
Heterostructure seed-mediated synthesis of zinc phosphide quantum dots for bright band-edge emission†
Ju ho
Kim
a,
Hyekyeong
Kwon
a,
Myoungho
Jeong
b and
Jiwon
Bang
 *a
*a
aDepartment of chemistry, Incheon National University Yeonsu-gu, Incheon 22012, Republic of Korea. E-mail: jwbang@inu.ac.kr
bSamsung Future Technology Campus, 130 Samsung-ro, Yeongtong-gu, Suwon, Korea Republic
First published on 4th September 2024
Abstract
This study explores the synthesis of colloidal zinc phosphide quantum dots (QDs) by a novel In(Zn)P cluster seed-mediated approach, addressing the challenge of achieving low-cost, high-quality, nontoxic QDs suitable for optoelectronic applications. By intentionally limiting the amount of In precursor added to a hot solvent containing Zn and P precursors, In-rich In(Zn)P cluster seeds were formed. Subsequently, these clusters served as seeds for the growth of zinc phosphide nanocrystals, effectively using the remaining Zn and P precursors for further crystal growth. The synthesized QDs exhibited a tetragonal-like Zn3P2 structure and exceptional optical properties, including band-edge photoluminescence (PL) emission under ambient conditions. A ZnS shell was applied to further enhance the PL intensity, achieving a PL quantum yield of 40% and an average PL decay lifetime of 74 ns, while significantly improving the stability of the QDs. Temperature-dependent PL spectroscopy revealed significant resistance to thermal quenching with an exciton dissociation energy of 62 meV, underscoring the potential of this approach for advancing the field of optoelectronics. This method provides a pathway to fabricate zinc phosphide-based QDs with controlled optical properties and highlights the effective use of earth-abundant materials in the development of environmentally benign photonic materials.
Introduction
Colloidal quantum dots (QDs) have garnered considerable attention in the field of optoelectronics owing to their unique optical properties, which can be tuned with the size, shape and composition of the QDs.1 Although Cd–chalcogenide and Pb–halide perovskite QDs have excellent optical properties in the visible range, their toxicity restricts their practical applications. As an alternative, heavy-metal-free InP-based QDs have been extensively studied owing to their excellent light emission properties, including intense and stable emissions with narrow bandwidths in the visible spectral range. These QDs have already been integrated as down-converting phosphors in displays and are being considered for other applications, such as biomedical imaging and sensors.2–5 However, the scarcity and high cost of In pose challenges to the widespread use of InP-based QDs, driving the search for more sustainable alternatives.Binary zinc phosphide crystals exist in various compounds such as Zn3P2, ZnP2, and ZnP4, with tetragonal-Zn3P2 being the most stable phase under zinc-rich conditions among the various Zn–P compounds with polymorphs.6,7 Tetragonal-Zn3P2, an earth-abundant semiconductor characterized by a large optical absorption coefficient,8 an ideal direct bandgap of 1.5 eV suited for solar light harvesting, and a long minority carrier diffusion length (up to 10 μm),9,10 which enables effective responses to visible and near-infrared light. Thus, Zn3P2 has emerged as a promising candidate for optoelectronic applications, particularly as a photovoltaic material.11 In addition to the light-absorbing capabilities of Zn3P2, quantum-confined Zn3P2 nanoparticles with controlled sizes can emit across the visible spectral range. However, the synthesis of nanosized colloidal Zn3P2 QDs is associated with considerable challenges. The process requires burst nucleation and subsequent crystal growth under high-temperature pyrolysis, which is critical for controlling the QD size and enhancing its optical properties.12,13 Furthermore, the limited availability of suitable precursors and challenges in managing the colloidal synthesis process often result in QDs with suboptimal excitonic absorption and emission properties.
Dialkyl zinc, the commonly used Zn source for preparing Zn3P2 QDs via the organometallic route,11,14–16 is highly pyrophoric, explosive, and costly. This highly reactive zinc precursor is difficult to control, does not produce reproducible results, and is prone to rapid deactivation via oxidation.17 Alternatively, zinc carboxylates have been studied as precursors; however, converting highly stable Zn–P molecular intermediates into monomers is challenging, which hinders their ability to enter the burst nucleation step.22,23 Additionally, they have yet to yield high-quality QDs with the desired emission properties,18 leaving scope for further research and development in this area. To the best of our knowledge, previous studies have not reported band-edge-like emissions for colloidal Zn3P2 QDs; instead, they have demonstrated large Stokes shifted emissions with low (<5%) emission quantum yields (QYs),16,18–20 which further limits their practical applications.
