
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multiscale study of the chiral self-assembly of cellulose nanocrystals during the frontal ultrafiltration process†
Samuel
Mandin
a,
Lorenzo
Metilli
b,
Mohamed
Karrouch
a,
Didier
Blésès
a,
Christine
Lancelon-Pin
b,
Pierre
Sailler
b,
William
Chèvremont
 c,
Erwan
Paineau
d,
Jean-Luc
Putaux
b,
Nicolas
Hengl
a,
Bruno
Jean
b and
Frédéric
Pignon
*a
c,
Erwan
Paineau
d,
Jean-Luc
Putaux
b,
Nicolas
Hengl
a,
Bruno
Jean
b and
Frédéric
Pignon
*a
aUniv. Grenoble Alpes, CNRS, Grenoble INP (Institute of Engineering Univ. Grenoble Alpes), LRP, F-38000 Grenoble, France. E-mail: frederic.pignon@univ-grenoble-alpes.fr
bUniv. Grenoble Alpes, CNRS, CERMAV, F-38000 Grenoble, France
cESRF, The European Synchrotron, CS 40220, F-38043 Grenoble, Cedex 9, France
dUniversité Paris Saclay, CNRS, Laboratoire de Physique des Solides, 1 rue Nicolas Appert, Orsay 91405, France
First published on 23rd September 2024
Abstract
The structural organization of cellulose nanocrystal (CNC) suspensions at the membrane surface during frontal ultrafiltration has been characterized, for the first time, at the nano- and microscale by in situ small-angle X-ray and light scattering (SAXS and SALS, respectively). During filtration, the particles assembled at the membrane surface and formed the so-called concentration polarization layer (CPL), which contains CNCs arranged in a chiral nematic (cholesteric) helicoidal structure, with the long axis of the CNCs oriented parallel to the membrane surface, and the helical axis of the cholesteric structure oriented perpendicular to the membrane surface. The self-organization of CNCs in the form of oriented cholesteric structures was further characterized by a pitch gradient in the CPL. The structure of the CPL was also investigated upon release of the transmembrane pressure. SAXS data revealed a relaxation process associated with a diffusion of the CNCs from the membrane surface towards the bulk, while SALS measurements revealed a re-organization of the cholesteric phase that was preserved all along the deposit. The preservation of the observed structure after 14 days of continuous filtration followed by air-drying was confirmed using scanning electron microscopy and wide-angle X-ray diffraction, demonstrating the feasibility of the process scale-up.
Introduction
Emerging climate issues are driving the search for alternatives to fossil resources, with an emphasis on the use of renewable resources.1 This concerns all sectors of industry, including those using nanoparticles. In this context, the focus has been on exploiting particles that are both biosourced and biodegradable.2 Among the variety of available particles, cellulose nanocrystals (CNCs) have seen renewed interest in recent years. CNCs are rods with a 3–20 nm width and a length ranging from 100 nm to several micrometers, depending on the cellulose source, resulting in an aspect ratio L/D ranging from 10 to 100.3 They are prepared by acid hydrolysis of cellulose fibers, generally using sulfuric acid that imparts negatively charged sulfate half-ester surface groups, which are responsible for the electrostatic repulsion at the origin of the colloidal stability in aqueous suspension.4 Furthermore, CNC suspensions exhibit a liquid-crystal behavior. Above a critical concentration and below the onset of physical gel formation, the CNCs self-organize into a chiral nematic (cholesteric) assembly. This results in a phase separation between an upper isotropic phase and a lower anisotropic phase.5–7 The latter phase contains CNCs in a left-handed, helicoidal arrangement. The helical pitch corresponds to a rotation of 360° of the director (common alignment axis of the anisotropic/anisomeric particles) and depends on the strength of the colloidal interactions between the CNCs.8,9 This helical organization imparts specific optical properties to the materials, especially when the pitch is of the order of the wavelength of visible light. This is particularly true in Nature, where the color of many living organisms comes from the interaction between visible light and their complex nanometric structure.10–15 Controlling the pitch is therefore a key factor in obtaining good mechanical and photonic properties.16,17 The pitch size can be tuned by varying parameters such as the intrinsic properties of the CNCs (source, treatment, surface modification),18–20 their volume fraction,21,22 or the ionic strength.23–26 The addition of other particles like dopants27 can also change the pitch value. The preparation and processing methods have some effect on the pitch value, including the drying conditions,28–31 the use of external fields (sonication,8 magnetic field32,33), blade coating,34,35 3D printing,36 electrospinning,37 or electrophoretic deposition.38An alternative approach based on vacuum membrane filtration (VASA) has been applied to CNC suspensions to produce iridescent films with a helical structure.39 Other studies have investigated the phenomena occurring during membrane filtration of CNC suspensions to optimize and control this structuring process. For example, the organization of CNC suspensions in the so-called concentration polarization layer (CPL) near the membrane surface was characterized by in situ small-angle X-ray scattering (SAXS).40–45 Jin et al. filtered a CNC suspension by tangential ultrafiltration, revealing the formation of a concentrated organized layer of CNCs near the membrane surface.41 Using a similar method and in situ SAXS characterization during the filtration of the CNC suspension, Semeraro et al. demonstrated an exponential relationship between concentration and anisotropy as a function of the distance from the membrane surface in the CPL.45 However, all these previous works have only focused on the filtration of CNC suspensions at the nanometer scale. A more comprehensive understanding of the structuring process is needed to promote and improve the properties of CNC-based materials.
Past experiments were carried out with a multiple lengthscale approach, either in static conditions to determine the angle of rotation of neighboring CNC pseudo-layers in cholesteric structures7 or under flow,46 which provided new insights on the interpretation of the typical three-regime rheological behavior of liquid crystalline CNC suspensions reported earlier.26,47–53 However, the in situ evolution of the CNC microstructure in the CPL during filtration, relevant for characterizing optical properties at larger lengthscales than nanometers, remains unexplored.
In this work, a multi-lengthscale approach was applied for the first time by combining in situ SAXS and small-angle light scattering (SALS). The analyses were first carried out on suspensions to establish their phase diagram and their intrinsic characteristics at rest and at equilibrium. In situ SAXS and SALS analyses were then implemented to characterize the evolution of the structural organization of the CNCs accumulated near the membrane surface during frontal ultrafiltration. Different conditions were explored by varying the filtration and/or relaxation times and the initial concentration of the filtered suspension. Based on these results, a concentrated deposit was prepared under optimized filtration conditions, dried, and characterized by scanning electron microscopy (SEM) and wide-angle X-ray diffraction (WAXD).
Materials and methods
Cellulose nanocrystals
CNCs were purchased from the UMaine Development Center (University of Maine, USA) as an aqueous suspension with a stock concentration of 11.5 wt%. This suspension was sonicated at 130 kJ g−1 L−1 (Branson Digital sonifier, Emerson Electric Company, USA) to break the CNC aggregates. The 11.5 wt% sonicated suspension was diluted with deionized water to reach the desired concentration. The dimensions of the CNCs were measured from transmission electron microscopy (TEM) images of negatively stained preparations with a JEOL JEM-2100 Plus microscope operating at 200 kV. Typical TEM images are available as ESI (Fig. S1†). In this study, the same CNC suspension as that used by Mandin et al.42 was used, i.e. with CNCs that were 127.4 ± 34.1 nm-long and 10.9 ± 2.6 nm-wide, corresponding to an aspect ratio of 12.3 ± 4.3 (Fig. S2†).Phase diagram
To determine the phase diagram, CNC suspensions at different concentrations were placed in 2 mm quartz cuvettes (Hellma Analytics, Germany) sealed with parafilm and left to rest for 48 h (a sufficient time to reach complete phase separation). Photographs were taken with a camera (KY-F55B, JVC, Japan) with the quartz cuvettes placed between crossed polarizers illuminated by white light (Fig. S3†). In this configuration, the anisotropic cholesteric phase at the bottom of the sample was birefringent. The volume occupied by the cholesteric phase was measured by image analysis using ImageJ. The percentage of anisotropic phase at each concentration ratio was calculated as the ratio of the thickness of the cholesteric phase to the total thickness of the suspension in the quartz cell.In situ characterization
 | ||
| Fig. 1 Scheme (A) and photographs of the filtration cells used for SAXS (B) and SALS (C) experiments. | ||
The samples were analyzed in situ in static conditions in quartz capillary cells and under frontal filtration in a dedicated ultrafiltration cell (Fig. 1B). In both cases, the incident beam passed through the cells. The two-dimensional scattered intensity patterns were recorded on a photon-counting pixel array detector (EIGER2-4M, Dectris, Switzerland). The scattering patterns were normalized to an absolute scale following a standard procedure.54 For all samples, background and solvent scattering were subtracted from the signal before analysis, thus obtaining the absolute scattered intensity I(q).
