
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mesoporous silica–amine beads from blast furnace slag for CO2 capture applications†
Baljeet
Singh
 *a,
Marianna
Kemell
a,
Juho
Yliniemi
b and
Timo
Repo
*a
*a,
Marianna
Kemell
a,
Juho
Yliniemi
b and
Timo
Repo
*a
aDepartment of Chemistry, University of Helsinki, FI-00014 Helsinki, Finland. E-mail: baljeet.singh@helsinki.fi; timo.repo@helsinki.fi
bUniversity of Oulu, Fibre and Particle Engineering Research Unit, FI-90014, Finland
First published on 8th August 2024
Abstract
Steel slag, abundantly available at a low cost and containing over 30 wt% silica, is an attractive precursor for producing high-surface-area mesoporous silica. By employing a two-stage dissolution-precipitation method using 1 M HCl and 1 M NaOH, we extracted pure SiO2, CaO, MgO, etc. from blast furnace slag (BFS). The water-soluble sodium silicate obtained was then used to synthesize mesoporous silica. The resulting silica had an average surface area of 100 m2 g−1 and a pore size distribution ranging from 4 to 20 nm. The mesoporous silica powder was further formed into beads and post-functionalized with polyethyleneimine (PEI) for cyclic CO2 capture from a mixture containing 15% CO2 in N2 at 75 °C. The silica-PEI bead was tested over 105 adsorption–desorption cycles, demonstrating an average CO2 capture capacity of 1 mmol g−1. This work presents a sustainable approach from steel slag to cost-effective mesoporous silica materials and making CO2 capture more feasible.
Introduction
In the quest for sustainable development, circular economy principles have gained significant attention in transforming waste into valuable products.1 This approach addresses the environmental challenges associated with slag and contributes to the design of innovative and more eco-friendly products. By repurposing blast furnace slag (BFS), we can reduce waste, minimize environmental impact, and create sustainable materials that support a greener economy.2 BFS to produce value-added products such as pure silica, CaO, and MgO, etc. creates closed-loop systems and prevents the accumulation of large volumes of waste in landfills. Transforming BFS into porous materials (silica, MOFs, metal oxides, etc.) will reduce environmental footprints and contribute to economic growth, fostering a sustainable and resilient future.3 Various porous functional materials with broad applications can be produced using waste from the steel industry, highlighting the potential for innovative and eco-friendly solutions.4–6In conventional steelmaking, molten pig iron is used to produce steel in a basic oxygen furnace. During this process, raw materials such as dolomite, limestone, and other additives are introduced into the furnace for the production of high-quality steel.7,8 In 2022, approximately 312 million tons (Mt) of granulated BFS, 104 Mt of air-cooled BFS, 143 Mt of basic oxygen furnace slag, and 68 Mt of electric arc furnace slag were generated worldwide. The gap between slag generated by steel industries and demand is significant. Storing such a large quantity of waste is neither sustainable nor environmentally friendly.9 Long deposition or storage can also lead to environmental issues, particularly due to the highly alkaline leachates (pH > 11) it produces.10 Currently, BFS is used in road construction, cement production, and structural construction; however, the demand for other types of slag is very limited.11
Hence, methods to recycle slag have been developed and explored for various applications, including metal separation, materials for CO2 mineralization, catalysts, zeolite synthesis, flue gas desulfurization, etc.12–26 For instance, Kim and colleagues successfully extracted 46% Mg and 35% Al using 2M HCl, while pH control allowed selective separation of 97% Ca.27,28 BFS was also employed to produce Ca-rich adsorbent materials for CO2 capture.29–31 Similarly, Tian et al. also utilized slag for CO2 capture, achieving an enhanced capture capacity of slag-derived adsorbents, exceeding 10 times compared to raw slag, with a maximum uptake of 0.50 g of CO2 g−1 adsorbent.32 Additionally, Chen et al. produced a series of Cu-exchanged zeolite NaX catalysts for selective catalytic reduction of NOx with NH3.33
Furthermore, Yamashita and co-workers used BFS to produce a layered double hydroxide (LDH) compound through a two-step dissolution-precipitation method and employed LDH as a heterogeneous catalyst for organic transformation.34 To further advancement of BFS application, Kuwahara et al. exploited BFS to synthesize calcium silicate hydrate for waste-water treatment.35 In another study, Yamashita and colleagues synthesized a mesoporous silica-CaO composite for CO2 capture.36 With BFS containing significant proportions of Ca (41 wt%), Si (34 wt%), Al (11 wt%), Mg (10 wt%), Ti (0.35 wt%), etc., which can be separated and utilized for various applications with growing market demand, it presents an excellent source of valuable metals.37–39
This study aims to extract Si (in the form of sodium silicate) from BFS and utilize it to synthesize mesoporous silica and design silica-supported solid amine beads for CO2 capture. We designed a 2-stage cyclic dissolution-precipitation method employing 1 M HCl and 1 M NaOH solutions to separate metals and silica (Fig. 1). Furthermore, as synthesized mesoporous silica powder was shaped into beads and post-functionalized by polyethylenimine (PEI) and hexamethylenediamine (HMDA) for CO2 capture. Overall, this study demonstrates a close circular economy loop by utilizing steel industrial waste to capture CO2 emissions of the same industrial outlets.
 | ||
| Fig. 1 Illustration of metals and silica separation from BFS. | ||
Experimental section
Materials
The chemicals used in this study were analytic grade. Hexamethylenediamine (HMDA), polyethylenimine branched (PEI, MW 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), cetyltrimethylammonium bromide (CTAB), and sodium alginate were purchased from Sigma Aldrich and used as received.
000), cetyltrimethylammonium bromide (CTAB), and sodium alginate were purchased from Sigma Aldrich and used as received.