Conversely, in the synthesis of colloidal InZnP alloy QDs, when both Zn and In cationic precursors are reacted with trimethylsilyl phosphine (TMS-P), the QDs typically exhibit an In-rich core with zinc predominantly on the surface,21–23 owing to the higher reactivity of In to form the InP lattice than for the Zn3P2 lattice formation.24 Inspired by this methodology, we adopted a novel approach using InP cluster seeds to facilitate the growth of Zn3P2 QDs. We reacted Zn and P precursors with a small amount of In precursor at a high temperature (approximately 270 °C). By precisely controlling the precursor concentrations and ratios, In precursors were rapidly consumed during the nucleation stage to form In(Zn)P cluster seeds. This was followed by the growth of Zn3P2 QDs, as residual Zn and P precursors were deposited on the InP seed template at a high growth temperature, resulting in high-optical-quality Zn3P2 QDs. The In(Zn)P seed-mediated synthesis of Zn3P2 QDs resulted in exceptional optical properties, including band-edge photoluminescence (PL) emission under ambient conditions. Furthermore, the shelling of Zn3P2 QDs with ZnS improved the PL QYs by approximately 40% and enhanced the stability of the QDs with resistance to thermally induced PL quenching, marking a significant advancement in the field of optoelectronic materials.
Experimental
Materials
Zinc acetate (Zn(OAc)2, 99.99%), indium acetate (In(OAc)3, 99.99%), palmitic acid (PA, >99%), and 1-octadecene (ODE, 90%) were purchased from Merck. S-pieces (99.5% purity) were purchased from Alfa Aesar. Oleylamine (OAm, >50%) and oleic acid (OA, >85%) were purchased from Alfa Aesar. Tris(trimethylsilyl)phosphine ((TMS)3P, >98%) was supplied by JI-tech, Korea. Trioctylphosphine (TOP, 97%) was purchased from Strem Chemicals. All chemicals were used as received.Synthesis of the In(Zn)P-Zn3P2 QDs
In a 50 ml three-necked round-bottom flask, 0.8 mmol of Zn(OAc)2, 0.08 mmol of In(OAc)3, and 1.8 mmol of PA were dissolved in a mixture of 9 ml ODE and 0.2 ml OAm. The reaction flask was degassed at 120 °C for 2 h. The temperature was then raised to 270 °C under a nitrogen atmosphere, and 0.16 mmol of (TMS)3P in 1 ml of TOP solution was rapidly injected into the flask. The solution was annealed at this temperature until In(Zn)P-Zn3P2 QDs of the desired size were formed. After being cooled to room temperature, the QDs were purified via centrifugation in a mixture of methyl acetate and ethanol (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 ratio) and redispersed in chloroform and hexane for optical and structural analyses, respectively.
:5 ratio) and redispersed in chloroform and hexane for optical and structural analyses, respectively.
Synthesis of the In(Zn)P-Zn3P2/ZnS QDs
Synthesis of the In(Zn)P-Zn3P2/ZnS QDs
In a 50 ml three-necked round-bottom flask, purified In(Zn)P-Zn3P2 QDs (approximately 5 mg) in chloroform were mixed with 0.8 mmol of Zn(OA)2 and 4 ml of ODE. The mixture was degassed at 100 °C for 2 h. The atmosphere was then replaced with nitrogen, and the temperature was raised to 300 °C. Subsequently, 1 ml of the TOP-S stock solution was added dropwise for 30 min, followed by additional annealing of the reaction solution at the same temperature for 30 min.Characterization
Ultraviolet-visible (UV-vis) and PL spectra were obtained using a Thermo Scientific UV-Vis spectrophotometer (Genesys 180) and Ocean Optics spectrometer (HR2000+), respectively. Absolute PL QYs were measured using an Otsuka QE 2000 instrument. Time-resolved PL measurements were performed using the time-correlated single-photon counting (TCSPC) technique. A picosecond pulsed 405 nm laser source (PicoQuant, LDH-P-C-405B) was used as the excitation source; emission photons were captured using a photon-counting photomultiplier tube (PMA 192, PicoQuant), and data processing was performed using the SPC-130 TCSPC module (PicoQuant, PicoHarp 300). X-ray diffraction(XRD) measurements were performed using a Smartlab (Rigaku) instrument with an X-ray source wavelength of 1.5412 Å. Computational XRD patterns were obtained using the Materials Project (MP)25 database version (v2024.05.02.) Transmission electron microscope (TEM) images were acquired using a Thermo Fisher Scientific FEI (Talos F200X) instrument capable of accelerating voltages of up to 200 kV. X-ray photoelectron spectroscopy (XPS) was performed using a PHI Versa Probe II (ULVAC-PHI) equipped with an Al Kα source. Temperature-dependent PL characterization was conducted using an open-cycle cryostat (ST-100, Janis) connected to a Hitachi spectrofluorometer (FS-2). Fourier-transform infrared (FTIR) spectra were obtained using a Vertex 80 V/Hyperion 2000. 1H nuclear magnetic resonance (NMR) spectra were acquired using a 400 MHz Bruker AVANCE III HD.Results and discussion
