
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Assembling Fe4 single-molecule magnets on a TiO2 monolayer†
Andrea Luigi
Sorrentino
 ab,
Lorenzo
Poggini
*bc,
Giulia
Serrano
ab,
Giuseppe
Cucinotta
b,
Brunetto
Cortigiani
b,
Luigi
Malavolti
d,
Francesca
Parenti
e,
Edwige
Otero
f,
Marie-Anne
Arrio
g,
Philippe
Sainctavit
fg,
Andrea
Caneschi
a,
Andrea
Cornia
e,
Roberta
Sessoli
bc and
Matteo
Mannini
*b
ab,
Lorenzo
Poggini
*bc,
Giulia
Serrano
ab,
Giuseppe
Cucinotta
b,
Brunetto
Cortigiani
b,
Luigi
Malavolti
d,
Francesca
Parenti
e,
Edwige
Otero
f,
Marie-Anne
Arrio
g,
Philippe
Sainctavit
fg,
Andrea
Caneschi
a,
Andrea
Cornia
e,
Roberta
Sessoli
bc and
Matteo
Mannini
*b
aDepartment of Industrial Engineering - DIEF - and INSTM Research Unit, University of Florence, Via Santa Marta 3, 50139 Florence, Italy
bDepartment of Chemistry “U. Schiff” - DICUS - and INSTM Research Unit, University of Florence, Via della Lastruccia 3-13, 50019 Sesto Fiorentino, FI, Italy. E-mail: lpoggini@iccom.cnr.it; matteo.mannini@unifi.it
cInstitute for Chemistry of Organo-Metallic Compounds (ICCOM-CNR), Via Madonna del Piano, 50019 Sesto Fiorentino, FI, Italy
dMax Planck Institute for Solid State Research, Heisenbergstr. 1, 70569 Stuttgart, Germany
eDepartment of Chemical and Geological Sciences and INSTM Research Unit, University of Modena and Reggio Emilia, Via G. Campi 103, 41125 Modena, Italy
fSynchrotron-SOLEIL, L'Orme des Merisiers, 91192 Saint-Aubin, France
gCNRS UMR7590, Institut de Minéralogie, de Physique des Matériaux et de Cosmochimie (IMPMC), Sorbonne Université/MNHN, 4 place Jussieu, 75252 Paris Cedex 5, France
First published on 1st July 2024
Abstract
The decoration of technologically relevant surfaces, such as metal oxides, with Single-Molecule Magnets (SMMs) constitutes a persistent challenge for the integration of these molecular systems into novel technologies and, in particular, for the development of spintronic and quantum devices. We used UHV thermal sublimation to deposit tetrairon(III) propeller-shaped SMMs (Fe4) as a single layer on a TiO2 ultrathin film grown on Cu(001). The properties of the molecular deposit were studied using a multi-technique approach based on standard topographic and spectroscopic measurements, which demonstrated that molecules remain largely intact upon deposition. Ultralow temperature X-ray Absorption Spectroscopy (XAS) with linearly and circularly polarized light was further employed to evaluate both the molecular organization and the magnetic properties of the Fe4 monolayer. X-ray Natural Linear Dichroism (XNLD) and X-ray Magnetic Circular Dichroism (XMCD) showed that molecules in a monolayer display a preferential orientation and an open magnetic hysteresis with pronounced quantum tunnelling steps up to 900 mK. However, unexpected extra features in the XAS and XMCD spectra disclosed a minority fraction of altered molecules, suggesting that the TiO2 film may be chemically non-innocent. The observed persistence of SMM behaviour on a metal oxide thin film opens new possibilities for the development of SMM-based hybrid systems.
Introduction
Single-Molecule Magnets (SMMs) are extremely promising building blocks for the development of molecular spintronic and quantum computing applications because of their magnetic bistability and unique quantum behavior.1–8 In this context, the assembling of SMMs on solid substrates is a key step for developing breakthrough technological devices.9–12 The persistence of SMM properties on a surface, including magnetic hysteresis and Quantum Tunneling of the Magnetization (QTM), was demonstrated in 2009 by sub-kelvin measurements.13,14 Since then, the operating temperature of surface-supported SMMs has increased considerably and the current record is 28 K for Tb2@C80(CH2Ph) assembled on graphene.15 More recently, the focus has moved to “active” substrates as a means to either directly control molecular quantum dynamics (e.g. superconductors),16,17 enhance SMM performances,18,19 or facilitate single-spin sensing.20–23 Thin decoupling layers of metal oxides, such as MgO, were found to dramatically enhance the magnetic remanence of TbPc2 SMM films (H2Pc = phthalocyanine).18,19 MgO layers20,21,24–26 were also crucial for sensing the local magnetic features of molecules or atoms by Scanning Tunneling Microscopy (STM),20,25,27,28 allowing the STM detection of EPR-like signals from paramagnetic species.20,27,29,30The vastness of available oxide materials and their widespread use for technological applications leaves additional space for the exploration of alternative materials which can support molecular spin functionalities. In this context, TiO2 has a high technological significance thanks to its photocatalytic31,32 and electron transport properties,33,34 which are of relevance for sensing,35 catalysis, and photovoltaics.36–38 The nature of the surface plays a decisive role in promoting these functionalities, and the richness of TiO2 structural phases that are accessible by finely tuning the preparation technique makes this material particularly versatile for multifunctional electronic devices.39 In fact, TiO2 can be nanostructured as a thin film grown on metals to control its electronic properties,40–42 which depend on the structural phase, the surface stoichiometry, and the presence of defects.43–45 For instance, some of us demonstrated that Cu(001) surface is a good playground for the growth of continuous TiO2 films having different structural phases and electronic properties depending on growth condition parameters.46 In parallel, we showed that SMM behaviour of TbPc2 molecules might persist on sub-monolayer TiO2 islands with a lepidocrocite-like structure grown on Ag(100).47
Here, we investigated the chemical, structural, and magnetic properties of tetrairon(III) propeller-like SMMs (Fe4) deposited on a single layer of TiO2 grown on Cu(001) (hereafter called TiO2/Cu). The Fe4 complexes are archetypal, low-temperature SMMs with general formula [Fe4(LR)2(dpm)6], where a central FeIII ion is surrounded by three peripheral FeIII ions arranged at the vertices of a triangle. The dipivaloylmethanido ligands (dpm−) bind exclusively to peripheral ions, while the magnetic core is held together by two tripodal ligands (LR)3− = [RC(CH2O)3]3−. The bridging oxygen atoms of the tripods promote antiferromagnetic interactions between the s = 5/2 spins of the central and peripheral FeIII ions, yielding a molecular spin S = 5 ground state.48 Complexes of this family exhibit slow magnetic relaxation only below 1 K, but have good chemical stability and can be easily functionalized by proper choice of the R substituent. In this way, derivatives suitable for deposition on surfaces by either a wet-chemistry approach, electrospray, or thermal sublimation in UHV have been designed.13,14,16,49–54 Recently, a Fe4 derivative with R = CH2SMe (hereafter called Fe4SMe) was sublimated on Pb(111), showing great chemical stability and enhanced organization on the surface promoted by the short S-functionalized tethering group.16 This derivative was indeed originally designed for deposition on metal surfaces (e.g., Au, Pb) to form a well-ordered and assembled monolayer. Although it is not specifically tailored for TiO2 substrates, it exhibits superior stability during thermal sublimation and improved magnetic behavior compared to all other Fe4 compounds.51,55
In this paper, we deposited Fe4SMe molecules on the TiO2/Cu surface and used both STM and X-ray Photoelectron Spectroscopy (XPS) to check their chemical and structural integrity after deposition. Furthermore, we used synchrotron-based X-ray Absorption Spectroscopy (XAS) methods, namely X-ray Natural Linear Dichroism (XNLD) and X-ray Magnetic Circular Dichroism (XMCD), to probe their electronic structure and magnetic properties. This spectroscopic study showed that the main fraction of Fe4SMe complexes are intact and feature magnetic hysteresis and QTM up to 900 mK, while a minority fraction of molecules contain reduced FeII metal centers and become paramagnetic.