Characterization
The surface morphology of the samples was studied using a field emission SEM (Hitachi S-4800). The samples were coated with carbon using a Cressington 108A Carbon coater. Elemental analysis and mapping were performed with an Oxford INCA 350 Energy dispersive X-ray spectrometer connected to the Hitachi S-4800. The EDX spectra were measured at 20 keV. Bruker ALPHA-T FTIR in transmittance mode was used to identify chemical species in samples.Thermogravimetric analysis (TGA) of samples was conducted using the Mettler-Toledo TGA/DSC-3 + thermal analysis system. The heating rate of 10 °C min−1 in a temperature range of 25–600 °C was used under the flow of air (50 mL min−1). The specific surface area and textural properties were measured by N2 physical adsorption at 77 K using a Quantachrome analyzer.
For CO2 capture (from 15% CO2 in N2), the Mettler-Toledo TGA/DSC-3 + thermal analysis system was used. Initially, samples were activated under the flow of N2 gas (50 mL min−1) for 15 min. Initially, CO2 flow was 50 mL min−1 (otherwise mentioned), and desorption was performed at 100 °C for 15 min, under N2 as a sweep gas (flow: 50 mL min−1).
| Qt = Qe[1 − exp(−k1t)] | (1) |
| Qt = k2Qe2t/(1 + k2Qet) | (2) |
| Qt = Qe[1 − exp(−kAt)nA] | (3) |
Results and discussion
Separation of metal and silica
All the experiments were conducted using the same BFS, and the SEM-EDX elemental analysis revealed that BFS contained 7 wt% Mg, 9 wt% Al, 21 wt% Ca, 1 wt% Ti, and 16 wt% Si (Fig. 2 and Table S1, entry 1†). SEM-EDX elemental mapping demonstrated that metals are uniformly distributed throughout the BFS particles (Fig. S1†), while SEM images indicated that slag particle size ranged from 3–20 μm (Fig. S2†). Elemental analysis revealed that the remaining solid mainly consisted of silica with a small amount of undissolved metal oxides such as Al and Mg (Fig. 2 and Table S1, entry 2†).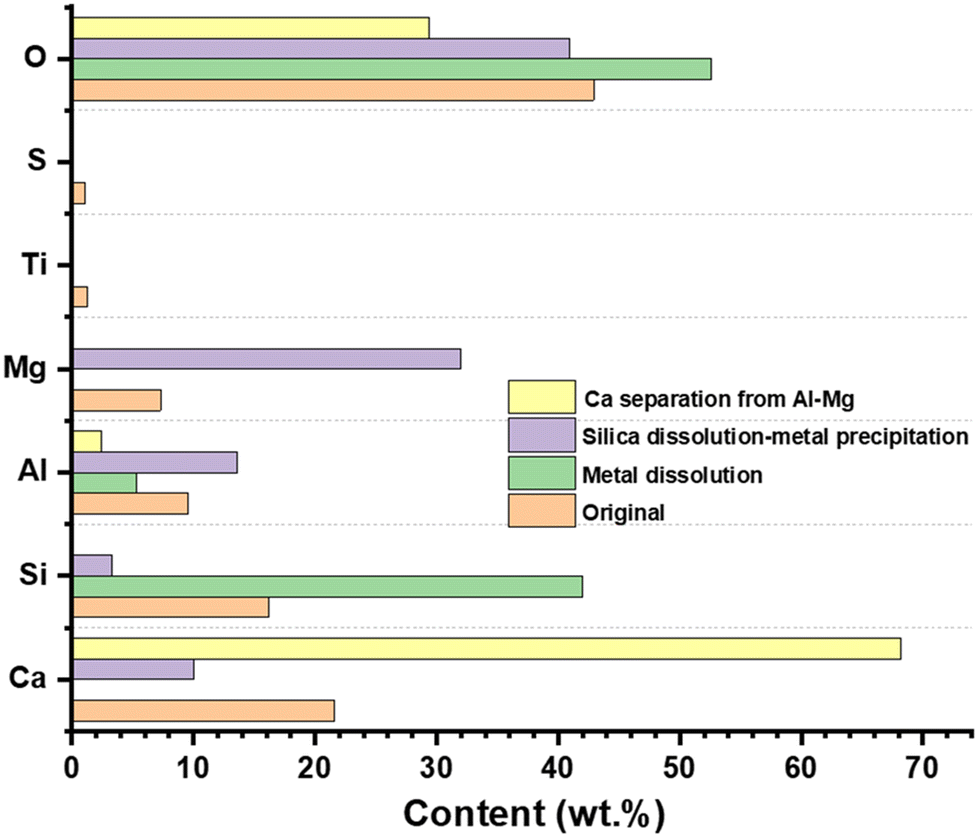 | ||
| Fig. 2 Chemical composition of original BFS and after dissolution-precipitation of metals and silica. | ||
To precipitate metals, the pH of the aqueous phase was adjusted using 1 M NaOH aq. solution. At pH 8–9, Al and Mg precipitated first, leaving the remaining aqueous phase containing only Ca (Fig. 2 and Table S1, entry 3†). After separating a mixture of metal (Al and Mg) hydroxides, the pH of the remaining aqueous phase was adjusted to 13–14, resulting in precipitation of Ca (confirmed by SEM-EDX analysis) (Fig. 2 and Table S1, entry 4†).
The remaining solid (mostly containing Si) was refluxed to improve separation in an aqueous NaOH solution (pH ∼ 14). The highly basic medium slowly hydrolyses the silica network (–Si–O–Si–) to produce sodium silicate.43–46 During this step, metals tend to precipitate out as corresponding hydroxides [M–(OH)x].47 Water-soluble sodium silicate was separated from metal hydroxide by filtration, and the filtrate was then used as a Si source for mesoporous silica synthesis.
