
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Continuous flow synthesis and simulation-supported investigation of tunable plasmonic gold patchy nanoparticles†
Julia S.
Seifert
ab,
Nico
Nees
 c,
Hamzah
Khan
d,
Nabi E.
Traoré
ab,
Dominik
Drobek
e,
Wolfgang
Peukert
ab,
Benjamin
Apeleo Zubiri
e,
Erdmann
Spiecker
e,
Michael
Stingl
cd,
Lukas
Pflug
d and
Robin N.
Klupp Taylor
*ab
c,
Hamzah
Khan
d,
Nabi E.
Traoré
ab,
Dominik
Drobek
e,
Wolfgang
Peukert
ab,
Benjamin
Apeleo Zubiri
e,
Erdmann
Spiecker
e,
Michael
Stingl
cd,
Lukas
Pflug
d and
Robin N.
Klupp Taylor
*ab
aInstitute of Particle Technology, Friedrich-Alexander-Universität Erlangen-Nürnberg, Cauerstr. 4, 91058 Erlangen, Germany. E-mail: robin.klupp.taylor@fau.de
bInterdisciplinary Center for Functional Particle Systems, Friedrich-Alexander-Universität Erlangen-Nürnberg, Haberstr. 9a, 91058 Erlangen, Germany
cDepartment of Mathematics, Chair of Applied Mathematics (Continuous Optimization), Friedrich-Alexander-Universität Erlangen-Nürnberg, Cauerstr. 11, 91058 Erlangen, Germany
dFAU Competence Unit for Scientific Computing (FAU CSC), Friedrich-Alexander-Universität Erlangen-Nürnberg, Martensstr. 5a, 91058 Erlangen, Germany
eInstitute of Micro- and Nanostructure Research (IMN) & Center for Nanoanalysis and Electron Microscopy (CENEM), Friedrich-Alexander-Universität Erlangen-Nürnberg, Interdisciplinary Center for Nanostructured Films (IZNF), Cauerstr. 3, 91058 Erlangen, Germany
First published on 13th September 2024
Abstract
Plasmonic nanoparticles have intriguing optical properties which make them suitable candidates for sensing or theranostic applications. Anisotropic patchy particles, where metal is locally deposited on the surface of a core particle, exhibit plasmon resonances that can be specifically adjusted for these applications. However, many existing synthesis routes are complex, yield too little material, or provide particles with limited optical tunability. In this work, we present a simple and scalable continuous flow synthesis of gold-on-polystyrene patchy particles with widely adjustable optical properties. By increasing the chloride concentration in the electroless deposition of gold, we slow down the redox reduction kinetics and obtain a dense patch morphology as well as a reduced nucleation rate. The latter is counteracted by introducing a low-level seeding approach where a small number of gold nanocrystals heterocoagulate with the core particles prior to patch growth. Seeding and patch growth are performed in a continuous flow set-up with two T-shaped milli-mixers. The resulting patchy particle samples exhibit a tunable dipolar plasmon peak between 600 nm and 1100 nm. We also investigate the structure–property relationship for our gold patchy particles using finite element method simulations. After identifying a suitable patch shape model, we elucidate the influence of individual geometric parameters on the optical properties and show that the relationship holds true for a large range of patch coverages. Finally, we apply the relationship to explain the time-dependent change in the optical properties of as-synthesized patches by correlating it with the patch shape transformation revealed by electron microscopy.
1. Introduction
Over the last few decades, nanoparticles have introduced new possibilities and functionalities in a plethora of application fields, including medicine1,2 or in the chemical industry.3 Specifically, the use of plasmonic metal nanoparticles has gained attention for potential applications in theranostics4 and sensing,2,5 due to their intriguing optical properties. These particles exhibit a localised surface plasmon resonance (LSPR), which is adjustable within the visible or near-infrared (IR) spectral range by altering the particle size or morphology, enhancing absorption and scattering properties.6,7 For the use in theranostic applications, for example, plasmonic particles with LSPR positions in the near-IR region need to be designed to efficiently use the human tissue's optically transparent window.4 As the achievable LSPR range of single component spherical nanoparticles excludes positions in this required spectral region,8 particle morphologies like nanostars,9 nanorods10 or nanoshells11 have been investigated.One specific promising type of anisotropic metal particle is the patchy particle, where the metal forms a partial coating, a so-called patch, on a non-metallic core particle's surface.12 Although seemingly related to the nanoshell system, such a metal patchy particle demands a fundamentally different and, until now, often quite complex synthetic approach. Thus, patchy particle synthesis has been generally conducted in small batch processes, following templated multistep synthesis routes or physical deposition methods.13–16 Moreover, the optical resonances achievable are often restricted to broad extinction peaks and/or negligible tunability.
Our group has developed a promising synthesis of silver patches on silica core nanospheres which combines both, scalability and tunability of the optical properties.17,18 These particles can be produced via a template-free, electroless plating reaction in a reproducible and gram-scale continuous flow process, whereby the patches grow from a small number of metal nanocrystals attached to the core particle. Traditionally, flow processes for wet colloidal synthesis of plasmonic particles are known to improve the mixing and, thus, the sample homogeneity.19–22 This advantage combined with a precise adjustment of the reaction parameters enables the tuning of narrow dipolar plasmon peaks across the entire visible and near-infrared spectral range of the silver patchy particles by a change in patch size, thickness and/or density.
Nevertheless, the use of silver as a plasmonic material has significant drawbacks, including oxidation23 and, especially relevant for applications in medicine, low compatibility with biological systems.4 Thus, particularly for the latter field of application, gold is preferred due to its chemical inertness and low toxicity.24
To meet this need, our group also developed an approach for the batch synthesis of gold patches on cationic polystyrene nanospheres.25 This relied on the electrostatic attraction of tetrachloroauric ions to the core particle's surface and subsequent reduction by ascorbic acid, leading to heterogeneous nucleation and lateral growth of highly dendritic gold patches with a broad size distribution. However, these morphologies are disadvantageous as their optical spectra exhibit broad extinction peaks due to the electromagnetic coupling of the dendritic branches, with negligible tunability. More recently, we successfully transferred the batch synthesis of gold patches to a continuous flow reactor.26 Through improved mixing, the patch size distribution was narrowed, and through the adjustment of the free diffusion paths of the involved molecules, some densification of the coatings could be achieved, resulting in more prominent plasmon peaks. Nonetheless, the dipolar plasmon resonance position could not be widely tuned at the same time as maintaining the prominence of corresponding extinction peaks.
Here, we show an alternative approach for the production of dense gold patches which is inspired by our work on the silver–silica system mentioned above. There, we observed that slowing down the metal reduction kinetics influences the patch shape and shifts the growth mode from a kinetically controlled growth regime, corresponding to dendritic patches, towards a thermodynamically controlled growth regime with dense, cup-like patches, which is the prerequisite for optical tunability.27 Consequently, we considered whether a switch in the growth mode of gold patches could be achieved in a similar fashion. In the literature, it has been reported that the nucleation kinetics for the homogeneous nucleation of gold nanoparticles can be altered by the addition of halide ions.28 For a increasing concentration of halide salts, Moiraghi et al. observed larger particle sizes and claimed a reduced nucleation rate, which they proved by the presence of unreduced, kinetically trapped Au(I) species. Along these lines, Luty-Błocho et al. reported lower observed reaction rate constants for the reduction of AuCl4− with ascorbic acid under higher chloride concentrations. The authors attributed the increase in stability of the gold complex to the presence of the halide anions, which form an electrostatic barrier and inhibit electron transfer during the redox reaction.29
In the present work, we aim to show how the addition of chloride ions in a scalable continuous flow synthesis of gold patches can lead to dense patches with pronounced and tunable optical properties. Since we anticipate that a reduction in redox kinetics will also lower the heterogeneous nucleation rate and hence the patch yield (fraction of core particles possessing at least one patch), it is a further aim to introduce low-level seeding into the procedure, similar to that already realized for the silver–silica system.18 In contrast to other work on the seeded growth of gold shells,11 we will only use a small number of seed nanocrystals. By combining the seeding and growth steps using two T-mixers, we will show how the added reagent amounts can be adjusted to produce a range of patch sizes with well-defined plasmon resonances stretching from the visible to the near-infrared spectral range.
