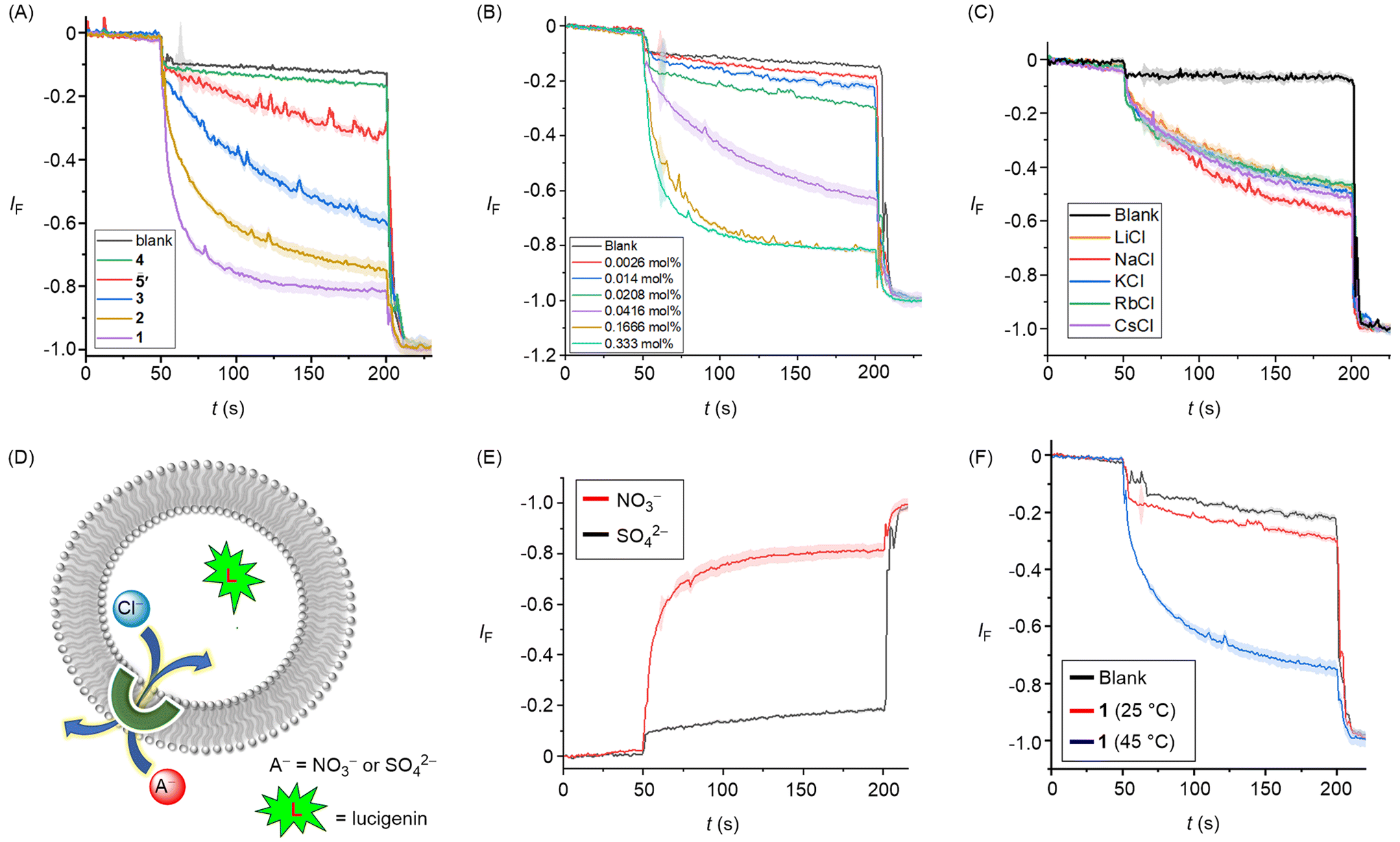 | ||
| Fig. 4 (A) Activity comparison of 1–5 (0.30 mol%) across POPC-LUVs⊃lucigenin. (B) Concentration dependent activity of 1 across POPC-LUVs⊃lucigenin. (C) Cation selectivity of 1 (0.035 mol%) by varying the external cations using different MCl external buffers (M+ = Li+, Na+, K+, Rb+, Cs+). (D) Schematic representation of lucigenin-based chloride efflux using either extravesicular SO42− or NO3− ions. (E) Ion transport activity of 1 (0.3 mol%) in presence of external SO42− and NO3− ions. (F) Ion transport activity of 1 (0.30 mol%) across DPPC-based vesicles at 25 °C and 45 °C temperatures, respectively. | ||
P, chloride binding Ka (M−1), EC50 (mM), and Hill coefficient (n) values
| Comp. | clogP |
K
aa (M−1)/Cl− |
EC50b (mol%) |
n |
|---|---|---|---|---|
|
a Association constants were obtained using the BindFit Program based on the 1:1 binding model for NH1 proton.