SEM image analysis revealed that silica particles were small, with an average size of 300 nm in size and agglomerated (Fig. 3c, d, and S4†). TGA analysis revealed two stages of weight loss. The initial weight loss of approximately 10 wt% (up to 200 °C) could be due to loss of adsorbed gases or moisture in a powder sample. The second stage, involving a weight loss of around 25 wt% between 200 and 600 °C, is attributed to the decomposition and combustion of CTAB, which acted as a structural directing agent in the production of mesoporous silica (Fig. S5†).51 The silica exhibited a moderate surface area of (100 m2 g−1) and a wide pore size distribution ranging from 4–20 nm (Fig. S6 and S7†), exhibiting that silica is a mesoporous characteristic. PXRD showed that as-synthesised silica is amorphous (Fig. S8†).
 | ||
| Fig. 3 SEM images of (a and b) BFS, (c and d) Silica. | ||
Spherical silica beads were post-functionalized with PEI and HMDA, respectively (Fig. S12†), and CO2 capture capacity was monitored using TGA. PEI and HMDA functionalized silica-bead exhibited CO2 capture capacity of 5.7 wt% (1.29 mmol g−1, N contents 12 wt%) and 4 wt% (0.90 mmol g−1, N-contents 9.9 wt%), respectively (Fig. 4 and Table S2†). Preliminary analysis indicated that PEI silica-bead showed higher CO2 capture capacity despite fewer primary amines compared to HMDA which could be due to better diffusion, and accessibility of active sites.
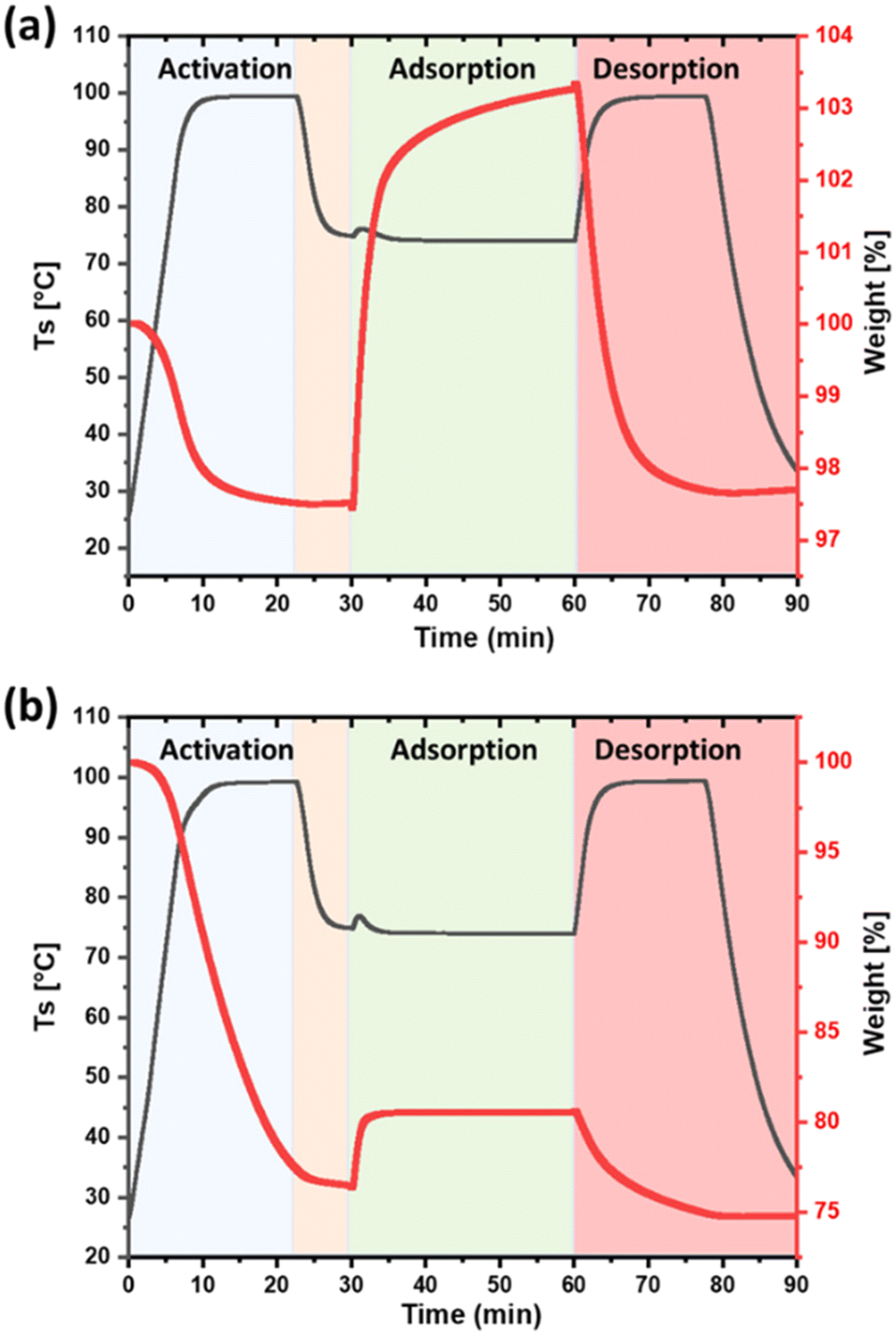 | ||
| Fig. 4 CO2 adsorption on (a) silica-PEI bead and (b) silica-HMDA bead. Adsorption at 75 °C for 30 min, using 15% CO2 in N2, desorption at 100 °C for 15 min. | ||
The adsorption of amine and formation of different CO2-amine chemical species in pellets were confirmed by FTIR. Before FTIR measurements, pellets are degassed at RT and saturated with CO2 overnight in a closed chamber. Powder samples were used to measure FTIR. The presence of PEI and HMDA in beads was confirmed by adsorption features in the range of 3000–2500, and 1500–1000 cm−1 (Fig. S13†). After CO2 adsorption, the spectrum indicated the presence of CO2 species in the samples, NH stretching was observed at 3280 cm−1, and with corresponding NH bending in the region of 1100–1800 cm−1.65 The absorption band on the region 1500–1200 cm−1 was attributed to chemical species formed when CO2 reacts with amines, the peak at 1560 cm−1 assigned to stretching band of carbamate (COO−). Peaks at 1404 cm−1, and 1376 cm−1 were attributed to NH2+ deformation of secondary amine and C–N stretching/NCOO− skeleton vibration of carbamate.65 In FTIR spectra of pellets, typical peaks of sodium alginates were much weaker than pure alginate powder, suppressed and superimposed by amine-CO2 chemical species in pellets.
