
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nanoparticle-enabled integration of air capture and conversion of CO2†
Huanqin
Guan
 a,
Ju Ye
Kim
b,
Kecheng
Wei
a,
Mayank
Agrawal
b,
Andrew A.
Peterson
*b and
Shouheng
Sun
*a
a,
Ju Ye
Kim
b,
Kecheng
Wei
a,
Mayank
Agrawal
b,
Andrew A.
Peterson
*b and
Shouheng
Sun
*a
aDepartment of Chemistry, Brown University, Providence, Rhode Island 02912, USA. E-mail: ssun@brown.edu
bSchool of Engineering, Brown University, Providence, Rhode Island 02912, USA. E-mail: Andrew_Peterson@brown.edu
First published on 9th September 2024
Abstract
Integrating air capture and conversion of CO2 is key to realizing energy sustainability. However, current integration approaches require high temperature and pressure, making them energy intensive. Here, we demonstrate a nanoparticle (NP) catalysis approach for the hydrogenation of alkyl carbonate, an intermediate obtained from the CO2 capture process, to formate, achieving one-pot air capture and conversion of CO2 under ambient conditions. The capture is realized in an ethylene glycol (EG) solution of KOH (EG–KOH) at room temperature, where CO2 is selectively converted into HO–CH2CH2–O–COOK (∼100% conversion). This carbonate is then hydrogenated using ammonia borane (under ambient pressure and at 50 °C) to formate (HCOOK) (>90% yield) in the presence of a stable Pd NP catalyst with EG being regenerated. Atomistic simulations suggest that the CO2 absorption process in the EG–KOH solution is energetically stable, and the catalyst surface provides the reaction site to break the C–O bond in the –O–COOK structure, enabling the hydrogenation of the alkyl carbonate to formate and the regeneration of EG. Our study provides a promising NP-catalysis approach for air capture and conversion of CO2 into value-added chemicals/fuels under ambient conditions.
Introduction
Energy-efficient air capture and conversion of CO2 is a promising solution to combat climate change and build an energy-sustainable future.1–5 The key to the success of this approach is the energy-efficient integration of the capture/conversion processes,6,7 a goal that has been extremely challenging to achieve. Past approaches for this integration include CO2 capture by an amine to form a carbamate, followed by catalytic hydrogenation of the carbamate to formate,8–11 or by an alkaline (KOH) solution to form alkyl carbonate, followed by catalytic hydrogenation to methanol.12–17 In all these studies, the hydrogenation step is performed under high pressure and temperature conditions and the conversion is catalyzed by homogeneous Ru-pincer hydride complexes.18–23 To realize the energy-efficient integration of CO2 capture and conversion, a stable and recyclable catalytic system should be present, which can activate and convert the alkyl carbonate into a valuable chemical or fuel under ambient conditions.Nanoparticles (NPs) have been studied extensively for catalytic applications. Herein, we report a Pd nanoparticle (NP)-enabled approach for air capture and conversion of CO2 under ambient conditions, as summarized in Scheme 1. The key idea of our approach is first to capture CO2 in an ethylene glycol (EG) solution of KOH to convert CO2 into alkyl carbonate and then to reduce this alkyl carbonate in situ to formate via Pd NP-catalyzed transfer hydrogenation of ammonia borane (NH3BH3, AB) with EG being regenerated. Previous studies have shown that pure CO2 may be hydrogenated by a metal borohydride or AB in the presence of a special pincer complex as a catalyst,24–28 but such a hydrogenation reaction is not suitable for the desired one-step air capture and conversion process. We found that Pd NPs were active in catalyzing the AB hydrogenation of alkyl carbonate to formate under ambient pressure. The reaction led to full conversion (100%) of alkyl carbonate and high yield (92%) formation of formate in a 6 h reaction period at 50 °C. The process can be readily applied to integrate air capture and conversion of CO2 into formate. The Pd NPs are stable in the one-pot capture/conversion process and can be reused for the next capture and conversion step. This NP-catalysis enabled one-pot approach demonstrates a potential energy-efficient integration of air capture and conversion of CO2 into value-added chemicals/fuels.