During filtration, the transmembrane pressure was applied for 49 min at ΔP = 1.2 × 105 Pa, then released in order to study the relaxation for 37 min. At the same time, 2D scattering patterns were recorded through the cell at different z positions and times. The vertical scan along z started at 100 μm, which corresponded to the minimal distance above which the SAXS data were available, as previously described.40,41,44 Vertical scans along z were performed during filtration, with the SAXS patterns collected every 25 μm from z = 100 to 500 μm and every 200 μm from z = 500 to 5600 μm. The 1D scattered intensity profile I(q) was calculated by azimuthal averaging of the normalized scattering patterns. Before the filtration of CNC suspensions, the cell was filled with deionized water. The normalized background scattering was first recorded and systematically subtracted from the scattering patterns of the CNC suspensions. The azimuthal averages of the scattering patterns were calculated after patching the gaps between the detector modules using the SAXS Utilities software.55 The anisotropy in the 2D patterns was analyzed using the MATLAB-based Small-Angle Scattering Evaluation Tool (SASET) software.56 Specifically, the model-free principal component analysis (PCA) method provided values for the anisotropy ranging from 0 for isotropic suspensions to 1 for fully aligned systems, quantitatively comparable to the commonly used order parameter.56 The anisotropy degree and the direction of maximum scattering ψ0 were calculated via the PCA method in the 0.02 to 2 nm−1q-ranges.
In order to measure the concentration profile in the CNC suspension as a function of filtration time and distance z from the membrane surface, the data were analyzed using the method described in a previous work.44 First, ten suspensions of known concentration from 1 to 10 wt% were probed by using a flow-through capillary cell with a diameter of 2 mm to establish the evolution of the scattered intensity as a function of CNC concentration. The scattered intensity I(q) was analyzed to measure the average interparticle distance d between the CNCs using the following formula:
| d = 2π/qpeak | (1) |
 | ||
| Fig. 2 (A) Phase diagram of the CNC suspensions (48 h at rest, preparation in ultrapure water, room temperature. (B) Evolution of CNC average interparticle distance as a function of concentration (C) evolution of pitch as a function of CNC concentration. | ||
| q = (4πn/λ) × sin(θ/2) | (2) |
During the experiments, the laser beam was directed perpendicular to the cell walls, through the sample that scattered on the Fresnel lens (Fig. S4†). For the filtration, the same CNC suspension at 6 wt% was filtered for 4.5 h at a transmembrane pressure of 1.2 × 105 Pa. At this concentration, the presence of a cholesteric anisotropic phase (83 vol%) made it possible to observe the characteristic pitch peak in the SALS profile throughout the filtration experiment. The laser beam spot size was about 250 μm, which allowed accessing the first measurement inside the deposit when the thickness of the deposit was at least thicker than 500 μm. Under the selected filtration conditions, this height limit was reached after 34 min of filtration. At this time, a concentration profile from z = 500 μm to larger z distances inside the filtration cell was accurately measured. The 2D scattering patterns were recorded vertically with 250 μm z-steps at different filtration times using a micrometric stage that allowed moving the filtration cell vertically in front of the laser beam.
The scattered intensity collected by the Fresnel lens was focused toward the camera and recorded. The azimuthal averages of the scattered intensities were calculated over the q-range from 6 × 10−4 to 4 × 10−3 nm−1.57 In addition to SAXS measurements, the SALS patterns allowed determining the evolution of the deposit structure as a function of the operating parameters in situ and during filtration. In order to measure the concentration profile in the CNC suspension as a function of filtration times and distance z from the membrane surface, nine suspensions of known concentrations from 2.0 to 10.4 wt% were measured in 2 mm-thick quartz cuvettes (Hellma Analytics, Germany). This calibration allowed establishing the evolution of the scattered intensity as a function of CNC concentration. The radial average scattered intensity I(q) was analyzed to measure the pitch value of the cholesteric P as:
| P = 4π/qpeak | (3) |
Ex situ characterization
Results and discussion
Characterization of the CNC suspensions
The CNC phase diagram is shown in Fig. 2A. Below Ci = 2.7 wt%, the CNCs were organized into an isotropic phase, while above Ca = 8 wt%, only the anisotropic cholesteric phase was present. Between these two concentrations, the samples were biphasic with an isotropic upper phase, and an anisotropic lower phase (Fig. S5A†).The CNC suspensions were analyzed by SAXS and SALS. The integration of both SAXS (Fig. S5B†) and SALS (Fig. S5C†) patterns allowed determining the evolution of the CNC interparticle distance (or swelling law, Fig. 2B) and the pitch of the cholesteric organization (Fig. 2C), respectively, as a function of concentration. The average interparticle distance decreased with increasing concentration, reaching a minimum of 30 nm at the highest measured concentration of 10.4 wt% (Fig. 2B). This swelling law followed a decreasing power law with concentration (C in wt%) given by:
| d = 97.45 × C−0.5 | (4) |
The power law exponent close to 0.5 is characteristic of a signal from long cylinders or rodlike colloids in a nematic arrangement.58 In addition, Fig. 2C shows that the pitch P of the cholesteric structures followed a decreasing power law with concentration C given by:
| P = 28.23 × C−0.9 | (5) |
The pitch decreased from 9.3 μm at the lowest concentration of 3 wt% to 2.9 μm at the highest measured concentration of 10.4 wt%. The coefficient of the power law is −0.9, close to −1 which is the coefficient found by Parker et al. under almost the same conditions (measurement of CNC suspension in flat capillaries using light diffraction).59d and P values were determined for each concentration from SAXS and SALS data and the rotation angle of CNCs in the cholesteric structures was determined by combining these values. The rotation angles increased with increasing CNC concentration (Fig. S7†), as previously shown by Schütz et al.7
Filtration monitored by in situ SAXS
 | ||
| Fig. 3 2D SAXS patterns (A) after a 23 min filtration (ΔP = 1.2 × 105 Pa, Tamb, SD = 10 m) (C) after a 37 min relaxation (ΔP = 0 Pa, Tamb, SD = 10 m). Concentration and anisotropy profiles deduced from the analysis of the scattering intensity collected from the deposits and eqn (4), under filtration (B) and relaxation (D). | ||
At the start of filtration (t = 0 min), the concentration was homogeneous over the entire height of the filtration cell, with a value of 5.7 wt% (close to the initial concentration of the suspension). When the transmembrane pressure was applied, the concentration near the membrane (z = 25 μm) increased by a factor of 4 during filtration, from 5.7 wt% at t = 0 min to 23 wt% at t = 49 min. The compactness of the near-membrane concentrated deposit was determined by plotting the interparticle distance as a function of the distance to the membrane surface for different filtration times (Fig. S9†). Typically, a 2-fold decrease in interparticle distance was observed during filtration. For example, at z = 25 μm, the initial value of d = 37.8 nm decreased to 19.2 nm after 49 min of filtration.
During filtration, the thickness of the concentrated deposit increased with filtration time, from 1200 μm at 23 min to 1400 μm at 49 min (Fig. 3B). In frontal filtration, the nanoparticles progressively accumulate, forming a thicker deposit, in contrast to tangential filtration, where shear forces near the membrane surface limit this steady growth of the deposit over time.40,44
The degree of anisotropy of the CNC structure during filtration followed the same trend as observed for the CNC concentration. Indeed, close to the membrane, the PCA value increased from 0.13 after 8 min of filtration to 0.31 at 49 min of filtration (Fig. 3B). For each filtration time, the degree of anisotropy decreased with increasing distance from the membrane surface until it reached almost 0 at the outlet of the concentrated deposit. This showed that the increase in anisotropy was correlated with an increase in concentration.45 Furthermore, the vertical orientation of the 2D scattering patterns (Fig. 3A) revealed that, in the concentrated deposit, the particles were oriented with their director parallel to the membrane surface, which was induced by the pressure forces imposed by filtration. A similar texturing effect was reported by Semeraro et al. who monitored the tangential filtration of CNCs by in situ SAXS.45 This orientation of CNCs parallel to the membrane surface can also be explained by the small interparticle distance of 19.2 nm at z = 25 μm. If this distance is compared with the width of the CNCs (10.9 ± 2.6 nm) plus their ionic cloud surrounding the particles, it can be concluded that the CNCs are forced to lie nearly parallel to one another and parallel to the membrane surface.
Fig. 3D shows the evolution of the deposit concentration profile during relaxation. After 9 min of relaxation, the shape of the profile was significantly altered, with a decrease in concentration from 23 to 16 wt% at the membrane surface, reflecting a diffusion of nanoparticles from the membrane surface towards the top of the deposit. This rapid relaxation of the deposit was also observed by Rey et al. in the profiles of relaxed LAPONITE® particle suspensions after tangential ultrafiltration.44 This relaxation in concentrated layers can be induced by the local compression/decompression properties of the deposit,60,61 as well as by the upward diffusion of CNCs from the deposit to the bulk, in order to restore a homogeneous osmotic pressure. The thickness of the deposit also increased by a factor of 2.5 with relaxation time (1400 μm under pressure and 4000 μm at 37 min relaxation). This disappearance of the concentration gradient within the CPL upon cessation of transmembrane pressure has already been reported in studies carried out on LAPONITE®62 and casein micelle63 suspensions.