Results and discussion
The TiO2 film was grown on Cu(001) single crystal (see Methods) and studied by XPS and STM to get chemical, morphological, and structural information. The Ti 2p XPS spectrum, reported in Fig. 1a, reflects an estimated coverage of 1 ML. It is dominated by the expected TiIV signal at 458.7 eV (Ti 2p3/2)46,56 and by its spin–orbit coupled component (Ti 2p1/2) shifted by 5.7 eV (Fig. 1a, in light magenta).46,57 Shake-up components are present at higher binding energies (460.8 and 466.5 eV, Fig. 1a, in wine) along with satellite features at 471.8 and 482.5 eV (Fig. 1a, in green).46,58–60 Furthermore, an additional component located at 457.6 eV, accompanied by its spin–orbit coupled peak, discloses a small amount of TiIII arising from the reductive annealing process (Fig. 1a, in blue).61–64 From the deconvolution analysis of the spectra this reduced fraction amounts to 10.5% of the overall titanium content (see Fig. S3 and Table S1†). The signals in the O 1s XPS spectrum (Fig. S1†) are typical for TiO2 films46,62,64 and are discussed in detail in the XPS characterization section of the ESI.†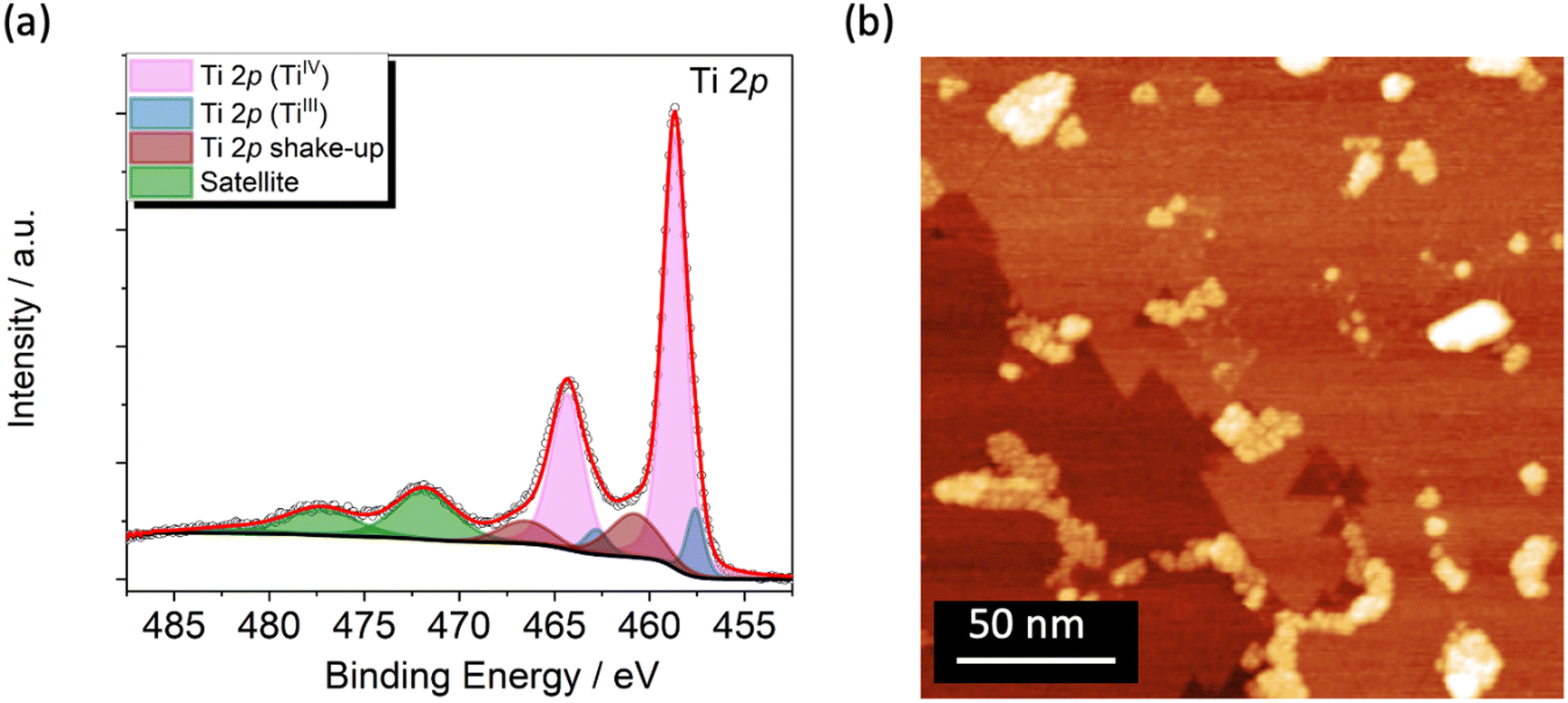 | ||
| Fig. 1 (a) Ti 2p XPS spectrum of the TiO2 single layer grown on Cu(001) single crystal. (b) STM image of the TiO2/Cu surface at RT (V = 1.5 V, It = 150 pA). | ||
STM images at Room Temperature (RT) were acquired to evaluate the film morphology and to confirm the TiO2 coverage estimated by XPS. The wide-area image in Fig. 1b shows a complete layer of TiO2, characterized by domain boundaries that suggest the presence of the quasi-hexagonal (QH) structure with squared bright spot ascribable to the growth of a second TiO2 layer.46 The formation of the single layer of TiO2-QH grown on Cu(001) structure is confirmed by Low Energy Electron Diffraction (LEED) showing two p(2 × 7) domains rotated by 30 degrees (Fig. S2†).46,65,66
A monolayer of Fe4SMe was deposited on TiO2/Cu following the protocol described in Methods. After molecular deposition, the XPS spectrum in the Ti 2p region (Fig. S3†) shows no significant variation as compared with that of the pristine substrate, thus confirming the stability of the TiO2 surface upon further processing.47,64,67 The small decrease (0.3%) of the TiIII fraction lies well within the limits of semiquantitative analysis by XPS (Table S1†). The C 1s spectrum reported in Fig. 2a reveals two main contributions attributed to the aliphatic and oxygen-bound carbon atoms of the molecular layer at 284.9 and 286.5 eV (Fig. 2a, in yellow and dark blue, respectively). An additional shake-up component is present at 289.6 eV (Fig. 2a, in cyan).16,68 The S 2p signal consists in two contributions located at 163.7 eV and at 164.9 eV (spin–orbit coupled component) (Fig. 2b), indicating the presence of intact CH2SMe functional groups.69–71 The C/S atomic ratio (46.0 ± 2.3) is in gross agreement with the expected value of 39 based on the molecular formula (C78H136Fe4O18S2), and suggests the overall integrity of Fe4SMe molecules on the TiO2/Cu surface. Unfortunately, a more comprehensive semiquantitative elemental analysis of the molecular species cannot be carried out due to the overlap of the molecular Fe 2p and O 1s signals with the Cu LMM Auger peaks and the oxygen signals, respectively, of the TiO2/Cu substrate.46,72 However, successful sublimation of Fe4SMe molecules has been demonstrated in a previous study.16
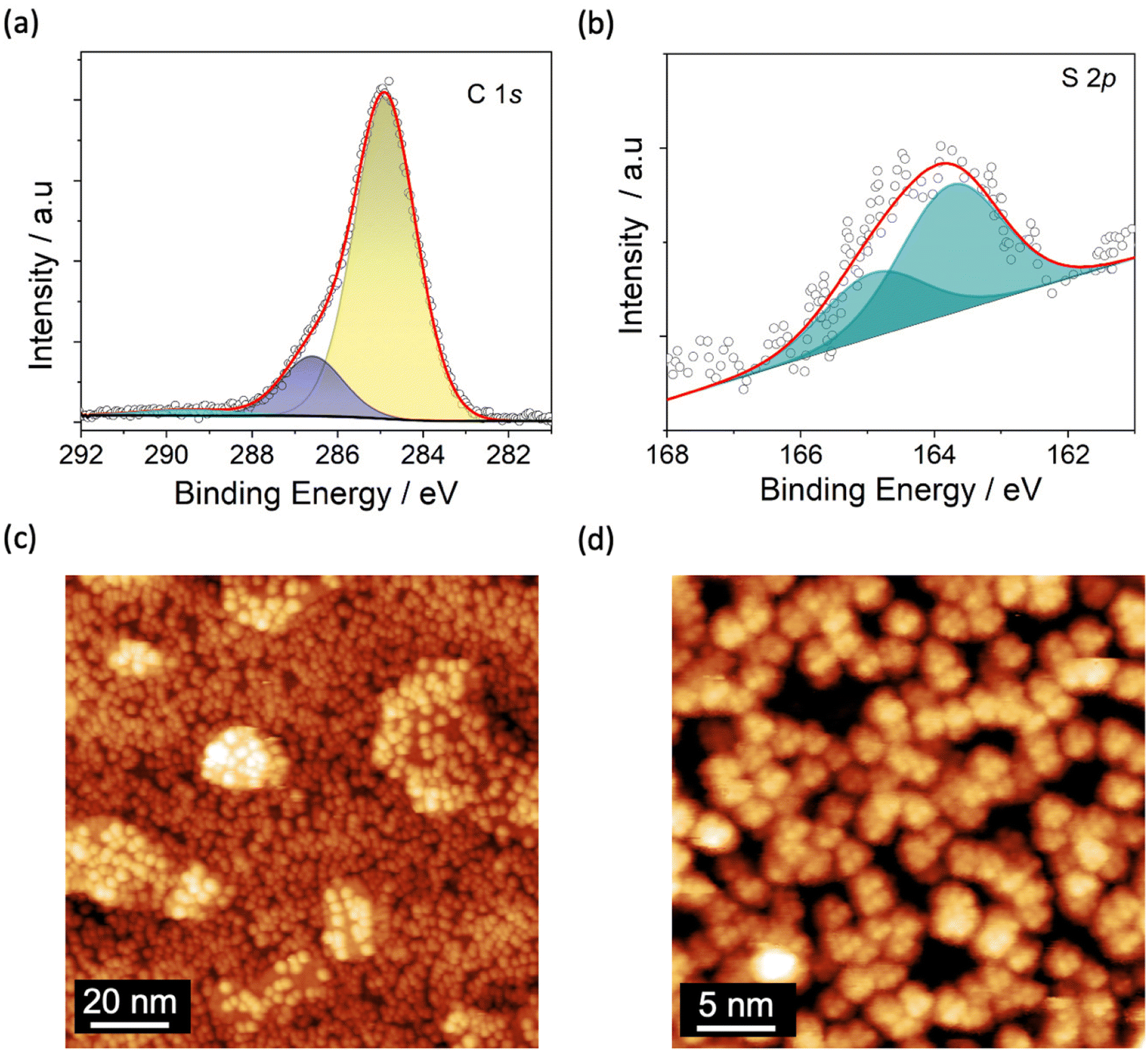 | ||
| Fig. 2 XPS spectra in the C 1s (a) and S 2p (b) regions after deposition of Fe4SMe on TiO2/Cu surface. (c) STM image at 30 K of Fe4SMe molecules distributed randomly on the TiO2/Cu surface (V = 2.6 V, It = 15 pA, 120 × 120 nm). (d) STM image at 30 K of the Fe4SMe layer showing the internal resolution (yellow dots) of single molecules (V = 2.6 V, It = 5 pA, 30 × 30 nm). | ||