 | ||
| Scheme 1 Illustration of the integrated air capture and conversion of CO2 into formate under ambient conditions. | ||
Methods and materials
Materials
Palladium acetylacetonate (Pd(acac)2, 99%) and hydrogen tetrachloroaurate(III) hydrate (HAuCl4·3H2O, 99.9%) were purchased from Strem Chemicals. Potassium hydroxide (99%), potassium bicarbonate (99%), potassium carbonate (99%), ammonia borane (90%), oleylamine (technical grade, 70%), borane-tert-butylamine (97%), triisopropylsilane (TIPS, 99%), ethylene glycol (anhydrous, 99.8%) and 1-methyl-2-pyrrolidinone (NMP, 99.5%) were purchased from Sigma Aldrich. d6-DMSO was used as received. Hexane (98.5%), isopropanol (100%), ethanol (100%), N,N-dimethylformamide (99.8%), and acetic acid (98%) were purchased from Fisher Scientific. The platinum wire (0.5 mm diameter, Premion, 99.997%) and Toray carbon paper (TGP-H-60) were from Alfa Aesar. Polyvinylidene fluoride (PVDF) was from MTI Corporation. Deionized water was from a Millipore Autopure system. All chemicals were used without further purification. Nitrogen (99.99%) and carbon dioxide (99.9999%) were purchased from Corp Brothers, Inc.Experimental procedures
Pd nanoparticle (NP) synthesis
5 nm Pd NPs were synthesized by modifying a previously reported method.29 0.150 g of Pd(acac)2 and oleylamine (15 mL) were added into a three-necked flask and then degassed with nitrogen at 60 °C for 30 min. 0.300 g of borane tert-butylamine was dispersed in 4 mL of oleylamine by vortexing and shaking. The temperature of the Pd precursor solution was lowered to and maintained at 40 °C. The solution of borane-tert-butylamine was injected into the reaction flask quickly, and within one minute the color of the solution changed to dark brown-black. The temperature was raised to 90 °C at a ramp rate of 3–5 °C min−1. The solution was maintained at 90 °C for 1 h. The NPs were precipitated by adding 100 mL of ethanol and separated by centrifugation at 9000 rpm for 8 min. The NPs were then washed twice with ∼75 mL of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 hexane:ethanol (9000 rpm for 8 min). The NPs were redispersed in hexane for storage. The subsequent catalyst loading and the activation step are presented in the ESI.†
:9 hexane:ethanol (9000 rpm for 8 min). The NPs were redispersed in hexane for storage. The subsequent catalyst loading and the activation step are presented in the ESI.†
Direct air capture of CO2 and in situ hydrogenation reaction
5 mmol of KOH was dissolved in 10 mL of EG. A water aspirator was used to create a partial vacuum in the reaction system so that ambient air (∼413 ppm CO2) could continuously bubble through the capture solution. The flow rate was controlled to be 0.2 L min−1. The solution pH was monitored during the capture process to determine the CO2 saturation point. After 48 hours of bubbling, the solution volume increased by about 3 mL due to moisture in the air. The capture solution was then purged with N2 to remove gas impurities. Next, 10 mg of Pd/C catalysts and 100 mg of ammonia borane were added to the solution, which was sealed using a balloon. The reaction mixture was maintained at 50 °C for 24 hours. Both gas and liquid products were subjected to GC and NMR analysis, respectively.The hydrogenation reaction could also be performed under air without N2 purging, yielding a similar reaction result.
All other electrochemical reaction procedures, characterization methods and computational details are given in the ESI.†
Results and discussion
To study the activation of alkyl carbonate, we first reacted pure CO2 with an ethylene glycol (EG) solution of KOH to form HO–CH2CH2–O–COOK. This alkaline chemistry leading to efficient CO2 capture has been well-known.7,12,13 In this study, we bubbled CO2 from its gas cylinder into the EG solution of KOH at room temperature. In the alkaline EG solution, EG can react with KOH to form a glycoxide anion and water, quickly reaching equilibrium. Upon exposure to CO2 at room temperature, this equilibrium is shifted to the right, as CO2 reacts with the glycoxide to form alkyl carbonate (Fig. 1A). This CO2 absorption process reaches its saturation point once KOH is consumed. We monitored solution pH and product formation to determine the endpoint of the capture process as well as the CO2 capture capacity. As shown in Fig. 1B, the initial pH of the EG solution of KOH (5 mmol KOH dissolved in 10 mL EG) is around 10.7. Upon CO2 bubbling, the pH gradually decreased and reached a plateau of 7.8 after 40 min. The CO2 capture capacity of this KOH solution at saturation was measured using the weight change of the solution before and after capture, which was determined to be 220 mg gravimetrically. The capture efficiency can reach nearly 100%, which corresponds to 1 mol CO2 per mol KOH (Fig. 1C). The alkyl carbonate formed in the EG solution is stable after the capture process and can be quantitatively characterized by both 1H and 13C nuclear magnetic resonance (NMR) spectroscopy, which is consistent with the weight change (Fig. S1†). In this capture process, the amount of KOH is the limiting factor, which determines the maximum amount of CO2 the system can capture.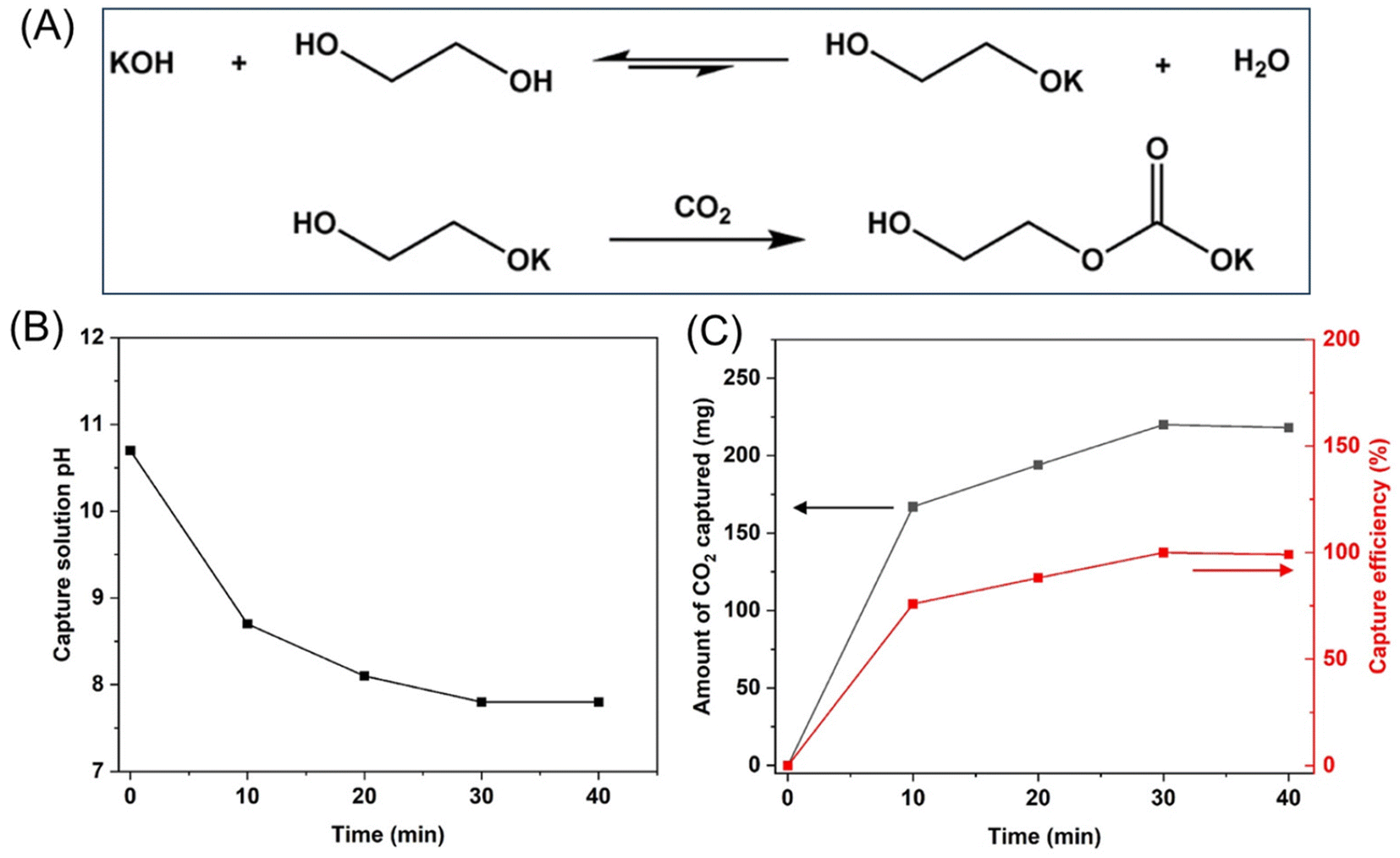 | ||