The Zn3P2 colloidal QDs were synthesized using an In(Zn)P cluster seed-mediated approach. This involved the injection of a TMS-P precursor into a solution containing Zn-palmitate and In-palmitate precursors 270 °C, while maintaining a Zn:In molar ratio in excess of 10 (Fig. 1). Notably, the formation enthalpy of the InP lattice (−88.7 kJ mol−1) is significantly lower than that of the Zn3P2 lattice (−31.6 kJ mol−1).24 This substantial difference in enthalpy, coupled with the higher reactivity of the molecular In–P intermediates compared with the Zn–P intermediates,21,26 ensures that the injected P precursor predominantly forms InP clusters via these molecular In–P intermediates. In contrast, the more stable Zn–P intermediates cannot convert to monomers with a sufficient concentration above the supersaturation level, despite the excess of Zn cations. This reaction leads to the immediate formation of tiny metal pnictide clusters with a predominant composition of In over Zn, referred to as In(Zn)P clusters,27 which serve as the seeds for Zn3P2 crystal growth. We carefully controlled the precursor concentrations to promote Zn3P2 crystal growth while inhibiting the expansion of In(Zn)P crystals on the cluster seeds. Our synthesis conditions, characterized by a relatively low concentration of the In cation precursor in the reaction vessel, primarily led to consumption of the In precursor during the initial stage of cluster seed formation. This approach effectively limited the transformation of cluster seeds into In(Zn)P QDs owing to a deficiency in the residual In precursor and an excess of the palmitate ligand. This finding was supported by control experiments conducted in the absence of the Zn precursor with all other parameters held constant. Following the injection of the P precursor into the solution containing In precursor at 270 °C, an absorption band below 400 nm emerged, indicating InP cluster formation.28 However, despite subsequent annealing at high temperatures, the absorption peak did not significantly shift into the visible range, suggesting a blockade of InP cluster growth or agglomeration into InP QDs under synthetic conditions characterized by a lower concentration of In precursor with a higher concentration of palmitic acid (Fig. S1a†). The coexistence of the preformed In(Zn)P cluster seeds with a sufficient quantity of Zn and P precursors in the reaction vessel facilitated Zn3P2 crystal growth on the In(Zn)P cluster seeds.
 | ||
| Fig. 1 Schematic of the In(Zn)P seed-mediated synthesis process for Zn3P2 QDs using In precursor as a limiting agent and the subsequent overgrowth of the ZnS shell. | ||
Fig. 2a illustrates the temporal evolution of the UV-vis absorption spectra of the Zn3P2 QD growth solution. Upon cooling to room temperature, aliquots of the reaction solution became turbid, presumably because of the presence of undissolved organic side products, which introduced a scattering background signal in the UV-vis spectra, as indicated by the absorption spectra in Fig. 2a. Despite this, the band-edge transitions of the aliquot samples were discernible over the scattering background. The band-edge absorption peak first appeared at 410 nm after the injection of the P precursor and gradually shifted to 480 nm upon annealing the solution for 5 min. This shift indicated the growth of Zn3P2 QDs on the In(Zn)P cluster seeds. In control experiments devoid of the In precursor, where the P precursor was introduced into a hot solvent containing only Zn-palmitate, the formation of Zn3P2 QDs or Zn3P2 clusters29 was not observed (Fig. S1b†). This observation suggests that the Zn–P molecular intermediates22,23 do not convert into a sufficient concentration of reactive monomer species to reach the supersaturation level, thereby failing to enter the burst nucleation phase. This finding emphasizes the necessity of a seed particle, i.e., the In(Zn)P cluster in this study, for the formation of Zn3P2 QD. Although Zn and P precursors alone cannot form the nuclei seed, they facilitate the growth of Zn3P2 crystals onto the In(Zn)P seed. The QDs, showing a 480 nm band-edge transition peak, exhibited strong quantum confinement effects because of their minute size, as evidenced by their absorption peak compared to the bulk Zn3P2 bandgap of 830 nm. Small-sized In(Zn)P-Zn3P2 (seed-core) QD samples tended to oxidize under ambient conditions, particularly during purification. This susceptibility to oxidation presents challenges for further optical and structural characterization. Moreover, no detectable emission was observed from the QDs. Hence, we prepared another batch of In(Zn)P-Zn3P2 QDs under identical experimental conditions, extending the reaction time to ensure sufficient growth of the Zn3P2 phase. For this batch, the growth solution was allowed to proceed for an extended period with minimal interruption, such as avoiding aliquot extraction from the inert reaction vessel at high temperatures to suppress oxidative layer formation during growth. Following the injection of the P precursor into the reaction vessel, the color of the solution transitioned to a yellowish-orange, which gradually turned to a reddish hue upon annealing at 270 °C. Fig. 2b presents the absorption and PL spectra of the QD samples in the growth solution. For optical analysis, crude aliquots were purified using methyl acetate as an antisolvent to remove the side products. After 30 min of growth, the samples exhibited an absorption onset at 600 nm, with a distinct band-edge absorption peak observed at approximately 520 nm. The absorption peak of In(Zn)P-Zn3P2 QDs shifted slightly to 530 nm after 2 h of annealing. Notably, aliquots of the QD growth solution demonstrated symmetrical PL emission spectra, with the PL peak gradually shifting toward red along with the absorption features as the growth time was extended (Fig. 2c). In(Zn)P-Zn3P2 QDs synthesized using In(Zn)P cluster seeds as a template maintained their optical properties under ambient air exposure for several months. The In(Zn)P-Zn3P2 QDs also preserved their colloidal stability over long periods. The surface ligands of the QDs incubated over a year were characterized by 1H NMR and FTIR spectroscopy methods. Both X-type palmitate and L-type OAm ligands were bound to the QD surface, with the palmitate ligand being the dominant species on the surface (Fig. S2†).