Next, we will explore the structure–property relationship for the LSPR of our gold patchy particles by defining different patch shape models for Finite Element Method (FEM) electrodynamic simulations. With this, we will study the influence of individual shape parameters on the optical properties and confirm trends comparing simulated and experimental spectral data. Finally, we will elucidate, with the support of microscopic analysis, the approximate shapes of the gold patches and investigate how these relate to an observed time-dependent change of the optical spectra in the days following patch synthesis.
2. Materials and methods
2.1 Materials
Styrene (≥99%), (vinylbenzyl)trimethylammonium chloride (VBTMAC, 99%), 2,2-azobis-2-methylpropionamidino dihydrochloride (AAPH, 97%) and gold(III) chloride trihydrate (HAuCl4, ≥99.9%) were purchased from Merck KGaA (Germany). Aluminium oxide 90 (Al2O3, neutral), sodium hydroxide (NaOH, ≥98%), L(+)-ascorbic acid (AA, ≥99%), sodium chloride (NaCl, >99.8%), potassium chloride (KCl, ≥99.5%), sodium lauryl sulfate (SDS, ≥99%), ethanol (EtOH, denatured, 96%), D(+)-sucrose (≥99.7%) and sodium borohydride (NaBH4, ≥97%) were obtained from Carl Roth GmbH & Co. KG. Ultrapure water with a resistivity of 18.2 MΩ cm was sourced from a PureLab Flex3 system by ELGA LabWater, Veolia Water Technologies Deutschland GmbH. Stock solutions of HAuCl4 (25 mM), sodium chloride (3 M) and SDS (0.6 M) were prepared and used over a timeframe of several months. All other solutions were freshly prepared for the synthesis.2.2 Synthesis of cationic core particles by emulsion polymerization
In this work, five batches of cationic polystyrene (PS) particles with different diameters were used as core particles. Their nomenclature consists of the particle material followed by the mean particle diameter obtained by scanning electron microscopy (SEM), e.g. PS74 are polystyrene core particles with a mean diameter of 74 nm. All particles were synthesized by a surfactant-free emulsion polymerization following a recipe from Meincke et al.30 The different particle sizes resulted from variations of the styrene and VBTMAC comonomer amounts which are listed in Table 1.| Core particle | Styrene content/vol% | C VBTMAC/mM | V water/mL |
|---|---|---|---|
| PS74 | 10.9 | 7.5 | 164 |
| PS115 | 10.9 | 5 | 170 |
| PS248 | 15.5 | 1 | 176 |
| PS352 | 15.5 | 0.25 | 168 |
| PS593 | 15.5 | 0 | 178 |
Before the synthesis, the inhibitor was removed from the styrene solution by mixing it with a 10 wt% NaOH solution in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio. After phase separation, the styrene was passed through an Al2O3 column until it appeared colourless. The washed styrene was stored at 8 °C before use.
:2 ratio. After phase separation, the styrene was passed through an Al2O3 column until it appeared colourless. The washed styrene was stored at 8 °C before use.
Emulsion polymerization of PS particles was performed in a round-bottom flask at 65 °C under vigorous stirring. First, ultrapure water was degassed with nitrogen for 40 min. Then, the required amount of styrene was added, followed by the comonomer dissolved in 5 mL of ultrapure water and, finally, a 5 mL aqueous solution of 0.15 M AAPH to start the reaction. The reaction solution was stirred for 24 h under nitrogen gas flow, before it was cooled down and filtered. The particles were then washed twice via centrifugation and redispersion in a 50/50 vol% water/ethanol mixture. PS74, PS248 and PS593 were synthesized in earlier work and washed according to the above-mentioned protocol before use. The PS particle stock concentrations were determined via drying of defined suspension volumes. Later, calibration curves were used applying Beer–Lambert's law.31
2.3 Synthesis of gold nanoparticle seeds
Spherical gold nanoparticles in the size range between 3 and 6 nm were used as seeds for subsequent patch growth. These particles, synthesized by a simple recipe from Deraedt et al.,32 exhibited long-term colloidal stability. First, an aqueous solution of 0.1 mM HAuCl4 was prepared. Then, an aliquot of a freshly prepared 0.05 M NaBH4 stock solution was added after one minute of dissolution under vigorous stirring. The molar ratio between HAuCl4 and NaBH4 was set to be 1:10. The solution was stirred for 15 min after the addition of NaBH4.
Due to a slow ageing phenomenon, the seed particle diameter was regularly estimated via extinction spectroscopy applying the empirical relationship from Haiss et al.33 For this, the extinction values at 450 nm (A450) and at the resonance peak position (Aspr) were obtained from the measured spectrum and used to calculate the particle diameter d following eqn (1):
 | (1) |
2.4 Non-seeded continuous flow synthesis of gold patchy particles
The initial experiments presented in this article were performed using a previously-reported single T-mixer set-up in which gold patches are grown on non-seeded cationic polystyrene particles.26 This set-up employed a single polypropylene T-mixer (2 mm maximum outer diameter) fed at a flow rate of 50 mL min−1 per inlet through polytetrafluoroethylene tubing (2 mm inner diameter) via a peristaltic pump (Reglo ICC, Cole-Parmer GmbH) from two equal volume reagent reservoirs (Fig. 1a). Under this condition, the mixing time was estimated according to be 319 ms. In the latter estimation we followed one of the established Villermaux–Dushman reaction protocols of Commenge and Falk34 (see reagent set 2b from Table 2 in addition to eqn (17) in ref. 34). The estimated mixing time is consistent with that reported in the literature for similar conditions.35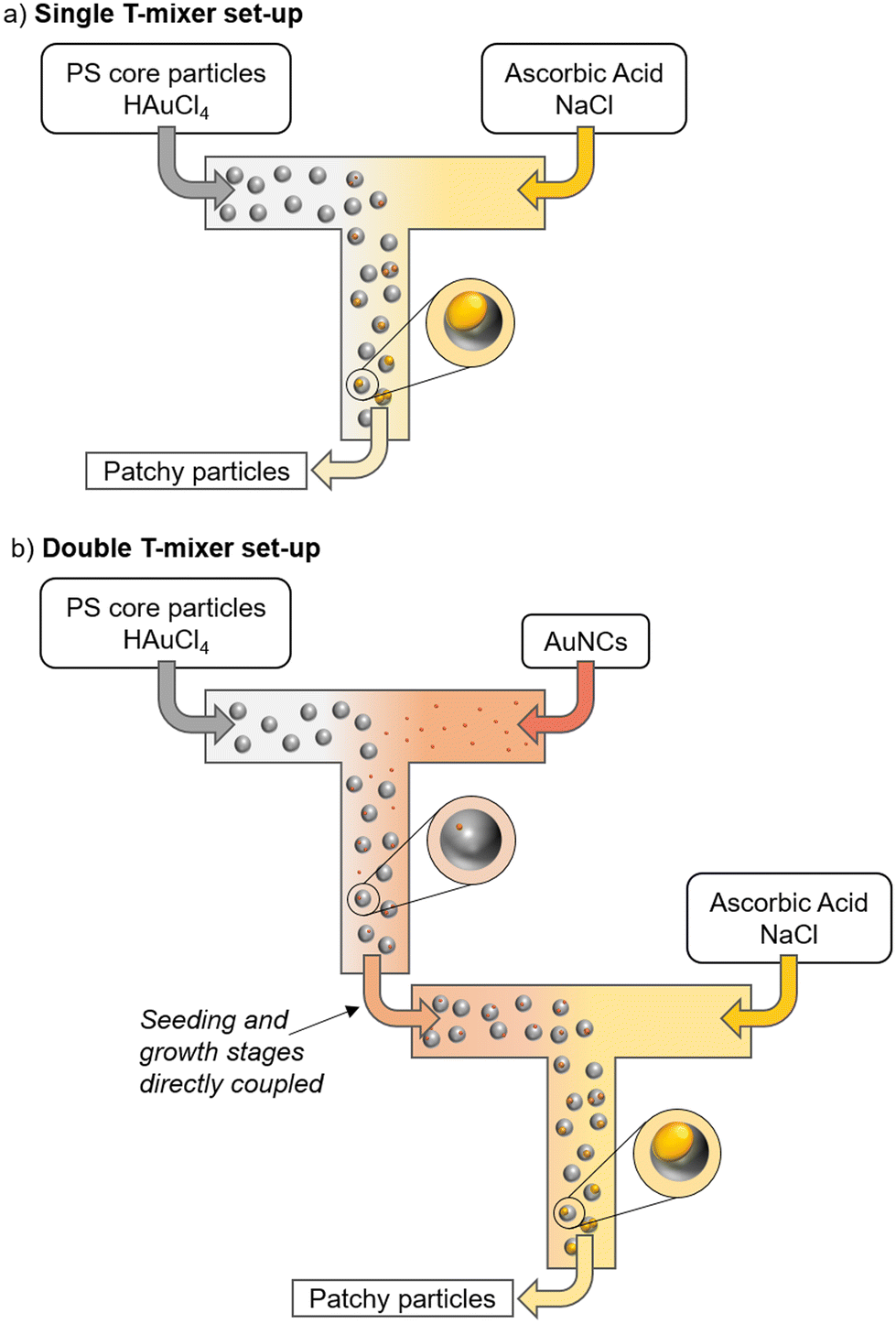 | ||
| Fig. 1 (a) Scheme of the single T-mixer set-up used for the non-seeded gold patch growth. (b) Scheme of the double T-mixer set-up used for the seeded gold patch growth. | ||