| Fig. 1 (A) CO2 capture by KOH–EG solution. Capture conditions: KOH (5 mmol), EG (10 mL), r.t., and t = 40 min. (B) pH change of capture solution during CO2 bubbling. (C) The amount of captured CO2 and capture efficiency during CO2 bubbling. | ||
The alkyl carbonate could not be activated and reduced by a common hydride agent (AB or sodium borohydride) under atmospheric pressure and at temperatures ranging from room temperature to 80 °C we screened. However, when we studied NP catalysis for transfer hydrogenation of AB, we found Pd NPs to be more active in catalyzing the hydrogenation of alkyl carbonate to formate at 50 °C. We prepared 5 nm Pd NPs via solution phase reduction of Pd(acac)2 with borane t-butylamine in oleylamine.29 Fig. S2A† shows the transmission electron microscopy (TEM) image of the 5 nm Pd NPs obtained from the synthesis. The NPs were then loaded onto carbon support, followed by an acetic acid wash to remove the surfactant oleylamine as reported.30 The TEM image showed that the Pd NPs are well-dispersed on the carbon support after this treatment, showing no sign of aggregation (Fig. S2B†). The Pd NPs have a typical fcc structure as characterized by X-ray diffraction (XRD) (Fig. S2C†). We studied the reduction of alkyl carbonate by AB in the capture solution in the presence of a Pd NP catalyst (Fig. 2A and ESI†). We used a balloon to seal the reaction vessel to ensure that the reaction proceeded under ambient pressure. The conversion of alkyl carbonate was monitored by 1H NMR using dimethylformamide as an internal standard in d6-DMSO solvent. A small amount of aliquot was taken for analysis at each hour time mark. The alkyl carbonate conversion increased with reaction time and reached 100% after 6 h of reaction (Fig. 2B). In the solution phase, formate was the only product characterized by both 1H and 13C NMR (Fig. S3†). The time-dependent formate yield during the reaction is shown in Fig. 2C, reaching its maximum of 92% after 6 h with a turnover frequency (TOF) of 0.059 s−1. The rest of the products contained small amounts of CO and CH4 as detected by gas chromatography, with CO at 6% and CH4 at 2%, which might be the result of the deep reduction of the adsorbed formate on the Pd surface, similar to what has been reported for CO2/formate reduction to CO/CH4 on a Pd–Al2O3 surface.31 We also detected the formation of H2 in the reaction system, which is the result of the competing reaction, alcoholysis of AB, under the transfer hydrogenation reaction conditions. Temperature-dependent studies showed that increasing the reaction temperature to 80 °C caused the formate yield to drop, which is due to the exothermic nature of the hydrogenation reaction,32 while decreasing the temperature to 20 °C also reduced the formate yield due to the slow kinetics (Fig. 2C). When the catalyst loading was doubled to 0.2 mol%, the formate yield did not change much under the same reaction conditions. Without the Pd NP catalyst, however, the formate yield was only 12% even after 48 h of reaction (Table S1†), suggesting that pure AB is not active enough to reduce alkyl carbonate. Our further control experiments showed that alkyl carbonate is stable in the reaction solution without AB and Pd NPs, even under heating at 120 °C (Fig. S4†). Additionally, without alkyl carbonate, the reaction of AB and Pd NPs in EG only yielded H2 that is generated from the Pd-catalyzed solvolysis of AB in EG, similar to what has been extensively reported regarding NP-catalyzed hydrolysis/alcoholysis of AB to generate H2.33–35 We further investigated the Pd-catalyzed hydrogenation reaction in the presence of NaBH4 and obtained a similar formate yield. However, atmospheric H2 was not active enough for the hydrogenation process as the formate yield was only 7% (Table S2†). This indicates that under our current reaction conditions, a hydride intermediate, not a free H2 molecule, is an active species in the Pd NP-catalyzed hydrogenation of alkyl carbonate to formate.