STM images of the TiO2/Cu surface decorated with Fe4SMe molecules acquired at 30 K and different magnifications are displayed in Fig. 2c and d. An almost complete coverage of the surface by Fe4SMe molecules is clearly visible. Molecules have a quasi-spherical shape but do not form densely packed and ordered islands, as observed on metals.16 Uncovered parts of the TiO2 surface and a few additional molecules sitting on top of the molecular film are visible, confirming that the desired monolayer coverage was reached. The statistical height distribution in the Fe4SMe monolayer is shown in Fig. S4† and provides a medium height of 0.65 ± 0.06 nm. This value is slightly lower than that observed for the same Fe4 derivative on Pb(111)16 and for the related complex with R = Ph (Fe4Ph) on Au(111)51 and Cu2N22 (∼0.8 nm). Such a slightly reduced molecular height could be attributed either to a different molecular orientation or to a stronger molecule/surface interaction, which would be consistent with the absence of dense and ordered molecular packing. Furthermore, within the limits of our STM investigation, the presence of molecular fragments can be excluded, confirming the enhanced stability of Fe4SMe16vs. Fe4Ph51,55 upon sublimation.
From the STM image at higher magnification (Fig. 2d) we estimated a lateral dimension of 1.80 ± 0.15 nm for individual molecules (Fig. S5†), a value in close agreement with the X-ray structure (1.7 nm) and consistent with literature data for Fe4 complexes on other surfaces.22,48,51 Additionally, the STM image in Fig. 2d evidences the internal resolution of single Fe4 units, with a triangle of bright features separated by 0.58 ± 0.06 nm (see line profile in Fig. S5a and S5b†). These features are similar to those observed on Pb(111)16 and can be attributed to the –CH2SMe group pointing out of the plane, a tert-butyl group of the topmost dpm− ligand, and the surrounding envelope of dpm− ligands.
The electronic and magnetic properties of the Fe4SMe deposit were investigated by synchrotron radiation at the DEIMOS beamline (SOLEIL, France) using a dilution cryostat to reach sub-kelvin temperatures.73 These experiments were carried out with linearly and circularly polarized light (Fig. 3) by monitoring the absorption at the Ti L2,3 and Fe L2,3 edges in the Total Electron Yield detection mode (TEY). Experiments were performed between 220 and 900 mK and in magnetic fields up to 30 kOe (see Methods). The XAS profile at the Ti L2,3 edges confirms the QH structure of the TiO2 monolayer deposited on Cu (see ESI and Fig. S6† for additional details). The XAS spectrum at the Fe L3 edge exhibits a fine structure with two main signals at 707.6 and 709.2 eV (Fig. 3a). We notice that the first peak has an additional shoulder approximately centered at 707.1 eV and marked with a green arrow in Fig. 3a. This extra XAS component was not detected in our previous investigations of Fe4 arrays at surfaces.16,51,74,75 Significantly, it was absent in monolayers of the same Fe4SMe complex prepared using identical thermal sublimation conditions but a different substrate.16 Therefore, the thermal sublimation process is unlikely to be responsible for this feature.55 Radiation damage is ruled out as a possible cause of the XAS profile alteration, since there is no evidence of time evolution of this feature under X-ray irradiation.71,76,77 A plausible explanation is the partial reduction of molecules interacting with TiIII sites on the TiO2 surface to give a minority fraction of FeII containing species55,64,78,79 and this aspect will be the subject of further discussion later.
