
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nanomechanical footprint of SARS-CoV-2 variants in complex with a potent nanobody by molecular simulations†
Luis F.
Cofas-Vargas
 *a,
Gustavo E.
Olivos-Ramirez
a,
Mateusz
Chwastyk
b,
Rodrigo A.
Moreira
c,
Joseph L.
Baker
d,
Siewert J.
Marrink
*e and
Adolfo B.
Poma
*a
*a,
Gustavo E.
Olivos-Ramirez
a,
Mateusz
Chwastyk
b,
Rodrigo A.
Moreira
c,
Joseph L.
Baker
d,
Siewert J.
Marrink
*e and
Adolfo B.
Poma
*a
aBiosystems and Soft Matter Division, Institute of Fundamental Technological Research, Polish Academy of Sciences, ul. Pawińskiego 5B, 02-106 Warsaw, Poland. E-mail: fcofas@ippt.pan.pl; apoma@ippt.pan.pl
bInstitute of Physics, Polish Academy of Sciences, al. Lotników 32/46, 02-668 Warsaw, Poland
cNEIKER, Basque Research and Technology Alliance (BRTA), Parque Científico y Tecnológico de Bizkaia, P812, E-48160 Derio, Spain
dDepartment of Chemistry, The College of New Jersey, 2000 Pennington Road, Ewing, NJ 08628, USA
eGroningen Biomolecular Sciences and Biotechnology Institute, University of Groningen, Nijenborgh 7, 9747 AG Groningen, The Netherlands. E-mail: s.j.marrink@rug.nl
First published on 1st October 2024
Abstract
Rational design of novel antibody therapeutics against viral infections such as coronavirus relies on surface complementarity and high affinity for their effectiveness. Here, we explore an additional property of protein complexes, the intrinsic mechanical stability, in SARS-CoV-2 variants when complexed with a potent antibody. In this study, we utilized a recent implementation of the GōMartini 3 approach to investigate large conformational changes in protein complexes with a focus on the mechanostability of the receptor-binding domain (RBD) from WT, Alpha, Delta, and XBB.1.5 variants in complex with the H11-H4 nanobody. The analysis revealed moderate differences in mechanical stability among these variants. Also, we identified crucial residues in both the RBD and certain protein segments in the nanobody that contribute to this property. By performing pulling simulations and monitoring the presence of specific native and non-native contacts across the protein complex interface, we provided mechanistic insights into the dissociation process. Force-displacement profiles indicate a tensile force clamp mechanism associated with the type of protein complex. Our computational approach not only highlights the key mechanostable interactions that are necessary to maintain overall stability, but it also paves the way for the rational design of potent antibodies that are mechanostable and effective against emergent SARS-CoV-2 variants.
1 Introduction
The global coronavirus disease (COVID-19) pandemic, sparked by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has led to more than 700 million infections worldwide, with a mortality rate of around 1%.1 Four structural and sixteen non-structural proteins comprise this virus2 and among them, the spike protein (S) plays a pivotal role in the infection mechanism of SARS-CoV-2. The S protein is composed of the N-terminal S1 (14–685 residues) and C-terminal S2 (686–1273 residues) subunits; the former mediates the binding to the human cells, and the latter is responsible for the viral-human membrane fusion process that enables the viral entry into cells. The S1 subunit can further be subdivided into two distinct domains: the N-terminal domain (NTD), comprising residues 14–305, and the RBD, comprising residues 319–541.3 The NTD is involved in the recognition of glycosaminoglycan chains that generally cover the cell surface.4–6 Furthermore, it is also important in the transition of the S protein from pre-fusion to post-fusion states.7 The RBD initiates the attachment of the virus to host cells by interacting with the human angiotensin-converting enzyme 2 (hACE2) receptor. The primary interface for binding to hACE2 is the receptor-binding motif (RBM), which is the most prominently exposed part of the RBD. Several mutations within the RBM have been shown to significantly influence the dynamics and functional properties of the RBD, impacting both affinity and immune evasion.8 Mutation N501Y, which is present in both the Alpha and Omicron variants, enhances the binding affinity between the spike protein and hACE2, increasing transmissibility. Additionally, mutations such as E484K, found in the Beta, Gamma, and certain Omicron sub-variants, along with L452R in the Delta variant, facilitate viral evasion of neutralizing antibodies by weakening or preventing key molecular interactions.3,9Viruses withstand diverse forces encountered during the infection process, including mechanical stress (e.g., cell movement and deformation), shear forces (resulting from the movement of cytoplasm and organelles within the cell), osmotic pressure, and molecular crowding.10 Several in vitro and in silico studies have been used to characterize the nanomechanics of the S protein and S/RBD/hACE2 complexes.8,10–15 Combination of single-molecule (SM) technique with all-atom molecular dynamics (AA-MD) simulation has shown clear evidence of mechanostability of the earlier Alpha variant who possessed the N501Y mutation while binding the hACE2 receptor.10 This is supported by the increased of the mechanostability of the RBD region from ∼200 pN in SARS-CoV-1 to ∼250 pN in the Alpha variant, followed by the S2 subunit and NTD.13,14 Studies by single-molecule force spectroscopy (SMFS) with the atomic force microscope has revealed that hACE2 forms more mechanostable complexes with the RBD from latest variants of concern (VoCs).8 The Alpha, Beta, Gamma, Delta, Kappa and Omicron variants withstand forces between 25 and 400 pN8 and this effect correlates with higher binding constants.15 These studies have demonstrated that the RBDOmicron/hACE2 complex is the most mechanically stable. The mutations in the VoCs are associated with increased stability of this complex, which correlates with higher transmissibility and improved immune evasion capabilities.8 While the mechanical stability of the RBD/hACE2 complexes has been studied, the mechanical stability of SARS-CoV-2 in combination with a potent antibody is less well understood. Exploring these interactions between VoCs and antibodies has the potential to reveal important interactions that may help improve the effectiveness of neutralizing antibodies. A current approach to the design of antibodies is to maximize the overlap between protein surfaces, which leads to high affinity. This approach achieves high-affinity binding with an apparent KD in range of 1–10 nM3. Recently, a novel consensus-based computational approach16 was developed for the accurate prediction of binding affinities of SARS-CoV-2 variants to neutralizing antibodies. This methodology, which also helps to identify the molecular footprints of variants, can be used with our nanomechanical studies to engineer more efficient nanobodies that retain potency against emerging variants and to some extent a higher mechanical stability.
