
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chirality induction to porphyrin derivatives co-confined at the air–water interface with silica nano-helices: towards enantioselective thin solid film surfaces†
Michela
Ottolini
a,
Zakaria
Anfar
b,
Nitika
Grover
c,
Gabriele
Magna
 d,
Manuela
Stefanelli
d,
Roberto
Paolesse
d,
Mathias O.
Senge
ce,
Simona
Bettini
*a,
Ludovico
Valli
a,
Reiko
Oda
*bf and
Gabriele
Giancane
g
d,
Manuela
Stefanelli
d,
Roberto
Paolesse
d,
Mathias O.
Senge
ce,
Simona
Bettini
*a,
Ludovico
Valli
a,
Reiko
Oda
*bf and
Gabriele
Giancane
g
aDepartment of Biological and Environmental Sciences and Technologies (DiSTeBA), Campus Ecotekne, University of Salento, Via per Monteroni, 73100 Lecce, Italy. E-mail: simona.bettini@unisalento.it
bUniv. Bordeaux, CNRS, Bordeaux INP, CBMN, UMR 5248, F-33600 Pessac, France
cSchool of Chemistry, Chair of Organic Chemistry, Trinity College Dublin, The University of Dublin, Trinity Biomedical Sciences Institute, 152-160 Pearse Street, Dublin, D02R590, Ireland
dDepartment of Chemical Science and Technologies, University of Rome Tor Vergata, Via Della Ricerca Scientifica 1, 00133 Rome, Italy
eInstitute for Advanced Study (TUM-IAS), Focus Group – Molecular and Interfacial Engineering of Organic Nanosystem, Technical University of Munich, Lichtenbergstrasse 2a, 85748 Garching, Germany
fWPI-Advanced Institute for Materials Research, Tohoku University, Katahira, Aoba-Ku, 980-8577 Sendai, Japan. E-mail: r.oda@cbmn.u-bordeaux.fr
gDepartment of Cultural Heritage, University of Salento, Via D. Birago 64, 73100 Lecce, Italy
First published on 1st August 2024
Abstract
A supramolecular approach based on self-assembled structures allows the formation of large structured co-assemblies based on chiral and achiral compounds with original physicochemical features. In this contribution, an achiral and hydrophobic porphyrin was co-assembled at the air–water interface with mesoscopic silica nano-helices dispersed in the water subphase of a Langmuir trough without covalent bond formation. This procedure allowed transferring the porphyrin/nano-helix co-assemblies on a solid support within a thin hybrid layer. The interaction between the two species was characterized using spectroscopic techniques and atomic force microscopy. As evidenced by the circular dichroism measurements performed directly on solid films, tunable chirality was induced to the porphyrin aggregates according to the chirality of the silica nano-helices. When the co-assemblies were transferred on surface plasmon resonance (SPR) slides and exposed to aqueous solutions of histidine enantiomers, selective chiral discrimination was observed which was determined by the matching/mismatching between the chirality of the analyte and the helicity of the nano-helical structure.
Introduction
Chirality is an essential characteristic of living matter and nature,1,2 which are present in an extensive size range, from the subatomic scale,3 molecular and supramolecular levels,4,5 nanoscale,6 and from the macroscopic7 up to galactic scale.8 The supramolecular chirality of molecular assemblies, which involves non-covalent intermolecular interactions, is particularly important for life as it is the basis for the formation of nano- and micro-sized self-assembled architectures.9,10 The structures and properties of chiral molecular assemblies are strongly influenced by the special spatial arrangement of the building blocks.11,12 Even though supramolecular chirality is often connected to the chirality of the constituent molecules, it is not a prerequisite that all molecules are chiral. Achiral molecules can be assembled to form supramolecular chiral systems. In this context, the interaction between the molecules must be accurately controlled and modulated. One of the known approaches to prepare chiral systems is to promote the aggregation of an achiral species and a chiral compound. For transferring the chirality to the assembled systems, non-covalent bonds such as hydrogen bonds and electrostatic and π-stacking can be used. Porphyrins13 are one of the most investigated systems among the π-conjugate compounds because they play crucial roles in essential biological processes and exhibit excellent self-assembling abilities.14,15 As observed in several organic π-conjugated systems,16 the functionalization of macrocycles with chiral groups induces the formation of hierarchical chiral superstructures.17–19 Furthermore, the widely developed chemistry of porphyrins allows for the facile modulation of tetrapyrrole aggregation and interaction with achiral and chiral chemical species.13,20 A strategy to tune aggregation can be the immobilization in thin solid molecular films of porphyrin-based-building blocks to obtain complex architectures using humid deposition techniques such as the Langmuir–Blodgett method.21–24 Indeed, Langmuir–Blodgett (LB) and its horizontal variation, the Langmuir–Schaefer (LS) methods allow the molecules soluble in solvents with different polarity to assemble at an interface.25–27 As the chemical–physical features of porphyrins are influenced by their aggregation state, the interactions between floating porphyrins and guest molecules can be finely tuned by controlling the properties of the Langmuir film. Additionally, LB and LS techniques allow the transfer of the floating film onto substrates without (or only slightly) altering their features.28 In this study, we formed a hybrid supramolecular system composed of a porphyrin derivative spread at the air–water interface with silica nano-helices suspended in the water subphase of a Langmuir trough, and then we deposited the obtained co-assemblies on solid supports using the LS approach. Silica nano-helices, left-handed and right-handed, were chosen as inert templates to induce chirality in the LS films of porphyrin aggregates; indeed, these nano-helical structures were demonstrated to efficiently induce chirality in molecules, including porphyrins, and inorganic nanoparticles when the latter are grafted on the silica surfaces.29–32 Porphyrin derivatives have been chosen as molecules commonly used for developing spectroscopic sensing devices33 and, also suitable for enantioselective sensor engineering.34 We chose a porphyrin derivative with a peripheral -COOH functionality capable of interacting with –NH2 groups present on the silica nano-helix surface upon functionalization with 3-aminopropyltriethoxysilane.35,36 Additionally, the utility of the compound with cubane isostere units37 (Fig. 1) to interact with the water dissolved nano-helices was shown. We aimed to create chiral thin films based on the co-assembly of achiral porphyrin and chiral inorganic helices capable of enantioselective discrimination of histidine enantiomers detected by surface plasmon resonance (SPR).38,39 | ||