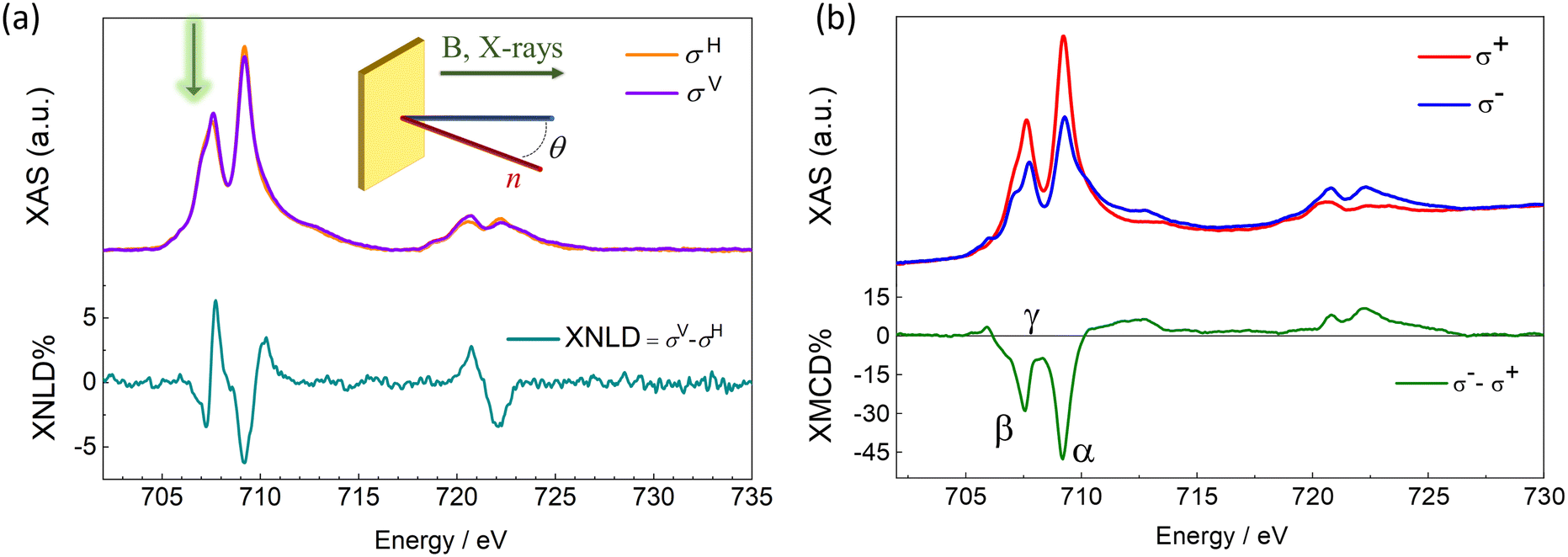 | ||
| Fig. 3 (a) XNLD and (b) XMCD spectra of Fe4SMe on TiO2/Cu acquired at the Fe L2,3 edges. (a) XAS (top) and XNLD (bottom) spectra recorded at θ = 45°, H = 30 kOe, and T = 220 mK. (b) XAS (top) and XMCD (bottom) spectra recorded at θ = 0, H = 30 kOe, and T = 220 mK. The inset in panel (a) depicts the experimental geometry used for the acquisition of the XAS spectra. The green arrow indicates the additional contribution at 707.1 eV, which is observable for all polarization, attributed to a minority fraction of Fe(II) containing species. | ||
The XNLD contribution (Fig. 3a) was extracted from the XAS signals recorded using linearly polarized X-rays with propagation vector directed at θ = 45° from the surface normal (see Methods and inset in Fig. 3a). The overall XNLD profile is essentially in line with that reported earlier for other Fe4 derivatives with short tethering groups.14,16,75 From the maximum XNLD amplitude, which reaches 6% of the average isotropic XAS signal, we conclude that the extent of preferential orientation on the surface is similar to that found for the same complex on Pb(111).16
The XMCD profile (Fig. 3b), obtained from the XAS signals recorded using circularly polarized X-rays at 220 mK and θ = 0, is dominated by two negative dichroic signals at the L3 edge. Although the position of the two XMCD minima (707.6 and 709.2 eV, β and α, respectively) agrees with that typically observed in Fe4 complexes, the exact shape and amplitude of the XMCD profile are slightly different. In the ground S = 5 state of Fe4 complexes, the opposing contribution of the central FeIII spin results in three important spectral fingerprints:16,48,51,74,75,80,81 (i) an intensity ratio of ca. 0.30 between the two XMCD minima at the L3 edge, (ii) saddle point between the two intense negative signals (708.3 eV, γ), and (iii) a maximum XMCD amplitude amounting to ca. 40% of the average isotropic XAS signal. In Fig. 3b, the intensity ratio between the two XMCD minima at the L3 edge is 0.55, hence higher than typically found in Fe4 complexes. In addition, the XMCD signal at 708.3 eV (−8%) remains significantly different from zero, and the normalized XMCD intensity at 709.2 eV is as large as 47%. It is worth stressing that heteronuclear CrIII-centered Fe3Cr complexes, in which the opposing spin contribution of the central FeIII ion is absent, also exhibit a non-zero XMCD signal at 708.3 eV and a much increased XMCD amplitude at 709.2 eV.74,80 Here, the Fe4 spectral fingerprints are presumably partly obscured by the surface-induced reduction of some FeIII centers to FeII.
To support this interpretation of the XMCD fine structure, we performed Ligand Field Multiplet (LFM) calculations. Starting from our previous knowledge of the XAS and XMCD signals for pristine Fe4 molecules, we replaced FeIII with variable amounts of FeII in the calculations. We reached a good agreement with the measured XAS spectra assuming that 30% of the Fe centers are reduced to FeII. With this percentage, the ratio between the first peak at 707.6 eV and the main peak at 709.2 eV in the XAS plots is nicely reproduced (Fig. S7†). In order to determine the speciation of FeII, we examined various situations where the amount of FeII was fixed to 30% of the total Fe ion content. It should be underlined that if one supposes that all the FeII ions are present as Fe oxyhydroxides at the TiO2 surface, the XMCD signal at 220 mK and 30 kOe would be much larger than measured. Thus, one can exclude this situation as the main location for FeII ions and suppose that FeII ions primarily occur in the Fe4 molecular structure. There are only five distinct distributions of oxidation states which are compatible with a 30% concentration of FeII ions, as fully detailed in the ESI (see Fig. S7†). Among these five different distributions, the one with 70% of pristine Fe4 molecules and 30% of FeII4 molecules yields by far the best agreement with the measured XAS and XMCD signals. Note that in the present fit we always considered that the central ion, whether it is an FeII or an FeIII ion, is coupled antiferromagnetically with the three peripheral Fe ions (see ESI† for the precise values of the FeII LFM parameters). The resulting simulation features an intensity ratio of ≈0.51 between the two XMCD minima at the L3 edge, a normalized XMCD intensity of ≈50% at 709.2 eV, and a negative XMCD signal of −4% at 708.3 eV. The three above features rather closely match the experimental spectra, strongly supporting the presence of a fraction of FeII ions in the molecular deposit, which however primarily comprises intact Fe4SMe molecules. An additional and strong indication that the monolayer contains a fraction of altered molecules was extracted by monitoring the magnetic field dependence of the XMCD signal at different energies to selectively address the magnetic behaviour of the different species on the surface. We expect that intact Fe4SMe molecules mainly contribute to the XMCD signal at 709.2 eV, and not at 708.3 eV. The temperature dependence of XMCD at 709.2 eV (α, Fig. 4a) mirrors the typical magnetic behaviour of Fe4 SMMs, whose hysteresis loop is open below 1 K and becomes wider with decreasing temperature.48 In particular, the sharp magnetization steps at 0 and 5 kOe show that Fe4SMe undergoes resonant QTM on TiO2/Cu, similarly to what has been observed on Pb(111).16 Additionally, in agreement with XNLD evidence, the angular-dependent experiment at 220 mK (Fig. 4b) confirms that the complex is preferentially oriented with the easy axis close to the surface normal. As the incidence angle θ increases, saturation is reached more slowly and the resonant condition for QTM at a nonzero field broadens and shifts to higher fields.1451
 | ||
| Fig. 4 Magnetic field-dependent XMCD curves acquired (a) as a function of temperature at 709.2 eV and normal incidence (θ = 0), (b) as a function of θ at 709.2 eV and 220 mK, and (c) as a function of photon energy at normal incidence and 220 mK (magnified from −10 to 10 kOe). All data were obtained at the same field sweep rate of 0.2 kOe s−1. The measurements at 220 mK, θ = 0°, E = 709.2 eV are plotted as dark blue dots with connecting line in (a), (b) and (c) and compared with the other measurements where one parameter (temperature (a), angle (b) and energy (c)) is varied. | ||
When the photon energy is decreased to 708.3 eV (γ), the intensity of the XMCD signal at 220 mK decreases considerably, primarily due to the contribution of the reduced species only (Fig. 4c). Crucially, its field dependence is markedly different from that of Fig. 4b, evidencing a closed hysteresis loop. We attribute this paramagnetic response at 708.3 eV to magnetic species containing FeII centres resulting from surface-induced reduction but still embedded in a Fe4-like structure, in accordance with STM measurements and LFM calculations. For the sake of completeness, setting the photon energy at 707.6 eV (β) yields a field-dependent XMCD signal with intermediate characteristics as compared with those recorded at 709.2 (α) and 708.3 (γ) eV. In this case, the hysteresis loop is still detectable but smeared out due to the additional paramagnetic contribution.
