
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Copper indium sulfide quantum dots enabling quantitative visible light photoisomerisation of (E)-azobenzene chromophores†
Zakaria
Ziani‡
 a,
Caterina
Bellatreccia‡
a,
Filippo Piero
Battaglia
a,
Giacomo
Morselli§
a,
Alessandro
Gradone
b,
Paola
Ceroni
*a and
Marco
Villa
*a
a,
Caterina
Bellatreccia‡
a,
Filippo Piero
Battaglia
a,
Giacomo
Morselli§
a,
Alessandro
Gradone
b,
Paola
Ceroni
*a and
Marco
Villa
*a
aDepartment of Chemistry Ciamician, University of Bologna, Via Selmi 2, 40126 Bologna, Italy. E-mail: marco.villa11@unibo.it
bIstituto per la Microelettronica ed i Microsistemi (IMM) – CNR Sede di Bologna, via Gobetti 101, 40129 Bologna, Italy
First published on 24th June 2024
Abstract
Azobenzene derivatives have long been studied for their photochromic behaviour. One of the greatest challenges in this field is the quantitative (E) to (Z) photoconversion triggered by visible light irradiation. In this work, the synthesis and characterization of CuInS2 quantum dots (CIS-QDs) appended with azobenzene units are reported: quantitative (E) → (Z) isomerisation is obtained by visible light (e.g., λex = 533 nm). Interestingly, catalytic amounts of CIS-QDs allow the full photoconversion of ungrafted (E)-azobenzene derivatives into the corresponding (Z)-isomers using visible light. This peculiar behaviour is associated with the direct complexation of the (Z)-isomer on the QD surface.
 Marco Villa | Marco Villa is a junior assistant professor at the University of Bologna, Italy. He received his PhD in 2018 through a joint agreement program between Aix-Marseille Université, under the supervision of Prof. Marc Gingras and the University of Bologna, with co-supervisor Prof. Paola Ceroni. After obtaining his doctoral degree, he returned to the University of Bologna, Italy, to work with Prof. Ceroni on a post-doctoral fellowship. His current work focuses on the synthesis and characterization of semiconducting nanomaterials and persulfurated aromatic compounds and the study of photocatalytic reactions. Villa's research has been recognized with the “Franco Scandola” Young Investigator Award by the Italian Photochemistry Group. |
Introduction
In 1959, Richard Feynman introduced his visionary miniaturization of objects at the nanoscale level during his talk, “There's Plenty of Room at the Bottom”.1 From his original dream, nanoscience has become a large and blossoming field in almost every discipline of modern science. As proof of this, Mongi G. Bawendi, Louis E. Brus and Alexei I. Ekimov were recently awarded the Nobel Prize for the discovery and development of quantum dots (QDs).QDs based on copper indium sulfide (CIS-QDs) are emerging as a valid alternative to conventional quantum dots based on II–VI semiconductors.2CIS-QDs have lower toxicity compared to the traditional cadmium or lead sulfide quantum dots and have excellent photophysical properties: (i) high molar absorption coefficients in the entire visible spectrum region (104–105 M−1 cm−1); (ii) high emission quantum yield, up to 70% in the red-NIR region of the spectrum (crucial for biological applications); and (iii) relatively long luminescence lifetimes (hundreds of nanoseconds).3–5 The chemical and photophysical properties of CIS-QDs can be tuned by ligand exchange, introducing appropriate capping agents to provide colloidal stability in solvents of different polarity and to protect the surface, preventing the formation of surface defects over time. Common capping agents are thiols,6 and, in this work, we will study the exchange of a single thiol (1-octanethiol) with a bidentate ligand appended to a photochromic azobenzene molecule.