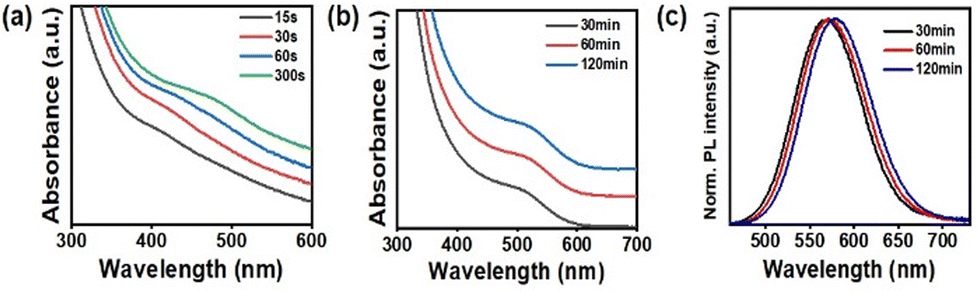 | ||
| Fig. 2 (a) Temporal evolution of the UV-vis absorption spectra of the In(Zn)P seed-mediated Zn3P2 QD growth solution during annealing at 270 °C up to 5 min. (b) UV-vis absorption spectra and (c) corresponding PL spectra of aliquots taken from the growth solution at 270 °C over extended periods up to 120 min. | ||
Structural examination of In(Zn)P-Zn3P2 QDs, with an absorption peak at 530 nm, was performed using TEM and XRD (Fig. 3). The QDs had a quasi-spherical shape with an average diameter of 2.65 ± 0.42 nm, based on measurements from 80 individual QDs (Fig. 3a). Energy-dispersive X-ray spectroscopy (EDS) (Fig. S3†) confirmed that the atomic ratio of Zn:In in the In(Zn)P-Zn3P2 QDs was 6.25:1, indicating the predominance of Zn cations rather than In cations in the composition of the QDs. Elemental analysis of surface-level elements by XPS also confirmed the dominance of Zn over In near the surface of the QDs (Fig. S4†). This finding contrasts with observations by Pietra et al., who noted an InZnP alloy structure without a dominant Zn3P2 phase for QDs, where the upper bound of the Zn/In ratio was 4, even when a feeding ratio of Zn/In of 20 was employed.30 In their study, the highly reactive TMS-P precursor, which was used as a limiting agent, led to the continuous consumption of the In precursor in competition with an excess amount of Zn precursor during QD growth, leading to InZnP alloyed structure. Conversely, in our experiments, the amount of In precursor was intentionally limited. This approach ensures that the In precursor, which is more reactive than the Zn precursor, is mostly consumed during the nucleation phase to form In(Zn)P clusters. These clusters subsequently served as seeds for the growth of Zn3P2 crystals, effectively utilizing the remaining Zn and P precursors for further crystal growth. The XRD profile of the In(Zn)P-Zn3P2 QDs showed broad diffraction patterns, which were attributed to the presence of the In(Zn)P-Zn3P2 heterostructure and their small size and imperfect crystallinity (Fig. 3d). The patterns did not reveal the cubic phase characteristics of InP and also differed significantly from those of control InZnP alloy QDs prepared using a similar method but with the Zn/In ratio adjusted to 2. This indicates that the crystal growth on the In(Zn)P seed in the reaction vessel is considerably different from InZnP alloy QD growth. Given the aforementioned discussion, an initially In-dominant In(Zn)P cluster seed was clearly formed before Zn3P2 QD growth. Previous study by Kwon et al. indicated that Zn-incorporated InP magic-sized clusters prefer a face-centered cubic (fcc) structure.27 We hypothesized that although direct nucleation and growth of Zn3P2 might be challenging, the preformed fcc phase of small In(Zn)P clusters served as the seeds for the heteroepitaxial growth of the tetragonal-like phase of Zn3P2.31,32 The observed diffraction patterns of the In(Zn)P-Zn3P2 QDs were not fully resolved and showed some discrepancies compared with those of bulk tetragonal Zn3P2; this discrepancy may be ascribed to the small physical size of 2.65 nm.19,33 Additionally, the grain size of the Zn3P2 phase should be smaller than the particle size of In(Zn)P-Zn3P2 heterostructured QDs. However, the XRD patterns of the In(Zn)P-Zn3P2 QDs showed peaks corresponding to the 2Θ reflection at 45° and the spread reflection between 50° and 60° of the tetragonal Zn3P2 structure. High-resolution TEM (HRTEM) images further supported the crystallinity of the particles (Fig. 3b). The d-spacing measured in the individual crystalline QDs, identified as 0.32 and 0.30 nm, correspond to the (202) and (212) planes, respectively, of the tetragonal Zn3P2 structure (Fig. 3c). Based on the Zn/In cation molar ratio of 6.25, as confirmed by EDS analysis, and assuming a cubic phase for the initial InP seed and a tetragonal Zn3P2 shell in the QD, the seed diameter was estimated to be 1.56 nm for the 2.65 nm-sized QD. Consequently, the volume fraction of Zn3P2 to InP in the QDs was approximately 3.9, indicating a dominant Zn3P2 lattice composition in these QDs.