For gold patchy particle synthesis in the above set-up, Reservoir 1 comprised an aqueous solution of 20 μg mL−1 PS core particles and 120 μM HAuCl4 and Reservoir 2 comprised an aqueous solution of 50 mM ascorbic acid. In addition to these reagents which were used at concentrations similar to our earlier work,26 here we also included various concentrations of NaCl (0 mM, 4.5 mM, 20 mM, 60 mM and 120 mM) in Reservoir 2. Following the T-mixer and a 10 m reaction tube (same inner diameter and material as above), the sample was collected after two residence times had elapsed since the start of pumping (63 s at 100 mL min−1).
2.5 Seeded continuous flow synthesis of gold patchy particles
Subsequent experiments in the present work were performed using a newly commissioned double T-set-up with three reagent reservoirs (Fig. 1b and S1†) and using the same pump, T-mixer and tubing specifications as described above. Reservoir 1 and 2 were connected to the first T-mixer via 10 cm tubes, followed by a 44 cm tube between the first and the second T-mixer. Reservoir 3, containing double the volume, was pumped with two channel inlets which were merged after a 10 cm tube and the resulting stream connected to the second T-mixer after a 44 cm tube. By the use of two inlets for Reservoir 3, it could be guaranteed that, with a constant channel flow rate of 25 mL min−1, the streams mixing at the second T-mixer had an identical flow rate. After the second T-mixer, a 10 m tube was mounted to ensure sufficient time for the reaction to complete. The product was collected at the tube outlet in a snap cap vial starting after two residence times had elapsed since the start of pumping (63 s at 100 mL min−1).For a standard reaction in the double T-set-up, Reservoir 1 contained a 20 mL aqueous solution of gold seeds corresponding to an approximate seed particle to PS core particle ratio of 1.22. This ratio was found to maximize the plasmonic response as well as minimize the number of multi-patch particles (see Results and discussion). Reservoir 2 consisted of a 20 mL aqueous solution of tetrachloroauric acid (240 μM) and the PS particles (40 μg mL−1 of PS115 corresponding to a particle number of 9.56 × 1011). A 40 mL aqueous solution of ascorbic acid (50 mM) and NaCl (120 mM) was used in the third reservoir. The as-prepared patchy particles were stabilised with a suitable amount of SDS to achieve 1000× excess of a theoretical monolayer surface coverage with 4.74 × 10−19 m2 per molecule36 or alternatively with 20 wt% sucrose. Within the different experimental rows, the concentrations of the reagents were varied (see Results and discussion).
2.6 Characterization
UV-Vis-NIR-extinction spectroscopy of gold seed nanoparticles or gold patchy particles was performed with a Lambda 35 or a Lambda 950 spectrometer from PerkinElmer Inc., respectively. Samples were referenced either against water or, in the case of sucrose stabilisation, against a 20 wt% sucrose solution. Patchy particle solutions with core particles larger than 115 nm were diluted prior to measurement. Peak positions in the smoothed extinction spectra were identified with the built-in PeakFinder function in Origin.37Zeta potential measurements of gold seed nanoparticles and PS core particles were performed on a Zetasizer Nano ZS instrument (Malvern Panalytical Ltd). Seed nanoparticles were measured as-prepared and the Hückel approximation was applied in the analysis. For the PS core particles, 1 mM KCl was used as background electrolyte and the Smoluchowski approximation was applied.38
Patchy particle samples for SEM were washed five times via centrifugation and redispersion in water and dried on silicon wafers under ambient conditions. SEM micrographs were obtained by a Gemini Ultra 55 instrument (Carl Zeiss AG) using the Inlens detector with 5 kV acceleration voltage. Patch thickness at the patch centre and patch diameter, which was defined as the chord length where a sidewards facing patch touches the core particle, were obtained from image analysis of SEM micrographs with ImageJ.39,40 From this, the patch coverage (cov), the area of gold coating related to the surface of the core particle, was calculated with eqn (2):
 | (2) |
Scanning transmission electron microscopy (STEM) and energy dispersive X-ray spectroscopy (EDX) of seeded core particles were performed using a probe-corrected Thermo Fisher Scientific Spectra 200 C-FEG transmission electron microscope operating in STEM mode with a high-angle annular dark-field (HAADF) detector, collecting angles ranging from 56 to 200 mrad, and an acceleration voltage of 200 kV. Prior to experimentation, the seeded core particles were deposited from solution onto a carbon-coated TEM grid with a 200-mesh specification (Plano GmbH). Velox version 3.0.0.815 software from Thermo Fisher Scientific was employed to evaluate the molar gold contents in the obtained EDX maps. This involved polynomial background correction and Cliff–Lorimer (K-factor) quantification and fitting of the relevant elements.
Patchy particle samples for shape analysis during ageing were washed five times via centrifugation and redispersion in water before performing scanning transmission electron microscopy. A single droplet of the washed particles solutions was drop cast onto a lacey carbon-supported copper grid with 200 mesh and dried under ambient conditions. Here, STEM analysis was carried out using a double aberration-corrected FEI Titan Themis3 300 transmission electron microscope operated at an acceleration voltage of 300 kV. For all samples, the HAADF mode was performed at a collection angle range of 61 to 200 mrad, a convergence angle of 15.7 mrad, a camera length of 91 mm, a dwell time of 1 μs with a resolution of 2048 × 2048 pixel and a screen current of 80–100 pA.
All micrographs, printed in this manuscript, received contrast enhancement as post-treatment.