In the concentrated deposit, relaxation resulted in a conservation of nanoparticle orientation over larger thicknesses than under frontal filtration, but at a lower degree of anisotropy. This can be seen from oriented scattering diffraction patterns up to 2825 μm (Fig. 3C), which became more isotropic at further distances from the membrane. A comparison of the 2D patterns in Fig. 3A and C again revealed an increase in anisotropy with increasing concentration. As long as the concentration remained above 10 wt%, the anisotropy remained almost constant with a PCA around 0.25 while below this value, it decreased to 0.1. In addition, the oriented deposit displayed a larger thickness under relaxation than under pressure. In fact, under pressure, the oriented deposit had a maximum thickness of 1250 μm, whereas, under relaxation, this thickness reached ca. 2500 μm, i.e. a 2-fold increase. It should also be noted that this organization remained very stable for 37 min, with PCA anisotropy values between 0.2 and 0.25. As the deposit relaxed, the CNC concentration near the membrane surface decreased, while that between 1000 and 2500 μm increased. This observation was likely caused by the particle reorganization between the pressurized and resting states. However, the relaxation of the CNCs took place without any change in their orientation in the deposit. Indeed, the 2D scattering patterns displayed the same degree and direction of anisotropy. Hence, we assumed that the particles kept their orientation during relaxation. The hypothesis was that the concentration was such that the particles did not have sufficient mobility (due to high viscosity6) in the relaxed deposit to change their orientation, which could also be related to the small interparticle distance between CNCs, as small as 20 to 38 nm (Fig. S9†), around 2 to 4 times the width of the CNCs (10.9 nm).
This homogenization and increasing deposit thickness with relaxation time can be explained by the cessation of pressure allowing colloidal forces between particles to govern the structuring of the system, resulting in a homogenization of osmotic pressure.62,64,65 The system tends to return to equilibrium throughout the volume, leading to homogenization of the concentration towards a given equilibrium value. Without pressure constraints in the dilute phase, CNCs acquire a higher degree of freedom, leading to a decrease in anisotropy.
However, none of the previous studies investigating the impact of relaxation during ultrafiltration studied the impact of this relaxation on particle orientation. Our results are thus essential to demonstrate that even after the transmembrane pressure has been switched off, the orientation induced by the increase in concentration under pressure in the vicinity of the membrane surface was largely retained during relaxation, certainly due to the high colloidal interaction forces at the reached concentration (23 wt%). Without a disturbing external field, when the pressure was released, the forces of re-swelling and osmotic pressure had no marked effect on the organization of the anisotropic structure achieved at these high concentrations.
Filtration monitored by in situ SALS
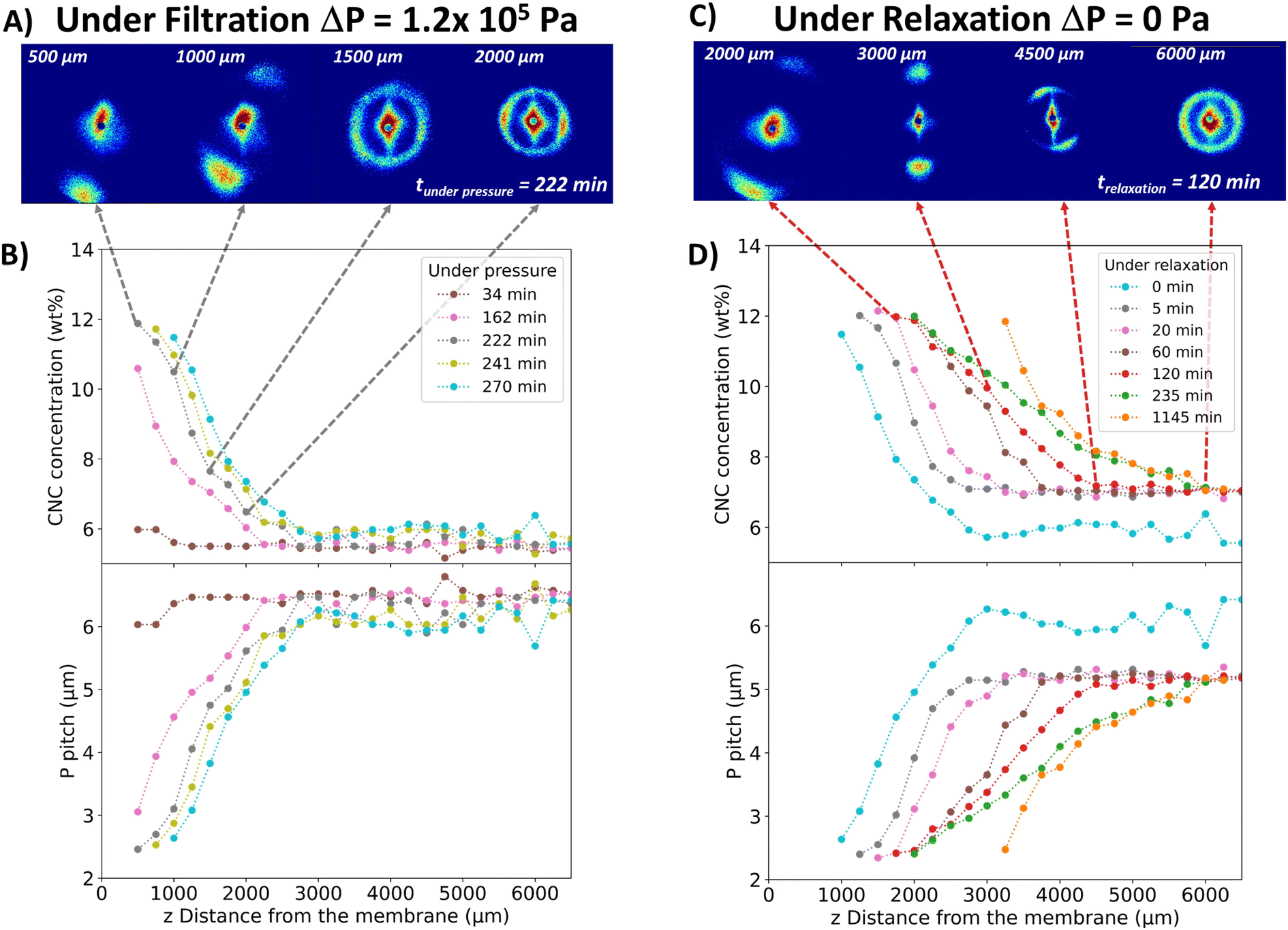 | ||
| Fig. 4 2D SALS patterns recorded under pressure (ΔP = 1.2 × 105 Pa, Tamb) at the filtration time t = 222 min (A) and evolution of the concentration and pitch profiles as a function of z and filtration time (B). 2D SALS patterns during relaxation at t = 120 min (C) and evolution of the concentration and pitch profiles as a function of z and relaxation time (D). | ||
The slight clockwise tilt of this cholesteric axis can be explained as follows. Compressed air was applied on one side of the cell only, which creates a longitudinal pressure drop along the membrane from the cell inlet to the cell outlet, inducing a slight clockwise tilt of the CNCs with respect to the membrane surface. Upon structuring induced by the transmembrane pressure and increasing concentration, this tilt of individual CNCs also induces a clockwise tilt of their cholesteric organization with respect to the vertical axis as observed in Fig. 4A near the membrane surface.
The degree of anisotropy of the 2D SALS patterns in Fig. 4A could not be determined due to the truncation of the scattering patterns recorded at z = 500 and 750 μm (closest to the membrane). However, the high anisotropy close to the membrane, which decreased as one moved away from it, correlated with the concentration decrease. This phenomenon is, therefore, analogous to that observed by SAXS, but on micrometric scale corresponding to the size of cholesteric structures.
The 2D scattering patterns were integrated to obtain the averaged azimuthal scattered intensity I(q) in order to quantify the concentration in the deposit. These curves contained a peak in intensity corresponding to the pitch value (Fig. S10†). As previously shown and since the pitch was directly related to concentration (Fig. 2C), the CNC concentration along the height of the deposit was determined (Fig. 4B). These results highlight the formation of a concentrated deposit close to the membrane with a concentration gradient, which is in line with the SAXS results. The deposit layers became denser over filtration time. For example, at z = 1250 μm, the concentration increased from 7.2 to 10.4 wt% in almost 2 h. The thickness of the concentrated deposit increased from 2250 to 3000 μm in 2 h, as deduced from SAXS data. Particle concentrations higher than 14 wt% could not be determined, since they correspond to scattering peaks outside of the detector range.