To summarize, our combined spectroscopic studies by XAS, XNLD, and XMCD converge in indicating that Fe4SMe monolayers on TiO2/Cu contain a dominant fraction of intact molecules and a minor fraction of molecules undergoing significant modification. XAS spectra suggest that such a modification consists of a reduction of FeIII centers to FeII. Theoretical calculations already predicted a similar effect for atoms on TiO2.82 A strong molecule–surface interaction was also observed when mono and bis-phthalocyaninato complexes are deposited on TiO2,64,79,83–86 leading in extreme circumstances to surface–molecule charge transfer processes.64,86–88 The latter could be favoured by TiO2 reactive sites, such as oxygen vacancies.89 We can tentatively associate the partial modification of Fe4SMe complexes on TiO2/Cu with the presence of TiIII active sites on the surface. Considering the overwhelming number of TiIII atoms compared to Fe4SMe molecules, such a hypothesis does not contradict the observation that the amount of TiIII remains substantially unaltered after Fe4SMe deposition (Fig. S3 and Table S1†).
Conclusions
We reported the chemical, structural, and magnetic properties of a monolayer of Fe4SMe complexes sublimated on an ultrathin film of TiO2-QH grown on Cu(001). The XPS and STM measurements evidenced the presence of chemically and structurally intact Fe4SMe molecules assembled into a disordered monolayer. This might imply that a significant molecule/surface interaction occurs, as previously observed for other molecules on TiO2 surfaces.47,64 An ultralow-temperature investigation by XNLD and XMCD at the Fe L2,3 edges revealed that Fe4SMe molecules display a preferential orientation in the monolayer and retain a bulk-phase-like behaviour (i.e., magnetic hysteresis up to 900 mK). However, the XAS spectra and the energy- and field-dependent XMCD signal disclosed a minor contribution from altered species containing FeII ions. These reduced species may originate from the reaction of Fe4SMe with active sites in the TiO2 monolayer, which hosts a detectable amount of TiIII ions. Additional experiments aimed at modifying the TiIII content and future DFT investigations will be required to clarify this aspect. Interestingly, the possibility to selectively address at the monolayer level the magnetic properties of the altered species combined with the SMM fingerprint can provide information on the underlying interface, as already observed for the intermediate superconducting state of a Pb substrate.16 Overall, our findings highlight the challenges associated with combining 2D oxide materials with SMM complexes into hybrid architectures suitable for new spintronic and quantum devices.Methods
A Cu(001) single crystal from the Surface Preparation Laboratory (SPL, The Netherlands) was cleaned by several cycles of Ar+ sputtering (1500 eV) and annealing (770 K) in UHV. The Cu(001) substrate, kept at the constant temperature of 570 K, was exposed to an oxygen partial pressure of ≈1 × 10−6 mbar to provide the oxygen amount necessary for growing the first TiO2 layer. Finally, titanium (purity 99.999%) was deposited employing an electron beam Omicron EFM3 micro-evaporator. The TiO2 deposition was achieved by consecutive steps following the procedure reported in the literature.46 After each step, the Cu(001) single crystal was annealed at ca. 700 K to promote a homogeneous growth on the metal surface. The deposition rate was monitored using the integrated flux monitor of the EFM3 and evaluated a posteriori by STM. The structure and the chemical composition of the TiO2 layer were studied by LEED and XPS, respectively. The Fe4SMe complex, prepared in pure crystalline form as reported elsewhere,16 was processed as described for deposition on Pb(111).16 It was heated at a temperature of around 490 K and evaporated with a homemade Knudsen cell in an UHV chamber (with a base pressure of 10−9 mbar). Molecular coverage was estimated by a Quartz Crystal Microbalance (QCM) and by STM measurements. The deposition rate was monitored by placing the QCM in front of the crucible. The molecular integrity after deposition was investigated by XPS. XPS data were acquired using a micro-focused monochromatic Al Kα radiation (λ = 1486.6 eV, SPECS mod. XR-MS focus 600) operating at a power of 100 W (13 kV and 7.7 mA) and a multichannel detector electron analyser, model SPECS Phoibos 150 1DLD. XPS spectra were recorded in normal emission with pass energy of 40 eV with the X-ray source mounted at an angle of 54.44° with respect to the analyser. XPS spectra were calibrated to the Cu 2p3/2 signal at 932.7 eV,46 and the background subtracted was adapted as a function of the elements, employing linear and Shirley method. A 70%–30% combination of Gaussian and Lorentzian functions was employed to fit all the spectra. LEED patterns were acquired using an Omicron three grid optics, model NG-LEED. STM measurements were carried out in UHV conditions at RT and at 30 K using a Variable Temperature (VT)-STM Omicron (model XA VT-STM) with a Pt/Ir tip.The electronic and magnetic characterization of the Fe4SMe complex on TiO2/Cu was carried out at the DEIMOS beamline (SOLEIL synchrotron).73,90 The XNLD was extracted as the difference between the cross sections recorded using vertically (σV) and horizontally (σH) polarized light (σV − σH) at θ = 45°, H = 30 kOe, and T = 220 mK. The XNLD contribution was normalized with respect to the L3 edge jump of the isotropic spectrum (1/3σV + 2/3σH) and expressed as percentage (XNLD%). Analogously, XMCD was obtained as the difference between the XAS spectra measured using negative (σ−) and positive (σ+) circular light polarizations (σ− − σ+) at θ = 0, H = 30 kOe, and T = 220 mK. The dichroic signal was normalized with respect to the L3 edge jump of (σ+ + σ−)/2 and expressed as percentage (XMCD%). The magnetic hysteresis measurements at specific photon energies were made at θ = 0 or 45° by cycling the magnetic field between −15 kOe and 15 kOe with a scan rate of 0.2 kOe s−1 and working at temperatures from 220 to 900 mK. All the samples were prepared in Florence and transferred to the beamline employing a home-made suitcase equipped with a D100 SAES Nextorr Neg-Ion Combination Pump that guarantees a pressure P < 10−10 mbar during the transport. All the steps from sample preparation to synchrotron measurements were accomplished without breaking the vacuum connection (P < 10−9 mbar).16 Data analysis was performed using pyDichroX software.91
The XAS and XMCD spectra were calculated within the Ligand Field Multiplet (LFM) theory using Quanty92 (see ESI† for details).