 | ||
| Fig. 2 (A) Integrated CO2 capture and conversion scheme. (B) Time dependent alkyl carbonate hydrogenation reaction at 50 °C showing carbonate conversion. (C) Temperature effect on the alkyl carbonate hydrogenation showing formate yield. Reaction conditions: solutions from the capture step were directly used for hydrogenation. Alkyl carbonate: 2.0 mmol, ammonia borane (AB): 1.0 mmol, Pd nanocatalyst: 0.1 mol%, and ambient pressure. Formate yields were determined by 1H NMR, and the average value was reported based on three independent experiments. | ||
The formation of potassium carbonate (K2CO3) and/or bicarbonate (KHCO3) is inevitable when water and KOH are present in the capture solution. We further studied how the formation of K2CO3/KHCO3 affects the AB transfer hydrogenation of alkyl carbonate. We found that both K2CO3 and KHCO3 could be dissolved in EG and further reduced by AB in the presence of Pd NPs to formate (87% and 91% yield, respectively) (Fig. 3). This finding is significant for the successful air capture and conversion of CO2, as it makes the hydrogenation of K2CO3/KHCO3 possible without high energy input (inorganic carbonate is generally considered inactive and can only be reduced under harsh conditions36,37). Under our reaction conditions, EG plays an important role in activating K2CO3/KHCO3 by forming alkyl carbonate, as confirmed by NMR of the precursor solution containing K2CO3/KHCO3 and EG (Fig. S5†). This suggests that there exists an equilibrium between carbonate and alkyl carbonate. To further support this point, we performed K2CO3/KHCO3 reduction in an aqueous solution without EG under the same conditions, and we could only detect a very small amount of formate (<6% yield). Therefore, we conclude that EG reacts with carbonates to form alkyl carbonate, reaching equilibrium. Under the Pd NP-catalyzed AB hydrogenation conditions, alkyl carbonate is reduced to formate, which shifts the equilibrium to the right until carbonates are consumed. This indicates that water in the EG/KOH solution only slows down the hydrogenation process, but does not affect the final reaction outcome, which is key to continuous air capture and conversion of CO2.
 | ||
| Fig. 3 Investigation of inorganic carbonate/bicarbonate hydrogenation under mild conditions. Reaction conditions: carbonate/bicarbonate (2.0 mmol), EG (10 mL), AB (1.0 mmol), Pd nanocatalyst (0.1 mol%), ambient pressure, 50 °C and 6 h. | ||
Our model study on pure CO2 capture and conversion in EG–KOH solution as well as the EG role in activating K2CO3/KHCO3 motivated us to further explore CO2 capture from air, where moisture is also ubiquitously present. The capture and conversion system and the related reactions are outlined in Scheme 1. A water aspirator was used as a pump to draw ambient air into the capture solution, in which ultra-dilute CO2 (∼413 ppm) was captured by KOH in EG.7 The solution pH was monitored throughout the capture process, and it took 48 h for the pH to stabilize (Fig. 4A). The 13C NMR analysis of the capture product shows an additional peak of CO32− (165.70 ppm) in addition to the presence of alkyl carbonate (Fig. 4B and C). The CO32− species must be formed from reactions among water (from the air), KOH, and CO2. Based on the quantitative 13C NMR analysis, the ratio between alkyl carbonate and K2CO3 was about 1:1. When the air capture solution was hydrogenated by AB at 50 °C in the presence of the Pd NP catalyst for 24 h, all carbonates were converted into formate (91% yield). Compared with the result observed from the pure CO2 capture and conversion demonstrated above, the hydrogenation of carbonates from air capture requires a longer reaction time due to the less efficient conversion of inorganic carbonate into alkyl carbonate in the presence of water. Another highlight of our reaction system is that the Pd NP-catalyzed transfer hydrogenation is O2 tolerant – the reaction can proceed without the need to purge the reaction system with N2 to remove O2. As a comparison, O2 must be removed from the reaction system under conventional hydrogenation reaction conditions.2,7,12 Finally, the Pd NPs showed excellent stability under the catalytic transfer hydrogenation reaction conditions and maintained a formate yield of over 90% after 5 reaction cycles (Fig. 4D) (the same amounts of AB and KOH were added in each reaction cycle). The Pd NPs retain their crystal structure (Fig. S6A†) and NP size/dispersity (Fig. S6B†) as confirmed by the XRD and TEM analyses of the NPs before and after the catalytic reactions, indicating the long-term stability of the Pd NPs as an efficient catalyst for continuous air capture and conversion.