Due to the experimental limitations in capturing the underlying mechanism of dissociation in protein complexes under mechanical forces, we employed MD simulation. This computational tool allows us to monitor atomic motion and the associated interactions on microsecond time scales and nanometer length scales with precision.17 Steered molecular dynamics simulations (denoted hereafter as SMD) have been instrumental in exploring the nanomechanical properties of protein complexes.18,19 However, these simulations have a high computational cost in systems that approach biological scales (i.e., hundreds of nm). The SMD protocol also often requires high pulling speeds and the use of position restraints to prevent protein unfolding.20–22 Hence, the results tend to overestimate the value of experimental properties in nanomechanical studies. The limitations inherent in investigating the nanomechanics of protein complexes can be addressed by coarse-grained MD (CG-MD), capable of describing larger systems and performing tests at lower speeds than AA-MD. In this study, we employed our recent GōMartini 3 approach23,24 and Martini 3 force field25 to describe the conformational changes in protein complexes during nanomechanical characterization of the interaction of RBD and an engineered potent nanobody. This approach is a promising solution to match experimental values.26–31 The GōMartini 3 approach employs virtual-sites implementation to define native contacts between amino acids, and this interaction is mapped via Lennard-Jones (12–6) potentials. The pair of native contacts are identified through a contact map that is based on van der Waals (vdW) radii overlap (OV) and repulsive chemical structural units (rCSU).23 Moreover, the GōMartini 3 approach preserves the tertiary structure of proteins without relying on harmonic restraints, and thus it is suitable to capture large conformational changes in proteins.24 The incorporation of virtual sites at protein–protein interfaces allows for detailed investigations into the nanomechanical properties of protein complexes using a CG approach. This approach provides crucial insights into the dynamics of mechanical stress impacts on protein stability and interactions by properly modeling how forces are distributed and transmitted across proteins. In this study, we explored the mechanical stability of the RBD/H11-H4 complex considering several variants of SARS-CoV-2, such as wild-type (WT), Alpha, Delta and XBB.1.5. The H11-H4 nanobody, which originates from llama, binds to a particular region on the RBD (Fig. 1A). This region partially overlaps the binding site of hACE2, preventing the spike protein from attaching to it in vitro. This inhibition is crucial for blocking viral entry into human cells. The affinity of H11-H4 for the RBD is high, with a KD of 12 ± 1.5 nM, indicating a strong interaction between the nanobody and the RBD.20,21,32 Three complementarity-determining regions (CDR) are critical for the interaction with the epitope on the RBD of the H11-H4 nanobody20,21,32 (Fig. 1B). The CDR1 region, which spans 26–32 residues, does not engage directly with the RBD. CDR2 includes 52–57 residues. Notably, residue R52 forms a salt bridge with E484 on the RBD, a π-cation interaction with F490, and establishes hydrogen bonds with the backbone of S103 and the side chain of Y109. The CDR3 region, comprising residues 99 to 115, is the longest and most flexible loop, engaging in multiple interactions with the RBD of several variants.32 Key interactions involve residues K444, F456, G482, and S494. Specifically, Y104 in H11-H4 interacts with a hydrophobic pocket formed by L455, F456, and Y489 of RBD. The CDR3 loop confers the specificity and high affinity of H11-H4, underscoring its potential in neutralizing the virus by blocking the ability of RBD to bind to hACE2.32
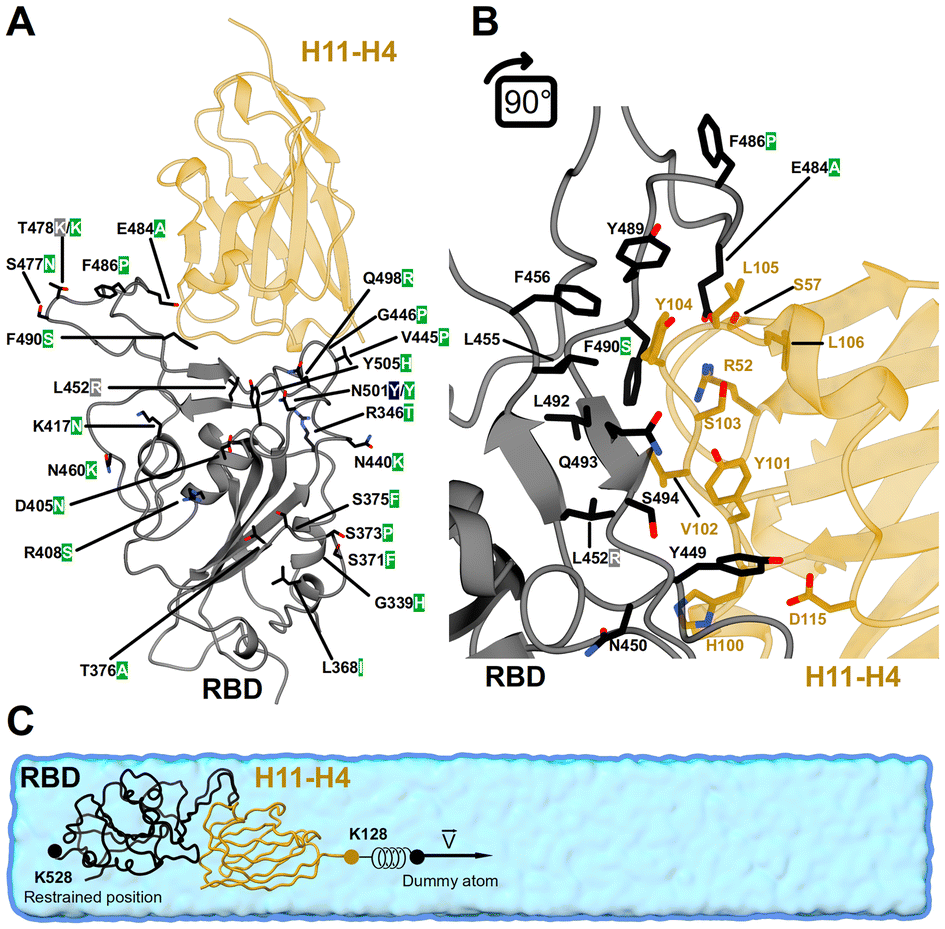 | ||
| Fig. 1 Atomistic structure of RBD in complex with a nanobody (H11-H4). (A) The RBD and H11-H4 structures are shown in black and orange color, respectively. A single letter code per residue responsible for a given mutation: WT (black), Alpha (blue), Delta (gray), and XBB.1.5 (green) is indicated next to the position of the residue. (B) Molecular insight into the interface of the RBD/H11-H4 complex. Some protein (native) contacts between RBD and H11-H4 are highlighted. Note that RBD of WT and Alpha share identical residues at the protein interface. Delta exhibits one mutation at position 452, whereas XBB.1.5 exhibits three mutations at positions 484, 486, and 490. (C) Coarse-grained representation of the RBD/H11-H4 complex. The protein complex is placed in a rectangular cuboid solvent box of dimensions 10 × 10 × 60 nm3, with a concentration of 150 mM NaCl. The last three residues from the C-terminus of RBD (G526, P527 and K528) were tethered along the pulling z-axis. Pulling simulations were performed on a virtual particle located at the center of mass of the last three residues (S126, S127, and K128) of the C-terminus of the H11-H4 nanobody. | ||
2 Methods
2.1 Molecular modelling
AA-MD simulations were necessary to relax the complexes and incorporate critical information about the most relevant interface contacts in the GōMartini 3 framework. These simulations were conducted using AMBER22 with the FF19SB force fields33 and the pmemd.cuda module for high performance. The initial structure of the RBD/H11-H4 complex was modeled from the ternary complex CR3022/H11-H4 and the WT RBD (PDB ID: 6ZH932), with the CR3022 antibody coordinates removed. To avoid artificial charges, the N- and C-termini residues of the RBD were capped with ACE and NME groups, respectively. Using the WT RBD as a template, several SARS-CoV-2 variants were modeled: (i) Alpha, (ii) Delta, (iii) XBB.1.5, (iv) BA.2.86, and (v) JN.1, using UCSF Chimera v1.17.334 with the Dunbrack rotamer library.35 A vacuum minimization was carried out for each complex to eliminate steric clashes around mutated residues. The protonation state, corresponding to a pH value of 7.4, was fixed with PDB-fixer.36 Each protein complex was solvated in a dodecahedral box initially extending 10 Å beyond the solute in each direction, using the four-site OPC water model,37 and system charges were neutralized with the appropriate number of counterions (9 Cl− ions for both the WT and Alpha variants, 9 Cl− ions for the Delta variant, 10 Cl− ions for the XBB.1.5 variant, 12 Cl− ions for the BA.2.86 variant, and 11 Cl− ions for the JN.1 variant). The systems underwent geometric optimization using the steepest descent algorithm for 5000 cycles to adjust the solvent orientation and remove local clashes.MD equilibration was carried out in multiple steps. Initial temperature equilibration of the complexes in the NVT ensemble involved a staged process where the temperature was incrementally raised through four steps: 150, 200, 250, 300, and finally to 310 K, with each step lasting 200 ps. Position restraints were applied to the heavy atoms of each protein, with decreasing spring constants of 5.0, 4.0, 3.0, and 1.0 kcal mol−1 Å−2 to allow for gradual relaxation of the complexes. This was followed by a 1 ns equilibration at 310 K in the NPT ensemble without position restraints. Production MD trajectories were performed in the NPT ensemble using periodic boundary conditions and PME38,39 with a grid spacing of 1.0 Å for long-range electrostatic interactions. Non-bonded interactions were described by the Lennard-Jones potential with a cutoff distance of 9 Å. Langevin dynamics40 was used for temperature control with a collision frequency of 4.0 ps−1, and the Berendsen barostat for pressure control41 with a relaxation time of 2.0 ps at 1 bar pressure. Bond constraints involving hydrogen atoms were maintained using the SHAKE algorithm.42 The hydrogen mass repartition scheme was applied using ParmEd,36 allowing the use of a 4 fs integration time step.43 Each protein complex was simulated for 1 μs, except for the BA.2.86 and JN.1 variants, where five replicas of 2 μs were run for each. During these simulations, both BA.2.86 and JN.1 complexes established initially 24 and 26 native contacts at the interface, respectively. In the course of the AA-MD simulations, these contacts were gradually lost, for instance about 9 native contacts remained at 1 μs for BA.2.86 and for JN.1 and by the end those numbers dropped on average to 4 and 6 respectively. Most of the RBD/H11-H4 complexes remained stable during the AA-MD simulation, except for the Omicron variants BA.2.86 and JN.1, which dissociated upon equilibration. These variants exhibit lower sensitivity to antibody neutralization, allowing them to bypass both therapeutic and vaccine-induced immune responses, as reported in.44,45 Therefore, these last variants were excluded from subsequent analysis.