Author contributions
A. L. S. – formal analysis, investigation (XPS, STM, large scale facilities experiments), writing – original draft; L. P. – formal analysis, investigation (XPS, large scale facilities experiments), supervision data discussion, writing – review & editing, data curation; G. S. – supervision, formal analysis, investigation (STM, large scale facilities experiments), data discussion, writing – review & editing; G. C. – investigation (large scale facilities) and software development; B. C. – technical assistance; L. M. – investigation (large scale facilities); F. P. investigation (synthesis); E. O. – investigation (large scale facilities) and technical assistance; M.-A. A. – data discussion and investigations (simulations); P. S. – data discussion and investigations (simulations); A. C. – supervision; A. Co. – conceptualization, investigation (synthesis); R. S. – conceptualization, funding acquisition, project administration, supervision, validation, writing – review & editing; M. M. – conceptualization, funding acquisition, project administration, supervision, validation, writing – review & editing, investigation (large scale facilities experiments).Data availability
Figures for this article are available at Zonodo repository website at https://doi.org/10.5281/zenodo.12598761.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the SOLEIL staff for smoothly running the facility. The work was supported by MUR Italy through the program Dipartimenti di Eccellenza 2023–2027 (DICUS 2.0 grant, assigned to the Department of Chemistry “Ugo Schiff” of the University of Florence, CUP: B97G22000740001) and a PNRR project (PE0000023-NQSTI). Financing by the Fondazione Ente Cassa di Risparmio di Firenze (project SPINE-2 2020.1634) and by ANR France (LabEx PALM project, ANR-10-LABX-0039-PALM) is also acknowledged. We thank the MatchLab Interdepartment Research Unit (Univ. of Florence). We acknowledge SOLEIL synchrotron (proposal #99180040 test) and DEIMOS Staff.References
- R. Sessoli, D. Gatteschi, A. Caneschi and M. A. Novak, Nature, 1993, 365, 141–143 CrossRef CAS.
- R. Clérac and R. E. P. Winpenny, Struct. Bonding, 2016, 172, 35–48 CrossRef.
- L. Bogani and W. Wernsdorfer, Nat. Mater., 2008, 7, 179–186 CrossRef CAS PubMed.
- C. A. Gould, K. R. McClain, D. Reta, J. G. C. Kragskow, D. A. Marchiori, E. Lachman, E.-S. Choi, J. G. Analytis, R. D. Britt, N. F. Chilton, B. G. Harvey and J. R. Long, Science, 2022, 375, 198–202 CrossRef CAS.
- F.-S. Guo, B. M. Day, Y.-C. Chen, M.-L. Tong, A. Mansikkamäki and R. A. Layfield, Science, 2018, 362, 1400–1403 CrossRef CAS.
- F.-S. Guo, B. M. Day, Y.-C. Chen, M.-L. Tong, A. Mansikkamäki and R. A. Layfield, Angew. Chem., Int. Ed., 2017, 56, 11445–11449 CrossRef CAS PubMed.
- C. A. P. Goodwin, F. Ortu, D. Reta, N. F. Chilton and D. P. Mills, Nature, 2017, 548, 439–442 CrossRef CAS PubMed.
- D. Gatteschi, R. Sessoli and J. Villain, Molecular Nanomagnets, Oxford University Press, 2006, vol. 54 Search PubMed.
- E. Coronado, Nat. Rev. Mater., 2020, 5, 87–104 CrossRef.
- D. Shimizu and A. Osuka, Chem. Sci., 2018, 9, 1408–1423 RSC.
- C. A. P. Goodwin, F. Ortu, D. Reta, N. F. Chilton and D. P. Mills, Nature, 2017, 548, 439–442 CrossRef CAS PubMed.
- F. Guo, B. M. Day, Y. Chen, M. Tong, A. Mansikkamäki and R. A. Layfield, Angew. Chem., Int. Ed., 2017, 56, 11445–11449 CrossRef CAS.
- M. Mannini, F. Pineider, P. Sainctavit, C. Danieli, E. Otero, C. Sciancalepore, A. M. Talarico, M.-A. Arrio, A. Cornia, D. Gatteschi and R. Sessoli, Nat. Mater., 2009, 8, 194–197 CrossRef CAS.
- M. Mannini, F. Pineider, C. Danieli, F. Totti, L. Sorace, P. Sainctavit, M.-A. Arrio, E. Otero, L. Joly, J. C. Cezar, A. Cornia and R. Sessoli, Nature, 2010, 468, 417–421 CrossRef CAS.
- L. Spree, F. Liu, V. Neu, M. Rosenkranz, G. Velkos, Y. Wang, S. Schiemenz, J. Dreiser, P. Gargiani, M. Valvidares, C.-H. Chen, B. Büchner, S. M. Avdoshenko and A. A. Popov, Adv. Funct. Mater., 2021, 31, 2105516 CrossRef CAS.
- G. Serrano, L. Poggini, M. Briganti, A. L. Sorrentino, G. Cucinotta, L. Malavolti, B. Cortigiani, E. Otero, P. Sainctavit, S. Loth, F. Parenti, A.-L. L. Barra, A. Vindigni, A. Cornia, F. Totti, M. Mannini and R. Sessoli, Nat. Mater., 2020, 19, 546–551 CrossRef CAS.
- G. Serrano, L. Poggini, G. Cucinotta, A. L. Sorrentino, N. Giaconi, B. Cortigiani, D. Longo, E. Otero, P. Sainctavit, A. Caneschi, M. Mannini and R. Sessoli, Nat. Commun., 2022, 13, 3838 CrossRef CAS.
- C. Wäckerlin, F. Donati, A. Singha, R. Baltic, S. Rusponi, K. Diller, F. Patthey, M. Pivetta, Y. Lan, S. Klyatskaya, M. Ruben, H. Brune and J. Dreiser, Adv. Mater., 2016, 28, 5195–5199 CrossRef.
- M. Studniarek, C. Wäckerlin, A. Singha, R. Baltic, K. Diller, F. Donati, S. Rusponi, H. Brune, Y. Lan, S. Klyatskaya, M. Ruben, A. P. Seitsonen and J. Dreiser, Adv. Sci., 2019, 6, 1901736 CrossRef CAS.
- X. Zhang, C. Wolf, Y. Wang, H. Aubin, T. Bilgeri, P. Willke, A. J. Heinrich and T. Choi, Nat. Chem., 2022, 14, 59–65 CrossRef CAS.
- F. Donati, S. Rusponi, S. Stepanow, L. Persichetti, A. Singha, D. M. Juraschek, C. Wäckerlin, R. Baltic, M. Pivetta, K. Diller, C. Nistor, J. Dreiser, K. Kummer, E. Velez-Fort, N. A. Spaldin, H. Brune and P. Gambardella, Phys. Rev. Lett., 2020, 124, 077204 CrossRef CAS.
- J. A. J. Burgess, L. Malavolti, V. Lanzilotto, M. Mannini, S. Yan, S. Ninova, F. Totti, S. Rolf-Pissarczyk, A. Cornia, R. Sessoli and S. Loth, Nat. Commun., 2015, 6, 8216 CrossRef.