| Fig. 1 Chemical structure of the achiral free base porphyrin (Pp) and its zinc(II) complex. | ||
Experimental
Materials
L-Histidine (99%) and D-histidine (98%) were purchased from Sigma-Aldrich and used as received without any further purification. Dichloromethane (99%) purchased from Carlo Erba Reagents was used as a spreading solvent. Water, used as the subphase, was purified with a Milli-Q/Elix3 Millipore system; in all experiments, water had a resistivity of 18.2 MΩ cm, pH ≈ 5.9. It was thermostated at 293 K by a Haake GH-D8 apparatus.An achiral free base porphyrin (Pp) and its zinc(II) metalated form (ZnPp) (Fig. 1), bearing a carboxylic group linked by a cubane bridge to the macrocycle, were used. Pp and ZnPp were synthesized according to procedures reported in the literature.40
Chiral silica helices were synthesized as previously reported.36 Briefly, the synthesis of right (RHH) and left-handed (LHH) silica nano-helices was achieved through the utilization of cationic bis-quaternary ammonium Gemini surfactants, which have a chemical formula of C2H4-1,2-((CH3)2N+C16H33)2 with a tartrate counterion.41 These surfactants can self-assemble in aqueous environments, forming nanometric helical structures when combined with tartrate counterions. With L- and D-tartrate, right-handed and left-handed nano-helices were formed respectively.35 This intriguing chirality switch in helix formation highlights the precise control over nanostructure synthesis achieved by manipulating the type of tartrate counterion. These self-assemblies were then used as templates to prepare silica nanostructures like helices through a sol–gel transcription procedure. After transcription, a mixture was washed with isopropyl alcohol to remove organic byproducts and excess tetraethyl orthosilicate (TEOS). This involved multiple washes through centrifugation. Typically, 5 mg of silica nano-helices were obtained from 0.36 mg of organic gel. The nano-helices underwent further processing, including centrifugation and redispersion in ethanol, followed by tip sonication to individualize and shorten them while eliminating aggregation. High-intensity ultrasonic processing was employed for dispersion and fragmentation, using a specific ultrasonic processor and specified parameters. Inorganic silica nano-helices were functionalized with 3-aminopropyltriethoxysilane (APTES). Therefore, 20 μL of APTES was added per 1 mg mL−1 of silica nano-helices in absolute ethanol. Then, the mixture was kept in an oil bath at 80 °C overnight. This procedure was repeated once to improve the grafting density of amines. The modified silica nanostructures, called LHH-NH2 and RHH-NH2 hereafter, were washed three times with absolute ethanol before further use.42
Instrumentation and characterization
Isotherms at the air–water interface were recorded using a NIMA-KSV trough equipped with a Brewster angle microscope (BAM). BAM images were acquired during barrier compression at different surface pressures. The surface pressure was monitored using 1 cm-wide filter paper as a Wilhelmy plate. Reflection spectra (RefSpec) were generated by measuring the difference in the reflection intensity of the subphase with and without the floating film during barrier compression. This difference is directly related to the absorbance of the thin floating film, providing valuable information about its properties.Pp and ZnPp molecules solubilized in dichloromethane solution (concentrations 1.24 × 10−4 M, spreading volume 250 μL) were spread on a subphase; 20 minutes after the spreading to allow solvent evaporation, surface compression was performed with the barrier speed set at 8 mm min−1. Floating films were transferred to solid supports (quartz and silicon dioxide) using the Langmuir–Schaefer method, a horizontal variation of the Langmuir–Blodgett technique. This technique is widely recognized for its distinct film transfer capabilities. The helices, in both enantiomeric forms, were suspended in the aqueous subphase at a concentration of 1 mg L−1, which appears to be, as will be discussed later, the ideal concentration for the formation of the adduct with the molecules that constitute the floating film.
UV-visible measurements were carried out using a PerkinElmer Lambda 650 spectrophotometer and electronic circular dichroism (ECD) spectra were recorded using a JASCO (J-1500 CD Spectrometer) with a scanning speed of 20 nm min−1. Si/SiO2 solid supports were used to deposit the LS films for morphological analysis using an atomic force microscopy (AFM) instrument (SmartSPM 1000 AIST-NT HORIBA). A PerkinElmer Spectrum One IR spectrometer was employed to assess the vibrational modes of immobilized porphyrin molecules on a solid substrate. This analysis was performed with a specialized multireflection accessory tailored for thin film examination. Each spectrum is an average of 32 scans (4 cm−1 resolution) from 1850 to 750 cm−1.
The interaction between the analytes and the transferred film was monitored using SPR using a Nanofilms Imaging EP4 apparatus (Accurion GmbH). Substrates used for SPR measurements were 2 cm × 2 cm SF-10 glass/Ti/Au slides and the SF-10 matching fluid (n = 1.725) was used for the optical coupling of the interfaces. The angle of incidence (AOI) was varied from 56° up to 66° with a step of 0.07° while the solutions were fluxed. The water solutions were fluxed using a peristaltic pump with a flux rate of 0.5 mL min−1. ΔAOI was calculated as the difference between the AOI measured for the SPR minimum recorded after ultrapure water flux and analyte solution flux on the same film. In a further experiment, ΔAOI was monitored during the time on the same film upon subsequent fluxes (10 min) of L-histidine, D-histidine, and racemic mixture (10−3 M) alternated with a washing step (5 min) in MilliQ grade water.