Similarly, the evolution of the pitch as a function of filtration time in the vicinity of the membrane showed a decrease with increasing filtration time, from 6 to 2.5 μm in 236 min (Fig. 4B). For all filtration times, the pitch increased with increasing distance from the membrane surface, following an inverse trend with the concentration.
This phenomenon involving the preservation of the cholesteric structural orientation during relaxation has not been reported before. Cholesteric structures that were vertically oriented close to the membrane by filtration diffuse with an overall upward movement in the cell without any significant change in orientation or organization. The system seemed to exhibit an elastic behavior that would stem from high interaction forces within the cholesteric organization rather than a fluid behavior, which would undergo more extensive reorganization under the effect of diffusive phenomena. Moreover, this occurred along the entire cell length, resulting in a homogeneous organization throughout the deposit, as discussed above.
From an energy point of view, the relaxation of a twisted cholesteric organization and its relaxation after compression by an external magnetic force have also been studied in detail by Dogic and Fraden.66 They mention that the formation of the cholesteric phase is understood as a competition between two elastic energies. On the one hand, the free energy of a chiral nematic is lowered in a twisted state because of the torque a chiral molecule exerts on its neighbor. Such a contribution to the free energy is characterized by the “twist” constant Kt. On the other hand, the creation of an elastically distorted state characterized by the usual twist elastic constant K22 increases the free energy. As a consequence, the wavelength of the cholesteric pitch is proportional to the Kt/K22 ratio. An evaluation of the twist constant Kt has been obtained from the measuring values of the twist elastic constant K22 and the corresponding pitch.
In the relaxation phenomena depicted in Fig. 4D, the evolution of the pitch shows systematically increase as a function of the distance z from the membrane surface followed by a constant value at the largest z distance. The corresponding SALS pattern in the first increase is vertically oriented and, as explained before, would suggest that the cholesteric phase exists as a single-oriented domain. For higher z-values, the SALS patterns became isotropic, while keeping a constant pitch value. This would be a signature of the reformation of isotropically distributed individual tactoids. Based on these observations and knowledge of the twist elastic constant of the cholesteric organization proposed in the literature, as well as the effect of osmotic pressure induced by the concentration gradient, it can be deduced that the relaxation in the first part where the pitch increases as a function of z, is associated with two simultaneous mechanisms: first, an elastic relaxation induced by the twist elastic constant of the cholesteric that prompts the unwinding of the cholesteric pitch and, second, an overall diffusion phenomenon from highly concentrated regions to lower concentrated regions due to osmotic effect.
In the upper part of the relaxed sample, where a constant pitch value was reached, tactoids separated from one another. The relaxation phenomena would be more associated to already existing processes well studied in the literature. For instance, Kim et al.67 have demonstrated the important role of surface anisotropy in morphogenesis of phase transitions in liquid crystals by studying the complex shapes and structure of tactoids and topological defects. The relaxation phenomena of the individual cholesteric tactoids have been very well described in the past. For example, Almohammadi et al.68 have shown that their relaxation fundamentally followed different paths, with first and second-order exponential decays, depending on the existence of splay/bend/twist orientation structures in the ground state. They proposed that the tactoids undergo relaxation on both shape and structure when the external flow field maintaining them in a non-equilibrium state is released. Convective currents can also affect the final deposition and even the orientation assemblies as shown by Davidson et al.69 during the drying of liquid crystal droplets. These authors evidenced that the drying process creates surface tension gradients accompanied by significant density and viscosity variations resulting in convective currents and the occurrence of a phase separation within the droplet.
In this section, we have shown that starting from a 6 wt% concentration with a high cholesteric phase fraction (80 vol%), cholesteric structures became oriented and denser in the concentrated layer close to the membrane surface. In order to investigate the behavior of the CNC suspension under filtration if its initial concentration was in the isotropic region of the phase diagram, a 2 wt% CNC suspension was subjected to filtration under identical conditions followed by some relaxation measurements.
Filtration at 2 wt% monitored by in situ SALS
The 2 wt% CNC suspension was filtered under the same conditions, but during much longer times. The 2D SALS patterns at 220 h of filtration are shown in Fig. 5A. The scattering rings showed the presence of a cholesteric structure even if, at the initial stage, the suspension was isotropic. Therefore, like with the filtration of the 6 wt% suspension, the concentration profiles were calculated from the integrated 2D scattering patterns (Fig. 5B). | ||
| Fig. 5 (A) 2D SALS patterns at 220 h of filtration (ΔP = 1.2 × 105 Pa, Tamb); (B) and evolution of the concentration and pitch profiles as a function of z and filtration time starting from an initial 2 wt% CNC suspension. | ||
Fig. 5B illustrates the evolution of the CNC concentration at different times during filtration as a function of distance z from the membrane surface. A concentrated deposit formed close to the membrane surface, characterized by a concentration gradient, in line with previous results. Moreover, the deposit layers became denser over time. For example, at a distance of 7000 μm from the membrane surface, the concentration increased from 6.91 to 10.26 wt% in 77 h (from t = 143 to 220 h). This densification was slower than the one observed for the 6 wt% CNC suspension. This was explained in part by the initial amount of particles, which was lower in the 2 wt% CNC suspension, meaning that the densification of the deposit formed by this sample took a longer time.
For the 2 wt% suspension, the same phenomenon of cholesteric structure orientation as with the 6 wt% sample was observed in the concentrated deposit. However, in the case of the 2 wt% suspension, the cholesteric structures in the concentrated deposit were well arranged with a more vertically oriented helical axis than in the case of the 6 wt% sample (Fig. 4A and 5A). This difference could be explained by the fact that at 6 wt%, there is already 80 vol% anisotropic phase at rest and higher colloidal interaction forces.
At low CNC concentration (2 wt%), the self-organization seems to be faster than at higher concentrations (6 wt%). This change in the speed of the densification was linked to the change in suspension viscosity (10-fold lower at 2 wt% than at 6 wt% (Fig. S11†)), and stems from weaker colloidal interaction forces at 2 than at 6 wt%.
Under the effect of an equivalent transmembrane pressure force, the system with a slower reorganization (2 wt%) concentrated more rapidly and more easily towards the membrane, inducing at the same time a higher permeation flux, promoting the accumulation of particles at the membrane. This is a well-known filtration phenomenon, for which it has been shown that, counter-intuitively, starting from a lower concentration, higher concentrations could be reached at the membrane surface at equivalent transmembrane pressure.62 This effect results in better organization and orientation of the particles in the vicinity of the membrane because colloidal forces are sufficiently weak compared with the pressure forces and local flow around the anisometric particles, which causes them to settle parallel to the membrane surface. Starting from a higher particle concentration, here at 6 wt%, colloidal forces and cholesteric phase reorganization kinetics are more strongly opposed to filtration-induced concentration and organization kinetics, and reduce the ability of local pressure and flow forces to deposit perfectly oriented particles on the membrane surface.
Another explanation of the better orientation of the CNCs from a lower initial concentration could be the confinement effect of the individual CNCs near the membrane surface. This effect has been studied in detail by Klop et al.70 on dispersions of fd-virus in microfluidic channels. The authors identified a nematic transition induced by the confinement between parallel plates. This effect was explained by the fact that when rods in an isotropic phase are confined between walls, aligning their major axis along the walls becomes entropically favorable, increasing the accessible space. This results in a positive adsorption of the rods on each wall forming a nematic layer, despite the overall concentration being below the isotropic-to-nematic transition in the bulk. In the case of the filtration process, this confinement effect is reinforced by the transmembrane pressure that induces a pressure force on the surface of the individual CNCs.
The 2D SALS patterns recorded at 7 mm from the membrane at different filtration times are shown in Fig. 6. After 70 h of filtration, no scattering was observed, which was explained by the fact that the 2 wt% suspension was isotropic (Fig. S5†). Subsequently, over time, the concentrated deposit grew, reaching 7 mm at 143 h, when the characteristic scattering ring of the cholesteric phase appeared without any particular orientation of the helical axis. The scattering patterns became anisotropic over time and the distance between scattering peaks increased, corresponding to a decrease in pitch value and, therefore, an increase in concentration with filtration time.