b Effective concentration to reach 50% of maximal activity across POPC-LUVs entrapped with 1 mM lucigenin, NaNO3 (200 mM), buffered at pH 6.5 using 10 mM phosphate buffer.
c Hill coefficient. clogP values were calculated using MarvinSketch software.
d EC50 could not be reliably determined due to precipitation at higher concentrations.
|
||||
| 1 | 6.4 | 168 M−1 ± 4% | 0.035 ± 0.0016 | 2.4 ± 0.21 |
| 2 | 4.9 | 165 M−1 ± 4% | 0.042 ± 0.0022 | 2.5 ± 0.34 |
| 3 | 4.6 | 150 M−1 ± 4% | 0.063 ± 0.0022 | 2.5 ± 0.24 |
| 4 | 4.3 | 167 M−1 ± 3% | >0.5d | —d |
| 5′ | 6.0 | 64 M−1 ± 1% | 0.092 ± 0.0012 | 1.8 ± 0.02 |
Mechanistically, chloride transport in the above studies could in principle occur either through M+/Cl− symport, Cl−/NO3− antiport, or H+/Cl− symport pathways. Variation of the cations in the external buffer using different MCl solutions (M+ = Li+, Na+, K+, Rb+, Cs+) did not affect the ion transport activity, ruling out the possible M+/Cl− symport pathway for chloride transport (Fig. 4C). To distinguish between the possible Cl−/NO3− antiport and H+/Cl− symport pathways, we performed a variation on the lucigenin assay, in which lucigenin was encapsulated inside the vesicles in the presence of NaCl (200 mM). The change in lucigenin fluorescence was monitored in the presence of 1 following addition of either Na2SO4 or NaNO3 salt solutions in the external buffer. Significant transport was observed only in the presence of nitrate, which suggests that a Cl−/NO3− antiport pathway dominates in this assay (Fig. 4E). For Cl−/X− antiport, NO3− or SO42− co-transport is requisite for chloride efflux, the latter being too hydrophilic to be readily transported. Conducting analogous experiments in dipalmitoylphosphatidylcholine (DPPC) LUVs provided evidence for a mobile carrier mechanism. Inactivity at 25 °C, and restoration of activity at 45 °C which is above the gel– liquid phase transition temperature for DPPC (Tm = 41 °C), is indicative of a mobile carrier process, rather than transport mediated by self-assembly into an ion channel, that activity of which would be typically expected to be invariant to lipid phase (Fig. 4F).