Results and discussion
Achiral Pp and silica nano-helix co-assembly formation
The Langmuir isotherm curve surface pressure (Π) vs. area per molecule of the floating film of Pp is shown in Fig. 2a. The curve's profile suggests that the Pp molecules form a strongly hydrophobic floating layer. A very long pseudo-gaseous phase was observed down to an area per molecule of about 75 Å2, at which surface area, an abrupt increase in the surface pressure was observed. It increased monotonically up to ∼45 mN m−1. The limiting area per molecule, calculated extrapolating the value of the area per molecule from the most pending part of the curve,43,44 was about 57 Å2. This is too small to suggest that the porphyrin derivatives lie flat on the water surface.45,46 Most likely, 3D aggregates of Pp are formed at the air–water interface. Indeed, the simultaneous presence of large Pp aggregates and the regions of the interface not covered by the organic molecules was observed by the BAM investigation (Fig. S1†).47,48 | ||
| Fig. 2 (a) Surface pressure variation (mN m−1) as a function of the area per molecule (Å2) of the Langmuir films of Pp spread on ultrapure water (black line), on the subphase containing the left-handed silica helices (blue line) and on the subphase containing the right-handed silica helices (red line); (b) reflection spectra of the Pp Langmuir film spread at the air–water interface (black line) and onto subphases containing RHH-NH2 (red line) and LHH-NH2 (blue line) recorded at 20 mN m−1 and the UV-visible spectrum (dashed line) of Pp dissolved in dichloromethane (1.24 × 10−4 M). | ||
The silica nano-helices in the subphase strongly affected the Langmuir curves’ profile, both for LHH-NH2 and for RHH-NH2.
The concentration of helices suspended in the aqueous subphase was selected by evaluating the profile of Langmuir curves by dissolving 0.1 mg L−1, 1 mg L−1, 6 mg L−1, 10 mg L−1, and 18 mg L−1 (Fig. S2†). For concentrations higher than 10 mg L−1, a weak surface activity of the helices was observed even in the absence of the Pp Langmuir film. For concentrations of 6 and 10 mg L−1, a short pseudo-gaseous phase was recorded as a consequence of the high number of helices reaching the aqueous subphase interface. In the case of the 0.6 mg L−1 concentration, an adequate number of helices were not obtained on the solid substrate (Fig. S3†) using the methods described in the Immobilization of the co-assemblies and their optical properties section.
The variation of the Langmuir curves’ profile suggested that the Pp molecules at the air–subphase interface interact with the nano-helices in the subphase, influencing the physicochemical properties of the floating film.49 We propose that the –COOH group attached to the porphyrin ring interacts with the –NH2 group at the surface of silica nano-helices. The limiting area per molecule of Pp increased from 57 Å2 to 95 Å2. The complex thus formed directly at the air–water interface (Pp@LHH-NH2 or @RHH-NH2) occupies a larger area.50 The morphology of the Langmuir films (reported in Fig. S4† for Pp@LHH-NH2 and S5 for @RHH-NH2) recorded through BAM images was different from that one observed in the absence of nano-helices (Fig. S1†), and the subphase surface was not uniformly covered by the floating films.
Even though the isotherm curves and BAM images confirm the interaction between the Pp molecules and the silica helices, no variation was induced by the inorganic structures on the Pp reflection spectrum when the co-assemblies were formed (Fig. 2b). The spectral profiles of the floating layers in the three different situations (spread on ultrapure water, subphase containing LHH-NH2 and RHH-NH2) were very similar both in the region of Q bands and in the position of the Pp Soret band that is located at 438 nm. It further suggested that the interaction between the silica nano-helices in the subphase and the Pp does not involve the macrocycle moieties. All three reflection spectra presented 18 nm red-shift when compared with the position of the Soret band of Pp dissolved in dichloromethane (420 nm, dashed line Fig. 2b). Such a shift is widely reported for Langmuir films of not completely amphiphilic compounds that are closely packed at the surface.51–53
Immobilization of the co-assemblies and their optical properties
The Langmuir–Schaefer method was used to transfer the film formed at the air–subphase interface on solid supports. This technique allows the formation of the mesoscopic complex of two species that are soluble in different chemical environments. In the present case, composite Pp and silica nano-helices were obtained without grafting approaches.12Pp and Pp@LHH-NH2 floating films and a cast film of amine-functionalized nano-helix aqueous suspension were transferred on gold slides and investigated using a FTIR spectrophotometer equipped with a multireflection tool. In Fig. S6,† the FTIR spectra of 8 layers of the Pp LS film (transferred@18 mN m−1, black line) and Pp@LHH-NH2 LS film (@20 mN m−1, blue line) were compared to the nano-helix spectrum (grey line). In particular, the spectra of the LS films based on Pp present the IR signals typical of porphyrin derivatives;33 whereas, the nano-helix cast film spectrum is dominated by the Si–O–Si stretching modes at about 1060 cm−1 (ref. 54) as well as the weak bands which correspond to the δNH2 and δCH APTES at about 1560 cm−1 and 1460 cm−1, respectively.55,56 When Pp@LHH-NH2 LS films are compared with the Pp LS film, the spectrum of the Pp@LHH-NH2 LS film presented slight modifications of IR bands localized at about 1740 cm−1 (labeled by the green arrow, νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and at about 1560 cm−1 (labelled by the red arrow, δNH2), which could be explained considering that these groups are indeed involved in the formation of the co-assemblies through weak interactions among –COOH and –NH2 pendant groups.
O) and at about 1560 cm−1 (labelled by the red arrow, δNH2), which could be explained considering that these groups are indeed involved in the formation of the co-assemblies through weak interactions among –COOH and –NH2 pendant groups.