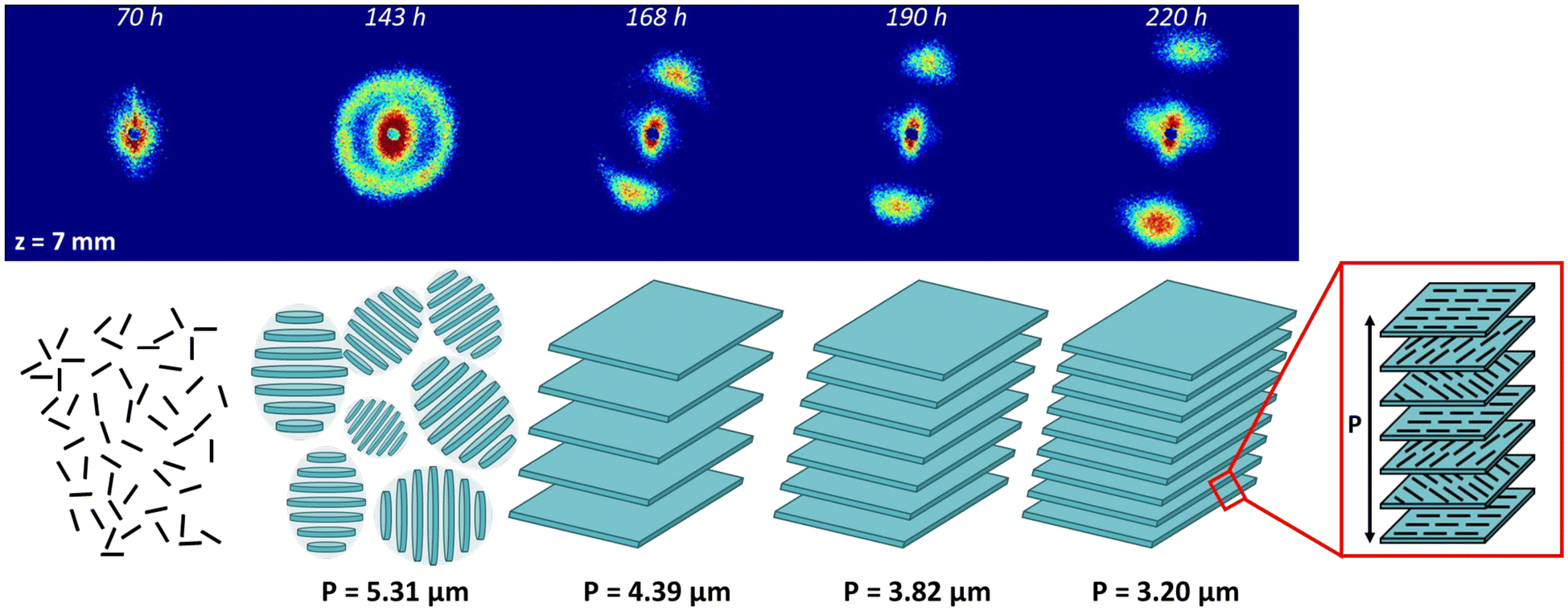 | ||
| Fig. 6 Evolution over time of the SALS pattern at z = 7 mm during filtration under ΔP = 1.2 × 105 Pa, starting from an initial CNC suspension at C = 2 wt% and corresponding schemes showing the appearance of the cholesteric structure with randomly oriented tactoids and then the formation of an oriented continue cholesteric organization with the evolution of its pitch over time. | ||
Fig. 6 shows how CPL is structured during filtration. The occurrence of the scattering ring between 70 and 143 h is the consequence of the increase of the CNC concentration during filtration, leading to the formation of randomly oriented tactoids. Then, between 143 and 168 h, as filtration time and concentration increased, the tactoids both enlarged by coalescence and oriented with their helical axes nearly perpendicular to the membrane surface, with a slight tilt due to the application of compressed air on one side. The tactoids thus formed an extended and continuous cholesteric region. Finally, as filtration time increased, the pitch of this continuous cholesteric region decreased with increasing concentration (168 to 220 h in Fig. 6). The coalescence of the tactoids to form a cholesteric continuous region with increasing CNC concentration has previously been reported during evaporative drying71 or VASA filtration72 of CNC/polymer suspensions.
The structuring of the CNCs under pressure depicted in Fig. 6 contrasts with what was observed by Pignon et al.46 under high shear followed by relaxation. In this work, a high shear was applied to the CNC suspension and, during the first step of relaxation, a fast build-up time was evidenced corresponding to a large-scale oriented nematic domain, possibly formed in this case because the previous high-shear forces have disrupted the cholesteric organization. The suspension, even at high concentration, was composed of oriented isolated particles. This situation is different from that of a structuring under pressure, which preserves the existence of a cholesteric organization, as demonstrated in Fig. 6. In the case of high-shear flow followed by relaxation, after this first step of relaxation and nematic organization, the system evolves by forming an oriented cholesteric organization after about 10 min. Then, an isotropic reorganization of the cholesteric phase occurred after 30 min.46 These relaxation phenomena have been associated with a nucleation and growth process, according to the work of Lettinga.73
Relaxation at 2 wt%
The relaxation of a suspension in a dilute domain (0.7 wt%) was studied by SAXS in previous works (ESRF report SC4177 and C. Rey thesis74). For the relaxation of a suspension in the dilute domain, these studies showed the same trend as the one for 6 wt% (Fig. 3) with a loss of the concentration gradient, and a homogenization of the concentration around a point (Fig. S12†). At the micrometric scale probed by SALS, a 2 wt% CNC filtration experiment was carried out with frontal filtration for 48 h followed by a 5 min relaxation. The results are presented in Fig. S12† and show the same relaxation trend as for the 6 wt% suspension, i.e. the diffusion of the concentrated deposit towards the top of the filtration cell.As explained before about the formation of the structuring under pressure, starting from the dilute domain (2 wt%) or concentrated domain (6 wt%) results in the same cholesteric phases without formation of an intermediate nematic organization, which could be the case if the relaxation appears after shear quenches from co-existing isotropic and nematic phases of a mixture of rod-like viruses and polymer in defined concentration regimes as described in the literature.75 To summarize, the relaxation mechanisms evidenced in a concentrated deposit obtained by filtration in the explored conditions of pressure and membrane porosity, do not differ between the case where the cholesteric structure was obtained from an isotropic dilute sample or from a biphasic sample with the co-existence of cholesteric and isotropic phases.
Ex situ characterization of CNC filtration deposits
In the previous sections, we have shown that the frontal ultrafiltration of CNC suspensions resulted in the formation of highly textured concentrated deposits exhibiting a cholesteric structure. A 2 wt% suspension was filtered for 14 days and the concentrated hydrogel formed on the membrane surface was air-dried for 48 h to form a thick self-standing deposit (Fig. S13†). Previous works reported that starting from a partial or complete cholesteric state (our case) avoided the formation of randomly oriented tactoids during drying, which would otherwise create defects in the structure of the final dry material.17,76The deposit was characterized by WAXD along two directions. Along the y-axis defined in Fig. 7A, hence parallel to the membrane, the pattern was strongly anisotropic and contained arcs associated with crystallographic planes of the CNCs (Fig. 7B). The reflections with hk0 and 00l Miller indices correspond to planes that were oriented along and perpendicular to the director of the rods, respectively. The distribution of arcs in the pattern was thus consistent with CNCs laying overall parallel to the membrane surface. The aspect of the WAXD pattern recorded along the x-axis was similar (not shown). Along the z-axis, hence perpendicularly to the membrane and parallel to the applied pressure, the pattern was isotropic, containing diffraction rings instead of arcs (Fig. 7C). This showed that, in projection, the CNCs adopted a wide range of orientations. Moreover, the fact that the 013 reflection, absent in Fig. 7B, was relatively stronger than what was expected in the powder pattern of randomly oriented CNCs,77 and that the thin 004 reflection was absent, suggests a significant uniplanarity of the CNCs. The combination of the information obtained along the different directions at the molecular scale is thus consistent with the cholesteric organization of CNCs previously deduced from the SAXS and SALS data.