 | ||
| Fig. 4 (A) Capture solution pH change in the direct air capture process. (B) 13C NMR spectra of CO2 captured from lab air in d6-DMSO. (C) Enlargement of Fig. 4B showing carbonyl peaks for alkyl carbonate and CO32−. (D) Catalytic stability of formate yields after 5 consecutive catalytic cycles. Reaction conditions: KOH (5 mmol), EG (10 mL), AB (2.0 mmol), Pd nanocatalyst (0.1 mol%), ambient pressure, and 50 °C. | ||
To understand why Pd NPs are efficient for the AB hydrogenation of alkyl carbonate, we used density functional theory (DFT) calculations to further investigate the reaction mechanism on Pd surfaces. We suggest a 5-step mechanism, starting with the incorporation of CO2 into a solution of KOH in EG, as shown in Fig. 1. Given that the Pd NPs are nano-sized polyhedra (∼5 nm) (Fig. S2†), which means that they consist of many step and terrace sites, both the planar (111) and the stepped (211) crystal facets of Pd were examined using DFT calculations, as shown in Fig. S9.† This comparison shows a more facile route on the (211) step edge than on the (111) terrace, and we highlight the (211) results herein.
The overall suggested reaction mechanism and the corresponding Gibbs free energies are summarized in Fig. 5, where “*” represents an adsorbed species on the Pd surface. State 1 → state 2 represents the formation of an EG–CO2 complex under the pH 7.8 environment; we considered the pH effect in state 1 because protons are involved in the reaction for forming the EG–CO2 complex.38 As expected from the experiments in which alkyl carbonate intermediates were formed immediately when CO2 was purged, the reaction from state 1 to state 2 proceeds spontaneously with a downhill energy path. While the reaction from state 1 to state 2 takes place without a Pd catalyst, after state 2, the reaction takes place on the surface of the Pd catalyst. Accordingly, each Gibbs energy of activation (reaction energy barrier) from state 3 was calculated to investigate the rate-determining step. T1 is an abbreviation of transition state 1, and a total of 3 transition states (T1, T2, and T3) and each Gibbs energy of activation ( , respectively) were calculated by the dynamic nudged elastic band (DyNEB) method.39 We modeled the process of H2 formation by AB as a process in which H* is adsorbed to Pd from H2 (state 2 → state 3), and this process is a thermodynamically easy reaction with a downhill energy profile. Considering the consistency in the calculation environment, we started the reaction with an H2 gas molecule represented by a black color bar, while also showing the reaction energy for generating H2 from NH3BH3 in a gray color bar to account for the use of NH3BH3 in the actual experiments. The theoretical Gibbs energy of activation
, respectively) were calculated by the dynamic nudged elastic band (DyNEB) method.39 We modeled the process of H2 formation by AB as a process in which H* is adsorbed to Pd from H2 (state 2 → state 3), and this process is a thermodynamically easy reaction with a downhill energy profile. Considering the consistency in the calculation environment, we started the reaction with an H2 gas molecule represented by a black color bar, while also showing the reaction energy for generating H2 from NH3BH3 in a gray color bar to account for the use of NH3BH3 in the actual experiments. The theoretical Gibbs energy of activation 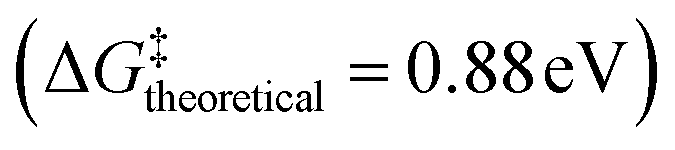 was calculated as the free energy change between state 3 (the lowest-energy state) and transition state 2 (the highest-energy barrier), which suggests that the rate-determining step of this reaction is the hydrogenation and release of EG (HOCH2CH2OCOO− + H* → HOCH2CH2OH). Considering that the experimental turnover frequency (TOF) is 0.059 s−1, we can estimate that
was calculated as the free energy change between state 3 (the lowest-energy state) and transition state 2 (the highest-energy barrier), which suggests that the rate-determining step of this reaction is the hydrogenation and release of EG (HOCH2CH2OCOO− + H* → HOCH2CH2OH). Considering that the experimental turnover frequency (TOF) is 0.059 s−1, we can estimate that  (Gibbs energy for the experimental activation) is around 0.95 eV from the Eyring equation:
(Gibbs energy for the experimental activation) is around 0.95 eV from the Eyring equation:

 | ||
Fig. 5 The schemes at the top of the graph show the reaction process, with the red, gray, white, and cyan atoms in the schemes representing O, C, H, and Pd, respectively. Bottom shows the free energy diagram for formic acid (HCOOH) formation by EG and CO2 at 50 °C and 1 atm. The y-axis represents the Gibbs free energy (G, eV), and the Gibbs free energy of activation  is shown (red color) inside the graph. Free energy diagram with error estimation (blue gradient box for each state) obtained from the BEEF ensembles. The free energy required for H2 production from NH3BH3 is shown in gray for state 1 and state 2. is shown (red color) inside the graph. Free energy diagram with error estimation (blue gradient box for each state) obtained from the BEEF ensembles. The free energy required for H2 production from NH3BH3 is shown in gray for state 1 and state 2. | ||
Although the DFT-calculated barriers were close to the experimental barrier, we wanted to understand if the deviation was reasonable. We predicted errors from the ensemble-based Bayesian error estimation functional (BEEF) (Fig. 5, see the blue and red gradient box),40 where the calculated 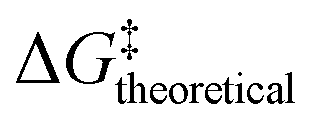 was in the range between 0.80 eV and 0.95 eV, as shown in Fig. 5. Given the error in any experimental measurement, we consider the experimental and calculated barriers to be indistinguishable.
was in the range between 0.80 eV and 0.95 eV, as shown in Fig. 5. Given the error in any experimental measurement, we consider the experimental and calculated barriers to be indistinguishable.