The experimental CO2 adsorption TGA profiles and corresponding fitting curves for silica-PEI and silica-HMDA are illustrated in Fig. 5 and relative parameters obtained after kinetic modelling are summarized in respective Table 1. The Pseudo first order kinetic model did not fit entirely the CO2 adsorption profile (Fig. 5a); however, it provides a good fit for the initial CO2 capture profile. Conversely, Pseudo second order (Fig. 5b) and Avrami kinetic models (Fig. 5c) demonstrated good fits across almost the entire CO2 adsorption profile.
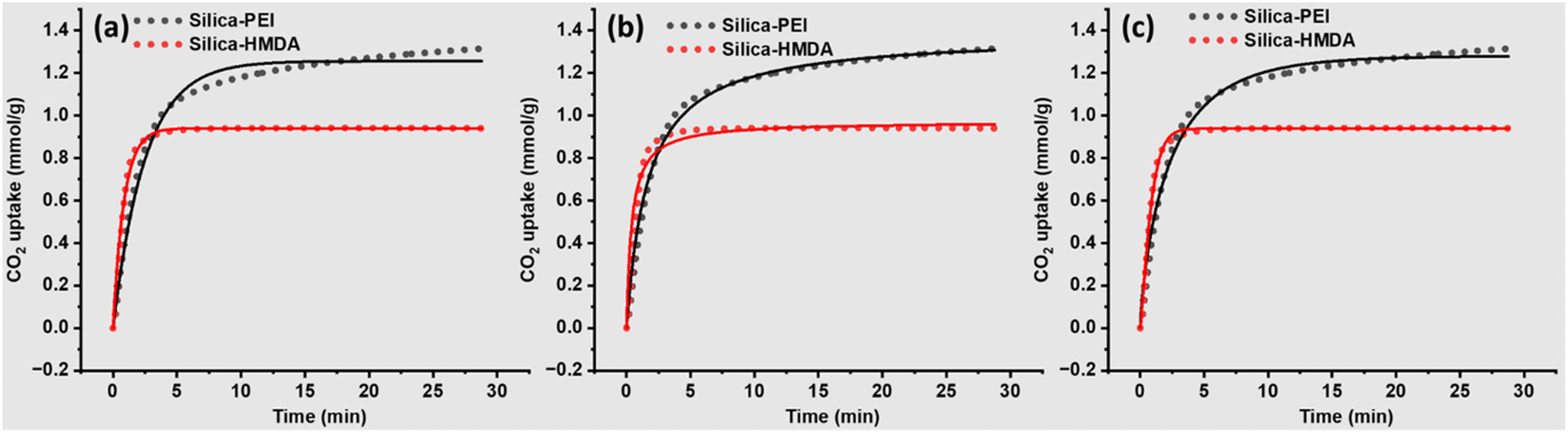 | ||
| Fig. 5 Fitting of CO2 adsorption profiles using three kinetic models, (a) Pseudo first order kinetic model, (b) Pseudo second order, and (c) Avrami kinetic model. | ||
| Kinetic Model | Parameters | Silica-PEI | Silica-HMDA |
|---|---|---|---|
| Pseudo first order | Q e | 1.25 ± 0.001 | 0.940 ± 0.0002 |
| k 1 | 0.400 ± 0.002 | 1.16 ± 0.004 | |
| R 2 | 0.9879 | 0.9899 | |
| Pseudo second order | Q e | 1.38 ± 0.001 | 0.972 ± 0.001 |
| k 2 | 0.432 ± 0.002 | 2.54 ± 0.042 | |
| R 2 | 0.9999 | 0.9999 | |
| Avrami | Q e | 1.28 ± 0.001 | 0.938 ± 0.0002 |
| k A | 0.396 ± 0.002 | 1.12 ± 0.002 | |
| n A | 0.767 ± 0.004 | 1.21 ± 0.005 | |
| R 2 | 0.9999 | 0.9999 |
Among the three kinetic models, the Avrami kinetic model accurately predicted Qe value of 1.28 which closely matched the experimental values of 1.29 for silica-PEI. Silica-HMDA showed a higher kA compared to silica-PEI despite having a lower CO2 adsorption capacity. This suggests that primary amine groups are highly reactive compared to secondary amines, as most of the amines in PEI are secondary. This difference in adsorption capacity could be due to the accessibility of CO2 capture sites, sites, and better diffusion of CO2 within the silica-PEI.