Synthesis and binding studies of protransporters 1a and 1b
Having demonstrated that compound 1 displayed the maximum ion transport activity, we selected this anionophore as the core scaffold for caging with stimulus-responsive groups (Fig. 5). In order to synthesize the ONB-based protransporter 1a, 4,6-dihydroxyisophthalate 5 was reacted with 1-(bromomethyl)-2-nitrobenzene 8 to form ONB-ester based compound 9. Treatment with hydrazine hydrate under reflux conditions led to the formation of hydrazine intermediate 10 (for further details, see ESI†). The hydrazine derivative 10, upon coupling with 4-trifluoromethyl benzaldehyde 7a, furnished the protransporter 1a in excellent yield. The sulfide-responsive protransporter 1b was synthesized using a different procedure due to incompatibility with the above method. Accordingly, compound 5 was reacted with 1-azido-4-(bromomethyl)benzene 11 to form azido-ester derivative 12, which upon hydrolysis led to the formation of acid derivative 13. This acid derivative was converted to the acid chloride with oxalyl chloride, followed by coupling with ((trifluoromethyl)benzylidene)hydrazine 15, which itself was prepared by reacting 4-trifluoromethylbenzaldehyde 7a with hydrazine hydrate, to afford protransporter 1b in good yield. | ||
| Fig. 5 Chemical synthesis of protransporters 1a and 1b. | ||
Following the synthesis of the pro-transporters, NMR chloride anion binding studies were performed on these caged derivatives. Both 1a and 1b displayed very weak anion binding with association constant values of 7 M−1 ± 1% for 1a and 9 M−1 ± 2% for 1b, respectively (Fig. 6C, S44–S47†). This demonstrates that hydrazone protons are locked through six-membered intramolecular hydrogen bonding which makes them unavailable for anion binding. Single crystal X-ray diffraction studies of 1a confirmed this hypothesis, revealing six-membered intramolecular hydrogen bonding interactions between the hydrazone protons with cage-functionalized attached oxygen atoms (Fig. 6A).
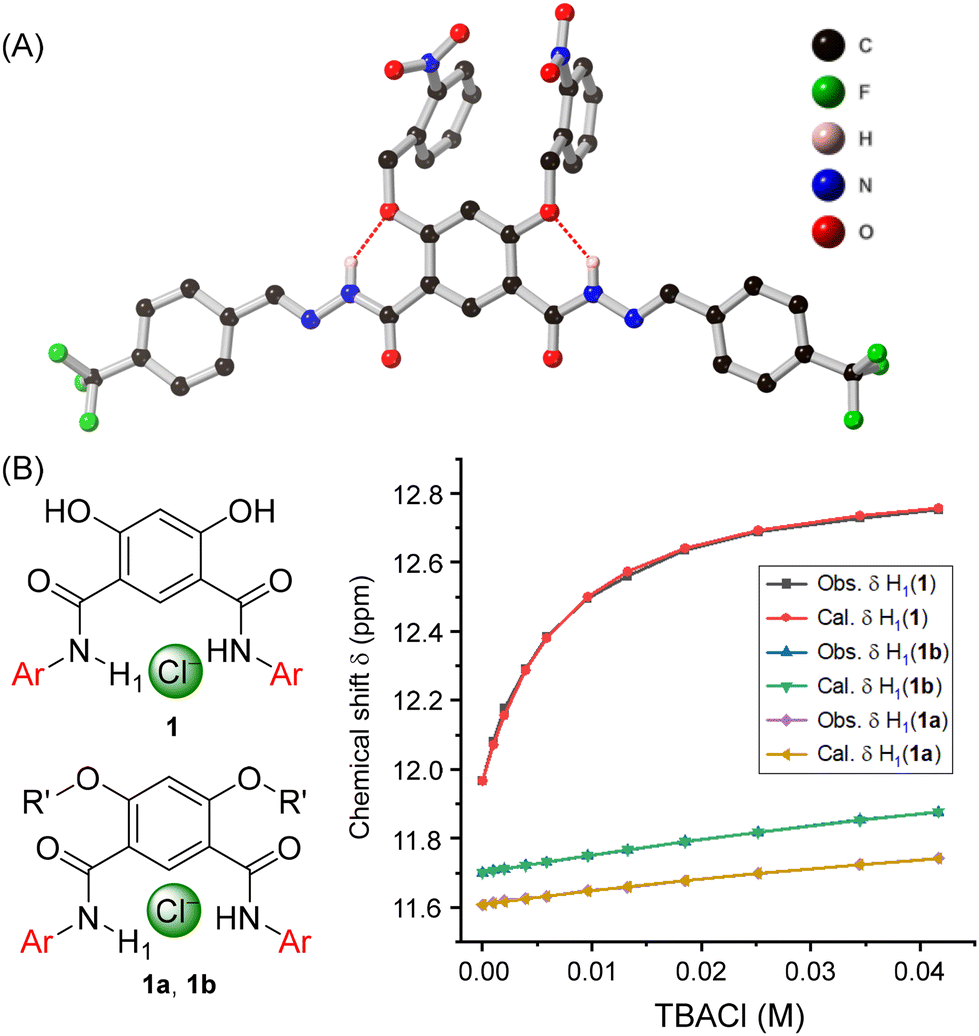 | ||
| Fig. 6 (A) Molecular structure of pro-transporters 1a from single crystal X-ray diffraction studies (selected hydrogen atoms and water of crystallisation omitted for clarity). (B) The plot of chemical shift (δ) of H1 proton vs. concentration of TBACl added, fitted to 1:1 binding model of BindFit v0.5 for active transporters 1 and caged-protransporters 1a and 1b, respectively. | ||