2.2 Coarse-grained (CG) MD simulation
The CG topology files for each protein complex were created with martinize246 and the Martini 3 force field.25 The secondary structure was identified using the DSSP v3.0 program.47 The GōMartini 3 approach23 was employed, substituting conventional harmonic bonds with Lennard-Jones (LJ) interactions based on contact maps obtained from AA-MD simulations. This method utilizes LJ potentials for virtual sites, which allows for the exploration of a broader conformational space, including critical unfolded states, enhancing our understanding of protein dynamics and functionality. A parameter study from standard values of 9.14 kJ mol−1 to 20 kJ mol−1 was conducted to determine the optimal value for the LJ potential depth (data not shown). The interaction energy of the contacts in GōMartini was set at 15.0 kJ mol−1. This effective value allowed us to recover agreement with the all-atom results of previously reported studies.21 The CG structures were initially minimized in vacuum during 5000 steps using the steepest descent algorithm. Subsequently, the complexes were solvated in a 10 × 10 × 60 nm3 box using the Martini water model. This solvation involved about 38![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 511 coarse-grained water beads, corresponding to around 154044 water molecules, with the addition of Na+ and Cl− ions to form a 0.15 M NaCl solution. The systems were then minimized using the same parameters as above. Positional restraints were imposed on the BB beads of each protein to prevent drifting during the equilibration phases. Both the NVT and NPT equilibrations, along with the production phase, utilized the V-rescale thermostat.48 The temperature coupling time constant was set at 1.0 ps for both protein and non-protein parts of the system, keeping the temperature at 300 K. The NVT equilibration was run for 2 ns, with an integration time of 20 fs. During the NPT equilibration and production, an isotropic pressure coupling was used with a compressibility set at 10−4 bar−1 and 1 bar pressure. The NPT equilibration was run for 5 ns using the C-rescale barostat49 with a pressure coupling time constant of 18 ps and an integration time of 10 fs. For the production phase, the Parrinello-Rahman barostat50 was used, with a pressure coupling time constant of 15 ps. The cutoff distances for Coulomb and van der Waals interactions were set at 1.2 nm across all equilibration and production phases. The pulling simulations (see Fig. 1C) were conducted over 1.2 microseconds, with a time step of 20 fs. For the RBD/H11-H4 complexes, specific constraints were applied: the positions of the heavy atoms of the final three residues from the C-terminus of RBD were frozen along the z-axis, and similarly, the coordinates of heavy atoms of residues S126, S127, and K128 in H11-H4 were fixed along the x- and y-axes. The center of mass (COM) of these coordinates was targeted for steered molecular dynamics simulation at a constant speed of 1 × 10−1 nm ps−1 and a spring constant of 37.6 kJ mol−1 nm−2. A total of 50 independent replicas were conducted for each system using GROMACS 2023.51
511 coarse-grained water beads, corresponding to around 154044 water molecules, with the addition of Na+ and Cl− ions to form a 0.15 M NaCl solution. The systems were then minimized using the same parameters as above. Positional restraints were imposed on the BB beads of each protein to prevent drifting during the equilibration phases. Both the NVT and NPT equilibrations, along with the production phase, utilized the V-rescale thermostat.48 The temperature coupling time constant was set at 1.0 ps for both protein and non-protein parts of the system, keeping the temperature at 300 K. The NVT equilibration was run for 2 ns, with an integration time of 20 fs. During the NPT equilibration and production, an isotropic pressure coupling was used with a compressibility set at 10−4 bar−1 and 1 bar pressure. The NPT equilibration was run for 5 ns using the C-rescale barostat49 with a pressure coupling time constant of 18 ps and an integration time of 10 fs. For the production phase, the Parrinello-Rahman barostat50 was used, with a pressure coupling time constant of 15 ps. The cutoff distances for Coulomb and van der Waals interactions were set at 1.2 nm across all equilibration and production phases. The pulling simulations (see Fig. 1C) were conducted over 1.2 microseconds, with a time step of 20 fs. For the RBD/H11-H4 complexes, specific constraints were applied: the positions of the heavy atoms of the final three residues from the C-terminus of RBD were frozen along the z-axis, and similarly, the coordinates of heavy atoms of residues S126, S127, and K128 in H11-H4 were fixed along the x- and y-axes. The center of mass (COM) of these coordinates was targeted for steered molecular dynamics simulation at a constant speed of 1 × 10−1 nm ps−1 and a spring constant of 37.6 kJ mol−1 nm−2. A total of 50 independent replicas were conducted for each system using GROMACS 2023.51
2.3 Atomistic contact map determination
We defined the interacting pairs of residues at the interface of each protein complex in the atomistic trajectories based on their spatial and chemical interactions,52 following the protocol outlined in ref. 13. The overlap (OV) contact map (CM), which is purely geometric, is based on the overlap of the van der Waals (vdW) radii of spheres associated with each heavy atom (i.e., N, C and O atoms) in the protein.53 To account for the attractive interaction between spheres, the vdW radius is multiplied by a factor of 1.24. The OV CM approach has been employed to determine the set of most relevant interactions in globular proteins, known as native contacts, and thus the OV CM provides a quantitative description of the folding mechanism as well as the thermal and mechanical unfolding in a single well-folded protein domain.13,23,27,29,54 The simple overlap between a pair of vdW spheres from different residues with a sequence distance larger than 4 residues defines a native contact. The other CM employed is the so-called rCSU method that integrates both attractive and repulsive electrostatic interactions. Further, it takes into account the types of interactions: hydrophobic, hydrophilic and aromatic interactions to provide a comprehensive assessment of contact dynamics within proteins. A contact is considered valid if the number of attractive interactions exceeds the number of repulsive ones, ensuring that the overall interaction contributes positively to the structural stability of the protein. To calculate protein contacts accurately, a sphere representing each atom is discretized into sections for detailed contact mapping. A Fibonacci grid is employed to generate an unbiased distribution of points across the surface of the sphere. The Fibonacci sequence is well suited for this purpose, as it minimizes clustering and achieves more uniform coverage compared to other distribution methods. This uniformity is crucial for ensuring accurate and representative sampling of the sphere's surface.52 The combination of the OV + rCSU CM yields a systematic tool for contact map determination in proteins and their complexes. Due to the dynamic character of contacts at the interface in protein complexes, we consider only high-frequency contacts (freq >0.7). In addition, each residue in a given contact is identified with one of the following types of chemical groups: polar, nonpolar, ionic and non-specific.2.4 Coarse-grained contact map determination
Enlarged vdW radii, also referred to as effective vdW spheres, enable a better approximation of the space occupied by atoms, incorporating the effect of their electron clouds.55 This approach allows for more accurate predictions of molecular interactions within proteins in CG representation. These enlarged vdW radii are particularly useful in cases where protein structures have a partially unsolved structure. The accurate reconstruction or modeling of protein conformations can be done by predicting pairs of interactions. The effective spheres are placed at the centers of the Cα atoms. A contact between amino acids is defined if these spheres overlap.We systematically tracked the native contacts at the interface of the complexes throughout each CG trajectory. For this, we calculated the distance between the backbone (BB) beads of amino acid pairs. These pairs were defined in the GōMartini gromacs itp file, which was constructed based on the OV + rCSU CM. For each native contact, a specific σ (the distance at which there is no intermolecular potential between two particles) is determined. To calculate Rmin (distance at which the potential energy of the LJ potential reaches its minimum), we multiplied σ by 21/6. During the simulations, we compared the Rmin value, scaled by a factor of 1.1, with the actual distance between the BB beads at each frame. A contact was considered established when the actual distance was less than or equal to Rmin. If the distance was greater than Rmin, the contact was considered broken.