2.7 Electrodynamic simulations
All numerical simulations in this study were based on a finite element discretization.41,42 The performed Finite Element Method (FEM) was used to simulate a single gold patchy particle with a PS core particle of 115 nm diameter and a defined patch shape. The surrounding medium was assumed to be water. For the absorbing boundary condition in the approximations, a perfectly matched layer was employed.43 Nédélec basis functions on tetrahedral elements were chosen for the discretization.44 Since the simulated particles are rotationally symmetric and the extinction cross-section is point symmetric, it was only necessary to approximate a 1D integral over incident directions distributed on a quarter circle with two polarizations each. This allowed for the calculation of the orientation and polarization averaged extinction cross-section (a 2D integral). Specifically, the quarter circle was discretized into 64 incident directions, each with two orthogonal polarizations of the incident light. This resulted in 128 complex-valued linear systems, each with approximately 105 degrees of freedom, for each considered wavelength. The resulting linear systems were solved using the open-source direct solver HSL_MA86.45,463. Results and discussion
3.1 Densification of gold patchy particles
To achieve a scalable synthesis of dense gold patchy particles with optical tunability, we took our previous work on the seedless continuous flow synthesis of gold patches on cationic polystyrene as a starting point26 and explored the effect of chloride addition during the synthesis on the resulting patch morphology. Here, we employed a single T-mixer (Fig. 1a) with a varying concentration of NaCl (0 mM, 4.5 mM, 20 mM, 60 mM and 120 mM) in Reservoir 2 as detailed described in Section 2.4.In Fig. 2, the extinction spectra of the synthesized samples are depicted together with SEM micrographs of individual patchy particles framed in the corresponding colour. The micrographs (see also wide-field micrographs in Fig. S2 in the ESI†) indicate that with increasing NaCl concentration, the dendritic patch structures densify starting from the centre of the patches. This densification is accompanied by a thickening of the dendritic branches, resulting in the formation of surface-conformally grown dense patches. At the same time, the optical spectrum of the patchy particles undergoes changes. The fully dendritic patches exhibit a non-zero extinction over a wide range of wavelengths, while the sample with a NaCl content of 20 mM shows a clear extinction peak. However, from the SEM micrographs, it can be seen that the densification for the 20 mM sample is incomplete compared to the patches synthesized with higher chloride concentrations. More prominent optical resonances would be expected for the completely densified patch structures. However, the extinction peak greatly diminishes and the overall level of the spectrum drops for the samples with higher chloride concentrations. This drop in the extinction could be explained by different phenomena. First, aggregation of the patchy particles can lead to a flattened spectrum due to plasmonic coupling. However, this is unlikely in the present case as the product dispersions were colloidally stable over the timescale of the synthesis and spectroscopic analysis. Second, a reduced yield, i.e. the fraction of core particles with patches in a given sample, could reduce the peak and overall extinction level of the spectrum. Taking into account that the size of the patches does not change qualitatively for the different added chloride concentrations (see SEM micrographs in Fig. 2 and S2†) and that more metal needs to be integrated per patch for the densified structures, for a constant amount of gold precursor, the number of nucleated patches must vary with the chloride concentration. Consequently, for higher chloride concentrations fewer patches have nucleated, implying a reduced yield in those samples. This is supported by the fact that during the microscopic analysis only few patchy particles could be found.
 | ||
| Fig. 2 Extinction spectra and corresponding SEM micrographs for patchy particle samples synthesized with varying NaCl concentrations in a single T-mixer set-up. Wide-field micrographs can be found in Fig. S2.† The scale bar represents 100 nm and applies to all micrographs. | ||
The reduction in heterogeneous nucleation events is likely related to a lower supersaturation due to a diminished availability of Au atoms. According to Luty-Błocho et al., the added chloride ions stabilize the gold precursor ion and reduce its rate of reduction.29 As fewer Au ions are reduced concurrently, fewer nuclei are formed due to the lower supersaturation. At the same time, the decrease in reduction rate leads to a slower supply of Au atoms for the growth. Hence, the Au atoms have time to diffuse and integrate at thermodynamically more stable positions, resulting in densified structures. This shift in growth mode from kinetic towards thermodynamic control on addition of chloride ions is consistent with our observations. For subsequent experiments in the present work, we fixed the NaCl concentration at 120 mM as this resulted in highly densified patch structures. Moreover, we did not attempt to further improve the yield of the patches via the single T-mixer approach e.g. by raising the metal precursor concentration, since our earlier studies showed that this results in an increase in homogeneously nucleated metal nanoparticles.26,47
3.2 Establishing a continuous flow double T-mixer set-up for the high yield synthesis of gold patchy particles
In order to improve the yield of patchy particles with dense patches and obtain well-defined plasmon resonances, we decided to perform low-level seeding which introduces growth sites on the core particle surface. This approach, which involves attaching a small number of gold nanocrystal seeds to the core particle, has already been shown to circumvent the challenges associated with simultaneous patch nucleation and growth in the silver–silica system.18 A key difference in the present work, however, is the fact that the core and seed particles have opposite charges, facilitating the adsorption of seeds onto the core particles by heterocoagulation. Gold seeds were synthesized by the reduction of tetrachloroauric acid with sodium borohydride (see Section 2.3).32 This results in spherical gold nanocrystals in a size range of 3 to 6 nm with a mean zeta potential of −43.6 ± 3.0 mV at neutral pH. Under similar conditions, the PS core particles exhibit a mean zeta potential of +41.0 ± 0.4 mV.To achieve sufficiently good mixing of the two colloids prior to irreversible heterocoagulation of the particles, seeding was performed in a continuous flow set-up. For this, a T-mixer was added directly before the already-established process for patch growth. Fig. 1b and S1† present this newly commissioned double T-mixer continuous flow set-up for the gold patchy particle synthesis. At the first T-mixer, two flows carrying the PS core particles and gold seed nanocrystals are combined. Moreover, to maintain process simplicity, we found that the gold precursor, needed for the second growth stage, could already be included in the reservoir supplying the PS core particle solution to the seeding T-mixer. Since the excess borohydride used to produce the gold seed nanoparticles would have lost its efficacy due to its self-hydrolysis by the time of patch synthesis,48 we assume the concentration of tetrachloroauric acid to remain constant during the seeding step. Additionally, we suppose that gold precursor anions accumulate close to the surface of the cationic PS particles due to their charge differences.
The outlet of the first T-mixer, which carries the heterocoagulating PS core particles and gold seed nanoparticles along with the gold precursor, is directly connected to a second T-mixer where the reducing agent for the patch reaction (ascorbic acid) and the morphology-densifying agent (NaCl) are added. The reducing agent is added in great excess (for standard reaction parameters as noted in Section 2.5: [AA]:[HAuCl4] ≈ 416), so that complete conversion of the gold precursor is assumed. The second T-mixer is followed by a 10 m long tube with a residence time of 19 seconds where the patch growth reaction takes place. Since our earlier studies of the corresponding batch process confirmed reaction completion within a few seconds,25 we assume the patches to be fully grown by the time of sample collection.