Stimuli-responsive activation studies on 1a and 1b were initially performed in solution phase experiments. For 1a, a solution of the pro-transporter in DMSO-d6 was subjected to photoirradiation using a 405 nm LED and analysed by 1H NMR experiments (Fig. S56†). For 1b, a solution of the pro-transporter in DMF was subjected to sodium hydrosulfide (NaSH, 20 eq.) as the H2S donor and subjected to HPLC analysis (Fig. S57†). In each case, efficient decaging to generate 1 was observed. With evidence of efficient activation of pro-transporters 1a and 1b with light and H2S in hand, the triggered off-on activation of ion transport was subsequently evaluated using lucigenin-containing LUVs in buffered NaNO3. Pro-transporters 1a and 1b were added in an aliquot of DMF (final concentration of 0.30 mol% with respect to lipid), followed by an external chloride pulse. No significant changes were observed in the lucigenin fluorescence, indicating that the pro-transporters are inactive at this concentration, thus achieving an effective off state. Pro-transporter 1a in buffer solution in a cuvette was then subjected to 405 nm light, before adding LUVs and initiating ion transport by addition of a chloride pulse. Photo-activation of 1a (0.30 mol%) using a 1 W 405 nm LED resulted in efficient activation, achieving comparable activity to a sample of 1 after 150 s of irradiation, indicative of near quantitative decaging under these conditions (Fig. 7A). For 1b, a sample in DMF (57 μM, 2.0 mL) was subjected to 10 eq. NaSH for different time intervals (10–40 min), and then aliquots (20 μL) of this solution were added to the cuvette containing the nitrate buffer and LUVs (final concentration of 1b = 0.30 mol%). Addition of NaSH generated comparable transport activity to a sample of 1 (Fig. 7B). These results demonstrate that by utilising acylhydrazones within the hydrogen bonding switchable framework, excellent OFF–ON transport activation profiles can be achieved, whilst also enhancing overall transport activity in comparison to the previously reported amide-based system 5′.
 | ||
| Fig. 7 Ion transport activities across POPC-LUVs⊃lucigenin, by (A) photo-irradiating 1a at 405 nm light using an LED (1 W), and (B) treating 1b with NaSH (10 eq.) for different time intervals (10–40 min). | ||
Computational analysis
To further understand the influence of H-bond (HB) interactions and preorganisation on chloride binding, we studied the best-performing transporter, 1, and its methylated analogue, 1m, using density functional theory (DFT) calculations.34–38An extensive conformational search confirmed that the minimal energy states 1 and 1m feature well defined O–H–O and O–H–N hydrogen bonding, respectively, as evidenced by second order perturbation NBO analysis (E2, Fig. 8A).39 As expected, based on the enhanced hydrogen bond donor ability of the more electronegative oxygen, the O–H–O HB in 1 are significantly stronger (80 kcal mol−1) than the O–H–N HB in 1m (20 kcal mol−1, see Fig. S63† for detailed analysis). Moreover, the shorter C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond in 1, compared to the C–N bond in 1m, results in hydrogen bonds that are 0.1–0.2 Å shorter for the intramolecular O–H–O HB in 1 compared to the O–H–N HB in 1m (Fig. S64–66†).