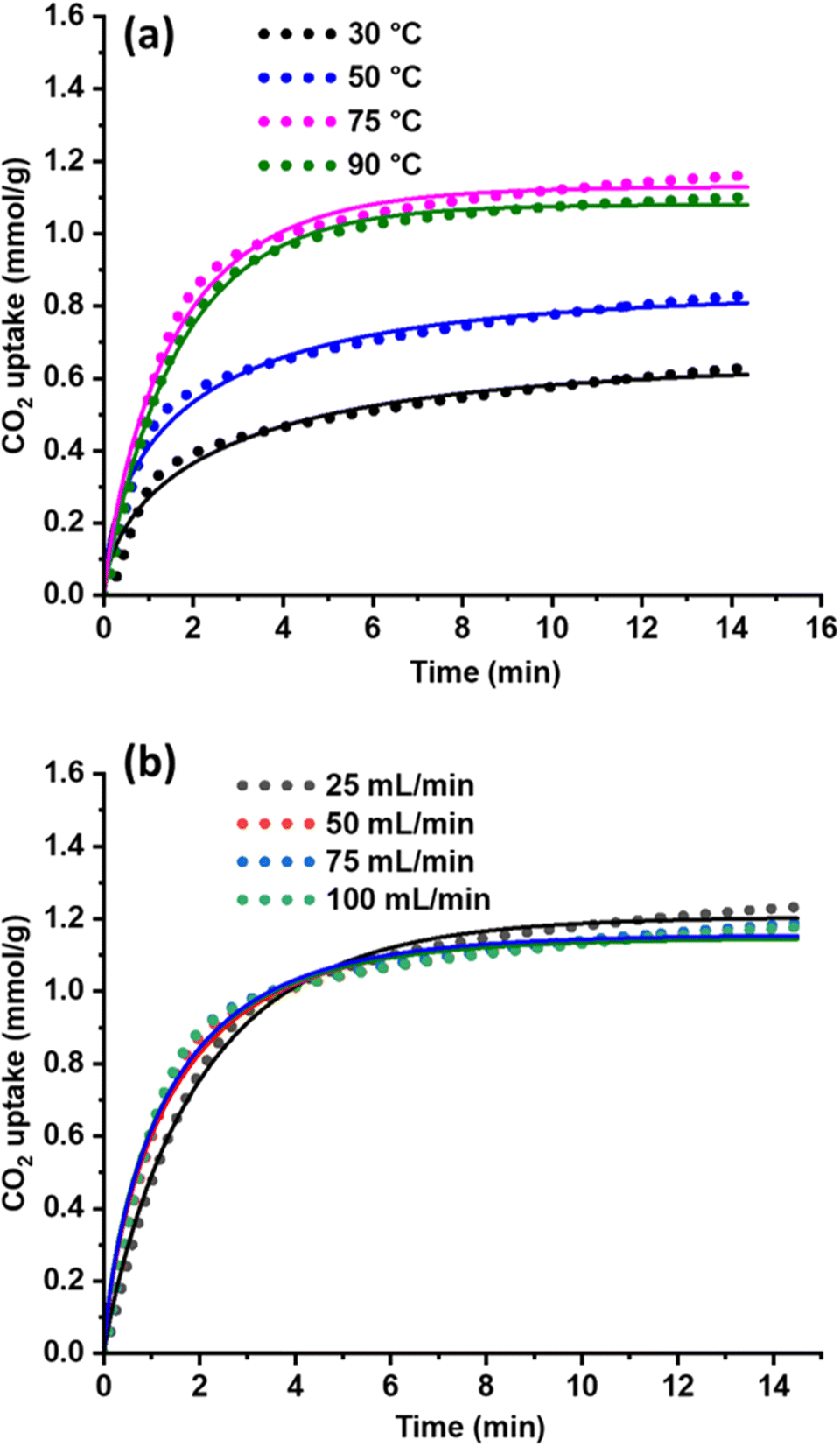 | ||
| Fig. 6 Effect of adsorption temperature (a) and CO2 flow rates (b) on CO2 capture capacity and their corresponding Avrami model fitted curves of silica-PEI bead. | ||
At 75 °C, a suitable adsorption temperature was identified for further analysis, and the effect of CO2 flow rate was optimized at 75 °C (Fig. S15†). Interestingly, the lowest flow (25 mL min−1) exhibited a higher CO2 adsorption capacity compared to flow rates of 50, 75, and 100 mL min−1, indicating that the bead achieved saturation within a short period (Fig. 6b and S15†). The rate constant kA increased with temperature and decreased at 90 °C (Table 2). This is supported by the higher kA values observed at various flow rates, indicating that the amine sites are highly accessible and saturate rapidly even at a low CO2 flow rate (25 mL min−1). Additionally, the nA value varied with different adsorption temperatures and flow rates, predicting that the CO2 adsorption mechanism was different for each system (Table 2). The CO2 diffusion rate and mass transfer rate significantly influence the adsorbent's CO2 adsorption performance, and rapid saturation and desorption are favourable for the large-scale deployment of such adsorbents.
| Parameters | 30 °C | 50 °C | 75 °C | 90 °C | |
|---|---|---|---|---|---|
| Effect of temperature | Q e | 0.64 ± 0.004 | 0.83 ± 0.003 | 1.13 ± 0.001 | 1.08 ± 0.001 |
| k A | 0.371 ± 0.008 | 0.510 ± 0.008 | 0.634 ± 0.003 | 0.599 ± 0.002 | |
| n A | 0.625 ± 0.009 | 0.596 ± 0.007 | 0.854 ± 0.007 | 0.935 ± 0.004 | |
| R 2 | 0.9999 | 0.9999 | 0.9999 | 0.9999 | |
| Effect of flow | 25 mL min−1 | 50 mL min−1 | 75 mL min−1 | 100 mL min−1 | |
| Q e | 1.20 ± 0.001 | 1.15 ± 0.001 | 1.15 ± 0.001 | 1.14 ± 0.001 | |
| k A | 0.489 ± 0.002 | 0.678 ± 0.004 | 0.708 ± 0.005 | 0.725 ± 0.005 | |
| n A | 0.904 ± 0.005 | 0.773 ± 0.006 | 0.771 ± 0.007 | 0.757 ± 0.006 | |
| R 2 | 0.9999 | 0.9999 | 0.9999 | 0.9999 | |
 | ||
| Fig. 7 (a) Desorption temperature optimization using Silica-PEI bead. (b) Kinetic parameters of four different desorption temperatures. | ||
As the desorption temperature increased, the rate of desorption (kA) also increased (Table S3†), and the bead desorbed quickly within 10 min (Fig. 7a). However, at 90 °C, the bead took a considerably longer time compared to the other desorption temperatures, reflecting the thermal stability of chemical species formed during the CO2 adsorption (Fig. 7a). These observations indicate that the beads were easily and fully regenerable within the range of 90–120 °C, as supported by the −Qe values (Fig. 7b). However, it is essential to note that higher temperatures could lead to amine degradation, leaching, and formation of stable chemical species.75–77 Therefore, 100 °C for 15 min was selected for further investigation to minimize thermal degradation and amine leaching.