 | ||
| Fig. 3 (a) TEM and (b) HRTEM images of In(Zn)P-Zn3P2 QDs. The inset in (a) presents a size distribution histogram of the QDs. (c) Line-intensity profiles extracted from individual QDs reveal several atomic planes, with lattice spacings of 0.32 nm and 0.30 nm indicated. (d) Powder XRD patterns of In(Zn)P-Zn3P2 QDs (blue) compared to control InZnP alloy QDs (black), with the blue and black bars at the bottom representing the diffraction patterns of bulk tetragonal Zn3P2 (mp-2071) and cubic InP (mp-20351) structures, respectively. | ||
The crystalline Zn3P2 QDs grown using the In(Zn)P seeds demonstrated exceptional PL characteristics, closely overlapping with the band-edge absorption energy range with symmetric features. (Fig. 2b and c) This contrasts the large Stokes shift of approximately 200 nm typically observed in solely synthesized Zn3P2 colloidal QDs, which is often attributed to mid-gap states20 or surface ZnO-induced indirect band gap emissions.19 We hypothesized that crystal growth at high temperatures (∼270 °C) on the In(Zn)P seed efficiently suppresses electronic defect sites, and can access band-to-band radiative recombination.13 The full width at half maximum (FWHM) of the PL emission reached approximately 80 nm, which can be primarily attributed to the broad size distribution of the QDs in a strong confinement regime, accounting for 16%, as shown in Fig. 2a. The band-edge excitonic excitation and radiative relaxation process in subsets of the QD ensemble were further explored by conducting photoluminescence excitation (PLE) spectroscopy on the polydispersed In(Zn)P-Zn3P2 QDs. The PLE spectra obtained at varying detection energies (Fig. 4) revealed distinctive band-edge transitions characteristic of the monodispersed subsets of the sample. The excitonic peak energies in the PLE spectra closely correlated with the detected PL energies, indicating that the PL emission of the In(Zn)P-Zn3P2 QDs was driven by a band-to-band transition. The broad band-edge PL feature in ensemble QDs results from size inhomogeneities within the sample rather than from trap state-involved relaxation processes that are commonly observed in Zn-doped InP QDs.21,34,35
 | ||
| Fig. 4 PLE spectra of the In(Zn)P-Zn3P2 QDs at various detection wavelengths, denoted by arrows in the corresponding PL spectrum (indicated in brown). The black dotted line represents the absorption spectrum of the ensemble of QDs. | ||
Dangling bonds on the surface of bare In(Zn)P-Zn3P2 QDs can trap charge carriers, resulting in a diminished PL intensity. Consequently, the PL QY of pristine QDs was less than 10%. Hence, to enhance their emission properties and photochemical stability, a wide bandgap ZnS shell layer was deposited onto the Zn3P2 QDs.18 For this procedure, an organic solution containing purified In(Zn)P-Zn3P2 QDs and Zn-oleate was heated to 300 °C. Subsequently, TOP-S, a sulfur source, was added dropwise, followed by annealing at the same temperature. The average particle size of the In(Zn)P-Zn3P2 core QDs, initially 2.65 ± 0.45 nm, increased to 3.78 nm ± 0.61 nm after ZnS shell coating (Fig. 5a), indicating that approximately 1.8 monolayers of the ZnS shell layer were deposited. Fig. 5b shows the optical absorption and PL spectra of the In(Zn)P-Zn3P2 core and In(Zn)P-Zn3P2/ZnS core/shell QDs. Both the band-edge absorption and PL peaks exhibited a slight redshift after ZnS shell coating, and the PL peak of In(Zn)P-Zn3P2/ZnS was 593 nm. Although a wide bandgap ZnS shell layer was coated onto the Zn3P2 QDs, the conduction band-offset between ZnS and Zn3P2 was reduced after band alignment,36 leading to a decreased optical bandgap as the wavefunction of the strongly confined photoexcited electron extended into the ZnS shell layer.37 The In(Zn)P-Zn3P2/ZnS core/shell QDs exhibited brighter emission, with the PL QY reaching up to 40%, as the ZnS shell layer effectively eliminated the surface trap states of the Zn3P2 core QDs. The PL decay profiles of the In(Zn)P-Zn3P2 and