We identified the non-native (NON) contacts at the interface of the complexes, mimicking the enlarged vdW radii approach. For this, we calculated the distance of all pairs of BB beads between the two proteins of each complex, excluding those pairs already identified as native contacts. Each BB pair was assigned enlarged vdW radii, using values reported in.55 To determine if a non-native contact is formed, we compared the actual distance between each bead pair to the sum of their corresponding VdW radii, scaled by a factor of 1.2. A contact was considered formed if the actual distance was less than or equal to this scaled sum.
These approaches enabled us to monitor the pattern of native and non-native contact stability and rupture across the distance of the virtual particle, providing insights into the dynamic stability of the complex and the mechanical forces exerted upon it.56,57
3 Results & discussion
MD simulations were performed to investigate the mechanical stability of the RBD/H11-H4 complex for several VoCs. For this purpose, we employed the GōMartini 3 approach to capture the molecular biomechanics of the protein complex at almost atomistic resolution. Such methodology allows us to mimic similar experimental conditions, as commonly used in single-molecule force spectroscopy (SMFS). For instance, the tethering of N- and C-termini residues, loading rates, and the atomic force microscopy (AFM) cantilever stiffness.8 Furthermore, GōMartini simulations allowed us to increase our statistics per variant and compute the interaction energy associated with the RBD/H11-H4 interface. In these simulations, the RBD/H11-H4 complexes were subjected to molecular constraints that avoided extension of the bonded parameters along residues G526, P527 and K528 of the RDB. On the other side of the complex, a velocity along the z-direction was applied on a dummy atom located at the COM of the residues S126, S127 and K128 of H11-H4.We report in Table S4† that the Delta and XBB.1.5 variants exhibited the highest and lowest total interface energies, respectively. Newer variants demonstrate greater affinity for the hACE2 receptor compared to the WT variant, correlating with increased transmissibility.8,12 Conversely, the affinity of monoclonal antibodies for the SARS-CoV-2 RBD has been decreasing58 as long as new variants appeared. Mutations in the Alpha and Delta variants do not fall within the H11-H4 nanobody binding region. However, two out of the 22 mutations in XBB.1.5 occur in this region. Specifically, the E484A mutation reduces the interaction with residue R52 in the nanobody. This unfavorable electrostatic interaction may in part account for the lowest interface energy of XBB.1.5.
The mechanical stability for each RBD/H11-H4 is reported in Fig. 2. This figure shows the force-displacement profiles based on the pulling protocol at constant velocity. Notably, all variants exhibited a single force peak that is associated with a characteristic Fmax. The average value of RMSD regarding the initial state (Force = 0 pN) is about 0.2–0.4 nm for the RBD and 0.1–0.3 for the H11-H4 (Fig. S1 and S2†). This simply shows that we did not observe significant conformational changes, such as partial unfolding events along the molecular trajectories during the pulling MD simulations. The average value of Fmax was determined in 50 independent MD trajectories for each variant: 359 ± 33 pN (WT), 328 ± 24 pN (Alpha), 461 ± 34 pN (Delta) and 391 ± 41 pN (XBB.1.5) and they fall within a Gaussian distribution (see inset in Fig. 2). The mechanical forces that RBD/H11-H4 complexes can withstand according to our simulation are in the range of 300–470 pN (Fig. S3†), with a statistically significant difference between WT and the other variants (Table S1†) with P-values in the range of 10−12–10−5. Overall, the in silico data suggests transitions from the bound to unbound state without unfolding events that are mediated by the interplay between stabilizing and destabilizing interactions located at the protein interface (Fig. 3).
 | ||
| Fig. 2 Nanomechanics of the RBD/H11-H4 complexes. Force-displacement profiles (n = 50) from GōMartini 3 pulling MD simulations for four RBD variants: WT (black), Alpha (blue), Delta (gray) and Omicron XBB.1.5 (green). The force profiles extend over displacement from the bound state (D = 0 nm) to the unbound state (D = 12 nm). The inset shows the distribution of Fmax for each variant case, and the solid red line shows the Gaussian fitting with the mean (μ) and the standard deviation (σ) values. | ||
 | ||
| Fig. 3 Transition profiles of the dissociation process of VoCs. The RBD from WT, Alpha, Delta and XBB are colored in black, blue, gray, and green, respectively. H11-H4 is shown in orange. Interface residues forming native contacts are depicted in balls-and-sticks (start). The residues that maintain contacts at Fmax (intermediate) are highlighted. The last column illustrates the dissociation of the complexes. | ||
We employed protein contact determination for the GōMartini 3 simulations (see method section). The contact map analysis allowed us to elucidate the characteristic interaction fingerprint per variant during the mechanical dissociation of the protein complexes. In summary, the RBDWT formed 24 stabilizing interactions at the protein interface, for Alpha, we reported 21 interactions, Delta established 22 interactions and XBB.1.5 only 16 protein contacts (see Fig. 4, S4 and Table S2†). The disruption of contacts at the protein interface across the studied variants followed a consistent three-stage sequence, which is defined by the displacement of the pulling (virtual) particle along the z-axis at intervals of below 2 nm, between 2–6 nm, and above 6 nm. The first two stages primarily involved the rupture of nonpolar and non-specific contacts. Notably, only one non-specific contact broke in both Delta (Y446-D115) and XBB.1.5 (A484-R52) variants. The contact between Y499 of RDB and Y101 of H11-H4, which is present in all variants, consistently disappeared during these two first stages. Another contact, L452-V102, also broke early but is exclusive to the WT and Alpha variants. The contact F490-Y104, found in the WT, Alpha and XBB.1.5 (where it is nonspecific because of the F490S mutation), dissociates early in these variants, but in Delta, it was one of the more persistent contacts. In the last (third) stage, the ionic interaction E484-R52 was lost in WT, Alpha, and Delta. Mutation E484A caused this contact to be absent from XBB.1.5. Other long-lasting contacts across all variants included the nonpolar pairs L492-Y104, Y489-Y104, Y489-L105 as well as the polar interaction Q483-S103, which were among the last to disappear in the pulling simulation. F456-Y104, present in the WT and Alpha, but not in Delta, also broke during this stage for those systems. However, it was also present in XBB.1.5, but it disappeared in the second stage. Y455-Y104, found in all variants except in the WT, remained one of the last interactions to break. Finally, the contact F486-L105, unique to WT, was among the most stable contacts during the mechanical dissociation.