A key parameter of this process is the seeding density ρ0 defined as the number ratio of seed nanocrystals NAuNC to PS core particles NPS added to the system (eqn (3)):
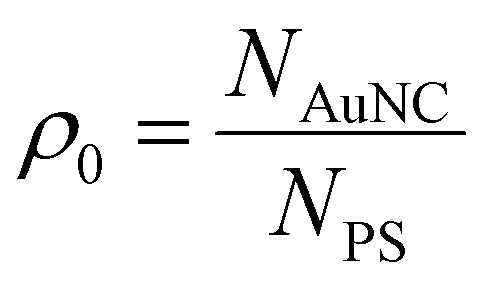 | (3) |
To verify that the gold seed nanoparticles indeed heterocoagulated with the PS cores, we collected the product after the first T-mixer. Here, the successful seeding of the core particles with the few nanometer sized gold nanocrystals was confirmed by HAADF-STEM combined with EDX analysis as shown in Fig. 3a. A corresponding HAADF-STEM micrograph at lower magnification is depicted in Fig. S3,† supporting the statistical significance of this observation. Additionally, Fig. S4† presents extinction spectra of typical gold seed nanocrystals as well as bare and seeded PS core particles.
 | ||
| Fig. 3 (a) HAADF-STEM micrographs of AuNC-seeded PS core particle (ρ0 = 1) with EDX map of a single gold nanocrystal. A wide-field micrograph of several seeded core particles can be found in Fig. S3.† (b) Extinction spectra and corresponding SEM micrographs for patchy particle samples synthesized with varying seeding density in the double T-mixer set-up. Wide-field micrographs can be found in Fig. S5.† The scale bar represents 100 nm and applies to all micrographs. | ||
Next, we operated the double T-mixer process combining both seeding and patch growth for five different values of ρ0 and, as a reference, without seeds. At the same time, the amount of gold precursor, ascorbic acid and sodium chloride were kept constant for all runs. The resulting extinction spectra with corresponding SEM micrographs of individual patchy particles are shown in Fig. 3b, with wide-field micrographs in Fig. S5.† Here, it can be observed that patch growth on seeded core particles, even at low seeding densities (ρ0 = 0.1), leads to a very strong increase in the extinction level of the spectra, with well-defined peaks compared to patch growth without seeds under the same reaction conditions (ρ0 = 0). The SEM micrographs reveal that the morphology of the patches stays dense for the seeded synthesis corresponding to the defined extinction features in the spectra. Moreover, the prominent dipolar plasmon peaks and evidence of a higher order resonance at around 700 nm in the extinction spectra of the two samples with the lowest ρ0 suggest a reasonably narrow patch size distribution, confirming that patches grow at similar rates from the previously attached seeds.
We can qualitatively infer from the increase in extinction level that the use of small numbers of seeds greatly increases the patch yield. While the SEM images in Fig. S5† appear to corroborate this, quantitative analysis of such images is not possible due to the uncertainties introduced by sample washing, drying effects, and the “hidden patch” error where small patches on the core hemisphere facing the substrate are obscured and the particle mistaken as bare.
An additional trend seen in Fig. 3b is that the peak position shifts to shorter wavelengths with increasing seeding density and the extinction maximum slightly decreases. This corresponds to the smaller patch sizes presented in the SEM micrographs. With increasing seed concentration, more patches grow and, therefore, the patch sizes decrease as the same amount of gold metal precursor has to be distributed over a larger number of seeds. Moreover, the average number of patches per particle qualitatively increases with seeding density (Fig. S5†). However, it must be stated here that the seeding density, and hence the number of seeds per core particle, is much higher than the visible number of patches per particle. This is partly due to errors that go into the calculation of ρ0. Furthermore, STEM measurements suggest that not every seed crystal grows into a patch (Fig. S6†). Unfortunately, due to the very weak contribution to the seeded PS extinction spectrum of the gold nanocrystals (Fig. S4b†) and the overlaying of the far more dominant patch resonance peaks (Fig. 3b), it is not possible to spectroscopically verify the presence of non-grown seeds in the patchy particle samples. Future studies will investigate whether seed nanocrystal defect structure or stabilization layer can result in their passivation. In order to guarantee that patchy particle samples have a low number of multipatch particles but also a high extinction level (Fig. 3b and S5†), we fixed the seeding density ρ0 in subsequent experiments to 1.22.
3.3 Tuning optical properties via reaction variables
Having demonstrated that we can synthesize dense gold patches at a high yield and adjust the size of the patches through the seeding density, we went on to explore to what extent the size of patches and, thereby, the optical properties can be controlled. The key parameter here is n*, which represents the ratio of metal precursor amount Nmetalprecursor to the number of seed nanocrystals NAuNC (eqn (4)): | (4) |
In the case of our earlier work on silver patches on silica core particles, where the continuous flow seeding and patch growth processes were carried out separately, we found that variation of the equivalent n* parameter led to a wide range of patch sizes and corresponding resonance positions being accessed.18 As shown in Fig. 4, an analogous result can be achieved with the presently discussed gold–polystyrene system. The graphs show plots of the dipolar plasmon resonance position for a large number of runs with n* spanning three orders of magnitude and variation of several other synthesis parameters. The plot in Fig. 4a depicts the trend in the dipolar plasmon peak for gold patches grown on PS115 core particles with defined NaCl (120 mM) and ascorbic acid (50 mM) concentration. Noting the logarithmic abscissa, a linear relationship between n* and the LSPR position exists up to an approximate value of 107 of the former, becoming roughly constant above this. Overall, through choice of n* the plasmon peak can be tuned in the range of 600 to 1100 nm for the chosen reactant concentrations. The achievable colour range of these patchy particle dispersions with n* values between 0.06 × 106 and 5 × 106 is depicted in the inset in Fig. 4a.
 | ||
| Fig. 4 Dipolar plasmon resonance position identified from extinction spectra plotted against n* for (a) gold patches on PS115 with 120 mM NaCl and 50 mM ascorbic acid. Exemplary raw spectra can be found in Fig. S7.† Insets: colour photograph of dispersions with n* values ranging from 0.06 × 106 to 5 × 106, SEM micrographs of distinct n* values of 0.15 × 106, 1.24 × 106 and 22.5 × 106. The scale bar represents 100 nm and applies to all micrographs. Wide-field micrographs can be found in Fig. S8;† (b) corresponding data plotted for different PS core particles sizes with 120 mM NaCl and 50 mM ascorbic acid; (c) corresponding data plotted for PS115 with 50 mM ascorbic acid and 120 mM as well as 300 mM NaCl; (d) corresponding data plotted for PS115 with 120 mM NaCl and 4 mM as well as 50 mM ascorbic acid. | ||
The SEM micrographs (Fig. 4a, further micrographs in Fig. S8†) suggest that for increasing n* the patches increase in size and the threshold n* value of 107 appears to correspond to the point at which patches cover half of the core particle surface i.e. a Janus particle morphology.49 This finding could be verified by carrying out similar synthesis campaigns with other core particle sizes. For core particles smaller than those used in Fig. 4a (PS115), the linear trend ceases at lower n* values, whereas for larger core particles the trend remains linear for the full range tested (Fig. 4b).
We also investigated the influence of the other two reagents, NaCl and ascorbic acid, on the relationship between n* and the plasmon resonance position. An increased NaCl or a decreased ascorbic acid concentration led to a roughly parallel shift of the trend to shorter wavelengths without losing the wide tunability (Fig. 4c and d). Fig. S9† shows that this corresponds in both cases to a reduced lateral growth of the patches with smoother edges, but also to an increase in patch thickness. In the case of higher chloride concentration, we correlate this change in the patch growth preference to a further reduction in the redox reaction rate due to the increase in chloride anion concentration. As we have seen above, this results in the reduced Au species having time to diffuse to thermodynamically more favourable integration points on the growing patches. Consequently, growth will not only occur at the patch edge, where the chloroauric ions are strongly attracted by the still-exposed cationic PS, but also on the solution-facing part of the patch, resulting in a thickening of the patch. In the case of a decreased ascorbic acid concentration, the effect on the reaction rate seems two-fold at first glance. Fewer reducing agent molecules are in solution leading to a reduction in reaction rate, while at the same time the pH value of the solution is increased. In more alkaline solutions, the portion of the single deprotonated form of ascorbic acid becomes more dominant and its reductive strength increases.29 Since the same trends in patch shape are observed for decreased ascorbic acid concentration and increased NaCl concentration, it can be concluded that the effect of concentration is dominating, leading to reduced supersaturation and slower patch growth. The lower reaction rate once again results in the gold patches preferentially growing away from the surface. Further investigations, based on in situ kinetic measurements, to deepen the understanding of the patch growth mechanism, are currently being performed within our group.