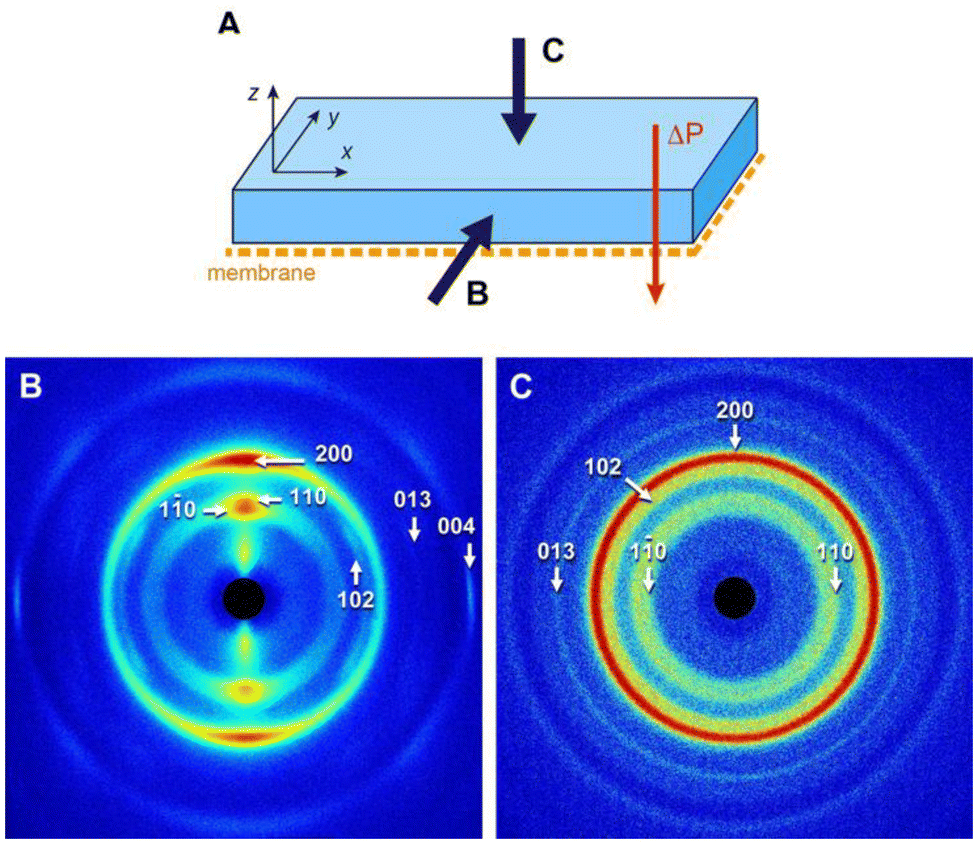 | ||
| Fig. 7 WAXD patterns recorded along two directions of the dry CNC deposit, as defined in (A) parallel (B) and perpendicular (C) to the surface. The indexing is that of the cellulose Iβ allomorph.77 | ||
SEM images of cross-sections of the fractured deposit are shown in Fig. S14† and 8. It must be noted that although the goal was to fracture the deposit in planes perpendicular to the y-axis (longitudinal view) and to the z-axis (planar view), as defined in Fig. 7A, the resulting surfaces were highly uneven, with a wide range of local surface orientations, due to a variety of paths of least resistance in the structure. Consequently, the selected images are those of regions that were assumed to be the closest to the intended orientation. The structure observed in the longitudinal cross-section revealed a layered organization (Fig. 8A and B). The orientation of these bands parallel to the membrane surface is consistent with the planar orientation adopted by CNCs under filtration, as was demonstrated by SAXS, SALS, and WAXD analyses. The period of the pseudo-layers in Fig. 8B was about 200 nm, which is smaller than the half-pitch of the cholesteric structure determined by SALS, due to the sample drying. Images recorded on a surface perpendicular to the x-axis were rather similar to the longitudinal views (not shown).
 | ||
| Fig. 8 SEM images of fragments prepared by fracturing the dry CNC deposit perpendicular (A and B) and parallel (C–F) to the surface. Low-magnification views of the fragments are shown in Fig. S15.† The images in (B), (D), and (F) are higher magnification views of the regions indicated by a black rectangle in (A), (C), and (E), respectively. In (A) and (B), the fracture surface was nearly perpendicular to the deposit surface, while in (C) and (D), it was nearly parallel to the deposit surface. (E) and (F) correspond to an oblique fracture. | ||
When the deposit was fractured perpendicularly to the z-axis, overlapping lamellae were observed in which the CNCs appeared to be fairly parallel over several micrometers (Fig. 8C and D), which is consistent with the CNC organization in an individual pseudo-layer of the cholesteric structure. Since the relative orientation of the CNCs in neighboring lamella appears to be similar, one lamella likely corresponds to one half-pitch of the structure and only a few top layers can be visible on the surface. However, within each lamella, the neighboring CNC layers rotate with respect to each other within a 180° angular range, which explains why the WAXD pattern in Fig. 7C, that integrated the whole thickness of the deposit, was isotropic.
The 3D model in Fig. 9A shows various sections of a cholesteric structure made of rods. The front surface contains bands that resemble the SEM image in Fig. 8B, with rods being perpendicular to the surface every half-pitch. The top surface is tilted by a 4° angle with respect to the z-axis and illustrates the parallel arrangement of rods in a single nematic layer, the layer below or above being rotated by a small angle. This view resembles the SEM image in Fig. 8D corresponding to a fracture plane more or less parallel to the membrane.
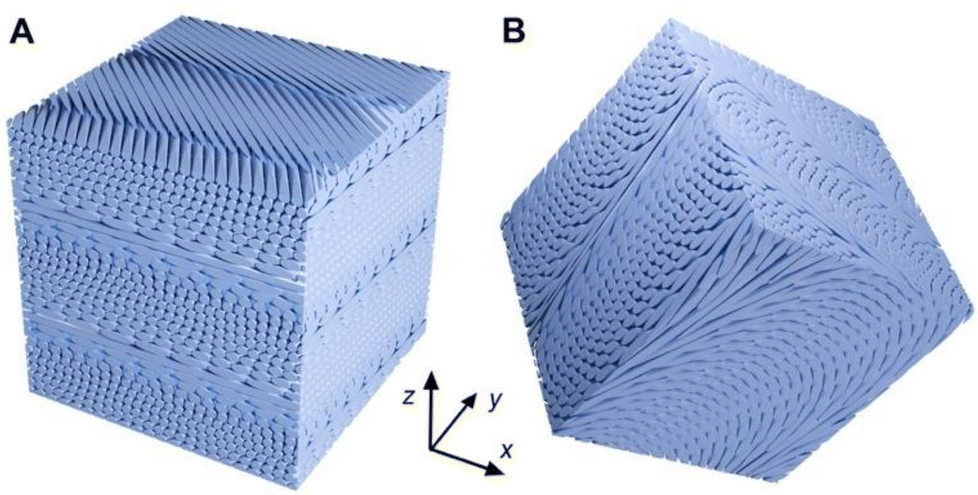 | ||
| Fig. 9 Tridimensional cubic models of a cholesteric structure cut along different planes using the Blender software.78 (A) The cube was cut with a 4° tilt angle around the x-axis; (B) the cube was cut with tilts of 30, 10, and 20° around the x, y, and z axes, respectively. | ||
The image in Fig. S15† shows a region of the deposit in which distinct fracture planes could be seen closely resembling those in Fig. 8A and C: a “cliff” with narrowly spaced layers corresponding to a longitudinal view of the material and overlapping lamellae corresponding to a more planar view of the cholesteric structure.
When the fracture plane was randomly oriented, regularly spaced so-called Bouligand arches were observed, typical of the cholesteric organization described in various works11,79,80 (Fig. 8E and F). The apparent spacing of the arches varied depending on the angle of the fracture plane with respect to the axis of the cholesteric structure, as illustrated by the 3D scheme in Fig. 9B.
The observation of the cholesteric structure of CNCs in SEM images correlates with the SALS analyses, showing that the cholesteric structure was oriented with the helicoidal axis nearly perpendicular to the membrane surface. The regularity of the periodic layered structure (Fig. 8A and B) showed that the CNC concentration was homogeneous over large regions of the deposit, as was demonstrated during the study of the CNC relaxation and amplified by drying (long relaxation time).
Conclusions
The mechanisms of orientation and organization of the CNCs during frontal ultrafiltration under pressure and during the release of the pressure were studied from the structural point of view over lengthscales ranging from nanometers to micrometers by in situ SAXS and SALS. Under pressure, the CNCs in the polarization layer were oriented with their director parallel to the membrane surface, which promoted the formation of cholesteric structures with their axis oriented nearly perpendicular to the membrane surface. Furthermore, inside the CPL, a concentration gradient was established as a function of the distance from the membrane, leading to a pitch gradient of the cholesteric structure. Finally, starting from a dilute suspension in the isotropic region of the phase diagram, the cholesteric structures formed under filtration were better organized and oriented than starting from the biphasic region. The self-organization of CNCs in the form of oriented cholesteric structures with a pitch gradient in the CPL was evidenced for the first time.Under relaxation, an overall diffusion of the cholesteric organizations from the concentrated layer near the membrane surface towards the more diluted upper region was observed. This resulted in a homogenization of the concentration while preserving the orientation induced by filtration. The novelty lies in the observation that the CNCs organized in a cholesteric structure by filtration retained their orientation even after cessation of the transmembrane pressure. This phenomenon was explained by an elastic behavior of the system due to the strong interaction forces at high concentration which would not be perturbed by the diffusion when pressure was released. Finally, the observation of Bouligand arches in SEM images of fractured solid deposits confirmed that the cholesteric structure was preserved upon drying.
This work provided a better understanding of the phenomena involved in the filtration of cellulose nanocrystals. By controlling filtration parameters, we could control the particle orientation and concentration, as well as the pitch of the cholesteric structure over a wide range of lengthscales. It would therefore be interesting to use this approach to manufacture functional cellulosic materials with controlled CNC orientations and organizations over several lengthscales.