Diffusion limitations within beads were also accessed by converting the bead into powder and using them for CO2 capture investigations. Corresponding PEI powder samples showed similar CO2 capture capacity as beads (Fig. S17†). The Avrami kinetic models of powder samples also revealed higher kA values for both adsorption (i.e. 0.90) and desorption (i.e. 0.65) compared to beads (i.e. 0.36, and 0.37 for adsorption and desorption, respectively) (Fig. S17, S18 and Tables S4, S5†). This indicates that both CO2 adsorption and desorption are diffusion-limited in beads.
 | ||
| Fig. 8 (a) 105 adsorption–desorption cycles of silica-PEI bead. (b) 20 adsorption–desorption cycles of silica-HMDA bead. Adsorption was performed at 75 °C, a CO2 flow rate of 25 mL min−1 for 15 min, and desorption at 100 °C for 15 min under the flow of N2 (50 mL min−1). | ||
Conclusion
We have successfully demonstrated the viability of utilizing BFS, a steel industrial waste abundant in supply, for the synthesizing mesoporous silica, employing a two-stage dissolution and precipitation using HCl and NaOH. The prepared mesoporous silica possesses a moderate surface area of 100 m2 g−1 and a wide pore size distribution ranging from 4–20 nm. Our investigation highlights the potential to explore BFS for the continuous production of mesoporous silica using dissolution-precipitation at a large scale.Furthermore, we converted the mesoporous silica powder into industrial-grade solid amine adsorbents and evaluated their performance for CO2 capture (15% CO2 in N2). PEI functionalized silica bead demonstrated superior performance compared to HMDA. After 105 adsorption–desorption cycles, the silica-PEI bead revealed no significant sign of performance degradation (approximately 8% decrease in adsorption capacity was observed). It's essential to note for the practical applications, making beads/pellets/monoliths a more feasible and scalable option for industries.
Author contributions
Conceptualization, analysis, methodology, validation, visualization, writing original draft and management: BS; SEM and elemental analysis: BS and MK; review, and editing: MK, JY, TR.Data availability
The Authors confirm that the data supporting the findings of this study are available within the article and ESI.†Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
B. S and T. R. are grateful for financial support from Business Finland 8205/31/2022. We would like to thank Dr. Risto Koivula for helping us with N2 sorption analysis.References
- M. Barreiro-Gen and R. Lozano, Bus. Strategy Environ., 2020, 29, 3484–3494 CrossRef.
- P. Schroeder, K. Anggraeni and U. Weber, J. Ind. Ecol., 2019, 23, 77–95 CrossRef.
- Y. Jiang, P. Tan, S.-C. Qi, X.-Q. Liu, J.-H. Yan, F. Fan and L.-B. Sun, Angew. Chem., Int. Ed., 2019, 58, 6600–6604 CrossRef CAS PubMed.
- N. Yuan, A. Zhao, Z. Hu, K. Tan and J. Zhang, Chemosphere, 2022, 287, 132227 CrossRef CAS PubMed.
- G. A. Khater, B. S. Nabawy, A. A. El-Kheshen, M. Abdel-Baki and M. M. Farag, Mater. Chem. Phys., 2022, 280, 125784 CrossRef CAS.
- W. Liu, Y. Wang, J. Li and B. Li, Int. J. Appl. Ceram. Technol., 2023, 20, 3584–3595 CrossRef CAS.
- D. M. Proctor, K. A. Fehling, E. C. Shay, J. L. Wittenborn, J. J. Green, C. Avent, R. D. Bigham, M. Connolly, B. Lee, T. O. Shepker and M. A. Zak, Environ. Sci. Technol., 2000, 34, 1576–1582 CrossRef CAS.
- A. J. Hobson, D. I. Stewart, A. W. Bray, R. J. G. Mortimer, W. M. Mayes, M. Rogerson and I. T. Burke, Environ. Sci. Technol., 2017, 51, 7823–7830 CrossRef CAS PubMed.
- Q. Zeng, X. Liu, Z. Zhang, C. Wei and C. Xu, Green Energy Resour., 2023, 1, 100012 CrossRef.
- W. M. Mayes, A. L. Riley, H. I. Gomes, P. Brabham, J. Hamlyn, H. Pullin and P. Renforth, Environ. Sci. Technol., 2018, 52, 7892–7900 CrossRef CAS PubMed.
- E. Özbay, M. Erdemir and H. İ. Durmuş, Constr. Build. Mater., 2016, 105, 423–434 CrossRef.
- X. Chen, Y. He, X. Cui and L. Liu, Fuel, 2023, 338, 127309 CrossRef CAS.
- H. Pullin, A. W. Bray, I. T. Burke, D. D. Muir, D. J. Sapsford, W. M. Mayes and P. Renforth, Environ. Sci. Technol., 2019, 53, 9502–9511 CrossRef CAS PubMed.
- M. Seggiani and S. Vitolo, Resour., Conserv. Recycl., 2003, 40, 71–80 CrossRef.
- L. Chen, S. Ren, H. Peng, J. Yang, M. Wang, Z. Chen and Q. Liu, Appl. Catal., A, 2022, 646, 118868 CrossRef CAS.
- T.-S. Tran, J. Yu, C. Li, F. Guo, Y. Zhang and G. Xu, RSC Adv., 2017, 7, 18108–18119 RSC.