The LS films were transferred onto a quartz slide (8 layers) and were characterized using UV-visible absorption spectroscopy (Fig. 3a). In all samples, the Soret band was located at about 430 nm with a slight shoulder at 415 nm, and no relevant shift was detected. This confirms that the interaction between the macrocycles spread onto the subphase interface and the nano-helices suspended in the subphase do not involve the macrocycle part of the porphyrin derivative, as previously observed by FTIR measurement.
 | ||
| Fig. 3 (a) UV-visible spectra of LS films transferred from ultrapure water (black line) from the subphase containing RHH-NH2 (red line) and LHH-NH2 (blue line); AFM images of the LS film of (b) Pp, (c) Pp@RHH-NH2 and (d) Pp@LHH-NH2 LS films. Inset: magnifications of the recorded images highlight the presence of the helices immobilized onto the solid slides. | ||
The presence of helices in Pp@ LHH-NH2 and Pp@RHH-NH2 was evidenced using atomic force microscopy (AFM) that clearly showed the unique helical structure transferred on ultra-flat substrates (Fig. 3). It should be noted that the silica nano-helices suspended in the ultrapure water subphase did not show any surface activity when compressed by Langmuir trough barriers, and it was not possible to transfer the suspended nanostructures onto solid substrates. This is further confirmation that the co-assemblies were directly formed at the air–subphase interface and the interaction was strong enough to ensure the transfer of the assembly formed onto solid supports. The morphology of the Pp LS film (Fig. 3b) is very different from those observed for the Pp@RHH-NH2 (Fig. 3c) or Pp@LHH-NH2 LS film (Fig. 3d) and the insets shown in Fig. 3c and d indicate the two different helicities of the transferred helices. The size of the observed tubular structures is in agreement with the data reported in the literature.57
Chirality induction on Pp aggregates
Circular dichroism spectroscopy was used to evaluate the chiral environment of Pp, Pp@LHH-NH2 or Pp@RHH-NH2 LS films in the 350–500 nm range. As expected and shown in Fig. 4, the dichroic spectrum of the LS film of the achiral Pp transferred from the ultrapure water subphase (black line) did not show any relevant signals.58 On the other hand, a clear signal was detected for both the Pp@LHH-NH2 (blue line, Fig. 4) or Pp@RHH-NH2 (red line, Fig. 4) LS films. Their CD signals are of opposite signs (Fig. 4), confirming that silica helicity governs the chirality of the supramolecular assembly.59 Circular dichroism spectra were also acquired for the Pp@RHH-NH2 and Pp@LHH-NH2 adducts using different concentrations of helices dispersed in the subphase (Fig. S7†). Again, the spectra indicate that the optimal concentration for obtaining the chiral adducts is 1 mg L−1. | ||
| Fig. 4 Circular dichroism spectra of 8 LS layers of Pp films transferred from the ultrapure water subphase (black line), from the subphase containing RHH-NH2 (red line), and LHH-NH2 (blue line); dotted lines refer to the corresponding absorbance spectra. | ||
The Pp@RHH-NH2 LS film showed a positive dichroic signal at around 415 nm and a weaker negative signal at about 395 nm. The Pp@LHH-NH2 LS film showed a mirror image signal. Such negative-positive or positive-negative dichroic bands (±) indicate that the porphyrins are arranged in a clockwise orientation (+) for RHH-NH2 or an anti-clockwise orientation (−) for LHH-NH2.60,61 The position of the dichroic bands in the hybrid films arises from the Siret band of Pp aggregates, since nano-helices did not show any absorption signals in the investigated wavelength range. Therefore, it is clear that the Langmuir method promoted the formation of the organic/inorganic composite chiral structures at the air–water interface, the immobilization of such co-assemblies within films, and the chirality of the obtained hybrid films was governed by the helicity of silica helices without any covalent bond formation.
As mentioned, the interaction between Pp and the silica chiral helices should involve the carboxylic acid group of the porphyrin and the amino groups on the nano-helices. This interaction was essential to allow the porphyrin to arrange according to the nano-helix chirality. Indeed, when the zinc complex of Pp was used (ZnPp), the chemical affinity between the central zinc ion and –NH2 groups of the silica is known to be stronger than the –COOH⋯NH2 affinity.62 Fig. S8† gives the FTIR spectra of ZnPp (black line), ZnPp@LHH-NH2 (grey line) and Pp@LHH-NH2 (blue line) LS films deposited on gold substrates. The arrows indicate the signals, arising from –CO and –NH2, which changed in the Pp@LHH-NH2 LS film FTIR spectrum upon Pp and nano-helix interaction (see above) but were not affected in the case of the Zn complex. Hence, the formation of the ZnPp@LHH-NH2 or ZnPp@RHH-NH2 complex involves ZnPp-NH2 axial interactions and not from the –COOH⋯NH2 affinity presumably and has a different spatial arrangement compared to the co-assemblies based on the Pp derivative. While the AFM characterization (Fig. S9†) also confirmed the nano-helices transfer promoted by the LS method in the presence of the ZnPp floating layer and the UV-visible spectra of ZnPp, ZnPp@LHH-NH2 and ZnPp@RHH-NH2 films (Fig. S10a†) showed that the Soret band profiles are different from Pp based films. In the case of ZnPp-based films, the Soret band was located at about 430 nm, but the shoulder at 415 nm was not seen (Fig. S10a†). Circular dichroism spectra of ZnPp, ZnPp@LHH-NH2 and ZnPp@RHH-NH2 (Fig. S10b†) showed no signal, highlighting the importance of the self-assembly process of the Pp at the surface of the nano-helices for the chirality induction.