O bond in 1, compared to the C–N bond in 1m, results in hydrogen bonds that are 0.1–0.2 Å shorter for the intramolecular O–H–O HB in 1 compared to the O–H–N HB in 1m (Fig. S64–66†).
 | ||
| Fig. 8 (A) Summary of DFT optimised structures of protected and deprotected variants of compound 1 with key features highlighted: sums of relevant second order perturbation energies between atoms X–Y–Z, ∑E(2)X–Y–Z, as well as chloride binding energies, of geometries calculated at the CPCM(DMSO)-É·B97X-D3/def2-QZVP//CPCM(DMSO)-É·B97X-D3/def2-SVP level. (B) Distortion–interaction analysis of the binding of Cl− and DMSO to 1 and Cl− to 1m. | ||
Chloride binding is favourable in 1 (−2.5 kcal mol−1, in agreement to that determined experimentally by titration: −3.0 kcal mol−1), but endergonic in 1m (+2.1 kcal mol−1). This can be understood by comparing the change in HB interactions upon binding. While both receptors bind chloride with similar strength (52 vs. 49 kcal mol−1 from NBO analysis), their intramolecular HBs are significantly different. In 1 the O–H–O HB remains almost intact upon chloride binding (78 vs. 80 kcal mol−1), while in 1m the O–H–N HB is completely broken. Moreover, 1m shows a significant conformational change upon chloride binding, evidenced by a heavy-atom Root Mean Square Deviation (RMSD) between unbound and bound states of 3.7 Å, which contrast with the small RMSD (0.47 Å) observed for 1 (Fig. S64–66†). To quantify the energetic cost of this conformational change we performed distortion–interaction analysis, which partitions ΔG into distortion (energy required to distort the geometry for binding) and interaction (accounting for interactions between host and guest upon binding, Fig. 8B).40 This analysis confirms that differences in ΔG arise almost completely from the greater distortion for 1m compared to 1 to bind chloride (5.7 vs. 1.6 kcal mol−1 for 1), while the interaction component is similar (−4.1 and −3.6 kcal mol−1 for 1 and 1m, respectively).
We also explored the role of DMSO solvent coordinating to the transporters, using both DFT and molecular dynamics (MD) simulations. DFT calculations suggest binding of a single DMSO molecule to 1 to be slightly unfavourable but thermally accessible (ΔG = 0.8 kcal mol−1, Table S3, and Fig. S65†). A simple Boltzmann weighting gives this [1·DMSO] complex in a 1:4 ratio to the unbound 1, but both the small difference in energy and the large concentration of the DMSO solvent suggested that this state would be populated under experimental conditions.
Indeed, unbiased MD simulations of 1 in explicit DMSO solvent confirmed a stable coordination of DMSO to the binding site. Moreover, it showed that the intramolecular O–H–O remains stable during the simulation time (Fig. S69†). To quantify the energy associated with chloride binding in explicit DMSO, we performed both Umbrella Sampling MD (US/MD) and Free Energy Perturbation (FEP) MD simulations (Fig. 9 and Table S4, for further details see ESI†).41–43
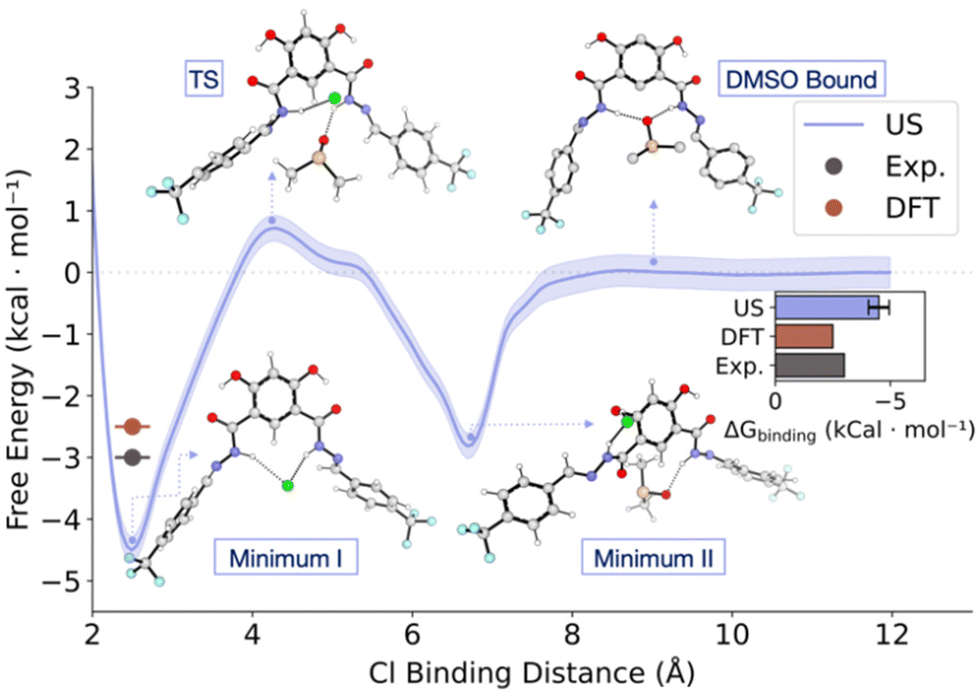 | ||
| Fig. 9 US/MD energy profile of chloride binding in explicit DMSO. Free energy is reported relative to the chloride-unbound state at a distance of 12 Å to the binding site, defined as the centre of mass (CoM) of the central atoms of the binding motif. This coordinate was used to generate 12 windows spaced 1 Å apart, with a 2000 kJ mol−1 nm−2 restraint; each window was sampled for 3 × 100 ns in the NPT ensemble, with the first 10% excluded from the analysis. The shaded areas indicating uncertainty from bootstrapping analysis. The bar plot insert compares the binding energies with DFT and experimental values from titration experiments calculated as −RTln(K). | ||