Author contributions
Samuel Mandin: writing review & editing, writing original draft, methodology, investigation, formal analysis. Lorenzo Metilli: writing review & editing, investigation, formal analysis. Mohamed Karrouch: investigation. Didier Blésès: investigation. Christine Lancelon-Pin: writing – review & editing, investigation, formal analysis. Pierre Sailler: investigation. William Chèvremont: investigation, formal analysis. Erwan Paineau: writing review & editing, investigation, formal analysis. Jean-Luc Putaux: writing – review & editing, investigation, formal analysis. Nicolas Hengl: writing review & editing, investigation, formal analysis. Bruno Jean: writing – review & editing, investigation, formal analysis. Frédéric Pignon: writing review & editing, writing original draft, validation, supervision, project administration, methodology, investigation, formal analysis.Data availability
Data from ESRF can be found online at https://doi.esrf.fr/10.15151/ESRF-ES-1030392887.Treated data (excel files, graphs): https://mycore.core-cloud.net/index.php/s/hBq1f0EbtGcEmxn.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
We thank Theyencheri Narayanan (ESRF, Grenoble) for fruitful discussions and for his kind help in scattering experiments, Frédéric Hugenell and Eric Faivre (Laboratoire Rhéologie et Procédés) for technical assistance. ESRF is acknowledged for the provision of synchrotron beamtime (proposal SC-5319). We thank also Fanny Bosson (Laboratoire Rhéologie et Procédés) and Thomas Gibaud (Laboratoire de Physique ENS de Lyon) for their help during the experiments at ESRF. The authors acknowledge the support of the French Agence Nationale de la Recherche (ANR), under grant agreement #ANR-20-CE43-0015 (ANISOFILM) for financial support of S. Mandin's PhD thesis and L. Metilli's postdoctoral position. The authors would like to thank the NanoBio-ICMG Platform (UAR 2607, Grenoble) for granting access to the electron microscopy facilities, and Yoshiharu Nishiyama (CERMAV) for advice in the indexing of XRD patterns. LRP is part of LabEx Tec21 (Investissements d'Avenir – grant agreement #ANR-11-LABX-0030). LRP and CERMAV are part of Institut Carnot PolyNat (Investissements d'Avenir – grant agreement #ANR-16-CARN-0025-01), and the Glyco@Alps programme (Investissements d'Avenir – grant agreement #ANR-15-IDEX-02).References
- M. N. Belgacem and A. Gandini, Monomers, Polymers and Composites from Renewable Resources, Elsevier, 2011 Search PubMed.
- K. De France, Z. Zeng, T. Wu and G. Nyström, Adv. Mater., 2021, 33, 2000657 CrossRef CAS PubMed.
- M. Y. Khalid, A. Al Rashid, Z. U. Arif, W. Ahmed and H. Arshad, J. Mater. Res. Technol., 2021, 14, 2601–2623 CrossRef CAS.
- R. H. Marchessault, F. F. Morehead and N. M. Walter, Nature, 1959, 184, 632–633 CrossRef CAS.
- J.-F. Revol, H. Bradford, J. Giasson, R. H. Marchessault and D. G. Gray, Int. J. Biol. Macromol., 1992, 14, 170–172 CrossRef CAS PubMed.
- E. E. Ureña-Benavides, G. Ao, V. A. Davis and C. L. Kitchens, Macromolecules, 2011, 44, 8990–8998 CrossRef.
- C. Schütz, M. Agthe, A. B. Fall, K. Gordeyeva, V. Guccini, M. Salajková, T. S. Plivelic, J. P. F. Lagerwall, G. Salazar-Alvarez and L. Bergström, Langmuir, 2015, 31, 6507–6513 CrossRef PubMed.
- T. G. Parton, R. M. Parker, G. T. van de Kerkhof, A. Narkevicius, J. S. Haataja, B. Frka-Petesic and S. Vignolini, Nat. Commun., 2022, 13, 2657 CrossRef CAS PubMed.
- S.-A. Jin, E. G. Facchine, S. A. Khan, O. J. Rojas and R. J. Spontak, J. Colloid Interface Sci., 2021, 599, 207–218 CrossRef CAS PubMed.
- R. M. Parker, G. Guidetti, C. A. Williams, T. Zhao, A. Narkevicius, S. Vignolini and B. Frka-Petesic, Adv. Mater., 2018, 30, 1704477 CrossRef PubMed.
- Y. Bouligand, Tissue Cell, 1972, 4, 189–217 CrossRef CAS.
- B. D. Wilts, H. M. Whitney, B. J. Glover, U. Steiner and S. Vignolini, Mater. Today: Proc., 2014, 1, 177–185 Search PubMed.
- M. Mitov, Soft Matter, 2017, 13, 4176–4209 RSC.
- D. Lee, in Nature's Palette, University of Chicago Press, 2010 Search PubMed.
- O. Karthaus, Biomimetics in Photonics, CRC Press, 2012 Search PubMed.
- S. Nikolov, M. Petrov, L. Lymperakis, M. Friák, C. Sachs, H.-O. Fabritius, D. Raabe and J. Neugebauer, Adv. Mater., 2010, 22, 519–526 CrossRef CAS.
- B. Frka-Petesic, T. G. Parton, C. Honorato-Rios, A. Narkevicius, K. Ballu, Q. Shen, Z. Lu, Y. Ogawa, J. S. Haataja, B. E. Droguet, R. M. Parker and S. Vignolini, Chem. Rev., 2023, 123, 12595–12756 CrossRef CAS PubMed.
- J. Araki, M. Wada and S. Kuga, Langmuir, 2001, 17, 21–27 CrossRef CAS.
- E. Kloser and D. G. Gray, Langmuir, 2010, 26, 13450–13456 CrossRef CAS PubMed.
- F. Azzam, L. Heux and B. Jean, Langmuir, 2016, 32, 4305–4312 CrossRef CAS PubMed.
- K. W. Klockars, B. L. Tardy, M. Borghei, A. Tripathi, L. G. Greca and O. J. Rojas, Biomacromolecules, 2018, 19, 2931–2943 CrossRef CAS PubMed.
- C. Honorato-Rios and J. P. F. Lagerwall, Commun. Mater., 2020, 1, 1–11 CrossRef.
- C. Browne, V. S. Raghuwanshi, G. Garnier and W. Batchelor, J. Colloid Interface Sci., 2023, 650, 1064–1072 CrossRef CAS PubMed.
- X.-M. Dong, T. Kimura, J.-F. Revol and D. G. Gray, Langmuir, 1996, 12, 2076–2082 CrossRef CAS.
- J. Pan, W. Hamad and S. K. Straus, Macromolecules, 2010, 43, 3851–3858 CrossRef CAS.
- S. Shafiei-Sabet, W. Y. Hamad and S. G. Hatzikiriakos, Cellulose, 2014, 21, 3347–3359 CrossRef CAS.
- H. Thérien-Aubin, A. Lukach, N. Pitch and E. Kumacheva, Angew. Chem., 2015, 127, 5710–5714 CrossRef.
- S. Beck, J. Bouchard and R. Berry, Biomacromolecules, 2011, 12, 167–172 CrossRef CAS PubMed.
- J. A. Kelly, A. M. Shukaliak, C. C. Y. Cheung, K. E. Shopsowitz, W. Y. Hamad and M. J. MacLachlan, Angew. Chem., Int. Ed., 2013, 52, 8912–8916 CrossRef CAS PubMed.
- A. Tran, W. Y. Hamad and M. J. MacLachlan, Langmuir, 2018, 34, 646–652 CrossRef CAS PubMed.
- B. Vollick, P.-Y. Kuo, M. Alizadehgiashi, N. Yan and E. Kumacheva, ACS Omega, 2017, 2, 5928–5934 CrossRef CAS.
- B. Frka-Petesic, G. Guidetti, G. Kamita and S. Vignolini, Adv. Mater., 2017, 29, 1701469 CrossRef PubMed.
- J.-F. Revol, L. Godbout, X.-M. Dong, D. G. Gray, H. Chanzy and G. Maret, Liq. Cryst., 1994, 16, 127–134 CrossRef CAS.
- J. A. Diaz, X. Wu, A. Martini, J. P. Youngblood and R. J. Moon, Biomacromolecules, 2013, 14, 2900–2908 CrossRef CAS PubMed.
- B. E. Droguet, H.-L. Liang, B. Frka-Petesic, R. M. Parker, M. F. L. De Volder, J. J. Baumberg and S. Vignolini, Nat. Mater., 2022, 21, 352–358 CrossRef CAS PubMed.