In(Zn)P-Zn3P2/ZnS core/shell QDs at room temperature are represented in Fig. 5c. Both the QD samples exhibited PL decay signals extending into the hundreds of nanoseconds range. However, in the case of the In(Zn)P-Zn3P2 QDs, the fast PL decay components were distinct, indicating nonradiative relaxation of charge carriers. After ZnS shell passivation, the PL signal showed a smoothly continuing decay profile, with no abrupt early-time decay following pulse excitation. This sustained decay supported the hypothesis of efficient radiative recombination over charge trapping. The average PL decay lifetime of the In(Zn)P-Zn3P2/ZnS QDs, which was calculated to be 74.3 ns, was determined by fitting the decay profile to a biexponential decay function after deconvolution with the instrument response function. The details of the fit and resulting parameters are provided in the ESI (Table S1†). The PL decay dynamics of colloidal Zn3P2 nanostructures have rarely been investigated. Previously, Miao et al. described a broad emission (∼200 nm FWHM) from colloidal Zn3P2 QDs with a ZnO thin-layer coating with a prolonged emission decay and an average lifetime extending to ∼400 ns, which was attributed to trap-related emissions or a type-II heterojunction between Zn3P2 and the surface oxide layer. Conversely, our In(Zn)P-Zn3P2/ZnS QDs demonstrated bright emission with a narrower spectral width and faster PL decay profile, confirming the interband radiative transitions of our QDs. To further analyze the PL properties of the QDs, we conducted temperature-dependent PL measurements of the InP-Zn3P2/ZnS core/shell QDs over a temperature range of 78–300 K. Fig. 5d displays the temperature-dependent PL spectra of the Zn3P2/ZnO QDs. Typically, the interaction between phonons and electrons, along with lattice expansion at elevated temperatures, leads to a reduction in the effective bandgap of bulk and nanocrystalline semiconductors as the temperature increases.38–40 Consistent with this, the QD sample exhibited a red-shift in the peak energy with increasing temperature, aligning with the behavior of bulk Zn3P2, which has a negative temperature coefficient (dEg/dT < 0).41,42 Additionally, the interaction between excitons and both optical and acoustic phonon modes became more pronounced at higher temperatures, leading to broadening of the line width of the QDs. As the temperature increased from 78 to 300 K, the PL intensity of the QDs decreased by approximately 30%. Temperature-dependent PL intensity changes are shown in Fig. 5e. The data were well-fitted using a simple Arrhenius model, as shown in eqn (1).
 | (1) |
 | ||
| Fig. 5 (a) TEM image and corresponding size distribution histogram of In(Zn)P-Zn3P2/ZnS core/shell QDs. (b) UV-vis absorption and PL emission spectra and (c) time-resolved PL decay curves of bare In(Zn)P-Zn3P2 core QDs (black) and In(Zn)P-Zn3P2/ZnS core/shell QDs (red). (d) Temperature-dependent PL emission spectra, and (e) PL peak intensity changes of In(Zn)P-Zn3P2/ZnS core/shell QDs as a function of temperature, ranging from 78 K to 300 K. | ||
From the plot, Ea, which reflects the energy required for the dissociation of band-edge excitons into nonradiative recombination channels, was determined to be 62 meV. This Ea value is higher than the reported values for Cd- and In-based visible-light-emitting QDs, which typically range from 10–30 meV.40,43–45 The higher energy required for the dissociation of bound excitons in the Zn3P2/ZnS QDs may imply that the ZnS shell layer effectively protects the excitons within the QD core, necessitating greater thermal energy for the charge carriers to escape to the surface states.46,47 This results indicate that the In(Zn)P-Zn3P2/ZnS QDs exhibit enhanced resistance to thermally induced PL quenching. Moreover, the exceptional PL properties of these QDs, achieved using both an In(Zn)P seed template and a ZnS passivation shell, covered the visible spectral range from 545 to 593 nm by varying the size of the In(Zn)P-Zn3P2 core (Fig. S5†).