For US/MD simulations we used the distance between chloride and the centre of mass (CoM) of the central atoms of the binding motif as the pulling coordinate to generate 12 windows spaced 1 Å apart (sampled 3 × 100 ns each using different initial velocities in the NPT ensemble). The resultant free energy profile shows two binding modes of chloride to 1. Minimum I (ΔG = −4.3 ± 0.5 kcal mol−1) corresponds to the structure with chloride bound to both hydrazides of 1, similar to the DFT optimised structures. Minimum II (ΔG = −2.3 ± 0.5 kcal mol−1) was observed during the dissociation of chloride, where it is bound to one of the hydrazide units as well as the neighbouring phenol, which disrupts the intramolecular O–H–O interaction. These minima are connected by a TS where one of the hydrazide units begins to interact with a DMSO molecule (ΔG‡Cl− dissociation = 5.2 ± 0.5 kcal mol−1). Complete dissociation of chloride from 1 leads to re-formation of the O–H–O hydrogen bond and bonding of DMSO by both hydrazides (DMSO bound, ΔG = 0 ± 0.2 kcal mol−1). The computed binding energy (−4.3 ± 0.5 kcal mol−1) is higher to the one computed by DFT, possibly due to hysteresis in umbrella sampling. To compute the binding energy in explicit solvent with a more accurate method, we performed FEP simulations, where we found the binding energy to be within error of the experimental value (−3.1 ± 1.1 kcal mol−1), building confidence in the parameters used in the MD simulations, as well as their results.
III. Conclusion
In conclusion, we have developed stimuli-responsive anion transporters utilizing the dynamic hydrogen bonding interactions of 4,6-dihydroxyisophthalohydrazone anionophores, displaying enhanced activity over previously reported amide-based derivatives. Caging of hydroxyl groups for the most active anionophore 1 with moieties that are responsive to light and H2S locks the hydrazone protons through six-membered intramolecular hydrogen bonding, rendering them unavailable for anion binding and hence for anion transport. This was confirmed by solid state X-ray crystallographic studies and further validated by NMR binding experiments. Decaging with light and H2S reverses the hydrogen bonding pattern to switch on anion binding and transport, with efficient off-on activation profiles. DFT calculations highlighted the energetic origins of this incredibly effective switching between an active and an inactive transporter. By using a model truncated system, and including explicit solvent, agreement with experimental binding energies was also obtained, and the inclusion of explicit solvent was validated using molecular dynamics. These results demonstrate the generality of using intra-molecular hydrogen bond switches for developing highly active, stimuli-responsive anionophores, including demonstrating the enhanced activity of systems employing arylhydrazone motifs compared to analogous amide hydrogen bond donors. We anticipate that this methodology should be amenable to integration of any stimuli-cleavable protecting group, such as those already optimised for drug delivery triggered by biological stimuli, providing a platform for future development of targeted therapeutics.Author contributions
M.J.L. conceived the project and directed the experimental studies. M.A. performed the synthesis and characterization of the compounds, as well as the anion binding and transport studies and stimuli-responsive investigations. M.F. and F.D. performed the theoretical studies. H.T. and A.L.T. performed the crystallographic studies. M.A. and M.F. wrote the first draft of the manuscript, which was edited by all authors.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. A. and M. J. L. acknowledge the Leverhulme Trust (RPG-2020-130) for financial support. M. F. thanks the Centre for Doctoral Training in Synthesis for Biology and Medicine for a studentship, generously supported by GlaxoSmithKline, MSD, Syngenta and Vertex. H. M. T acknowledges the Clarendon Fund and Oxford Australia Scholarships Fund for a post-graduate research scholarship. M. J. L. is a Royal Society University Research Fellow.References
- G. Nagel, D. Ollig, M. Fuhrmann, S. Kateriya, A. M. Musti, E. Bamberg and P. Hegemann, Science, 2002, 296, 2395 CrossRef CAS PubMed.