 | ||
| Fig. 4 Dissociation profiles of protein (native) contacts in RBD/H11-H4 complexes. Each panel displays the list of protein contacts at the interface. The z-axis shows all protein contacts identified by the OV + rCSU approach that are necessary to stabilize the complex interface and involved in the dissociation process. The y-axis denotes the displacement (D) of the pulling particle along the z-direction. The average displacement per contact is given next to the standard deviation (as an error bar). Red dashed lines indicate the rupture distance for each protein complex. Each contact is colored according to the interaction type: nonpolar (brown), polar (blue), ionic (purple), and no-specific (white). | ||
Fig. 5 reports on the contact map analysis of new interactions (also known as non-native contacts, NON) that are formed during the dissociation process in GōMartini 3 pulling simulations. Such protein contacts are short-lasting interactions that are necessary to capture the nanomechanics of the protein complex. Some of these protein contacts were lost gradually (Fig. S5–8 and Table S3†). It is worth noting that contacts were not observed to disappear sequentially (one after another) or linearly during the pulling simulations. This is an intrinsic feature of the interplay between the GōMartini 3 approach and the Martini 3 force field. The correct energetic balance between the former and latter allows the conformational changes associated with non-equilibrium processes in protein complexes. We noticed that protein contacts at the interface were collectively lost at ∼7 nm in WT, Alpha, and XBB.1.5 variants, whereas in Delta, those contacts were lost at ∼9 nm. This shift in the rupture distance indicates an enhanced mechanical stability in the Delta variant that may be attributed to the higher plasticity while forming a complex with H11-H4 as a result of the T478K mutation.59 This increased flexibility enables the Delta variant to not only bind more effectively to hACE2, but also to withstand higher external forces.8 The analysis of (native) contacts persisting at Fmax revealed the conserved presence of some contact pairs, Y489-Y104, Y489-L105, L482-Y104, and Q493-S103, across all variants (Fig. S5†). The L455-Y104 contact was absent in XBB.1.5, but it was observed in other variants. Conversely, F486-L105 was exclusive to WT. Likewise, the formation and disruption of short-lasting NON contacts along the pulling CG trajectories were tracked by statistical analysis (Fig. 5, S5–8 and Table S3†). Multiple NON contacts were identified, demonstrating a consistent and gradual breakdown pattern across all analyzed complexes. In contrast to the abundance of native contacts observed at Fmax, only a minimal number of NON contacts were detected (Fig. S9†).
 | ||
| Fig. 5 Dissociation profiles of non-native contacts (NON) in RBD/H11-H4 complexes. Each panel displays the NON contact as a result of the Martini 3 force field. The x-axis shows all protein contacts identified by the OV + rCSU approach that are necessary to stabilize the complex interface and involved in the dissociation process. The y-axis denotes the displacement (D) of the pulling particle along the z-direction. The average displacement per contact is given next to the standard deviation (as an error bar). Red dashed lines indicate the rupture distance for each protein complex. Each contact is colored according to the interaction type: nonpolar (brown) and polar (blue). | ||
The present work provides insights into the mechanostability of SARS-CoV-2 variants binding nanobody H11-H4, forming stable complexes, revealing distinct variations on the force-displacement profiles, and characterizing a set of relevant residues at the protein interface that support mechanical stability. Previous investigations using AA-MD simulations of RBD/H11-H4 complexes showed force–distance profiles similar to ours, but Fmax was sensitive to the pulling protocol and no systematic analysis could be extracted. Such studies were limited by position restraints on the RBD heavy atoms and neglected the intrinsic backbone fluctuations that play a major role during protein dissociation.20–22 Anchoring one terminal position of one of the proteins while the other one is pulled apart at constant velocity from another terminal residue, are similar conditions as used in AFM-SMFS studies. Such a pulling approach allows us to gain a deeper comprehension of mechanical stability in protein complexes and capture the dissociation mechanisms under mechanical loads. Our studies show a resilient protein complex that does not present unfolding events during the dissociation process in the range of forces of 300–470 pN. This observation is consistent with previous findings suggesting that the RBD in SARS-CoV-2 has a higher mechanical stability13 than its predecessors (Fmax = 250 ± 11 pN for SARS-CoV-2 and Fmax = 200 ± 13 pN for SARS-CoV-1). It also suggests that the engineered nanobody (H11-H4 nanobody) is mechanostable.
Our study sheds light on the profile of high-frequency native contacts that are originally present at zero applied force and their evolution during the dissociation process. Furthermore, we identified several NON contacts using our enlarged vdW radii protocol in CG trajectories. The latter analysis supports the need of Martini 3 interaction for correct description of the process. Most of the native contacts were established between the RBD and the CDR3 part of the H11-H4. By tracking protein contact formation and dissociation along pulling MD trajectories, we identified both stable and transient interactions within the complexes, thereby providing valuable insights into its structural dynamics under mechanical stress.
The dynamic patterns of NON contacts closely resemble those of native contacts. While the former contacts are only present during conformational changes, the latter are necessary to maintain complex stability. The transient (NON) interactions, described by the Martini 3 force field, are fundamental for the correct nanomechanical characterization of protein complexes. A significant proportion of these transient interactions were found to be nonpolar in nature, highlighting the importance of hydrophobic interactions in stabilizing the complex interface. This observation is consistent with previous AA-MD reports that showed that nonpolar interactions play a major role in the mechanical stability of SARS-CoV-2/nanobody complexes.20,21 The mechanical dissociation profile of the RBD/H11-H4 complexes displayed one common force peak that corresponds to a tensile mechanical clamp without involving shear of protein chains. The main process occurs at the cost of stretching the length of NAT contacts within the CDR3 and CDR2 regions of H11-H4. Most contacts that were broken first corresponded to CDR2, and in some cases, breaking contacts involved CDR1, but most of them broke at the very early stage of the process. Specifically, the RBDWT and RBDDelta exhibited three NAT contacts with CDR2, whereas the RBDAlpha and RBDXBB.1.5 showed four contacts with CDR2. The Alpha variant presented the most NON contacts along the pulling trajectories, whereas Delta had more NON contacts than WT. The XBB.1.5 variant displayed a decaying number of these NON contacts along the MD trajectories (see Fig. S9†). Notably, none of the complexes formed native contacts with CDR1. Both NAT and NON contacts contributed to the dissociation pattern. In the WT, Alpha, and Delta variants, residues Y104, V102, and L105 in the H11-H4 were conserved and involved in establishing the highest number of NAT contacts, suggesting their key role in stabilizing the complex interface. Residues E484, Y499, and Q493 within the RBD were primarily involved in interactions with H11-H4. However, these interactions were notably reduced in the XBB.1.5 variant, particularly at position 484, where the residue is mutated to alanine. This decrease in contacts suggests a significant alteration in the binding dynamics of this variant compared with the others.