- A. J. Heinrich, W. D. Oliver, L. M. K. Vandersypen, A. Ardavan, R. Sessoli, D. Loss, A. B. Jayich, J. Fernandez-Rossier, A. Laucht and A. Morello, Nat. Nanotechnol., 2021, 16, 1318–1329 CrossRef CAS PubMed.
- S. Loth, K. von Bergmann, M. Ternes, A. F. Otte, C. P. Lutz and A. J. Heinrich, Nat. Phys., 2010, 6, 340–344 Search PubMed.
- S. Baumann, W. Paul, T. Choi, C. P. Lutz, A. Ardavan and A. J. Heinrich, Science, 2015, 350, 417–420 CrossRef CAS.
- W. Paul, K. Yang, S. Baumann, N. Romming, T. Choi, C. P. Lutz and A. J. Heinrich, Nat. Phys., 2017, 13, 403–407 Search PubMed.
- F. Donati and A. J. Heinrich, Appl. Phys. Lett., 2021, 119, 160503 CrossRef CAS.
- P. Willke, Y. Bae, K. Yang, J. L. Lado, A. Ferrón, T. Choi, A. Ardavan, J. Fernández-Rossier, A. J. Heinrich and C. P. Lutz, Science, 2018, 362, 336–339 CrossRef CAS PubMed.
- J. Kim, K. Noh, Y. Chen, F. Donati, A. J. Heinrich, C. Wolf and Y. Bae, Nano Lett., 2022, 22, 9766–9772 CrossRef CAS PubMed.
- P. Willke, T. Bilgeri, X. Zhang, Y. Wang, C. Wolf, H. Aubin, A. Heinrich and T. Choi, ACS Nano, 2021, 15, 17959–17965 CrossRef CAS PubMed.
- A. L. Linsebigler, G. Lu and J. T. Yates, Chem. Rev., 1995, 95, 735–758 CrossRef CAS.
- Y. Sari, P. L. Gareso, B. Armynah and D. Tahir, Int. J. Hydrogen Energy, 2024, 55, 984–996 CrossRef CAS.
- S. A. Chambers, S. Thevuthasan, R. F. C. Farrow, R. F. Marks, J. U. Thiele, L. Folks, M. G. Samant, A. J. Kellock, N. Ruzycki, D. L. Ederer and U. Diebold, Appl. Phys. Lett., 2001, 79, 3467–3469 CrossRef CAS.
- A. Omar, M. S. Ali and N. Abd Rahim, Sol. Energy, 2020, 207, 1088–1121 CrossRef CAS.
- J. Zhao, H. Wang, Y. Cai, J. Zhao, Z. Gao and Y.-Y. Song, ACS Sens., 2024, 9, 1644–1655 CrossRef CAS.
- S. Hussain, C. Cao, Z. Usman, Z. Chen, G. Nabi, W. S. Khan, Z. Ali, F. K. Butt and T. Mahmood, Thin Solid Films, 2012, 522, 430–434 CrossRef CAS.
- D. Joshy, S. B. Narendranath, Y. A. Ismail and P. Periyat, Nanoscale Adv., 2022, 4, 5202–5232 RSC.
- I. Ali, M. Suhail, Z. A. Alothman and A. Alwarthan, RSC Adv., 2018, 8, 30125–30147 RSC.
- U. Diebold, Surf. Sci. Rep., 2003, 48, 53–229 CrossRef CAS.
- F. Sedona, G. A. Rizzi, S. Agnoli, F. X. Llabrés i Xamena, A. Papageorgiou, D. Ostermann, M. Sambi, P. Finetti, K. Schierbaum and G. Granozzi, J. Phys. Chem. B, 2005, 109, 24411–24426 CrossRef CAS PubMed.
- S. Agnoli, T. Orzali, M. Sambi, A. Vittadini, M. Casarin and G. Granozzi, J. Phys. Chem. C, 2008, 112, 20038–20049 CrossRef CAS.
- G. T. Harrison, M. C. Spadaro, C. L. Pang, D. C. Grinter, C. M. Yim, P. Luches and G. Thornton, Mater. Sci. Technol., 2016, 32, 203–208 CrossRef CAS.
- S. Tosoni and G. Pacchioni, J. Phys. Chem. C, 2019, 123, 7952–7960 CrossRef CAS.
- K. G. Reeves, J. Ma, M. Fukunishi, M. Salanne, S. Komaba and D. Dambournet, ACS Appl. Energy Mater., 2018, 1, 2078–2086 CrossRef CAS.
- F. Rossella, P. Galinetto, M. C. Mozzati, L. Malavasi, Y. Diaz Fernandez, G. Drera and L. Sangaletti, J. Raman Spectrosc., 2009, 41, 558–565 CrossRef.
- A. L. Sorrentino, G. Serrano, L. Poggini, B. Cortigiani, K. E. El-Kelany, M. D'Amore, A. M. Ferrari, A. Atrei, A. Caneschi, R. Sessoli and M. Mannini, J. Phys. Chem. C, 2021, 125, 10621–10630 CrossRef CAS.
- A. L. Sorrentino, I. Cimatti, G. Serrano, L. Poggini, B. Cortigiani, L. Malavolti, E. Otero, P. Sainctavit, M. Mannini, R. Sessoli and A. Caneschi, J. Mater. Chem. C, 2021, 9, 15011–15017 RSC.
- A. Cornia, M. Mannini, R. Sessoli and D. Gatteschi, Eur. J. Inorg. Chem., 2019, 2019, 552–568 CrossRef CAS.
- L. Margheriti, M. Mannini, L. Sorace, L. Gorini, D. Gatteschi, A. Caneschi, D. Chiappe, R. Moroni, F. B. de Mongeot, A. Cornia, F. M. Piras, A. Magnani and R. Sessoli, Small, 2009, 5, 1460–1466 CrossRef CAS.
- S. Ninova, V. Lanzilotto, L. Malavolti, L. Rigamonti, B. Cortigiani, M. Mannini, F. Totti and R. Sessoli, J. Mater. Chem. C, 2014, 2, 9599–9608 RSC.
- L. Malavolti, V. Lanzilotto, S. Ninova, L. Poggini, I. Cimatti, B. Cortigiani, L. Margheriti, D. Chiappe, E. Otero, P. Sainctavit, F. Totti, A. Cornia, M. Mannini and R. Sessoli, Nano Lett., 2015, 15, 535–541 CrossRef CAS.
- P. Erler, P. Schmitt, N. Barth, A. Irmler, S. Bouvron, T. Huhn, U. Groth, F. Pauly, L. Gragnaniello and M. Fonin, Nano Lett., 2015, 15, 4546–4552 CrossRef CAS PubMed.
- L. Gragnaniello, F. Paschke, P. Erler, P. Schmitt, N. Barth, S. Simon, H. Brune, S. Rusponi and M. Fonin, Nano Lett., 2017, 17, 7177–7182 CrossRef CAS.
- F. Paschke, P. Erler, V. Enenkel, L. Gragnaniello and M. Fonin, ACS Nano, 2019, 13, 780–785 CrossRef CAS.
- V. Lanzilotto, L. Malavolti, S. Ninova, I. Cimatti, L. Poggini, B. Cortigiani, M. Mannini, F. Totti, A. Cornia and R. Sessoli, Chem. Mater., 2016, 28, 7693–7702 CrossRef CAS.
- U. Diebold and T. E. Madey, Surf. Sci. Spectra, 1996, 4, 227–231 CrossRef CAS.
- W. S. Oh, C. Xu, D. Y. Kim and D. W. Goodman, J. Vac. Sci. Technol., A, 1997, 15, 1710–1716 CrossRef CAS.