- W. A. Catterall, Annu. Rev. Biochem., 1995, 64, 493–531 CrossRef CAS PubMed.
- M. Jaiteh, A. Taly and J. Hénin, PLoS One, 2016, 11, e0151934 CrossRef PubMed.
- J. Y. Choi, D. Muallem, K. Kiselyov, M. G. Lee, P. J. Thomas and S. Muallem, Nature, 2001, 410, 94–97 CrossRef CAS.
- S. B. H. Hladky and D. A. Ion, Movement in GramicidinChannels, in Current Topics in Membranes and Transport, ed. F. Bronner, Academic Press, New York, 1984, Vol. 21, pp. 327–372 Search PubMed.
- R. D. Vaughan-Jones, K. W. Spitzer and P. Swietach, J. Mol. Cell. Cardiol., 2009, 46, 318–331 CrossRef CAS.
- V. Soto-Cerrato, P. Manuel-Manresa, E. Hernando, S. Calabuig-Fariñas, A. Martínez-Romero, V. Fernández-Dueñas, K. Sahlholm, T. Knöpfel, M. García-Valverde, A. M. Rodilla, E. Jantus-Lewintre, R. Farràs, F. Ciruela, R. Pérez-Tomás and R. Quesada, J. Am. Chem. Soc., 2015, 137, 15892–15898 CrossRef CAS PubMed.
- S.-K. Ko, S. K. Kim, A. Share, V. M. Lynch, J. Park, W. Namkung, W. Van Rossom, N. Busschaert, P. A. Gale, J. L. Sessler and I. Shin, Nat. Chem., 2014, 6, 885–892 CrossRef CAS.
- N. Busschaert, M. Wenzel, M. E. Light, P. Iglesias-Hernández, R. Pérez-Tomás and P. A. Gale, J. Am. Chem. Soc., 2011, 133, 14136–14148 CrossRef CAS PubMed.
- T. Saha, A. Gautam, A. Mukherjee, M. Lahiri and P. Talukdar, J. Am. Chem. Soc., 2016, 138, 16443–16451 CrossRef CAS PubMed.
- E. N. W. Howe, N. Busschaert, X. Wu, S. N. Berry, J. Ho, M. E. Light, D. D. Czech, H. A. Klein, J. A. Kitchen and P. A. Gale, J. Am. Chem. Soc., 2016, 138, 8301–8308 CrossRef CAS PubMed.
- A. Roy, O. Biswas and P. Talukdar, Chem. Commun., 2017, 53, 3122–3125 RSC.
- Y. R. Choi, G. C. Kim, H.-G. Jeon, J. Park, W. Namkung and K.-S. Jeong, Chem. Commun., 2014, 50, 15305–15308 RSC.
- E. B. Park and K.-S. Jeong, Chem. Commun., 2015, 51, 9197–9200 RSC.
- M. M. Tedesco, B. Ghebremariam, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 1999, 38, 540–543 CrossRef CAS.
- G. A. Woolley, M. K. Kapral and C. M. Deber, FEBS Lett., 1987, 224, 337–342 CrossRef CAS.
- N. Akhtar, N. Pradhan, A. Saha, V. Kumar, O. Biswas, S. Dey, M. Shah, S. Kumar and D. Manna, Chem. Commun., 2019, 55, 8482–8485 RSC.