- Z. Lu, Z. Lei, S. Hao, J. Yang, Z. Lei, B. Fang and C. Xiaosheng, ACS Omega, 2020, 5, 32216–32226 CrossRef CAS PubMed.
- L. Zhang, Y. Han, H. Shu, L. Zhang, Z. Han, X. Yang and Y. Chen, J. Chem. Technol. Biotechnol., 2023, 98, 949–957 CrossRef CAS.
- Y. Kuwahara and H. Yamashita, J. CO2 Util., 2013, 1, 50–59 CrossRef CAS.
- Z. Lei, L. Xi, Q. Lingbo, S. Hao, J. Yang, L. Zhang, Y. Yao and B. Fang, RSC Adv., 2021, 11, 15036–15043 RSC.
- C. Chen, S.-R. Wu and C.-T. Kao, ACS Omega, 2023, 8, 47075–47085 CrossRef CAS PubMed.
- F. Qi, J. Sun, G. Zhu, H. Li, Y. Wu, S. Li, C. Yang, J. Zheng and Y. Zhang, Process Saf. Environ. Prot., 2022, 165, 1–12 CrossRef CAS.
- M. S. Barbarey, M. M. El-Sayed Seleman, A. A. El Kheshen and M. F. Zawrah, Constr. Build. Mater., 2024, 411, 134226 CrossRef CAS.
- W. Ren, P. Zhou, Y. Tian, W. Wang, Y. Dong, T. Wang, L. Zhang, C. Ma and X. Zhao, Appl. Catal., A, 2020, 606, 117810 CrossRef CAS.
- G. Hu, S. Rohani, X. Jiang, J. Li, Q. Liu and W. Liu, ACS Sustainable Chem. Eng., 2021, 9, 13963–13971 CrossRef CAS.
- C.-F. Liu and S.-M. Shih, Environ. Sci. Technol., 2004, 38, 4451–4456 CrossRef CAS PubMed.
- Y. H. Lee, H. Eom, S. M. Lee and S. S. Kim, RSC Adv., 2021, 11, 8306–8313 RSC.
- V. Trinkel, O. Mallow, C. Thaler, J. Schenk, H. Rechberger and J. Fellner, Ind. Eng. Chem. Res., 2015, 54, 11759–11771 CrossRef CAS.
- Y. Kuwahara, A. Hanaki and H. Yamashita, ACS Sustainable Chem. Eng., 2022, 10, 372–381 CrossRef CAS.
- M. Owais, M. Järvinen, P. Taskinen and A. Said, J. CO2 Util., 2019, 31, 1–7 CrossRef CAS.
- J. Yu and K. Wang, Energy Fuels, 2011, 25, 5483–5492 CrossRef CAS.
- S. Tian, J. Jiang, F. Yan, K. Li and X. Chen, Environ. Sci. Technol., 2015, 49, 7464–7472 CrossRef CAS PubMed.
- L. Chen, S. Ren, X. Xing, J. Yang, J. Yang, M. Wang, Z. Chen and Q. Liu, ACS Sustainable Chem. Eng., 2022, 10, 7739–7751 CrossRef CAS.
- Y. Kuwahara, K. Tsuji, T. Ohmichi, T. Kamegawa, K. Mori and H. Yamashita, ChemSusChem, 2012, 5, 1523–1532 CrossRef CAS PubMed.
- Y. Kuwahara, S. Tamagawa, T. Fujitani and H. Yamashita, J. Mater. Chem. A, 2013, 1, 7199–7210 RSC.
- Y. Kuwahara, A. Hanaki and H. Yamashita, Green Chem., 2020, 22, 3759–3768 RSC.
- N. Shekhar Samanta, P. P. Das, S. Dhara and M. K. Purkait, Ind. Eng. Chem. Res., 2023, 62, 9006–9031 CrossRef CAS.
- M. Oge, D. Ozkan, M. B. Celik, M. Sabri Gok and A. Cahit Karaoglanli, Mater. Today: Proc., 2019, 11, 516–525 CAS.
- W. Duan, Q. Yu, J. Liu, L. Hou, H. Xie, K. Wang and Q. Qin, Appl. Therm. Eng., 2016, 98, 936–943 CrossRef CAS.
- Y. W. Chiang, R. M. Santos, J. Elsen, B. Meesschaert, J. A. Martens and T. Van Gerven, Chem. Eng. J., 2014, 249, 260–269 CrossRef CAS.
- J. Xiao, F. Li, Q. Zhong, H. Bao, B. Wang, J. Huang and Y. Zhang, Hydrometallurgy, 2015, 155, 118–124 CrossRef CAS.
- E. A. Gorrepati, P. Wongthahan, S. Raha and H. S. Fogler, Langmuir, 2010, 26, 10467–10474 CrossRef CAS PubMed.
- B. Rao, H. Dai, L. Gao, H. Xie, G. Gao, K. Peng, M. Zhang, F. He and Y. Pan, J. Cleaner Prod., 2022, 341, 130779 CrossRef CAS.
- R. Dupuis, R. Pellenq, J.-B. Champenois and A. Poulesquen, J. Phys. Chem. C, 2020, 124, 8288–8294 CrossRef CAS.
- M. Fertani-Gmati, K. Brahim, I. Khattech and M. Jemal, Thermochim. Acta, 2014, 594, 58–67 CrossRef CAS.
- F. K. Crundwell, ACS Omega, 2017, 2, 1116–1127 CrossRef CAS PubMed.
- A. Pohl, Water, Air, Soil Pollut., 2020, 231, 503 CrossRef CAS.
- J. Hwang, J. H. Lee and J. Chun, Mater. Lett., 2021, 283, 128765 CrossRef CAS.
- K. Kosuge, N. Kikukawa and M. Takemori, Chem. Mater., 2004, 16, 4181–4186 CrossRef CAS.
- M. Hessien and E. Prouzet, ChemistrySelect, 2021, 6, 1440–1447 CrossRef CAS.