SPR enantioselective discrimination of histidine enantiomers
We evaluated the possibility of selectively discriminating the two enantiomers of histidine63 using the chiral co-assemblies Pp@LHH-NH2 or RHH-NH2 by SPR. This technique was already used to evaluate the presence of histidine in water mixtures using porphyrin-based active layers.64 First, the SPR curves were recorded using the 8-layer LS film of Pp subjected to ultrapure water flux and then to the flux of two histidine enantiomer solutions at 10−3 M concentration (Fig. S11†). The Pp active layer is sensitive without preferential interaction towards the two enantiomers and a ΔAOI of about 0.18° was recorded both for L- and D-histidine flux.A clear amino acid enantiomer effect was observed with the Pp@RHH-NH2 LS film (8 layers). A relevant shift of the SPR minimum was observed when the L-histidine was fluxed (ΔAOI = 0.47° at 10−3 M), whereas the shift was negligible with D-histidine (about 0.08°). This demonstrated that the supramolecular arrangement of Pp induced by the presence of chiral silica nanostructures influenced the interaction with the chiral analyte (Fig. S12a†). SPR sensitivity towards the histidine's enantiomers is reversed when Pp@LHH-NH2 was used (8 layers LS film, Fig. S12b†). L-Histidine might influence the Pp packing of the porphyrin derivative arranged on RHH-NH2, affecting the refractive index of the composite complex, which results in a variation of the AOI detected by SPR. As expected, specular behaviour was found in the case of the D-histidine flux onto the SPR slide covered by Pp@LHH-NH2.
The AOI shifts of Pp, Pp@RHH-NH2 and Pp@LHH-NH2 LS films were checked during the time upon L-histidine, D-histidine and a racemic solution at 10−3 M concentration injection (see the Materials and methods section). The responses of the active Pp@silica helix layer were markedly influenced by the fluxed enantiomer, while the Pp-only layer showed the same AOI shifts for the racemic and enantiopure histidine solutions (Fig. 5a). Very interestingly, all the tested active layers demonstrated a reversible interaction with analytes upon fluxing MilliQ grade water for 5 min. It was also possible to calculate the equilibrium dissociation constant (KD) values for the two co-assembled films, which exhibited enantioselective behaviour. Specifically, for the Pp@RHH-NH2 film, a KD value of 2 × 10−3 M was obtained for L-histidine, and 1.2 × 10−2 M for D-histidine. These values clearly indicate that the interactions fall within the weak interaction range65 (presumably hydrogen bonds and van der Waals interactions) and that the chiral hybrid film shows a higher affinity towards the L-enantiomer. Similarly, the same reasoning applies to the Pp@LHH-NH2 film, which demonstrated higher affinity towards the D-enantiomer. In this case, the calculated KD values are 4.6 × 10−3 M for L-histidine and 1.9 × 10−3 M for D-histidine. Fig. 5(b and c) showed that we obtained a linear relationship between the angle of incidence variations and the histidine concentration in the range 10−6–10−3 M for Pp@silica nano-helices and this is a fundamental result for the development of an active layer. Additionally, L-histidine and D-histidine were fluxed over the LS films of Pp, Pp@LHH-NH2, and Pp@RHH-NH2 in different ratios (Fig. 5d), maintaining a total histidine concentration of 10−3 M. It is observed that the variations in AOI are influenced by the ratio between the two enantiomers, and the recorded values are in good agreement with what can be predicted using the ΔAOI reported in Fig. 5b and c.
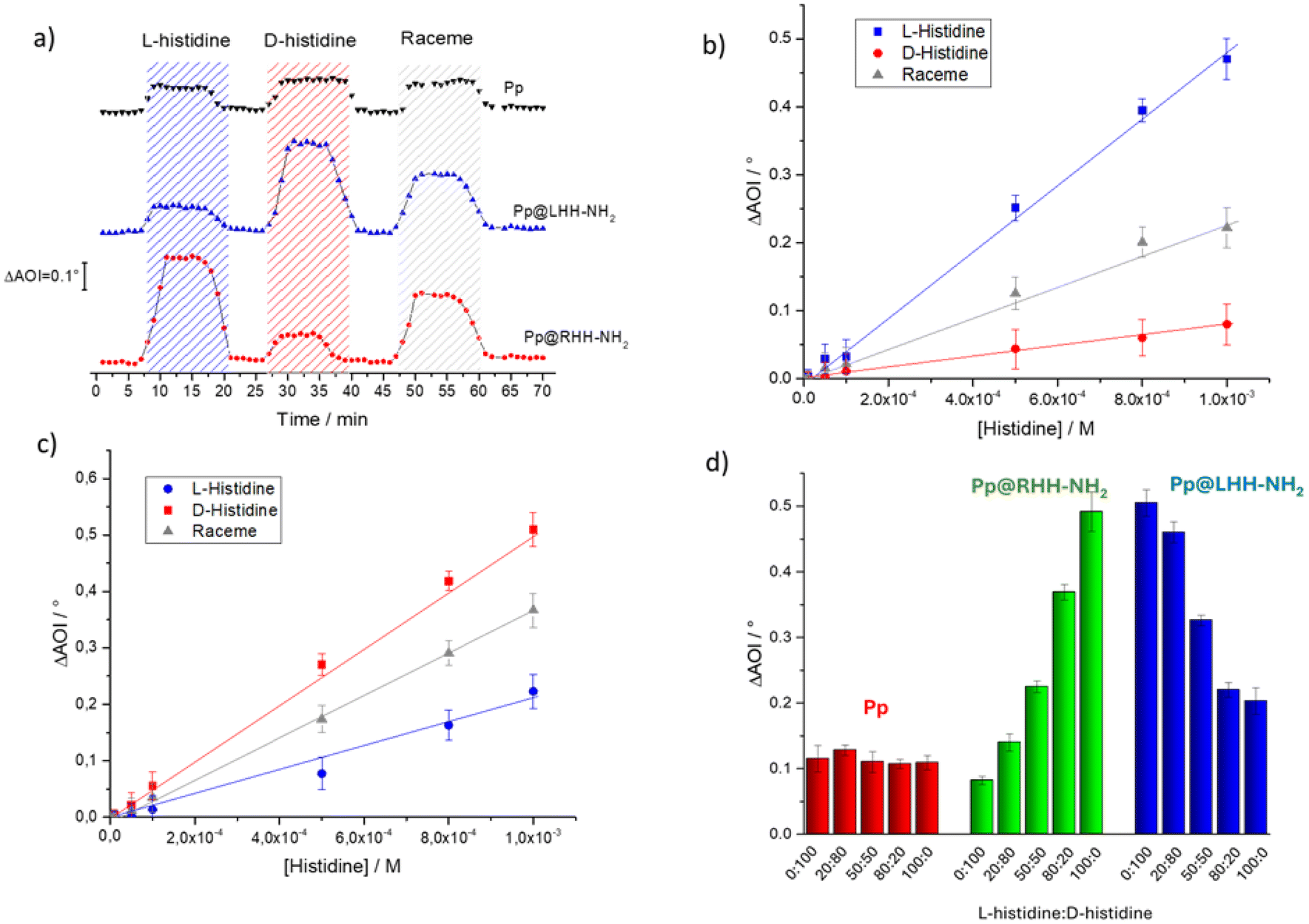 | ||
| Fig. 5 (a) AOI time variations of Pp, Pp@RHH-NH2 and Pp@LHH-NH2 when exposed to analyte fluxes are compared; the angle of incident variations as a function of D- L- and racemic histidine concentrations using as an active layer the LS film of (b) Pp@RHH-NH2 and (c) Pp@LHH-NH2 co-assembly. (d) ΔAOI of Pp, Pp@RHH-NH2 and Pp@LHH-NH2 exposed to different molar ratios of L- and D-histidine (histidine total concentration was kept at 10−3 M). | ||