After the breakage of the intermediate product and adsorption on the Pd surface (state 4), a hydrogenation reaction proceeds to produce EG and HCOOH. State 4 → state 5 represents the production of recycled EG, and state 5 → state 6 is an HCOOH production step. As a result, it was confirmed that the CO2 absorption process in the solution of KOH in EG is energetically stable (state 1 → state 2), and the major role of the Pd catalyst is to break the C–O bond of the alkyl carbonate intermediate (state 3 → state 4), as well as to provide a reservoir of hydride for product release.
We also tested if the direct air capture system can be coupled with electrocatalysis to reduce alkyl carbonate without the use of AB. In our preliminary studies, Pd NPs were active in catalyzing the electrochemical reduction of alkyl carbonate to formate, but the reduction at the catalyst surface produced formate at too low a concentration to detect. Instead of pursuing the reduction to a liquid product, we tested the reduction to a gaseous product that can be easily separated from the reaction system and characterized. Since Au nanowires are known to be active to electro-catalyze CO2 to CO,41 we prepared 2 nm ultra-thin Au nanowires (Fig. S7†) and studied their catalysis for the electrochemical reduction of alkyl carbonate in the capture solution (EG/H2O v/v ∼ 5/1) (Fig. S8†). At −1.4 V (vs. Ag/AgCl), the CO faradaic efficiency reached 21% with H2 as the side product. An important point to note is that carbonate/bicarbonate cannot be reduced under common electrochemical CO2 reduction conditions,42 and some studies suggest that the electrochemical conversion of carbonates from direct air capture solution could be attributed to the acidification of the carbonates to CO2 at the bipolar membrane interface.43 We are currently working on demonstrating nanocatalysts for efficient electro-reduction of alkyl carbonate as a new approach for the integration of direct air capture and conversion of CO2 into value-added chemicals.
Conclusions
This report demonstrates an NP-catalyzed hydrogenation of carbonates to realize the integration of air capture and conversion of CO2 into formate. The key concept is to fast capture CO2 from the air via a strong alkaline solution of EG–KOH, converting the CO2 into alkyl carbonate HOCH2CH2O–COOK. This carbonate is stable in the capture medium and can be characterized and quantified. More importantly, the alkyl carbonate can be further activated by Pd NPs and then hydrogenated by AB (100% conversion) to formate (>90% yield) under ambient pressure and 50 °C with EG being regenerated. The formation of carbonate/bicarbonate salts from the air (moisture) capture process only affects the reaction kinetics, not the overall reaction outcome due to the established equilibrium among carbonates, EG, and alkyl carbonate. Without EG, these carbonates cannot be easily hydrogenated, demonstrating the important role of EG in promoting the hydrogenation of carbonates. DFT calculations suggest that the (211) step edge exposure on the Pd NP surface is especially active for promoting the carbonate hydrogenation reaction. The Gibbs free energy diagram shows a 5-step reaction mechanism for the CO2 capture and conversion process, and the rate-determining step is the hydrogenation and release of EG. After incorporating the solvation effect, the calculated theoretical activation barriers match well with the experimental ones. Our studies provide a promising approach for the continuous air capture of CO2 and subsequent formate synthesis. Our preliminary tests also show that it is possible to use an NP catalyst to activate the alkyl carbonate electrochemically. We are working to develop a robust electrocatalyst system to selectively convert this alkyl carbonate into value-added chemicals/fuels and to realize energy-efficient integration of direct air capture and conversion of CO2 for green and sustainable energy applications.Data availability
The data supporting this article are available within the main article as well as its ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the NSF under Grants CHE-2102290 and CBET-2324345 and by Brown University's Office of Vice President for Research. Electronic structure calculations were carried out at the Brown University Center for Computation and Visualization (CCV).References
- E. S. Sanz-Pérez, C. R. Murdock, S. A. Didas and C. W. Jones, Direct Capture of CO2 from Ambient Air, Chem. Rev., 2016, 116(19), 11840–11876 CrossRef PubMed.
- M. Erans, E. S. Sanz-Pérez, D. P. Hanak, Z. Clulow, D. M. Reiner and G. A. Mutch, Direct air capture: process technology, techno-economic and socio-political challenges, Energy Environ. Sci., 2022, 15(4), 1360–1405 RSC.
- S. De, A. Dokania, A. Ramirez and J. Gascon, Advances in the Design of Heterogeneous Catalysts and Thermocatalytic Processes for CO2 Utilization, ACS Catal., 2020, 10(23), 14147–14185 CrossRef CAS.
- X. Jiang, X. Nie, X. Guo, C. Song and J. G. Chen, Recent Advances in Carbon Dioxide Hydrogenation to Methanol via Heterogeneous Catalysis, Chem. Rev., 2020, 120(15), 7984–8034 CrossRef CAS PubMed.
- K. Wei, H. Guan, Q. Luo, J. He and S. Sun, Recent advances in CO2 capture and reduction, Nanoscale, 2022, 14(33), 11869–11891 RSC.
- I. Sullivan, A. Goryachev, I. A. Digdaya, X. Li, H. A. Atwater, D. A. Vermaas and C. Xiang, Coupling electrochemical CO2 conversion with CO2 capture, Nat. Catal., 2021, 4(11), 952–958 CrossRef CAS.