Photochromic compounds exhibit a light-induced change in physico-chemical properties, not only in terms of absorption spectra, but also in geometry and polarity. They can be incorporated into systems of increasing complexity, from molecular, supramolecular and macromolecular systems up to biological systems and bulk materials, affecting the properties of the overall architecture.7–17
Azobenzene is one of the most popular molecular photoswitches because of the ease of synthesis, functionalization and its good fatigue resistance. (E) → (Z) isomerisation of the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond of azobenzene photoswitches is accompanied by a large change in properties such as geometry and polarity (e.g., the distance between the para carbon atoms of azobenzene decreases from 9 to 5.5 Å and the dipole moment increases from 0 to 3.0 D).13 In the case of unsubstituted azobenzene, the conversion from the (E)-isomer to the metastable (Z)-isomer is usually accomplished by π → π* excitation (e.g., λex = 365 nm), resulting in a photostationary state highly enriched in the (Z)-isomer (PSS365 nm = 91%). On the other hand, visible light excitation (e.g., λex = 436 nm) leads to a photostationary state containing only 14% of the (Z)-isomer.18 The development of efficient visible light operating photoswitches is crucial for a variety of applications from biology to materials science, but it still remains challenging. (E) → (Z) photoisomerization with visible light yielding a photostationary state with a high percentage of (Z)-isomer (>90%) has rarely been reported.19–21
N bond of azobenzene photoswitches is accompanied by a large change in properties such as geometry and polarity (e.g., the distance between the para carbon atoms of azobenzene decreases from 9 to 5.5 Å and the dipole moment increases from 0 to 3.0 D).13 In the case of unsubstituted azobenzene, the conversion from the (E)-isomer to the metastable (Z)-isomer is usually accomplished by π → π* excitation (e.g., λex = 365 nm), resulting in a photostationary state highly enriched in the (Z)-isomer (PSS365 nm = 91%). On the other hand, visible light excitation (e.g., λex = 436 nm) leads to a photostationary state containing only 14% of the (Z)-isomer.18 The development of efficient visible light operating photoswitches is crucial for a variety of applications from biology to materials science, but it still remains challenging. (E) → (Z) photoisomerization with visible light yielding a photostationary state with a high percentage of (Z)-isomer (>90%) has rarely been reported.19–21
Azobenzene has been grafted onto several classes of nanoparticles, both metallic and semiconducting ones, to induce electronic, optical and structural changes in the resulting nanomaterial. For example, a photoinduced electron transfer has been reported for CdS and CdTe quantum dots, with significantly effective quenching of the QD emission,22–24 while for Si nanocrystals, a photoinduced energy transfer from the Si nanocrystals to the azobenzene was observed, which resulted in both quenching of the Si-QD emission and a photosensitized (Z) → (E) conversion by direct excitation at 660 nm where azobenzene does not absorb.25
The coupling of azobenzene with CIS-QDs has not been reported in the literature. In the present paper, the synthesis, functionalization, and interaction of the organic azobenzene photoswitch and inorganic quantum dots are studied to evaluate two effects: the effect of azobenzene on the luminescence properties of the CIS-QDs and the effect of the CIS-QDs on the photochemical and thermal isomerization of azobenzene. This study has been performed for azobenzene units either grafted onto the quantum dot surface or dispersed in a solution of the CIS-QDs.
Experimental procedures
General procedures and instrumentation
All chemicals were purchased from Merck KGaA while solvents from ACS and were used without further purification. Water was purified by reverse osmometry with an Elga Purelab Classic purification system (13 MΩ cm). Size-exclusion chromatography (SEC) was performed over BioBeads™ S-X1 support (200–400 mesh) using chloroform as the eluent. The syntheses of the nanoparticles were performed by using a J-KEM Scientific Gemini 230 temperature controller with two stainless steel thermocouples connected to heating mantles. Nuclear magnetic resonance (1H-NMR and 13C-NMR) spectra were recorded on an ARX Varian INOVA 400 or a Bruker Advanced 600 MHz spectrometer in CDCl3. Chemical shifts were calibrated to the residual solvent peak.26 Coupling constant values (J) are given in hertz, chemical shifts (δ) in ppm and multiplicity data are reported as follows: s: singlet, d: doublet, and m: multiplet. High-resolution mass spectrometry (HRMS) analyses were conducted using a Waters Xevo G2-XS QTof spectrometer in positive electrospray ionization (ESI+) mode.HR-TEM and high angle annular dark field (HAADF)-STEM micrographs were recorded via a Tecnai F20T TEM FEI equipped with a 200 kV Schottky emitter. A chloroform solution of the material was deposited by drop casting on an Au TEM grid with a lacey ultrathin continuous carbon film. The solvent was evaporated at 80 °C for 10 min on a hot plate.
Powder XRD measurements were performed on a Panalytical X'Pert Pro powder diffractometer equipped with a Cu X-ray tube (Kα radiation, 1.54184 Å, 40 mA, 40 kV), with a Bragg–Brentano configuration and an X′celerator detector.
The elemental composition of CIS-QDs was determined via an Agilent 4210 MP-AES atomic emission spectrometer. For this purpose, 0.2 mL of the purified CIS-QDs was digested into 3 mL of 7 M nitric acid.