These results evidence that the composite thin films of Pp@silica helices can be successfully deposited on a plethora of different solid supports while preserving the imprinted physicochemical features capable of enantioselective recognition. The use of inert but purely morphologically chiral templates, such as the silica nano-helices, to induce chirality by the Langmuir trough approach is a promising and versatile strategy, adding up the enantioselectivity according to the chirality of the nano-helices.
Moreover, this represents a starting point of paramount importance since it is challenging to develop and engineer enantioselective sensors. This may represent a successful strategy to easily produce chiral sensing materials. The proposed composite films show a good enantioselectivity that can be further improved, for example, using a sensor array approach and proper normalization or multivariate data analysis, aiming to increase the enantiomer discrimination.34
Conclusions
A Langmuir trough was used to promote the formation of composite thin films at the air–subphase interface between chiral silica helices functionalized with –NH2 groups dispersed in the aqueous subphase and the floating film formed by the porphyrin derivative. The deposition of such co-assemblies by the Langmuir–Schaefer method was confirmed using AFM and FTIR, which clearly showed the interaction between the organic and the inorganic species at the interface. In particular, atomic force microscopy, carried out on solid films, showed the presence of helices within the film and that the morphology of the porphyrin film is drastically different from that of the hybrid layer. Circular dichroism investigations showed that a chiral arrangement of the Pp assembly is induced by the presence of the chiral helices, promoting the formation of chiral co-assemblies. Hybrid layers of Pp@LHH-NH2 and Pp@RHH-NH2 were deposited onto SPR slides and ΔAOI was monitored when exposed to L- and D-histidine enantiomers. A clear chiral discrimination was observed for Pp@RHH-NH2 towards L-histidine and specular behavior was observed for the Pp@LHH-NH2 system that appears more sensitive towards D-histidine than L-histidine.Author contributions
Michela Ottolini: investigation and writing – original draft; Zakaria Anfar: investigation and conceptualization; Nitika Grover: investigation and conceptualization; Gabriele Magna: conceptualization; Manuela Stefanelli: conceptualization; Roberto Paolesse: conceptualization; Mathias O. Senge: writing – review & editing, and conceptualization; Simona Bettini: conceptualization, methodology, investigation, and writing – review & editing; Ludovico Valli: supervision, funding acquisition, and writing – review & editing; Reiko Oda: writing – review & editing and conceptualization; Gabriele Giancane: conceptualization, methodology, supervision, writing – review & editing.Data availability
All data generated or analysed during this study are included in the main manuscript and in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by PRIN PNRR 2022 SupraPhotoChem (P2022WLAY7), MISE ANASTASIA (Prog n. F/310253/05/X56), the Science Foundation Ireland (SFI award 21/FFP-A/9469), the Technical University of Munich – Institute for Advanced Study (Hans Fischer Senior Fellowship, MOS) and the European Union's Horizon 2020 Research and Innovation Program under the FET-OPEN project INITIO, grant agreement 828779.References
- R. A. Hegstrom and D. K. Kondepudi, Sci. Am., 1990, 262, 108–115 CrossRef.
- D. G. Blackmond, Cold Spring Harbor Perspect. Biol., 2019, 11, a032540 CrossRef CAS PubMed.
- J. J. L. M. Cornelissen, A. E. Rowan, R. J. M. Nolte and N. A. J. M. Sommerdijk, Chem. Rev., 2001, 101, 4039–4070 CrossRef CAS PubMed.
- M. A. Mateos-Timoneda, M. Crego-Calama and D. N. Reinhoudt, Chem. Soc. Rev., 2004, 33, 363 RSC.
- Y. Lin and C. Mao, Front. Mater. Sci., 2011, 5, 247–265 CrossRef PubMed.
- M. Sun, X. Wang, X. Guo, L. Xu, H. Kuang and C. Xu, Chem. Sci., 2022, 13, 3069–3081 RSC.
- L. Wu, Y. Xu, T. Hou, J. Jia, X. Huang, G. Weng, S. Bao and L. Zheng, Chem. – Eur. J, 2021, 27, 16722–16734 CrossRef CAS PubMed.
- C. Bai and M. Liu, Angew. Chem., Int. Ed., 2013, 52, 2678–2683 CrossRef CAS PubMed.
- G. M. Whitesides and M. Boncheva, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4769–4774 CrossRef CAS PubMed.
- M. Liu, L. Zhang and T. Wang, Chem. Rev., 2015, 115, 7304–7397 CrossRef CAS PubMed.