Conclusions
In this study, we successfully synthesized colloidal Zn3P2 based QDs through a hot-injection approach. By incorporating small amounts of the In precursor with the Zn and P precursors, we achieved the initial formation of In(Zn)P cluster seeds, followed by the crystalline growth of Zn3P2 on these seeds. Structural analyses confirmed that the resulting In(Zn)P-Zn3P2 QDs exhibit a predominantly tetragonal Zn3P2 structure on the In(Zn)P cluster seeds. These QDs demonstrated exceptional optical properties, including band-edge PL emission under ambient conditions. Furthermore, the ZnS shell coating significantly enhanced the PL intensity of the In(Zn)P-Zn3P2 core QDs, achieving a PL QY of 40% and an average PL decay time of 74 ns. The band-edge PL peaks were tunable by controlling the core size of the Zn3P2. Temperature-dependent PL spectroscopy, spanning from 78 to 300 K, revealed high resistance to thermal quenching, with an exciton dissociation energy of 62 meV. This innovative approach utilizing InP cluster seeds represents a significant advancement in the synthesis of Zn3P2 QDs with exceptional optical properties, allowing for new developments in the field of optoelectronics.Author contributions
The study was conceptualized by J. H. K and J. B. The materials were synthesized by J. H. K and H. K. The structure analyses were conducted by M. J and J. B. Data analysis was done by J. H. K. and J. B. All authors have contributed to original draft writing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF, grant no. NRF-2022R1C1C1003667). This research was also supported by the Technology Innovation Program (20016350) funded by the Ministry of Trade, Industry & Energy (MOTIE) of Korea.References
- F. P. García de Arquer, D. V. Talapin, V. I. Klimov, Y. Arakawa, M. Bayer and E. H. Sargent, Science, 2021, 373, eaaz8541 CrossRef PubMed.
- E. Jang, Y. Kim, Y.-H. Won, H. Jang and S.-M. Choi, ACS Energy Lett., 2020, 5, 1316–1327 CrossRef CAS.
- S. M. Click and S. J. Rosenthal, Chem. Mater., 2023, 35, 822–836 CrossRef CAS.
- A. M. Saeboe, A. Y. Nikiforov, R. Toufanian, J. C. Kays, M. Chern, J. P. Casas, K. Han, A. Piryatinski, D. Jones and A. M. Dennis, Nano Lett., 2021, 21, 3271–3279 CrossRef CAS.
- P.-R. Chen, K.-Y. Lai, C.-W. Yeh and H.-S. Chen, ACS Appl. Mater., 2021, 4, 3977–3988 CAS.
- J. Dutkiewlcz, J. Phase Equilib., 1991, 12, 435–438 CrossRef.
- M. Ghasemi, E. Stutz, S. E. Steinvall, M. Zamani and A. Fontcuberta i Morral, Materialia, 2019, 6, 100301 CrossRef CAS.
- E. A. Fagen, J. Appl. Phys., 1979, 50, 6505–6515 CrossRef CAS.
- G. M. Kimball, A. M. Müller, N. S. Lewis and H. A. Atwater, Appl. Phys. Lett., 2009, 95, 112103 CrossRef.
- N. C. Wyeth and A. Catalano, J. Appl. Phys., 1979, 50, 1403–1407 CrossRef CAS.
- E. J. Luber, M. H. Mobarok and J. M. Buriak, ACS Nano, 2013, 7, 8136–8146 CrossRef CAS.
- S. G. Kwon and T. Hyeon, Small, 2011, 7, 2685–2702 CrossRef CAS PubMed.
- C. B. Murray, D. J. Norris and M. G. Bawendi, J. Am. Chem. Soc., 1993, 115, 8706–8715 CrossRef CAS.
- M. Q. Ho, R. J. A. Esteves, G. Kedarnath and I. U. Arachchige, J. Phys. Chem. C, 2015, 119, 10576–10584 CrossRef CAS.
- M. H. Mobarok, E. J. Luber, G. M. Bernard, L. Peng, R. E. Wasylishen and J. M. Buriak, Chem. Mater., 2014, 26, 1925–1935 CrossRef CAS.
- B. A. Glassy and B. M. Cossairt, Chem. Commun., 2015, 51, 5283–5286 RSC.
- L. Qu, Z. A. Peng and X. Peng, Nano Lett., 2001, 1, 333–337 CrossRef CAS.
- I. J. Paredes, C. Beck, S. Lee, S. Chen, M. Khwaja, M. R. Scimeca, S. Li, S. Hwang, Z. Lian, K. M. McPeak, S.-F. Shi and A. Sahu, Nanoscale, 2020, 12, 20952–20964 RSC.
- S. Miao, T. Yang, S. G. Hickey, V. Lesnyak, B. Rellinghaus, J. Xu and A. Eychmüller, Small, 2013, 9, 3415–3422 CrossRef CAS.
- R. A. Swain, B. F. P. McVey, H. Virieux, F. Ferrari, Y. Tison, H. Martinez, B. Chaudret, C. Nayral and F. Delpech, Chem. Commun., 2020, 56, 3321–3324 RSC.
- N. Kirkwood, A. De Backer, T. Altantzis, N. Winckelmans, A. Longo, F. V. Antolinez, F. T. Rabouw, L. De Trizio, J. J. Geuchies, J. T. Mulder, N. Renaud, S. Bals, L. Manna and A. J. Houtepen, Chem. Mater., 2020, 32, 557–565 CrossRef CAS.
- S. Koh, T. Eom, W. D. Kim, K. Lee, D. Lee, Y. K. Lee, H. Kim, W. K. Bae and D. C. Lee, Chem. Mater., 2017, 29, 6346–6355 CrossRef CAS.