- A. Docker, T. G. Johnson, H. Kuhn, Z. Zhang and M. J. Langton, J. Am. Chem. Soc., 2023, 145, 2661–2668 CrossRef CAS PubMed.
- X. Wu, J. R. Small, A. Cataldo, A. M. Withecombe, P. Turner and P. A. Gale, Angew. Chem., Int. Ed., 2019, 58, 15142–15147 CrossRef CAS PubMed.
- M. Ahmad, S. Chattopadhayay, D. Mondal, T. Vijayakanth and P. Talukdar, Org. Lett., 2021, 23, 7319–7324 CrossRef CAS PubMed.
- M. Ahmad, D. Mondal, N. J. Roy, T. Vijayakanth and P. Talukdar, ChemPhotoChem, 2022, e202200002 CrossRef CAS.
- M. Ahmad, S. Metya, A. Das and P. Talukdar, Chem. – Eur. J., 2020, 26, 8703–8708 CrossRef CAS PubMed.
- A. Kerckhoffs, Z. Bo, S. E. Penty, F. Duarte and M. J. Langton, Org. Biomol. Chem., 2021, 19, 9058–9067 RSC.
- A. Kerckhoffs and M. J. Langton, Chem. Sci., 2020, 11, 6325–6331 RSC.
- T. G. Johnson, A. Sadeghi-Kelishadi and M. J. Langton, J. Am. Chem. Soc., 2022, 144, 10455–10461 CrossRef CAS PubMed.
- C. Wang, S. Wang, H. Yang, Y. Xiang, X. Wang, C. Bao, L. Zhu, H. Tian and D. H. Qu, Angew. Chem., Int. Ed., 2021, 60, 14836–14840 CrossRef CAS PubMed.
- S. J. Wezenberg, L.-J. Chen, J. E. Bos, B. L. Feringa, E. N. W. Howe, X. Wu, M. A. Siegler and P. A. Gale, J. Am. Chem. Soc., 2022, 144, 331–338 CrossRef CAS PubMed.
- M. Ahmad, N. J. Roy, A. Singh, D. Mondal, A. Mondal, T. Vijayakanth, M. Lahiri and P. Talukdar, Chem. Sci., 2023, 14, 8897–8904 RSC.
- S. B. Salunke, J. A. Malla and P. Talukdar, Angew. Chem., Int. Ed., 2019, 58, 5354–5358 CrossRef CAS PubMed.
- C. Bao, M. Ma, F. Meng, Q. Lin and L. Zhu, New J. Chem., 2015, 39, 6297–6302 RSC.
- L. E. Bickerton and M. J. Langton, Chem. Sci., 2022, 13, 9531–9536 RSC.
- Q. Zhong, Y. Cao, X. Xie, Y. Wu, Z. Chen, Q. Zhang, C. Jia, Z. Wu, P. Xin, X. Yan, Z. Zeng and C. Ren, Angew. Chem., Int. Ed., 2024, 63, e202314666 CrossRef CAS PubMed.
- M. Ahmad, T. G. Johnson, M. Flerin, F. Duarte and M. J. Langton, Angew. Chem., Int. Ed., 2024, 63, e202403314 CrossRef CAS PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- E. D. Glendening, C. R. Landis and F. Weinhold, J. Comput. Chem., 2013, 34, 1429–1437 CrossRef CAS.
- S. Grimme, J. Chem. Theory Comput., 2019, 15, 2847–2862 CrossRef CAS PubMed.
- F. M. Bickelhaupt and K. N. Houk, Angew. Chem., Int. Ed., 2017, 56, 10070–10086 CrossRef CAS PubMed.
- A. Lindahl, Hess and van der Spoel, GROMACS 2021.3 Source code (version 2021.3), Zenodo, 2021 Search PubMed.
- J. S. Hub, B. L. de Groot and D. van der Spoel, J. Chem. Theory Comput., 2010, 6, 3713–3720 CrossRef CAS.
- C. H. Bennett, J. Comput. Phys., 1976, 22, 245–268 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2371936 and 2371937. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4nr03220a |
| This journal is © The Royal Society of Chemistry 2024 |