- S. He, Z. Jiang, X. Dou, L. Gao and C. Feng, ChemPlusChem, 2023, 88, e202300226 CrossRef CAS PubMed.
- A. R. A. Palmans and E. W. Meijer, Angew. Chem., Int. Ed., 2007, 46, 8948–8968 CrossRef CAS PubMed.
- Fundamentals of porphyrin chemistry: a 21st century approach, ed. P. Brothers and M. Senge, Wiley, Hoboken, NJ, 2022 Search PubMed.
- G. Magna, D. Monti, C. Di Natale, R. Paolesse and M. Stefanelli, Molecules, 2019, 24, 4307 CrossRef CAS PubMed.
- T. Fukui, S. Kawai, S. Fujinuma, Y. Matsushita, T. Yasuda, T. Sakurai, S. Seki, M. Takeuchi and K. Sugiyasu, Nat. Chem., 2017, 9, 493–499 CrossRef CAS PubMed.
- H. Liu, J. Xu, Y. Li and Y. Li, Acc. Chem. Res., 2010, 43, 1496–1508 CrossRef CAS PubMed.
- F. Helmich, C. C. Lee, M. M. L. Nieuwenhuizen, J. C. Gielen, P. C. M. Christianen, A. Larsen, G. Fytas, P. E. L. G. Leclère, A. P. H. J. Schenning and E. W. Meijer, Angew. Chem., Int. Ed., 2010, 49, 3939–3942 CrossRef CAS PubMed.
- H. Jiang, L. Zhang, J. Chen and M. Liu, ACS Nano, 2017, 11, 12453–12460 CrossRef CAS PubMed.
- M. F. J. Mabesoone, A. J. Markvoort, M. Banno, T. Yamaguchi, F. Helmich, Y. Naito, E. Yashima, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2018, 140, 7810–7819 CrossRef CAS PubMed.
- M. O. Senge, N. N. Sergeeva and K. J. Hale, Chem. Soc. Rev., 2021, 50, 4730–4789 RSC.
- A. Buccolieri, M. Hasan, S. Bettini, V. Bonfrate, L. Salvatore, A. Santino, V. Borovkov and G. Giancane, Anal. Chem., 2018, 90, 6952–6958 CrossRef CAS PubMed.
- A. J. Al-Alwani, V. N. Mironyuk, M. V. Pozharov, M. V. Gavrikov and E. G. Glukhovskoy, Soft Mater., 2022, 20, 310–321 CrossRef CAS.
- C. M. Drain, A. Varotto and I. Radivojevic, Chem. Rev., 2009, 109, 1630–1658 CrossRef CAS PubMed.
- T. Da Ros, M. Prato, M. Carano, P. Ceroni, F. Paolucci, S. Roffia, L. Valli and D. Guldi, J. Organomet. Chem., 2000, 599, 62–68 CrossRef CAS.
- S. A. Hussain, B. Dey, D. Bhattacharjee and N. Mehta, Heliyon, 2018, 4, e01038 CrossRef PubMed.
- G. Brezesinski and H. Möhwald, Adv. Colloid Interface Sci., 2003, 100–102, 563–584 CrossRef CAS PubMed.
- M. C. Petty, Langmuir-Blodgett Films: An Introduction, Cambridge University Press, 1st edn, 1996 Search PubMed.
- M. M. Velázquez, T. Alejo, D. López-Díaz, B. Martín-García, M. D. Merchán, M. M. Velázquez, T. Alejo, D. López-Díaz, B. Martín–García and M. D. Merchán, in Two-dimensional Materials - Synthesis, Characterization and Potential Applications, IntechOpen, 2016 Search PubMed.
- J. Cheng, G. Le Saux, J. Gao, T. Buffeteau, Y. Battie, P. Barois, V. Ponsinet, M.-H. Delville, O. Ersen, E. Pouget and R. Oda, ACS Nano, 2017, 11, 3806–3818 CrossRef CAS PubMed.
- M. J. Álvaro-Martins, J. Garcés-Garcés, A. Scalabre, P. Liu, F. Fernández-Lázaro, Á. Sastre-Santos, D. M. Bassani and R. Oda, ChemPhysChem, 2023, 24, e202200573 CrossRef PubMed.
- A. Scalabre, Y. Okazaki, B. Kuppan, T. Buffeteau, F. Caroleo, G. Magna, D. Monti, R. Paolesse, M. Stefanelli, S. Nlate, E. Pouget, H. Ihara, D. M. Bassani and R. Oda, Chirality, 2021, 33, 494–505 CrossRef CAS PubMed.
- G. Duroux, L. Robin, P. Liu, E. Dols, M. D. S. L. Mendes, S. Buffière, E. Pardieu, A. Scalabre, T. Buffeteau, S. Nlate, R. Oda, M. S. Raju, M. Atzori, C. Train, G. L. J. A. Rikken, P. Rosa, E. A. Hillard and E. Pouget, Nanoscale, 2023, 15, 12095–12104 RSC.
- S. Bettini, R. Pagano, L. Valli and G. Giancane, J. Phys. Chem. C, 2014, 118, 12384–12390 CrossRef CAS.
- G. Magna, M. Šakarašvili, M. Stefanelli, G. Giancane, S. Bettini, L. Valli, L. Ustrnul, V. Borovkov, R. Aav, D. Monti, C. Di Natale and R. Paolesse, ACS Appl. Mater. Interfaces, 2023, 15, 30674–30683 CrossRef CAS PubMed.
- T. Delclos, C. Aimé, E. Pouget, A. Brizard, I. Huc, M.-H. Delville and R. Oda, Nano Lett., 2008, 8, 1929–1935 CrossRef CAS PubMed.
- N. Ryu, Y. Okazaki, K. Hirai, M. Takafuji, S. Nagaoka, E. Pouget, H. Ihara and R. Oda, Chem. Commun., 2016, 52, 5800–5803 RSC.