3.4 Investigation of gold patch morphology via FEM simulations
Having established the synthesis of dense gold patchy particles with tunable optical properties, we explored the structure–property relationship for the given system. For this, we developed a FEM model for the electrodynamic simulation of the optical properties of the gold patchy particles.During model development, simulations with three different patch shapes were performed and compared to experimental data. An example of the latter is shown in Fig. 5 for a sample with a patch thickness, defined at the patch centre, and coverage of 15 ± 6 nm and 14 ± 5%, respectively. The patch diameter and thickness were obtained from image analysis of SEM micrographs (example in Fig. 5b) and eqn (2) was then used to calculate the patch coverage. The accuracy of this analysis is limited due to the random orientations of the patches in the SEM micrographs, leading to errors in the measured patch diameter and thickness.
 | ||
| Fig. 5 (a) Comparison of the experimental extinction spectra of a patchy particle sample with 14 ± 5% coverage and a thickness of 15 ± 6 nm (black) and the simulated extinction spectra of a patch with 13% coverage and 15 nm central thickness of different models: constant thickness (dashed green), elliptical (dotted green) and curved cone (solid green); (b) SEM micrograph of the experimental sample (upper left, scale bar: 100 nm) and 3D models of the simulated patch shapes: constant thickness (upper right), elliptical (lower left) and curved cone (lower right). | ||
The simulations presented in Fig. 5a were performed with a thickness (defined at the patch centre) of 15 nm and a coverage of 13% for all shapes. The simplest shape implemented was a gold patch with constant thickness (Fig. 5b). The resulting spectrum (dashed green line in Fig. 5a) shows a peak which does not agree with the experimentally obtained one (black line). For the second, elliptical model, we took into account that the patches appear to taper towards their edges (Fig. 5b and Fig. S8†). This was incorporated in the constant thickness model by an additional parameter, the patch edge curvature rs (Fig. S10†). For the simulations presented in this work, rs was taken as 2 nm unless stated differently. By choosing an edge curvature much smaller than the central patch thickness, we obtain an elliptical patch cross-section which shifts the dipolar resonance position towards longer wavelengths (dotted green line in Fig. 5a) compared to the constant thickness model. The third model was based on the observation that the prevalent patch cross-section appears more conical than elliptical (see SEM micrographs in Fig. 5b and Fig. S8†). Therefore, we introduced a curved cone model with a tip curvature radius rw (here: rw = 16.64 nm). The tip curvature rw is defined by the length rt, which is the distance from the position of a sharp cone tip to where a smoothening tip sphere with radius rw tangentially touches the patch side length (Fig. S10b†). rt is set to 25% of the sharp cone patch side length in all simulations unless stated differently. Simulations of this curved cone model (solid green line in Fig. 5a) further red-shifted the dipole peak and increased its separation from the shorter wavelength quadrupole peak compared to the other shape models. Consequently, the curved cone model shows the best agreement with the experimental spectrum for the presented shapes. Nevertheless, the simulated spectra clearly exhibit narrower peaks compared to the experimental spectrum. This can be attributed, on the one hand, to the fact that the simulations were performed for a single, monodisperse patch size rather than a distribution of patch sizes and on the other hand that size-dependent damping due to electron surface dispersion was not taken into account.50
With the curved cone model identified as the most representative for the observed optical properties, the influence of the individual shape parameters, including patch coverage, thickness, edge radius and tip curvature radius could be systematically studied. For this, the individual parameters of interest were varied, while all other parameters in the simulations were held constant (13% coverage, H = 15 nm, rs = 2 nm, rt = 25% of patch side length). In Fig. 6 and Fig. S11,† the results of these studies are presented. Increasing coverage leads to a red shift of the dipolar and quadrupolar plasmon resonance positions (Fig. 6a), while increasing patch thickness causes a blue shift (Fig. 6b). Even more striking is that a variation of a few nanometers in the edge radius rs results in strong variations in the LSPR peak positions, with increasing radius leading to a blue shift (Fig. 6c). In contrast, variations of a few nanometers in the tip curvature rt were found to have no influence on the LSPR peak positions (Fig. S11†). As a result of these systematic variations of patch shape parameters in the curved cone model, we could find good agreement between the simulated and experimentally obtained spectral data of patches with different coverages and thicknesses (Fig. S12†).
 | ||
| Fig. 6 Extinction spectra simulated with the curved cone model for different parameters (a–c): (a) varied coverage between 2 and 13% and H = 15 nm, rs = 2 nm, rt = 25%; (b) varied patch thickness between 10 and 20 nm and 13% coverage, rs = 2 nm, rt = 25%; (c) varied edge radius rs between 2 and 5 nm and 13% coverage, H = 15 nm, rt = 25%; (d) dipolar plasmon resonance position plotted over the patch coverage for patchy particle samples synthesized with n* values up to 4.5 × 106 (black) and for simulated patches with the curved cone model (2, 4, 6 and 13% coverage, H = 15 nm, rs = 2 nm and rt = 25% (green)). | ||
Moreover, we could revisit the relationship between the dipolar plasmon resonance wavelength and n* (Fig. 4) to explore if the model results in a similar trend. As n* is a process parameter, rather than a geometric parameter, we had to first estimate the patch coverage for the corresponding experimental samples. This was achieved by analysing the diameter of patches synthesized at n* values up to 4.5 × 106 from SEM micrographs and then applying eqn (2). This led to the experimental patch coverage–LSPR wavelength-relationship shown in Fig. 6d. Corresponding simulation data for a range of patch coverages between 2 and 13% was obtained by extracting the dipolar peak positions from the spectra in Fig. 6a. This revealed a linear trend which corresponds well to the experimental data. Deviations from this trend are attributed to the measurement errors mentioned above as well as variations in patch thickness and edge radius, parameters which cannot be individually adjusted in the patch synthesis and which vary from sample to sample.
We were also able to show how the curved cone model can explain, the shift of the LSPR position-n* relationship to smaller wavelengths for an increased NaCl or a reduced ascorbic acid concentration (Fig. 4c and d respectively). As noted above, we observed in SEM micrographs (Fig. S9†) a reduced lateral spread of the patches on the surface of the core particles and subsequent thickening. Implementation of these shape changes in the curved cone model reflects in a smaller patch diameter, an increased patch thickness and an increased edge radius. As seen in Fig. 6(a–c), such variations in these parameters lead to a blue shift of the resonance wavelength, corroborating the experimentally-observed shift of the linear relationship in Fig. 4c and d.
3.5 Elucidation of the effect of post-synthesis ageing on patch shape and optical properties
As we have seen, patch densification, as afforded by chloride addition, and high-yield synthesis promoted by low-level seeding, enables the production of strongly coloured dispersions (see the inset of Fig. 4a). However, we observed throughout the work that visible changes in the appearance of the samples occurred over the course of several days following synthesis. Fig. 7a shows the results of a systematic investigation of this phenomenon for patches produced at a rather low value of n* of 0.06 × 106. Here, the dipolar plasmon resonance peak position is plotted over time for an as-prepared patchy particle sample in one case and with 20 wt% sucrose added directly after synthesis in the other. The addition of sucrose, first established by Meincke and Klupp Taylor for gold–polystyrene26 and silver–silica patchy particles,47 emerged as the most promising means to achieve long-term morphological stability. The graph in Fig. 7a shows that over time, the main dipole peak position shifts towards shorter wavelengths for both the as-prepared and sucrose-containing samples. However, the as-prepared sample shows a more significant shift within the first few days. Additionally, assuming the initial peak to be around 680 nm, by the time of the first measurement, a few hours after synthesis, the resonance of the as-prepared sample had already undergone a nearly 80 nm blue shift. In comparison, the change in the patch resonance position of the sucrose-containing sample is less pronounced. | ||
| Fig. 7 (a) Dipolar resonance position plotted over time for a sucrose-stabilized and as-prepared patchy particle sample. Below the graph: STEM micrographs of the corresponding samples after one week. The scale bar applies to both micrographs and represents 50 nm. (b) Comparison of the extinction spectra after one week of the as-prepared (dotted black) and sucrose-stabilized (solid black) patchy particle samples and simulated spectra of an elliptical (dotted green) and curved cone (solid green) patch with the same dimensions as the experimental samples. Below the graph: 3D models of the simulated curved cone (H = 10 nm, 0.6% coverage, rs = 4 nm, rw = 9 nm) and elliptical patches (H = 9.19 nm, 0.46% coverage, rs = 4 nm). | ||
To obtain information on the corresponding patch shapes, we conducted STEM (Fig. 7a) and SEM (Fig. S13†) analysis of our samples. Due to the rapid change in optical properties of the non-stabilized sample and the challenge of observing the patches in their as-prepared state by electron microscopy, these measurements were made on patches that had already been aged for a week. After this duration, the sample without sucrose exhibited elliptical, rounded patch shapes with a contact angle to the polystyrene greater than 90° (see right micrograph in Fig. 7a). In contrast, the sample with sucrose as a stabilizer maintained patches with a rounded cone morphology similar to their shape on the day of the synthesis (see left micrograph in Fig. 7a and Fig. S13†).