- M. Oku, K. Wagatsuma and S. Kohiki, Phys. Chem. Chem. Phys., 1999, 1, 5327–5331 RSC.
- K. S. Kim and N. Winograd, Chem. Phys. Lett., 1975, 31, 312–317 CrossRef CAS.
- S. K. K. Sen, J. Riga and J. Verbist, Chem. Phys. Lett., 1976, 39, 560–564 CrossRef CAS.
- J. T. Mayer, U. Diebold, T. E. Madey and E. Garfunkel, J. Electron Spectrosc. Relat. Phenom., 1995, 73, 1–11 CrossRef CAS.
- M. J. Jackman, A. G. Thomas and C. Muryn, J. Phys. Chem. C, 2015, 119, 13682–13690 CrossRef CAS.
- W. S. Epling, C. H. F. Peden, M. A. Henderson and U. Diebold, Surf. Sci., 1998, 412–413, 333–343 CrossRef.
- G. Serrano, A. L. Sorrentino, L. Poggini, B. Cortigiani, C. Goletti, R. Sessoli and M. Mannini, Phys. Chem. Chem. Phys., 2021, 23, 12060–12067 RSC.
- A. Atrei, A. M. Ferrari, P. Finetti, A. Beni and G. Rovida, J. Phys. Chem. C, 2009, 113, 19578–19584 CrossRef CAS.
- P. Finetti, M. Caffio, B. Cortigiani, A. Atrei and G. Rovida, Surf. Sci., 2008, 602, 1101–1113 CrossRef CAS.
- G. Liu, A. Klein, A. Thissen and W. Jaegermann, Surf. Sci., 2003, 539, 37–48 CrossRef CAS.
- L. Rigamonti, M. Piccioli, L. Malavolti, L. Poggini, M. Mannini, F. Totti, B. Cortigiani, A. Magnani, R. Sessoli and A. Cornia, Inorg. Chem., 2013, 52, 5897–5905 CrossRef CAS PubMed.
- P. Gobbo, M. C. Biesinger and M. S. Workentin, Chem. Commun., 2013, 49, 2831–2833 RSC.
- H. Bennettand, G. J. O. Wiley, A. Benninghoven, K. T. F. Janssen, J. Tumpner and H. W. Wer, J. Chem. Educ., 1993, 70, A25 Search PubMed.
- L. Poggini, A. Lunghi, A. Collauto, A. Barbon, L. Armelao, A. Magnani, A. Caneschi, F. Totti, L. Sorace and M. Mannini, Nanoscale, 2021, 13, 7613–7621 RSC.
- C. J. Powell, J. Electron Spectrosc. Relat. Phenom., 2012, 185, 1–3 CrossRef CAS.
- J.-P. Kappler, E. Otero, W. Li, L. Joly, G. Schmerber, B. Muller, F. Scheurer, F. Leduc, B. Gobaut, L. Poggini, G. Serrano, F. Choueikani, E. Lhotel, A. Cornia, R. Sessoli, M. Mannini, M.-A. Arrio, Ph. Sainctavit and P. Ohresser, J. Synchrotron Radiat., 2018, 25, 1727–1735 CrossRef CAS PubMed.
- M. Mannini, E. Tancini, L. Sorace, P. Sainctavit, M.-A. Arrio, Y. Qian, E. Otero, D. Chiappe, L. Margheriti, J. C. Cezar, R. Sessoli and A. Cornia, Inorg. Chem., 2011, 50, 2911–2917 CrossRef CAS.
- L. Poggini, E. Tancini, C. Danieli, A. L. Sorrentino, G. Serrano, A. Lunghi, L. Malavolti, G. Cucinotta, A. L. Barra, A. Juhin, M. A. Arrio, W. Li, E. Otero, P. Ohresser, L. Joly, J. P. Kappler, F. Totti, P. Sainctavit, A. Caneschi, R. Sessoli, A. Cornia and M. Mannini, Adv. Mater. Interfaces, 2021, 2101182, 1–9 Search PubMed.
- N. Giaconi, A. L. Sorrentino, L. Poggini, M. Lupi, V. Polewczyk, G. Vinai, P. Torelli, A. Magnani, R. Sessoli, S. Menichetti, L. Sorace, C. Viglianisi and M. Mannini, Angew. Chem., Int. Ed., 2021, 60, 15276–15280 CrossRef CAS.
- K. Kuepper, C. Taubitz, D. Taubitz, U. Wiedwald, A. Scheurer, S. Sperner, R. W. Saalfrank, J.-P. Kappler, L. Joly, P. Ziemann and M. Neumann, J. Phys. Chem. Lett., 2011, 2, 1491–1496 CrossRef CAS.
- P. Palmgren, S. Yu, F. Hennies, K. Nilson, B. Åkermark and M. Göthelid, J. Chem. Phys., 2008, 129, 074707 CrossRef CAS.
- P. Palmgren, B. R. Priya, N. P. P. Niraj and M. Göthelid, Sol. Energy Mater. Sol. Cells, 2006, 90, 3602–3613 CrossRef CAS.
- E. Tancini, M. Mannini, P. Sainctavit, E. Otero, R. Sessoli and A. Cornia, Chem. – Eur. J., 2013, 19, 16902–16905 CrossRef CAS PubMed.
- M. J. Rodriguez-Douton, M. Mannini, L. Armelao, A.-L. Barra, E. Tancini, R. Sessoli and A. Cornia, Chem. Commun., 2011, 47, 1467–1469 RSC.
- S. Tosoni and G. Pacchioni, J. Phys. Chem. C, 2020, 124, 20960–20973 CrossRef CAS.
- H. Yanagi, S. Chen, P. A. Lee, K. W. Nebesny, N. R. Armstrong and A. Fujishima, J. Phys. Chem., 1996, 100, 5447–5451 CrossRef CAS.
- P. Palmgren, K. Nilson, S. Yu, F. Hennies, T. Angot, J. M. Layet, G. Le Lay and M. Gothelid, J. Phys. Chem. C, 2008, 112, 5972–5977 CrossRef CAS.
- S. Yu, S. Ahmadi, P. Palmgren, F. Hennies, M. Zuleta and M. Göthelid, J. Phys. Chem. C, 2009, 113, 13765–13771 CrossRef CAS.
- S. Yu, S. Ahmadi, C. Sun, P. Palmgren, F. Hennies, M. Zuleta and M. Göthelid, J. Phys. Chem. C, 2010, 114, 2315–2320 CrossRef CAS.
- L. Cao, Y. Wang, J. Zhong, Y. Han, W. Zhang, X. Yu, F. Xu, D. C. Qi and A. T. S. Wee, J. Phys. Chem. C, 2011, 115, 24880–24887 CrossRef CAS.
- V. Lanzilotto, G. Lovat, G. Fratesi, G. Bavdek, G. P. Brivio and L. Floreano, J. Phys. Chem. Lett., 2015, 6, 308–313 CrossRef CAS.
- S. C. Li, L. N. Chu, X. Q. Gong and U. Diebold, Science, 2010, 328, 882–884 CrossRef CAS PubMed.
- P. Ohresser, E. Otero, F. Choueikani, K. Chen, S. Stanescu, F. Deschamps, T. Moreno, F. Polack, B. Lagarde, J.-P. Daguerre, F. Marteau, F. Scheurer, L. Joly, J.-P. Kappler, B. Muller, O. Bunau and P. Sainctavit, Rev. Sci. Instrum., 2014, 85, 013106 CrossRef CAS PubMed.
- G. Cucinotta, pyDichroX, Available at: https://github.com/BeppeC/pyDichroX.
- M. W. Haverkort, M. Zwierzycki and O. K. Andersen, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 165113 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr02234c |
| This journal is © The Royal Society of Chemistry 2024 |