- G. M. Locke, S. S. R. Bernhard and M. O. Senge, Chem. – Eur. J., 2019, 25, 4590–4647 CrossRef CAS PubMed.
- P. Arora, E. Talker, N. Mazurski and U. Levy, Sci. Rep., 2018, 8, 9060 CrossRef PubMed.
- M. G. Manera, E. Ferreiro-Vila, J. M. García-Martín, A. Cebollada, A. García-Martín, G. Giancane, L. Valli and R. Rella, Sens. Actuators, B, 2013, 182, 232–238 CrossRef CAS.
- N. Grover, G. M. Locke, K. J. Flanagan, M. H. R. Beh, A. Thompson and M. O. Senge, Chem. – Eur. J., 2020, 26, 2405–2416 CrossRef CAS PubMed.
- Y. Okazaki, N. Ryu, T. Buffeteau, S. Pathan, S. Nagaoka, E. Pouget, S. Nlate, H. Ihara and R. Oda, Chem. Commun., 2018, 54, 10244–10247 RSC.
- A. Amestoy, A. Rangra, V. Mansard, D. Saya, E. Pouget, E. Mazaleyrat, F. Severac, C. Bergaud, R. Oda and M.-H. Delville, ACS Appl. Mater. Interfaces, 2023, 15, 39480–39493 CrossRef CAS PubMed.
- J. M. Kroon, E. J. R. Sudhoelter, A. P. H. J. Schenning and R. J. M. Nolte, Langmuir, 1995, 11, 214–220 CrossRef CAS.
- A. Ulman, An Introduction to Ultrathin Organic Films: From Langmuir–Blodgett to Self–Assembly, Academic Press, San Diego, 1991 Search PubMed.
- G. Giancane, V. Borovkov, Y. Inoue, S. Conoci and L. Valli, Soft Matter, 2013, 9, 2302 RSC.
- A. Colombelli, M. G. Manera, V. Borovkov, G. Giancane, L. Valli and R. Rella, Sens. Actuators, B, 2017, 246, 1039–1048 CrossRef CAS.
- S. Hénon and J. Meunier, Rev. Sci. Instrum., 1991, 62, 936–939 CrossRef.
- D. Hoenig and D. Moebius, J. Phys. Chem., 1991, 95, 4590–4592 CrossRef CAS.
- A. Buccolieri, S. Bettini, L. Salvatore, F. Baldassarre, G. Ciccarella and G. Giancane, Sens. Actuators, B, 2018, 267, 265–271 CrossRef CAS.
- R. Flores-Sánchez, M. Bigorra-Mir, F. Gámez, T. Lopes-Costa, P. G. Argudo, M. T. Martín-Romero, L. Camacho and J. M. Pedrosa, Chem. Phys. Lett., 2023, 819, 140450 CrossRef.
- D.-J. Qian, C. Nakamura and J. Miyake, Thin Solid Films, 2001, 397, 266–275 CrossRef CAS.
- F. Armand, P.-A. Albouy, F. Da Cruz, M. Normand, V. Huc and E. Goron, Langmuir, 2001, 17, 3431–3437 CrossRef CAS.
- D.-J. Qian, H.-T. Chen, B. Liu, X.-M. Xiang, T. Wakayama, C. Nakamura and J. Miyake, Colloids Surf., A, 2006, 284–285, 180–186 CrossRef.
- B. C. Smith, Infrared spectral interpretation: a systematic approach, CRC Press, Boca Raton, 1999 Search PubMed.
- R. Nag, Y. Okazaki, A. Scalabre, Z. Anfar, S. Nlate, T. Buffeteau, R. Oda and E. Pouget, Chem. Commun., 2022, 58, 13515–13518 RSC.
- E. J. Cueto-Díaz, A. Castro-Muñiz, F. Suárez-García, S. Gálvez-Martínez, M. C. Torquemada-Vico, M. P. Valles-González and E. Mateo-Martí, Nanomaterials, 2021, 11, 2893 CrossRef PubMed.
- A. Brizard, D. Berthier, C. Aimé, T. Buffeteau, D. Cavagnat, L. Ducasse, I. Huc and R. Oda, Chirality, 2009, 21(1E), E153–E162 CrossRef CAS PubMed.
- S. Wu, Z.-Z. Yin, D. Wu, Y. Tao and Y. Kong, Langmuir, 2019, 35, 16761–16769 CrossRef CAS PubMed.
- M. A. Castriciano, A. Romeo, G. De Luca, V. Villari, L. M. Scolaro and N. Micali, J. Am. Chem. Soc., 2011, 133, 765–767 CrossRef CAS PubMed.
- M. Stefanelli, F. Mandoj, G. Magna, R. Lettieri, M. Venanzi, R. Paolesse and D. Monti, Molecules, 2020, 25, 4544 CrossRef CAS PubMed.
- P. Liu, Y. Battie, Y. Okazaki, N. Ryu, E. Pouget, S. Nlate, T. Sagawa and R. Oda, Chem. Commun., 2021, 57, 12024–12027 RSC.
- B. Liu, D.-J. Qian, H.-X. Huang, T. Wakayama, S. Hara, W. Huang, C. Nakamura and J. Miyake, Langmuir, 2005, 21, 5079–5084 CrossRef CAS PubMed.
- S. Bettini, N. Grover, M. Ottolini, C. Mattern, L. Valli, M. O. Senge and G. Giancane, Langmuir, 2021, 37, 13882–13889 CrossRef CAS PubMed.
- S. Bettini, R. Pagano, V. Borovkov, G. Giancane and L. Valli, J. Colloid Interface Sci., 2019, 533, 762–770 CrossRef CAS PubMed.
- L. Verdier, J. Gharbi-Benarous, G. Bertho, N. Evrard-Todeschi, P. Mauvais and J.-P. Girault, J. Chem. Soc., Perkin Trans. 2, 2000, 2363–2371 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr02344g |
| This journal is © The Royal Society of Chemistry 2024 |