The RBDAlpha/H11-H4 complex, characterized by a single mutation at position 501, displayed reduced mechanical stability compared to the other variants. Interestingly, this mutation decreased the mechanostability of the complex without directly interacting with the nanobody. It is suggested that this mutation alone does not compromise the neutralization effect of antibodies targeting the WT.60 Furthermore, AA-MD simulations have shown that in the presence of hACE2, H11-H4 dissociates from RBDAlpha, whereas it remains bound to the WT variant, indicating enhanced affinity for hACE2 compared to the WT variant.20 Additionally, SMFS experiments have shown that the RBDAlpha/hACE2 complex is more mechanostable than its WT counterpart.10 The RBDDelta/H11-H4 complex, containing mutations L452R and T478K in the RBD, displayed increased mechanical stability compared to all other variants. Despite the presence of the L452R mutation, the complex maintained contact with residue V102 in the nanobody. Although this contact is already not present at Fmax, it provides us with important information about this characteristic mutation. Several recent VoCs, such as BA.1, BA.2, BA.3, BA.4, and BA.5, have retained leucine at this position,61 suggesting that whereas the Delta variant may exhibit enhanced affinity for hACE2, it could also be susceptible to antibodies or engineered nanobodies targeting this binding region. These mutations, which likely enhance the overall stability, may be responsible for the observed rise in mechanostability in this variant, allowing the RBD to adapt more effectively under external forces. The RBDXBB.1.5/H11-H4 complex exhibited fewer native contact pairs and reduced mechanical stability. The F486P mutation did not form NAT or NON contacts. It has been reported that the introduction of proline at position 486 increases the rigidity of the loop region spanning residues 475–487.62,63 This reduced flexibility may account for the decreased efficacy of this variant in establishing dynamic contacts with the H11-H4 nanobody. The contact pattern was perturbed, as evidenced by the absence or weakening of NAT contacts in RBD positions 455–456, 484 and 493–494. Interestingly, the A484 mutation led to the establishment of a new contact with residue A48, located within CDR2 of the H11-H4 nanobody. Furthermore, the recurrence of conserved residues L455 and E484 across diverse SARS-CoV-2 variants, including WT, Alpha, Delta, BA.2, BA.4, and BA.5, suggests their evolutionary significance and impact on antibody recognition.61 Mutations at positions 484, 486, and 490 have the potential to diminish or abolish the neutralizing effect of several anti/nanobodies such as Bebtelovimab and Sotrovimab.62,64–66 Additionally, mutations L455F and F456L, generally present in XBB lineages but absent in XBB.1.5, are linked to increased antigenicity and immune evasion.67 The data indicated that among all studied SARS-CoV-2 RBD variants, the residues Y489, L492, Q493, and S494 are consistently key for the mechanostability of the complex. These residues are particularly conserved across the variants studied, highlighting their crucial importance for the structural integrity, transmissibility, and/or recognition of hACE2. Notably, in the BA.1 variant, the mutation Q493R was observed; however, this mutation was reverted in subsequent variants. This reversion underscores the importance of the glutamine residue at this position for enhanced interaction with hACE2.68 The conservation of these residues suggests a critical role in maintaining the correct conformation of the RBD to facilitate receptor recognition and subsequent viral entry into host cells.69,70 In the H11-H4 nanobody, the residues S57, V102, S103, Y104, and L105 consistently exhibited high mechanostability across all studied variants. Particularly, V102 and Y104 presented the highest numbers of contacts with the RBD in all variants, indicating their important role in binding efficacy. However, in the XBB.1.5 variant, the number of contacts involving these two residues was reduced, suggesting that these mutations might affect binding stability. Residue S57, despite forming a non-specific interaction with E484 in the WT, Alpha, Delta, and XBB.1.5 variants, showed significant mechanostability in our studies. Interestingly, this interaction was retained in the presence of the mutation E484A in XBB.1.5. These results unveiled that these residues are in part responsible for the effectiveness of this nanobody across different variants and showcased their importance in the mechanostable nature of this family of nanobodies. The design of therapeutic antibodies should focus on preserving and enhancing these interactions. In this manner, therapeutics can consider mechanical stability in the formulation of novel compounds and remain effective against a wider range of variants, including emerging ones.
4 Conclusions
The present work relies on a CG methodology that uses the popular Martini 3 force field and as some limitation as reported regarding the description of specific side-chain interactions71 or subtle conformational changes that are critical in some nanomechanical studies, such as the emergence of catch-bonds in bacterial adhesion systems.19 The structure-dependence of the GōMartini 3 approach for the construction of the native contacts in protein interfaces is another limitation in cases where a large mechanical contribution comes from the reorganisation of the interface. In this regard, our protein complexes do not fall in that category and together with the improvement in the determination of the contact map via the high-frequency contacts, we can construct an optimal set of interaction for each protein complex that captures the underlying interaction as in AA-MD. The present work provides valuable insights into the mechanical stability of SARS-CoV-2 variants in complex with a potent nanobody. For instance, the Delta variant displayed a higher degree of mechanostability, which could potentially impact its viral fitness and transmissibility. The mechanostability of XBB.1.5 was intermediate between the WT and Delta variants. Nonetheless, both native and non-native contacts decreased, suggesting a reduced susceptibility to antibody neutralization. These insights emphasize the importance of targeting conserved epitopes or stabilizing RBD/antibody interactions to improve vaccine effectiveness and counteract immune escape. This study combines both AA-MD and state-of-the-art CG-MD simulations for the study of large conformational changes in protein complexes. Our findings indicate that specific mutations significantly affect the formation and rupture of native and non-native contacts under high mechanical loads, affecting the overall mechanical stability of virus-antibody complexes. We observed significant stability in the RBD and H11-H4, with no protein unfolding during pulling MD simulations at high load rates. In contrast with the WT, the Alpha variant showed decreased mechanostability, whereas the Delta variant displayed an opposite effect. These differences highlight the significance of mechanical stability in the immune evasion mechanisms of the virus and emphasize the significance of incorporating this information into the design of therapeutic antibodies. A thorough understanding of the nanomechanical aspects of these interactions can guide the development of novel monoclonal antibodies, optimizing them not only by surface complementarity and thus high binding affinity, but also for enhanced stability under mechanical stress. Furthermore, the methods and insights derived from this study can be extended to enhance therapies against other coronaviruses. Including mechanical stability considerations in the design process may enhance the efficacy of treatments and preventive measures against both current and emerging variants of SARS-CoV-2.Author contributions
L. F. C. V: writing – review & editing, writing – original draft, methodology, investigation, formal analysis. M. Ch., R. A. M., S. J. M.: review & editing, scientific discussion, methodology. G. E. O. R., J. L. B.: review & editing, scientific discussion A. B. P: writing – review & editing, writing – original draft, supervision, methodology, investigation, funding acquisition, conceptualization.Data availability
The data supporting the findings of this study are openly available in the Zenodo repository.Citation for dataset.72
The repository includes all input files and simulations utilized in this research.
Conflicts of interest
The authors declare that they have no known financial or personal relationships that could have influenced the work reported in this paper.Acknowledgements
A. B. P. acknowledges financial support from the National Science Center, Poland, under grant 2022/45/B/NZ1/02519 and gratefully acknowledges Polish high-performance computing infrastructure PLGrid (HPC Centers: ACK Cyfronet AGH) for providing computer facilities and support within computational grant no. PLG/2023/016519 and PLG/2024/017332. M. Ch. acknowledges financial support from the National Science Centre (NCN), Poland, under grant No. 2018/31/B/NZ1/00047.References
- R. Savan and M. Gale Jr, Immunity, 2023, 56, 1443–1450 CrossRef CAS PubMed.
- M.-Y. Wang, R. Zhao, L.-J. Gao, X.-F. Gao, D.-P. Wang and J.-M. Cao, Front. Cell. Infect. Microbiol., 2020, 10, 587269 CrossRef CAS PubMed.
- N. A. Valdez-Cruz, E. García-Hernández, C. Espitia, L. Cobos-Marín, C. Altamirano, C. G. Bando-Campos, L. F. Cofas-Vargas, E. W. Coronado-Aceves, R. A. González-Hernández, P. Hernández-Peralta, D. Juárez-López, P. A. Ortega-Portilla, S. Restrepo-Pineda, P. Zelada-Cordero and M. A. Trujillo-Roldán, Microb. Cell Fact., 2021, 20, 88 CrossRef CAS PubMed.
- X. Chi, R. Yan, J. Zhang, G. Zhang, Y. Zhang, M. Hao, Z. Zhang, P. Fan, Y. Dong, Y. Yang, Z. Chen, Y. Guo, J. Zhang, Y. Li, X. Song, Y. Chen, L. Xia, L. Fu, L. Hou, J. Xu, C. Yu, J. Li, Q. Zhou and W. Chen, Science, 2020, 369, 650–655 CrossRef CAS PubMed.
- L. Casalino, Z. Gaieb, J. A. Goldsmith, C. K. Hjorth, A. C. Dommer, A. M. Harbison, C. A. Fogarty, E. P. Barros, B. C. Taylor, J. S. McLellan, E. Fadda and R. E. Amaro, ACS Cent. Sci., 2020, 6, 1722–1734 CrossRef CAS PubMed.