To elucidate if the shape change during ageing determined by electron microscopy corresponds to the observed spectral change, our newly developed structure–property model was applied. We performed FEM simulations for an elliptical (H = 9.19 nm, 0.46% coverage, rs = 4 nm) and a curved cone (H = 10 nm, 0.6% coverage, rs = 4 nm, rw = 9 nm) patch shape with dimensions derived from the STEM measurements of the samples. In the case of the as-prepared patches, the elliptical model mentioned above was modified, to represent the higher contact angle observed, by shifting the patch normal to the polystyrene surface (see 3D models in Fig. 7b). Fig. 7b depicts the experimentally determined extinction spectra of the as-prepared and sucrose-stabilized patchy particle samples after one week (black lines), where the resonance peak position of the as-prepared, degraded sample is blue-shifted compared to the stabilized sample. The simulations show good agreement with the experimental data regarding the peak positions of the spectra (green lines in Fig. 7b), indicating the morphological change as the likely reason for the larger blue shift in samples without added sucrose. As mentioned above, the differences in the peak width between the experimental and simulated spectra arise due to the latter ignoring the effect of patch size distribution and electron surface dispersion.
Based on these experimental and simulated results, we conclude that in the absence of sucrose, patches become rounded over time, leading to a blue shift of the resonance position. While this can be postulated to arise from the rearrangement of surface atoms in order to reduce interfacial energy, the exact mechanism for the morphological changes and their hindrance by sucrose are currently under investigation.
4. Conclusion
This work demonstrates the scalable continuous flow synthesis of dense gold-on-polystyrene patchy particles with widely adjustable optical properties. It also shows how a newly developed structure–property relationship based on FEM simulations can elucidate shape-related changes reflected in the optical properties.First, we showed that the addition of chloride ions enabled the synthesis of gold patches with a dense morphology by slowing down the redox reaction kinetics. However, this approach also reduced the patch yield, the fraction of coated particles in a sample. To address this, we introduced a low-level seeding step. This involved the heterocoagulation of negatively charged gold nanoparticles, synthesized by the reduction of tetrachloroauric acid with sodium borohydride, with the cationic PS core particles. This, and the subsequent patch growth reaction were realised in a double T-mixer continuous flow set-up. By adjusting the metal precursor to seed ratio (n*), we could control the patch size and, thereby, the optical properties. We found that the main dipolar peak position scales linearly with the log of n* across the visible and near-infrared range. This relationship holds true for different core particle sizes and varied reaction conditions, shifting the LSPR peak position to shorter wavelengths for higher chloride or lower ascorbic acid concentrations.
Next, we developed a structure–property relationship for our gold patchy particles. Using FEM electrodynamic simulations, we tested different patch models, including constant thickness, elliptical and curved cone patch shapes. We found that the curved cone model fitted best to our experimentally observed spectral data. Using this model, we investigated the influence of individual shape parameters, such as coverage, thickness, edge and tip curvature radius, on the resulting optical properties. Here we could show that the trend observed for the LSPR position-n* relationship can be replicated with the model for a large range of patch coverages.
Finally, we used the developed structure–property relationship to explain the change in the optical properties of our patchy particle samples during post-synthesis ageing. We observed a continuous blue shift in the resonance position of the patches over several days, which could be mitigated by the addition of sucrose. We used STEM and SEM analysis to visually confirm that the patch developed from a rounded cone to a more elliptical shape for samples without sucrose addition. By performing FEM simulations for these morphologies, we confirmed that the observed blue shift in the plasmon resonance position was indeed correlated with a rounding of the patches.
This study presents a widely tunable synthesis of anisotropic plasmonic nanoparticles, which can be easily scaled-up, using high volume reservoirs or through numbering up, to meet the requirements of applications, e.g. in sensing, labelling or catalysis. Moreover, our newly developed structure–property relationship facilitates the study of inaccessible shape parameters and reduces the necessary experimental time. Current studies in our group make use of in situ kinetic measurements and advanced electron microscopy (4D STEM) in order to elucidate in more detail the patch growth mechanism as well as the influence of the seed morphology on the patch shape and size distribution. These are key steps towards a process–structure–property relationship which will in future be inverted to identify process conditions which lead to high quality, narrowly distributed patches with a sharp LSPR peak at an arbitrarily desired wavelength.
Author contributions
Julia S. Seifert: conceptualization, data curation, formal analysis, investigation, methodology, visualization, writing – original draft, writing – review and editing. Nico Nees: investigation, visualization, writing – original draft, writing – review and editing. Hamzah Khan: investigation, methodology. Nabi E. Traoré: investigation, writing – original draft, writing – review and editing. Dominik Drobek: investigation, writing – original draft. Wolfgang Peukert: funding acquisition, resources, supervision. Benjamin Apeleo Zubiri: resources, supervision. Erdmann Spiecker: funding acquisition, resources, supervision, writing – review and editing. Michael Stingl: funding acquisition, resources, supervision. Lukas Pflug: funding acquisition, methodology, resources, supervision. Robin N. Klupp Taylor: funding acquisition, resources, supervision, conceptualization, writing – original draft, writing – review and editing.Data availability
Data for this article, including all data included in the figures, are available at Zenodo.org (https://doi.org/10.5281/zenodo.11264121).Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge Marc Bürner for contributing to the establishment of the continuous flow process and image analysis, Gia-Han Tong and Lisa Büttner for experimental support, Maximilian Jordan for synthesis of polystyrene core particles and Andreas Völkl for the support in the graphic design of the flow set-up schematics. The authors further acknowledge the support of the Center for Nanoanalysis and Electron Microscopy (CENEM, Friedrich-Alexander-Universität Erlangen-Nürnberg) and the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) for funding of the Collaborative Research Centre 1411 “Design of Particulate Products” (Project-ID 416229255) as well as the Research Training Group GRK1896 “In situ Microscopy with Electrons, X-rays and Scanning Probes” (Project-ID 218975129).References
- K. McNamara and S. A. M. Tofail, Phys. Chem. Chem. Phys., 2015, 17, 27981–27995 RSC.
- J. Wang, A. J. Drelich, C. M. Hopkins, S. Mecozzi, L. Li, G. Kwon and S. Hong, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2022, 14, 1754–1767 Search PubMed.
- W. J. Stark, P. R. Stoessel, W. Wohlleben and A. Hafner, Chem. Soc. Rev., 2015, 44, 5793–5805 RSC.
- P. Di Pietro, G. Strano, L. Zuccarello and C. Satriano, Curr. Top. Med. Chem., 2016, 3069–3102 CrossRef CAS PubMed.
- M. De, P. S. Ghosh and V. M. Rotello, Adv. Mater., 2008, 20, 4225–4241 CrossRef CAS.