- M. K. Hausmann, P. A. Rühs, G. Siqueira, J. Läuger, R. Libanori, T. Zimmermann and A. R. Studart, ACS Nano, 2018, 12, 6926–6937 CrossRef CAS PubMed.
- Y. Liu and P. Wu, Adv. Funct. Mater., 2020, 30, 2002193 CrossRef CAS.
- S. Atifi, M.-N. Mirvakili, C. A. Williams, M. M. Bay, S. Vignolini and W. Y. Hamad, Adv. Mater., 2022, 34, 2109170 CrossRef CAS PubMed.
- Q. Chen, P. Liu, F. Nan, L. Zhou and J. Zhang, Biomacromolecules, 2014, 15, 4343–4350 CrossRef CAS PubMed.
- Y. Jin, N. Hengl, S. Baup, F. Pignon, N. Gondrexon, A. Magnin, M. Sztucki, T. Narayanan, L. Michot and B. Cabane, J. Membr. Sci., 2014, 453, 624–635 CrossRef CAS.
- Y. Jin, N. Hengl, S. Baup, F. Pignon, N. Gondrexon, M. Sztucki, A. Romdhane, A. Guillet and M. Aurousseau, Carbohydr. Polym., 2015, 124, 66–76 CrossRef CAS PubMed.
- S. Mandin, L. Metilli, M. Karrouch, C. Lancelon-Pin, J.-L. Putaux, W. Chèvremont, E. Paineau, N. Hengl, B. Jean and F. Pignon, Carbohydr. Polym., 2024, 122162 CrossRef CAS PubMed.
- L. Metilli, H. Ugo, W. Chèvremont, C. Picard and F. Pignon, Soft Matter, 2023, 19, 8228–8239 RSC.
- C. Rey, N. Hengl, S. Baup, M. Karrouch, E. Gicquel, A. Dufresne, H. Djeridi, R. Dattani, Y. Jin and F. Pignon, ACS Sustainable Chem. Eng., 2019, 7, 10679–10689 CrossRef CAS.
- E. F. Semeraro, N. Hengl, M. Karrouch, L. J. Michot, E. Paineau, B. Jean, J.-L. Putaux, C. Lancelon-Pin, L. Sharpnack and F. Pignon, Colloids Surf., A, 2020, 584, 124030 CrossRef CAS.
- F. Pignon, M. Challamel, A. De Geyer, M. Elchamaa, E. F. Semeraro, N. Hengl, B. Jean, J.-L. Putaux, E. Gicquel, J. Bras, S. Prevost, M. Sztucki, T. Narayanan and H. Djeridi, Carbohydr. Polym., 2021, 260, 117751 CrossRef CAS PubMed.
- S. Onogi and T. Asada, Rhéology, Springer, 1980, 127–147 Search PubMed.
- W. J. Orts, L. Godbout, R. H. Marchessault and J.-F. Revol, Macromolecules, 1998, 31, 5717–5725 CrossRef CAS.
- M. Alizadehgiashi, A. Khabibullin, Y. Li, E. Prince, M. Abolhasani and E. Kumacheva, Langmuir, 2018, 34, 322–330 CrossRef CAS PubMed.
- B. Derakhshandeh, G. Petekidis, S. Shafiei-Sabet, W. Y. Hamad and S. G. Hatzikiriakos, J. Rheol., 2013, 57, 131–148 CrossRef CAS.
- A. D. Haywood, K. M. Weigandt, P. Saha, M. Noor, M. J. Green and V. A. Davis, Soft Matter, 2017, 13, 8451–8462 RSC.
- T. Ebeling, M. Paillet, R. Borsali, O. Diat, A. Dufresne, J.-Y. Cavaillé and H. Chanzy, Langmuir, 1999, 15, 6123–6126 CrossRef CAS.
- S. Shafiei-Sabet, W. Y. Hamad and S. G. Hatzikiriakos, Langmuir, 2012, 28, 17124–17133 CrossRef CAS PubMed.
- T. Narayanan, M. Sztucki, T. Zinn, J. Kieffer, A. Homs-Puron, J. Gorini, P. Van Vaerenbergh and P. Boesecke, J. Appl. Crystallogr., 2022, 55, 98–111 CrossRef CAS PubMed.
- M. Sztucki, SAXSutilities2 Zenodo, 2021 Search PubMed.
- M. Muthig, S. Prévost, R. Orglmeister and M. Gradzielski, J. Appl. Crystallogr., 2013, 46, 1187–1195 CrossRef CAS.
- J.-M. Piau, M. Dorget, J.-F. Palierne and A. Pouchelon, J. Rheol., 1999, 43, 305–314 CrossRef CAS.
- C. Baravian, L. J. Michot, E. Paineau, I. Bihannic, P. Davidson, M. Impéror-Clerc, E. Belamie and P. Levitz, Europhys. Lett., 2010, 90, 36005 CrossRef.
- R. M. Parker, B. Frka-Petesic, G. Guidetti, G. Kamita, G. Consani, C. Abell and S. Vignolini, ACS Nano, 2016, 10, 8443–8449 CrossRef CAS PubMed.
- M. Loginov, F. Doudiès, N. Hengl, F. Pignon and G. Gésan-Guiziou, J. Membr. Sci., 2020, 595, 117498 CrossRef CAS.
- M. Loginov, F. Doudiès, N. Hengl, M. Karrouch, N. Leconte, F. Garnier-Lambrouin, J. Pérez, F. Pignon and G. Gésan-Guiziou, J. Membr. Sci., 2021, 630, 119289 CrossRef CAS.
- F. Pignon, M. Abyan, C. David, A. Magnin and M. Sztucki, Langmuir, 2012, 28, 1083–1094 CrossRef CAS PubMed.
- M. Loginov, F. Doudiès, N. Hengl, M. Karrouch, N. Leconte, F. Garnier-Lambrouin, J. Pérez, F. Pignon and G. Gésan-Guiziou, J. Membr. Sci., 2021, 630, 119289 CrossRef CAS.
- Y. Bessiere and P. Bacchin, Recents Prog. Genie Procedes, 2007, 1–8 Search PubMed.
- Y. Bessiere, D. F. Fletcher and P. Bacchin, J. Membr. Sci., 2008, 313, 52–59 CrossRef CAS.
- Z. Dogic and S. Fraden, Langmuir, 2000, 16, 7820–7824 CrossRef CAS.
- Y.-K. Kim, S. V. Shiyanovskii and O. D. Lavrentovich, J. Phys.: Condens. Matter, 2013, 25, 404202 CrossRef PubMed.
- H. Almohammadi, S. A. Khadem, M. Bagnani, A. D. Rey and R. Mezzenga, Nat. Commun., 2022, 13, 2778 CrossRef CAS PubMed.
- Z. S. Davidson, Y. Huang, A. Gross, A. Martinez, T. Still, C. Zhou, P. J. Collings, R. D. Kamien and A. G. Yodh, Nat. Commun., 2017, 8, 15642 CrossRef CAS PubMed.
- K. E. Klop, R. P. A. Dullens, M. P. Lettinga, S. A. Egorov and D. G. A. L. Aarts, Mol. Phys., 2018, 116, 2864–2871 CrossRef CAS.
- P.-X. Wang, W. Y. Hamad and M. J. MacLachlan, Nat. Commun., 2016, 7, 11515 CrossRef PubMed.
- Z. Wang, Y. Yuan, J. Hu, J. Yang, F. Feng, Y. Yu, P. Liu, Y. Men and J. Zhang, Carbohydr. Polym., 2020, 245, 116459 CrossRef CAS PubMed.
- M. P. Lettinga, K. Kang, A. Imhof, D. Derks and J. K. G. Dhont, J. Phys.: Condens. Matter, 2005, 17, S3609 CrossRef CAS.
- C. Rey, Mécanique des fluides [physics.class-ph], Université Grenoble Alpes, 2017, Français, 〈NNT : p. 2017GREAI124〉, 〈tel-01820653〉 Search PubMed.
- M. P. Lettinga, Z. Dogic, H. Wang and J. Vermant, Langmuir, 2005, 21, 8048–8057 CrossRef CAS PubMed.
- J. H. Park, J. Noh, C. Schütz, G. Salazar-Alvarez, G. Scalia, L. Bergström and J. P. F. Lagerwall, ChemPhysChem, 2014, 15, 1477–1484 CrossRef CAS PubMed.
- A. D. French, Cellulose, 2014, 21, 885–896 CrossRef CAS.
- B. Foundation blender.org – Home of the Blender project – Free and Open 3D Creation Software, https://www.blender.org/, (accessed July 8, 2024).
- Y. Bouligand, C. R. Chim., 2008, 11, 281–296 CrossRef CAS.
- J. Majoinen, E. Kontturi, O. Ikkala and D. G. Gray, Cellulose, 2012, 19, 1599–1605 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr02840f |
| This journal is © The Royal Society of Chemistry 2024 |