- H. Kwon, S. Kim, B.-S. Cheong, J. Song and J. Bang, J. Alloys Compd., 2024, 989, 174337 CrossRef CAS.
- S. Baek, J.-S. Ha, S. Kim and S.-W. Kim, Nanoscale, 2021, 13, 13368–13374 RSC.
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder and K. A. Persson, APL Mater., 2013, 1, 011002 CrossRef.
- D. Shin, H. J. Lee, D. Jung, J. A. Chae, J. W. Park, J. Lim, S. Im, S. Min, E. Hwang, D. C. Lee, Y.-S. Park, J. H. Chang, K. Park, J. Kim, J.-S. Park and W. K. Bae, Adv. Mater., 2024, 2312250 CrossRef PubMed.
- Y. Kwon, J. Oh, E. Lee, S. H. Lee, A. Agnes, G. Bang, J. Kim, D. Kim and S. Kim, Nat. Commun., 2020, 11, 3127 CrossRef CAS.
- Y. Kwon and S. Kim, NPG Asia Mater., 2021, 13, 37 CrossRef CAS.
- H. Weller, A. Fojtik and A. Henglein, Chem. Phys. Lett., 1985, 117, 485–488 CrossRef CAS.
- F. Pietra, L. De Trizio, A. W. Hoekstra, N. Renaud, M. Prato, F. C. Grozema, P. J. Baesjou, R. Koole, L. Manna and A. J. Houtepen, ACS Nano, 2016, 10, 4754–4762 CrossRef CAS.
- H. S. Im, K. Park, D. M. Jang, C. S. Jung, J. Park, S. J. Yoo and J.-G. Kim, Nano Lett., 2015, 15, 990–997 CrossRef CAS PubMed.
- D. Yu, L. Gao, T. Sun, J. Guo, Y. Yuan, J. Zhang, M. Li, X. Li, M. Liu, C. Ma, Q. Liu, A. Pan, J. Yang and H. Huang, Nano Lett., 2021, 21, 1003–1010 CrossRef CAS PubMed.
- B. A. Glassy and B. M. Cossairt, Chem. Mater., 2016, 28, 6374–6380 CrossRef CAS.
- E. M. Janke, N. E. Williams, C. She, D. Zherebetskyy, M. H. Hudson, L. Wang, D. J. Gosztola, R. D. Schaller, B. Lee, C. Sun, G. S. Engel and D. V. Talapin, J. Am. Chem. Soc., 2018, 140, 15791–15803 CrossRef CAS.
- H. Cho, S. Jung, M. Kim, H. Kwon and J. Bang, J. Lumin., 2022, 245, 118647 CrossRef.
- J. P. Bosco, S. B. Demers, G. M. Kimball, N. S. Lewis and H. A. Atwater, J. Appl. Phys., 2012, 112, 093703 CrossRef.
- B. O. Dabbousi, J. Rodriguez-Viejo, F. V. Mikulec, J. R. Heine, H. Mattoussi, R. Ober, K. F. Jensen and M. G. Bawendi, J. Phys. Chem. B, 1997, 101, 9463–9475 CrossRef CAS.
- A. Narayanaswamy, L. F. Feiner and P. J. van der Zaag, J. Phys. Chem. C, 2008, 112, 6775–6780 CrossRef CAS.
- Y. P. Varshni, Physica, 1967, 34, 149–154 CrossRef CAS.
- B. Chon, J. Bang, J. Park, C. Jeong, J. H. Choi, J.-B. Lee, T. Joo and S. Kim, J. Phys. Chem., 2011, 115, 436–442 CAS.
- V. V. Sobolev and N. N. Syrbu, Phys. Status Solidi B, 1974, 64, 423–429 CrossRef CAS.
- E. Z. Stutz, M. Zamani, D. A. Damry, L. Buswell, R. Paul, S. Escobar Steinvall, J.-B. Leran, J. L. Boland, M. Dimitrievska and A. Fontcuberta i Morral, Mater. Adv., 2022, 3, 1295–1303 RSC.
- D. Kushavah, P. K. Mohapatra and P. Ghosh, Phys. E, 2018, 102, 58–65 CrossRef CAS.
- C. Wang, Q. Wang, Z. Zhou, W. Wu, Z. Chai, Y. Gao and D. Kong, J. Lumin., 2020, 225, 117354 CrossRef CAS.
- D. Valerini, A. Cretí, M. Lomascolo, L. Manna, R. Cingolani and M. Anni, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 235409 CrossRef.
- P. Jing, J. Zheng, M. Ikezawa, X. Liu, S. Lv, X. Kong, J. Zhao and Y. Masumoto, J. Phys. Chem., 2009, 113, 13545–13550 CrossRef CAS.
- S. S. Savchenko, A. S. Vokhmintsev and I. A. Weinstein, J. Lumin., 2022, 242, 118550 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr02524e |
| This journal is © The Royal Society of Chemistry 2024 |