- N. Li, P. Zhao and D. Astruc, Angew. Chem., Int. Ed., 2014, 53, 1756–1789 CrossRef CAS PubMed.
- S. Eustis and M. A. El-Sayed, Chem. Soc. Rev., 2006, 35, 209–217 RSC.
- J. Kimling, M. Maier, B. Okenve, V. Kotaidis, H. Ballot and A. Plech, J. Phys. Chem. B, 2006, 110, 15700–15707 CrossRef CAS PubMed.
- S. Atta, M. Beetz and L. Fabris, Nanoscale, 2019, 11, 2946–2958 RSC.
- B. Nikoobakht and M. A. El-Sayed, Chem. Mater., 2003, 15, 1957–1962 CrossRef CAS.
- S. J. Oldenburg, R. D. Averitt, S. L. Westcott and N. J. Halas, Chem. Phys. Lett., 1998, 288, 243–247 CrossRef CAS.
- K. J. Lee, J. Yoon and J. Lahann, Curr. Opin. Colloid Interface Sci., 2011, 16, 195–202 CrossRef CAS.
- D. Mann, S. Voogt, R. van Zandvoort, H. Keul, M. Möller, M. Verheijen, D. Nascimento-Duplat, M. Xu, H. P. Urbach, A. J. L. Adam and P. Buskens, Chem. Commun., 2017, 53, 3898–3901 RSC.
- D. Kohler, N. Madaboosi, M. Delcea, S. Schmidt, B. G. de Geest, D. V. Volodkin, H. Möhwald and A. G. Skirtach, Adv. Mater., 2012, 24, 1095–1100 CrossRef CAS PubMed.
- A. A. Shah, B. Schultz, K. L. Kohlstedt, S. C. Glotzer and M. J. Solomon, Langmuir, 2013, 29, 4688–4696 CrossRef CAS PubMed.
- P. van Dorpe and J. Ye, ACS Nano, 2011, 5, 6774–6778 CrossRef CAS PubMed.
- H. Bao, W. Peukert and R. N. Klupp Taylor, Adv. Mater., 2011, 23, 2644–2649 CrossRef CAS PubMed.
- A. Völkl, J. Toutouly, D. Drobek, B. Apeleo Zubiri, E. Spiecker and R. N. Klupp Taylor, ACS Appl. Nano Mater., 2023, 6, 10126–10137 CrossRef.
- M. Biegel, T. Schikarski, P. Cardenas Lopez, L. Gromotka, C. Lübbert, A. Völkl, C. Damm, J. Walter and W. Peukert, RSC Adv., 2023, 13, 18001–18013 RSC.
- J. Wagner and J. M. Köhler, Nano Lett., 2005, 5, 685–691 CrossRef CAS PubMed.
- S. Watanabe, Y. Asahi, H. Omura, K. Mae and M. T. Miyahara, Adv. Powder Technol., 2016, 27, 2335–2341 CrossRef CAS.
- A. Knauer, A. Thete, S. Li, H. Romanus, A. Csáki, W. Fritzsche and J. M. Köhler, J. Chem. Eng., 2011, 166, 1164–1169 CrossRef CAS.
- W. Cai, H. Zhong and L. Zhang, J. Appl. Phys., 1998, 83, 1705–1710 CrossRef CAS.
- M. Kus-Liśkiewicz, P. Fickers and I. Ben Tahar, Int. J. Mol. Sci., 2021, 22, 10952 CrossRef PubMed.
- H. Bao, T. Bihr, A.-S. Smith and R. N. Klupp Taylor, Nanoscale, 2014, 6, 3954–3966 RSC.
- T. Meincke and R. N. Klupp Taylor, Particuology, 2023, 75, 137–150 CrossRef CAS.
- F.-Z. Sadafi, C. Sauerbeck, B. Braunschweig and R. N. Klupp Taylor, CrystEngComm, 2018, 20, 6214–6224 RSC.
- R. Moiraghi, O. A. Douglas-Gallardo, E. A. Coronado, V. A. Macagno and M. A. Pérez, RSC Adv., 2015, 5, 19329–19336 RSC.
- M. Luty-Błocho, K. Pacławski, M. Wojnicki and K. Fitzner, Inorg. Chim. Acta, 2013, 395, 189–196 CrossRef.
- T. Meincke, M. Jordan, N. Vogel and R. N. Klupp Taylor, Macromol. Chem. Phys., 2018, 219, 1700457 CrossRef.
- M. Quinten, Optical Properties of Nanoparticle Systems. Mie and Beyond, Wiley VCH, Weinheim, 2011 Search PubMed.
- C. Deraedt, L. Salmon, S. Gatard, R. Ciganda, R. Hernandez, J. Ruiz and D. Astruc, Chem. Commun., 2014, 50, 14194–14196 RSC.
- W. Haiss, N. T. K. Thanh, J. Aveyard and D. G. Fernig, Anal. Chem., 2007, 79, 4215–4221 CrossRef CAS PubMed.
- J.-M. Commenge and L. Falk, Chem. Eng. Process., 2011, 50, 979–990 CrossRef CAS.
- J. M. Reckamp, A. Bindels, S. Duffield, Y. C. Liu, E. Bradford, E. Ricci, F. Susanne and A. Rutter, Org. Process Res. Dev., 2017, 21, 816–820 CrossRef CAS.
- J. R. Lu, A. Marrocco, T. J. Su, R. K. Thomas and J. Penfold, J. Colloid Interface Sci., 1993, 158, 303–316 CrossRef CAS.
- OriginLab Corporation, Origin, Version 2021, Northampton, MA, USA Search PubMed.
- T. F. Tadros, Handbook of Colloid and Interface Science. Volume 1: Basic Principles of Interface Science and Colloid Stability, De Gruyter, Berlin, 2018 Search PubMed.
- C. T. Rueden, J. Schindelin, M. C. Hiner, B. E. DeZonia, A. E. Walter, E. T. Arena and K. W. Eliceiri, BMC Bioinf., 2017, 18, 529 CrossRef PubMed.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J.-Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed.
- M. Distaso, O. Zhuromskyy, B. Seemann, L. Pflug, M. Mačković, E. Encina, R. K. Taylor, R. Müller, G. Leugering, E. Spiecker, U. Peschel and W. Peukert, J. Quant. Spectrosc. Radiat. Transfer, 2017, 189, 369–382 CrossRef CAS.
- U. Frank, D. Drobek, A. Sánchez-Iglesias, S. E. Wawra, N. Nees, J. Walter, L. Pflug, B. Apeleo Zubiri, E. Spiecker, L. M. Liz-Marzán and W. Peukert, ACS Nano, 2023, 17, 5785–5798 CrossRef CAS PubMed.
- J.-P. Berenger, J. Comput. Phys., 1994, 114, 185–200 CrossRef.
- J. Schöberl and S. Zaglmayr, COMPEL – Int. J. Comput. Math. Electr. Electron. Eng., 2005, 24, 374–384 CrossRef.
- The HSL Mathematical Software Library, A Collection of Fortran Codes for Large Scale Scientific Computation, https://www.hsl.rl.ac.uk/catalogue/hsl_ma86.html, accessed 10 January 2015.
- J. D. Hogg, J. K. Reid and J. A. Scott, SIAM J. Sci. Comput., 2010, 32, 3627–3649 CrossRef.
- T. Meincke, H. Bao, L. Pflug, M. Stingl and R. N. Klupp Taylor, Chem. Eng. J., 2017, 308, 89–100 CrossRef CAS.
- L. Yu and M. A. Matthews, Int. J. Hydrogen Energy, 2011, 36, 7416–7422 CrossRef CAS.
- P. G. de Gennes, Rev. Mod. Phys., 1992, 64, 645–648 CrossRef.
- C. Noguez, Opt. Mater., 2005, 27, 1204–1211 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr02516d |
| This journal is © The Royal Society of Chemistry 2024 |