- S. Kar, A. Goeppert and G. K. S. Prakash, Integrated CO2 Capture and Conversion to Formate and Methanol: Connecting Two Threads, Acc. Chem. Res., 2019, 52(10), 2892–2903 CrossRef CAS PubMed.
- Y.-N. Li, L.-N. He, A.-H. Liu, X.-D. Lang, Z.-Z. Yang, B. Yu and C.-R. Luan, In situ hydrogenation of captured CO2 to formate with polyethyleneimine and Rh/monophosphine system, Green Chem., 2013, 15(10), 2825–2829 RSC.
- H.-C. Fu, F. You, H.-R. Li and L.-N. He, CO2 Capture and in situ Catalytic Transformation, Front. Chem., 2019, 7(525), 1–15 Search PubMed.
- S. Zhang, Y.-N. Li, Y.-W. Zhang, L.-N. He, B. Yu, Q.-W. Song and X.-D. Lang, Equimolar Carbon Absorption by Potassium Phthalimide and In Situ Catalytic Conversion Under Mild Conditions, ChemSusChem, 2014, 7(5), 1484–1489 CrossRef CAS PubMed.
- D. Wei, H. Junge and M. Beller, An amino acid based system for CO2 capture and catalytic utilization to produce formates, Chem. Sci., 2021, 12(17), 6020–6024 RSC.
- R. Sen, A. Goeppert, S. Kar and G. K. S. Prakash, Hydroxide Based Integrated CO2 Capture from Air and Conversion to Methanol, J. Am. Chem. Soc., 2020, 142(10), 4544–4549 CrossRef CAS PubMed.
- R. Sen, C. J. Koch, A. Goeppert and G. K. S. Prakash, Tertiary Amine-Ethylene Glycol Based Tandem CO2 Capture and Hydrogenation to Methanol: Direct Utilization of Post-Combustion CO2, ChemSusChem, 2020, 13(23), 6318–6322 CrossRef CAS PubMed.
- J. Kothandaraman, A. Goeppert, M. Czaun, G. A. Olah and G. K. Surya Prakash, CO2 capture by amines in aqueous media and its subsequent conversion to formate with reusable ruthenium and iron catalysts, Green Chem., 2016, 18(21), 5831–5838 RSC.
- M. Scott, B. Blas Molinos, C. Westhues, G. Franciò and W. Leitner, Aqueous Biphasic Systems for the Synthesis of Formates by Catalytic CO2 Hydrogenation: Integrated Reaction and Catalyst Separation for CO2-Scrubbing Solutions, ChemSusChem, 2017, 10(6), 1085–1093 CrossRef CAS PubMed.
- S. Kar, R. Sen, A. Goeppert and G. K. S. Prakash, Integrative CO2 Capture and Hydrogenation to Methanol with Reusable Catalyst and Amine: Toward a Carbon Neutral Methanol Economy, J. Am. Chem. Soc., 2018, 140(5), 1580–1583 CrossRef CAS PubMed.
- D. Wei, R. Sang, P. Sponholz, H. Junge and M. Beller, Reversible hydrogenation of carbon dioxide to formic acid using a Mn-pincer complex in the presence of lysine, Nat. Energy, 2022, 7(5), 438–447 CrossRef CAS.
- K. Sordakis, C. Tang, L. K. Vogt, H. Junge, P. J. Dyson, M. Beller and G. Laurenczy, Homogeneous Catalysis for Sustainable Hydrogen Storage in Formic Acid and Alcohols, Chem. Rev., 2018, 118(2), 372–433 CrossRef CAS PubMed.
- M. D. Porosoff, B. Yan and J. G. Chen, Catalytic reduction of CO2 by H2 for synthesis of CO, methanol and hydrocarbons: challenges and opportunities, Energy Environ. Sci., 2016, 9(1), 62–73 RSC.
- D. B. Lao, B. R. Galan, J. C. Linehan and D. J. Heldebrant, The steps of activating a prospective CO2 hydrogenation catalyst with combined CO2 capture and reduction, Green Chem., 2016, 18(18), 4871–4874 RSC.
- S. Kar, A. Goeppert, V. Galvan, R. Chowdhury, J. Olah and G. K. S. Prakash, A Carbon-Neutral CO2 Capture, Conversion, and Utilization Cycle with Low-Temperature Regeneration of Sodium Hydroxide, J. Am. Chem. Soc., 2018, 140(49), 16873–16876 CrossRef CAS PubMed.
- S. Kostera, S. Weber, M. Peruzzini, L. F. Veiros, K. Kirchner and L. Gonsalvi, Carbon Dioxide Hydrogenation to Formate Catalyzed by a Bench-Stable, Non-Pincer-Type Mn(I) Alkylcarbonyl Complex, Organometallics, 2021, 40(9), 1213–1220 CrossRef CAS PubMed.
- R. Bhardwaj, A. Kumar and J. Choudhury, An all-aqueous and phosphine-free integrated amine-assisted CO2 capture and catalytic conversion to formic acid, Chem. Commun., 2022, 58(82), 11531–11534 RSC.
- T. Zhao, C. Li, X. Hu, F. Liu and Y. Wu, Base-assisted transfer hydrogenation of CO2 to formate with ammonia borane in water under mild conditions, Int. J. Hydrogen Energy, 2021, 46(29), 15716–15723 CrossRef CAS.