- S. Olofsson, M. Bally, E. Trybala and T. Bergström, Annu. Rev. Virol., 2023, 10, 283–304 CrossRef CAS PubMed.
- C. B. Jackson, M. Farzan, B. Chen and H. Choe, Nat. Rev. Mol. Cell Biol., 2022, 23, 3–20 CrossRef CAS PubMed.
- A. Ray, T. T. Minh Tran, R. d. Santos Natividade, R. A. Moreira, J. D. Simpson, D. Mohammed, M. Koehler, S. J. L. Petitjean, Q. Zhang and F. Bureau, et al. , ACS Nanosci. Au, 2024, 4, 136–145 CrossRef CAS PubMed.
- W. Ma, H. Fu, F. Jian, Y. Cao and M. Li, Nat. Ecol. Evol., 2023, 7, 1457–1466 CrossRef PubMed.
- M. S. Bauer, S. Gruber, A. Hausch, M. C. Melo, P. S. Gomes, T. Nicolaus, L. F. Milles, H. E. Gaub, R. C. Bernardi and J. Lipfert, Nat. Nanotechnol., 2024, 19, 399–405 CrossRef CAS PubMed.
- R. Zhu, D. Canena, M. Sikora, M. Klausberger, H. Seferovic, A. R. Mehdipour, L. Hain, E. Laurent, V. Monteil and G. Wirnsberger, et al. , Nat. Commun., 2022, 13, 7926 CrossRef CAS PubMed.
- M. S. Bauer, S. Gruber, A. Hausch, P. S. Gomes, L. F. Milles, T. Nicolaus, L. C. Schendel, P. L. Navajas, E. Procko and D. Lietha, et al. , Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2114397119 CrossRef CAS PubMed.
- R. A. Moreira, M. Chwastyk, J. L. Baker, H. V. Guzman and A. B. Poma, Nanoscale, 2020, 12, 16409–16413 RSC.
- R. A. Moreira, H. V. Guzman, S. Boopathi, J. L. Baker and A. B. Poma, Materials, 2020, 13, 5362 CrossRef CAS PubMed.
- M. Koehler, A. Ray, R. A. Moreira, B. Juniku, A. B. Poma and D. Alsteens, Nat. Commun., 2021, 12, 6977 CrossRef CAS PubMed.
- N. Kumar, R. Kaushik, K. Y. Zhang, V. N. Uversky, U. Sahu, R. Sood and S. Bhatia, Proteins: Struct., Funct., Bioinf., 2023, 91, 798–806 CrossRef CAS PubMed.
- R. C. Bernardi, E. Durner, C. Schoeler, K. H. Malinowska, B. G. Carvalho, E. A. Bayer, Z. Luthey-Schulten, H. E. Gaub and M. A. Nash, J. Am. Chem. Soc., 2019, 141, 14752–14763 CrossRef CAS PubMed.
- A. B. Poma, T. T. M. Thu, L. T. M. Tri, H. L. Nguyen and M. S. Li, J. Phys. Chem. B, 2021, 125, 7628–7637 CrossRef CAS PubMed.
- Z. Liu, H. Liu, A. M. Vera, R. C. Bernardi, P. Tinnefeld and M. A. Nash, Nat. Commun., 2020, 11, 4321 CrossRef CAS PubMed.
- M. Golcuk, A. Hacisuleyman, B. Erman, A. Yildiz and M. Gur, J. Chem. Inf. Model., 2021, 61, 5152–5160 CrossRef CAS PubMed.
- M. Golcuk, A. Hacisuleyman, S. Z. Yilmaz, E. Taka, A. Yildiz and M. Gur, J. Chem. Inf. Model., 2022, 62, 2490–2498 CrossRef CAS PubMed.
- H. Nguyen and M. S. Li, Sci. Rep., 2022, 12, 1–15 CrossRef PubMed.
- A. B. Poma, M. Cieplak and P. E. Theodorakis, J. Chem. Theory Comput., 2017, 13, 1366–1374 CrossRef CAS PubMed.
- P. C. T. Souza, L. Borges-Araújo, C. Brasnett, R. A. Moreira, F. Grünewald, P. Park, L. Wang, H. Razmazma, A. C. Borges-Araújo, L. F. Cofas-Vargas, L. Monticelli, R. Mera-Adasme, M. N. Melo, S. Wu, S. J. Marrink, A. B. Poma and S. Thallmair, bioRxiv, 2024, preprint, DOI:10.1101/2024.04.15.589479.
- P. C. T. Souza, R. Alessandri, J. Barnoud, S. Thallmair, I. Faustino, F. Grünewald, I. Patmanidis, H. Abdizadeh, B. M. H. Bruininks, T. A. Wassenaar, P. C. Kroon, J. Melcr, V. Nieto, V. Corradi, H. M. Khan, J. Domański, M. Javanainen, H. Martinez-Seara, N. Reuter, R. B. Best, I. Vattulainen, L. Monticelli, X. Periole, D. P. Tieleman, A. H. de Vries and S. J. Marrink, Nat. Methods, 2021, 18, 382–388 CrossRef CAS PubMed.
- X. Chu, Y. Wang, P. Tian, W. Li and D. Mercadante, Advanced Sampling and Modeling in Molecular Simulations for Slow and Large-Scale Biomolecular Dynamics, Frontiers Media SA, 2022 Search PubMed.
- A. B. Poma, T. T. M. Thu, L. T. M. Tri, H. L. Nguyen and M. S. Li, J. Phys. Chem. B, 2021, 125, 7628–7637 CrossRef CAS PubMed.
- Z. Liang, A. Poma, K. Kremer, K.-I. Okazaki, S. Pantano and S. Poblete, Recent advances in computational modelling of biomolecular complexes, Frontiers Media SA, 2023 Search PubMed.
- Z. Liu, R. A. Moreira, A. Dujmović, H. Liu, B. Yang, A. B. Poma and M. A. Nash, Nano Lett., 2022, 22, 179–187 CrossRef CAS PubMed.
- L. F. Cofas-Vargas, R. A. Moreira, S. Poblete, M. Chwastyk and A. B. Poma, Acta Phys. Pol., A, 2024, 145, S9–S9 CrossRef CAS.
- W. Pezeshkian, F. Grünewald, O. Narykov, S. Lu, V. Arkhipova, A. Solodovnikov, T. A. Wassenaar, S. J. Marrink and D. Korkin, Structure, 2023, 31, 492–503 CrossRef CAS PubMed.
- J. Huo, A. Le Bas, R. R. Ruza, H. M. E. Duyvesteyn, H. Mikolajek, T. Malinauskas, T. K. Tan, P. Rijal, M. Dumoux, P. N. Ward, J. Ren, D. Zhou, P. J. Harrison, M. Weckener, D. K. Clare, V. K. Vogirala, J. Radecke, L. Moynié, Y. Zhao, J. Gilbert-Jaramillo, M. L. Knight, J. A. Tree, K. R. Buttigieg, N. Coombes, M. J. Elmore, M. W. Carroll, L. Carrique, P. N. M. Shah, W. James, A. R. Townsend, D. I. Stuart, R. J. Owens and J. H. Naismith, Nat. Struct. Mol. Biol., 2020, 27, 846–854 CrossRef CAS PubMed.
- R. Salomon-Ferrer, A. W. Götz, D. Poole, S. Le Grand and R. C. Walker, J. Chem. Theory Comput., 2013, 9, 3878–3888 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- M. V. Shapovalov and R. L. Dunbrack Jr, Structure, 2011, 19, 844–858 CrossRef CAS PubMed.
- P. Eastman, J. Swails, J. D. Chodera, R. T. McGibbon, Y. Zhao, K. A. Beauchamp, L.-P. Wang, A. C. Simmonett, M. P. Harrigan, C. D. Stern, R. P. Wiewiora, B. R. Brooks and V. S. Pande, PLoS Comput. Biol., 2017, 13, e1005659 CrossRef PubMed.