- N. Bayal, B. Singh, R. Singh and V. Polshettiwar, Sci. Rep., 2016, 6, 24888 CrossRef CAS PubMed.
- A. Samanta, A. Zhao, G. K. H. Shimizu, P. Sarkar and R. Gupta, Ind. Eng. Chem. Res., 2012, 51, 1438–1463 CrossRef CAS.
- J. Wang, L. Huang, R. Yang, Z. Zhang, J. Wu, Y. Gao, Q. Wang, D. O'Hare and Z. Zhong, Energy Environ. Sci., 2014, 7, 3478–3518 RSC.
- M. Bisht, Bhawna, B. Singh and S. Pandey, J. Mol. Liq., 2023, 384, 122203 CrossRef CAS.
- B. Singh and V. Polshettiwar, J. Mater. Chem. A, 2016, 4, 7005–7019 RSC.
- B. Singh, J. Na, M. Konarova, T. Wakihara, Y. Yamauchi, C. Salomon and M. B. Gawande, Bull. Chem. Soc. Jpn., 2020, 93, 1459–1496 CrossRef CAS.
- B. Singh and V. Polshettiwar, Pure Appl. Chem., 2023, 95, 451–462 CrossRef CAS.
- B. Singh and V. Polshettiwar, Nanoscale, 2019, 11, 5365–5376 RSC.
- B. Singh, A. Maity and V. Polshettiwar, ChemistrySelect, 2018, 3, 10684–10688 CrossRef CAS.
- C. Chen, S.-T. Yang, W.-S. Ahn and R. Ryoo, Chem. Commun., 2009, 3627–3629 RSC.
- D. Zabiegaj, M. Caccia, M. E. Casco, F. Ravera and J. Narciso, J. CO2 Util., 2018, 26, 36–44 CrossRef CAS.
- S. Yang, L. Peng, O. A. Syzgantseva, O. Trukhina, I. Kochetygov, A. Justin, D. T. Sun, H. Abedini, M. A. Syzgantseva, E. Oveisi, G. Lu and W. L. Queen, J. Am. Chem. Soc., 2020, 142, 13415–13425 CrossRef CAS PubMed.
- K. Maresz, A. Ciemięga, J. J. Malinowski and J. Mrowiec-Białoń, Chem. Eng. J., 2020, 383, 123175 CrossRef CAS.
- M. A. Sakwa-Novak, C.-J. Yoo, S. Tan, F. Rashidi and C. W. Jones, ChemSusChem, 2016, 9, 1859–1868 CrossRef CAS PubMed.
- J. Yu and S. S. C. Chuang, Energy Fuels, 2016, 30, 7579–7587 CrossRef CAS.
- E. D. Revellame, D. L. Fortela, W. Sharp, R. Hernandez and M. E. Zappi, Clean. Eng. Technol., 2020, 1, 100032 CrossRef.
- V. K. Singh and E. A. Kumar, Energy Procedia, 2016, 90, 316–325 CrossRef CAS.
- Q. Liu, J. Shi, S. Zheng, M. Tao, Y. He and Y. Shi, Ind. Eng. Chem. Res., 2014, 53, 11677–11683 CrossRef CAS.
- M. Songolzadeh, M. Soleimani and M. Takht Ravanchi, J. Nat. Gas Sci. Eng., 2015, 27, 831–841 CrossRef CAS.
- B. Guo, Y. Wang, X. Qiao, X. Shen, J. Guo, J. Xiang and Y. Jin, Chem. Eng. J., 2021, 421, 127865 CrossRef CAS.
- H. Chen, S. Dong, Y. Zhang and P. He, Energy, 2022, 239, 122348 CrossRef CAS.
- F. Rezaei, R. P. Lively, Y. Labreche, G. Chen, Y. Fan, W. J. Koros and C. W. Jones, ACS Appl. Mater. Interfaces, 2013, 5, 3921–3931 CrossRef CAS PubMed.
- Y. Fan and X. Jia, Energy Fuels, 2022, 36, 1252–1270 CrossRef CAS.
- J. J. Lee, C. Sievers and C. W. Jones, Ind. Eng. Chem. Res., 2019, 58, 22551–22560 CrossRef CAS.
- A. I. Balabanovich, I. A. Klimovtsova, V. P. Prokopovich and N. R. Prokopchuk, Thermochim. Acta, 2007, 459, 1–8 CrossRef CAS.
- G. T. Rochelle, Curr. Opin. Chem. Eng., 2012, 1, 183–190 CrossRef CAS.
- A. Azarpour and S. Zendehboudi, ACS Omega, 2023, 8, 26850–26870 CrossRef CAS PubMed.
- R. A. Khatri, S. S. C. Chuang, Y. Soong and M. Gray, Energy Fuels, 2006, 20, 1514–1520 CrossRef CAS.
- J. S. A. Carneiro, G. Innocenti, H. J. Moon, Y. Guta, L. Proaño, C. Sievers, M. A. Sakwa-Novak, E. W. Ping and C. W. Jones, Angew. Chem., Int. Ed., 2023, 62, e202302887 CrossRef CAS PubMed.
- Q. T. Vu, H. Yamada and K. Yogo, Ind. Eng. Chem. Res., 2021, 60, 4942–4950 CrossRef CAS.
- C. S. Srikanth and S. S. C. Chuang, ChemSusChem, 2012, 5, 1435–1442 CrossRef CAS PubMed.
- C. Xu, Y. Zhang, Y.-L. Peng, T. Yang, Z. Liu, F. Qiu, A. Oloruntoba and S. Jiang, Sep. Purif. Technol., 2024, 331, 125621 CrossRef CAS.
- Q. T. Vu, H. Yamada and K. Yogo, Energy Fuels, 2019, 33, 3370–3379 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr02495h |
| This journal is © The Royal Society of Chemistry 2024 |