- I. Knopf and C. C. Cummins, Revisiting CO2 Reduction with NaBH4 under Aprotic Conditions: Synthesis and Characterization of Sodium Triformatoborohydride, Organometallics, 2015, 34(9), 1601–1603 CrossRef CAS.
- L. Lombardo, Y. Ko, K. Zhao, H. Yang and A. Züttel, Direct CO2 Capture and Reduction to High-End Chemicals with Tetraalkylammonium Borohydrides, Angew. Chem., Int. Ed., 2021, 60(17), 9580–9589 CrossRef CAS PubMed.
- W. Zhao, H. Li, H. Zhang, S. Yang and A. Riisager, Ammonia borane-enabled hydrogen transfer processes: Insights into catalytic strategies and mechanisms, Green Energy Environ., 2023, 8, 948–971 CrossRef CAS.
- Z.-W. Qu, H. Zhu, R. Streubel and S. Grimme, Catalyst-free CO2 hydrogenation with BH3NH3 in water: DFT mechanistic insights, Phys. Chem. Chem. Phys., 2022, 24(23), 14159–14164 RSC.
- V. Mazumder and S. Sun, Oleylamine-Mediated Synthesis of Pd Nanoparticles for Catalytic Formic Acid Oxidation, J. Am. Chem. Soc., 2009, 131(13), 4588–4589 CrossRef CAS PubMed.
- D. Li, C. Wang, D. Tripkovic, S. Sun, N. M. Markovic and V. R. Stamenkovic, Surfactant Removal for Colloidal Nanoparticles from Solution Synthesis: The Effect on Catalytic Performance, ACS Catal., 2012, 2(7), 1358–1362 CrossRef CAS.
- X. Wang, H. Shi, J. H. Kwak and J. Szanyi, Mechanism of CO2 Hydrogenation on Pd/Al2O3 Catalysts: Kinetics and Transient DRIFTS-MS Studies, ACS Catal., 2015, 5(11), 6337–6349 CrossRef CAS.
- J. Su, M. Lu and H. Lin, High yield production of formate by hydrogenating CO2 derived ammonium carbamate/carbonate at room temperature, Green Chem., 2015, 17(5), 2769–2773 RSC.
- C. Yu, X. Guo, M. Shen, B. Shen, M. Muzzio, Z. Yin, Q. Li, Z. Xi, J. Li, C. T. Seto and S. Sun, Maximizing the Catalytic Activity of Nanoparticles through Monolayer Assembly on Nitrogen-Doped Graphene, Angew. Chem., Int. Ed., 2018, 57(2), 451–455 CrossRef CAS PubMed.
- M. Muzzio, H. Lin, K. Wei, X. Guo, C. Yu, T. Yom, Z. Xi, Z. Yin and S. Sun, Efficient Hydrogen Generation from Ammonia Borane and Tandem Hydrogenation or Hydrodehalogenation over AuPd Nanoparticles, ACS Sustainable Chem. Eng., 2020, 8(7), 2814–2821 CrossRef CAS.
- H. Guan, M. Shen, C. Harris, H. Lin, K. Wei, M. Morales, N. Bronowich and S. Sun, Cu2O nanoparticle-catalyzed tandem reactions for the synthesis of robust polybenzoxazole, Nanoscale, 2022, 14(16), 6162–6170 RSC.
- S. Coufourier, Q. Gaignard Gaillard, J.-F. Lohier, A. Poater, S. Gaillard and J.-L. Renaud, Hydrogenation of CO2, Hydrogenocarbonate, and Carbonate to Formate in Water using Phosphine Free Bifunctional Iron Complexes, ACS Catal., 2020, 10(3), 2108–2116 CrossRef CAS.
- C. Rebreyend, E. A. Pidko and G. A. Filonenko, Homogeneous hydrogenation of saturated bicarbonate slurry to formates using multiphase catalysis, Green Chem., 2021, 23(22), 8848–8852 RSC.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode, J. Phys. Chem. B, 2004, 108(46), 17886–17892 CrossRef.
- P. Lindgren, G. Kastlunger and A. A. Peterson, Scaled and Dynamic Optimizations of Nudged Elastic Bands, J. Chem. Theory Comput., 2019, 15(11), 5787–5793 CrossRef CAS PubMed.
- J. J. Mortensen, L. B. Hansen and K. W. Jacobsen, Real-space grid implementation of the projector augmented wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71(3), 035109 CrossRef.
- W. Zhu, Y.-J. Zhang, H. Zhang, H. Lv, Q. Li, R. Michalsky, A. A. Peterson and S. Sun, Active and Selective Conversion of CO2 to CO on Ultrathin Au Nanowires, J. Am. Chem. Soc., 2014, 136(46), 16132–16135 CrossRef CAS PubMed.
- O. Gutiérrez-Sánchez, N. Daems, W. Offermans, Y. Y. Birdja, M. Bulut, D. Pant and T. Breugelmans, The inhibition of the proton donor ability of bicarbonate promotes the electrochemical conversion of CO2 in bicarbonate solutions, J. CO2 Util., 2021, 48, 101521 CrossRef.
- O. Gutiérrez-Sánchez, B. de Mot, N. Daems, M. Bulut, J. Vaes, D. Pant and T. Breugelmans, Electrochemical Conversion of CO2 from Direct Air Capture Solutions, Energy Fuels, 2022, 36(21), 13115–13123 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr02925a |
| This journal is © The Royal Society of Chemistry 2024 |