- S. Izadi, R. Anandakrishnan and A. V. Onufriev, J. Phys. Chem. Lett., 2014, 5, 3863–3871 CrossRef CAS PubMed.
- A. C. Simmonett and B. R. Brooks, J. Chem. Phys., 2021, 154, 054112 CrossRef CAS PubMed.
- D. S. Cerutti, R. E. Duke, T. A. Darden and T. P. Lybrand, J. Chem. Theory Comput., 2009, 5, 2322 CrossRef CAS PubMed.
- D. J. Sindhikara, S. Kim, A. F. Voter and A. E. Roitberg, J. Chem. Theory Comput., 2009, 5, 1624–1631 CrossRef CAS PubMed.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. DiNola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- J.-P. Ryckaert, G. Ciccotti and H. J. C. Berendsen, J. Comput. Phys., 1977, 23, 327–341 CrossRef CAS.
- C. W. Hopkins, S. Le Grand, R. C. Walker and A. E. Roitberg, J. Chem. Theory Comput., 2015, 11, 1864–1874 CrossRef CAS PubMed.
- D. Planas, I. Staropoli, V. Michel, F. Lemoine, F. Donati, M. Prot, F. Porrot, F. Guivel-Benhassine, B. Jeyarajah, A. Brisebarre, O. Dehan, L. Avon, W. H. Bolland, M. Hubert, J. Buchrieser, T. Vanhoucke, P. Rosenbaum, D. Veyer, H. Péré, B. Lina, S. Trouillet-Assant, L. Hocqueloux, T. Prazuck, E. Simon-Loriere and O. Schwartz, Nat. Commun., 2024, 15, 1–17 Search PubMed.
- L. Zhang, A. Kempf, I. Nehlmeier, A. Cossmann, A. Richter, N. Bdeir, L. Graichen, A.-S. Moldenhauer, A. Dopfer-Jablonka, M. V. Stankov, E. Simon-Loriere, S. R. Schulz, H.-M. Jäck, L. Čičin-Šain, G. M. N. Behrens, C. Drosten, M. Hoffmann and S. Pöhlmann, Cell, 2024, 187, 596–608 CrossRef CAS PubMed.
- P. Kroon, F. Grunewald, J. Barnoud, M. van Tilburg, P. Souza, T. Wassenaar and S. Marrink, eLife, 2023, 12, 33 Search PubMed.
- W. G. Touw, C. Baakman, J. Black, T. A. H. te Beek, E. Krieger, R. P. Joosten and G. Vriend, Nucleic Acids Res., 2015, 43, D364–D368 CrossRef CAS PubMed.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- M. Bernetti and G. Bussi, J. Chem. Phys., 2020, 153, 114107 CrossRef CAS PubMed.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- K. Wołek, À. Gómez-Sicilia and M. Cieplak, J. Chem. Phys., 2015, 143, 243105 CrossRef PubMed.
- J. Tsai, R. Taylor, C. Chothia and M. Gerstein, J. Mol. Biol., 1999, 290, 253–266 CrossRef CAS PubMed.
- P. S. F. C. Gomes, M. Forrester, M. Pace, D. E. B. Gomes and R. C. Bernardi, Front. Chem., 2023, 11, 1107427 CrossRef CAS PubMed.
- M. Chwastyk, A. P. Bernaola and M. Cieplak, Phys. Biol., 2015, 12, 046002 CrossRef PubMed.
- M. Chwastyk, A. Galera-Prat, M. Sikora, À. Gómez-Sicilia, M. Carrión-Vázquez and M. Cieplak, Proteins: Struct., Funct., Bioinf., 2014, 82, 717–726 CrossRef CAS PubMed.
- M. Gunnoo, P.-A. Cazade, A. Orlowski, M. Chwastyk, H. Liu, D. T. Ta, M. Cieplak, M. Nash and D. Thompson, Phys. Chem. Chem. Phys., 2018, 20, 22674–22680 RSC.
- M. Cox, T. P. Peacock, W. T. Harvey, J. Hughes, D. W. Wright, B. J. Willett, E. Thomson, R. K. Gupta, S. J. Peacock and D. L. Robertson, et al. , Nat. Rev. Microbiol., 2023, 21, 112–124 CrossRef CAS PubMed.
- A. A. Elfiky, I. M. Ibrahim and A. M. Elgohary, Int. J. Pept. Res. Ther., 2022, 28, 146 CrossRef CAS PubMed.
- R. A. A. Pondé, Virology, 2022, 572, 44–54 CrossRef PubMed.
- Z.-S. Yang, T.-S. Li, Y.-S. Huang, C.-C. Chang and C.-M. Chien, Sci. Rep., 2024, 14, 2753 CrossRef CAS PubMed.
- F. Scarpa, I. Azzena, C. Locci, M. Casu, P. L. Fiori, A. Ciccozzi, S. Angeletti, E. Imperia, M. Giovanetti, A. Maruotti, A. Borsetti, R. Cauda, A. Cassone, A. Via, S. Pascarella, D. Sanna and M. Ciccozzi, Microorganisms, 2023, 11, 13 Search PubMed.
- Q. Chen, J. Zhang, P. Wang and Z. Zhang, iScience, 2022, 25, 105044 CrossRef CAS PubMed.
- G. Verkhivker, M. Alshahrani and G. Gupta, Viruses, 2023, 15, 36 Search PubMed.
- C. Yue, W. Song, L. Wang, F. Jian, X. Chen, F. Gao, Z. Shen, Y. Wang, X. Wang and Y. Cao, Lancet Infect. Dis., 2023, 23, 278–280 CrossRef CAS PubMed.
- L. Wu, C. Peng, Y. Yang, Y. Shi, L. Zhou, Z. Xu and W. Zhu, Briefings Bioinf., 2022, 23, 9 Search PubMed.
- F. Jian, L. Feng, S. Yang, Y. Yu, L. Wang, W. Song, A. Yisimayi, X. Chen, Y. Xu, P. Wang, L. Yu, J. Wang, L. Liu, X. Niu, T. Xiao, R. An, Y. Wang, Q. Gu, F. Shao, R. Jin, Z. Shen, Y. Wang, X. Wang and Y. Cao, PLoS Pathog., 2023, 19, e1011868 CrossRef CAS PubMed.
- A. M. Philip, W. S. Ahmed and K. H. Biswas, Comput. Struct. Biotechnol. J., 2023, 21, 1966–1977 CrossRef CAS PubMed.
- C. Yi, X. Sun, J. Ye, L. Ding, M. Liu, Z. Yang, X. Lu, Y. Zhang, L. Ma, W. Gu, A. Qu, J. Xu, Z. Shi, Z. Ling and B. Sun, Cell. Mol. Immunol., 2020, 17, 621–630 CrossRef CAS PubMed.
- S. Borkotoky, D. Dey and Z. Hazarika, Mol. Biol. Rep., 2023, 50, 2713–2721 CrossRef CAS PubMed.
- R. Alessandri, P. C. Souza, S. Thallmair, M. N. Melo, A. H. De Vries and S. J. Marrink, J. Chem. Theory Comput., 2019, 15, 5448–5460 CrossRef CAS PubMed.
- L. F. Cofas-Vargas, G. E. Olivos Ramirez, M. Chwastyk, R. A. Moreira, J. L. Baker, S. J. Marrink and A. B. Poma, 2024, Zenodo/dataset: 13150300, DOI:10.5281/zenodo.13150300.
Footnote |
| † Electronic supplementary information (ESI) available: Coarse-grained pulling simulations snapshots and contact maps analysis scripts for SARS-CoV-2 RBD variants in complex with H11-H4 nanobody. See DOI: https://doi.org/10.5281/zenodo.13150300. |
| This journal is © The Royal Society of Chemistry 2024 |