Photophysical measurements were carried out in air-equilibrated chloroform at 298 K. UV/vis absorbance spectra were recorded with a PerkinElmer λ650 or with a Cary 50 Bio spectrophotometer, using 1 cm Hellma quartz Suprasil cells or two-chamber Yankeelov cuvette. Kinetic studies of back thermal isomerization (Z) → (E) were performed on a Cary 50 Bio spectrophotometer equipped with a Julabo F12 temperature controller unit. Emission spectra were obtained with an Edinburgh FS5 instrument equipped with PMT980 and InGaAs detectors for visible and NIR spectral range observations, respectively. Correction of the emission spectra for detector sensitivity in the 550–1000 nm spectral region was performed using a calibrated lamp.27 Emission quantum yields were measured following the method of Demas and Crosby28 (standard used: [Ru(bpy)3]2+ in air-equilibrated aqueous solution, Φ = 0.0407).29 PL lifetime measurements in the range 0.5 ns to 1 μs were performed using an Edinburgh FLS920 spectrofluorometer equipped with a TCC900 card for data acquisition in time-correlated single-photon counting experiments (0.2 ns time resolution) with a 340 nm pulsed diode and an LDH-P-C-405 pulsed diode laser. The estimated experimental errors are as follows: 2 nm on the band maxima, 15% on the molar absorption coefficient and luminescence lifetime, and 10% on the fluorescence and photoisomerization quantum yields. The molar absorption coefficient of CIS-QDs (εCIS) was determined following the procedure reported by Booth et al. in 2012, which correlates the photophysical properties of CIS-QDs to their dimension.30 The band gap of pristine CIS-QDs as well as that of CIS@n_Azo was determined through the second derivative method.30 Irradiation experiments were performed in air-equilibrated solution. The concentrations used for UV/vis and NMR spectroscopy were classically ∼10−5 M and ∼10−3 M, respectively, unless otherwise stated. The samples were kept at room temperature during the irradiation. NMR tube irradiation was performed with a 365 nm Kessil lamp (40 W) and UV/vis cells were irradiated using a mid-pressure Hg lamp equipped with a 365 or 436 nm band pass-filter (Edmund Optics or Andover Corporation) or with LEDs from LED Engin centered at 365 nm (12 W) or 533 nm (3 W) powered with an AimTTi EX355R power supply. The isomerization process was monitored either by UV/vis or by 1H NMR. The PSS was determined by integration of residual 1H NMR peaks. Quantum yields for the photoinduced isomerization process (φ(E)→(Z) and φ(Z)→(E)) were determined by illumination of solutions of (E) and (Z)-isomers at room temperature using monochromatic light (i.e., λex = 365 and 436 nm). Typically, samples were placed 5 cm from the irradiation source. The photon flux was determined using a ferrioxalate actinometer. Quantum yields were then determined by fitting the results using the photo-kinetic model reported by Maafi and Brown.31,32
Synthesis of 4 nm CuInS2 quantum dots (CIS-QDs)
Copper(I) iodide (0.25 mmol) and indium(III) acetate (0.25 mmol) were suspended in a mixture of 1-octanethiol (4 mL) and 1-octadecene (6 mL) in a flushed three-necked round-bottomed flask equipped with a condenser. The temperature was raised to 150 °C until complete dissolution of the solids and then it was increased again to 210 °C. The solution was stirred continuously for 1.5 h before quenching it by dipping the flask in a cold-water bath. Aliquots (6 × 1 mL) of the reaction mixture were diluted with chloroform (6 × 1 mL) and methanol (6 × 40 mL) was added until complete precipitation of the nanoparticles. The suspensions were centrifuged (2 times, 8000 rpm, 10 minutes each) and the supernatants were discarded. The precipitates were dissolved in chloroform (10 mL) yielding a colloidally stable dispersion of CIS-QDs.Synthesis of AzoLA
To a mixture of 4-aminoazobenzene (500 mg, 2.53 mmol) with α-lipoic acid (628 mg, 3.04 mmol, 1.2 equiv.), and DMAP (618 mg, 5 mmol, 2 equiv.) in dichloromethane (25 mL), DCC (1.04 g, 5 mmol, 2 equiv.) was added. The solution was stirred at room temperature for 24 h. The reaction mixture was filtered and the solid was washed with 1 M HCl (2 × 30 mL), 1 M NaOH (2 × 30 mL), and brine (30 mL), dried over Na2SO4, and filtered and evaporated under reduced pressure. The crude product was purified by recrystallisation in ethanol to afford the desired compound as an orange powder (620 mg, 1.59 mmol) with 63% yield. 1H NMR (400 MHz, chloroform-d) δ: 8.06–7.82 (m, 4H), 7.69 (d, J = 8.6 Hz, 2H), 7.61–7.40 (m, 3H), 7.30 (s, 1H), 3.64–3.52 (m, 1H), 3.24–3.05 (m, 2H), 2.55–2.33 (m, 3H), 2.02–1.87 (m, 1H), 1.87–1.70 (m, 4H), 1.65–1.46 (m, 2H). 13C NMR (150 MHz, chloroform-d) δ: 171.21, 152.80, 149.13, 140.54, 130.92, 129.22, 124.16, 122.88, 119.85, 56.52, 40.41, 38.64, 37.68, 34.78, 29.00, 25.28.Synthesis of AzoDHLA
To a solution of AzoLA (100 mg, 0.26 mmol) in a mixture of MeOH (3 mL) and THF (3 mL), NaBH4 (118 mg, 3.1 mmol, 12 equiv.) was slowly added at 0 °C. The resulting suspension was stirred for 1.5 h before being quenched with 1 M HCl (5 mL). The resulting mixture was washed with toluene (2 × 30 mL), dried over Na2SO4, filtered and evaporated under reduced pressure. The desired compound was obtained as an orange powder (97 mg, 0.25 mmol) with 96% yield. Caution: Note that this compound is not stable over time due to the oxidation of the thiol moieties and should be used immediately after this synthetic step. 1H NMR (400 MHz, chloroform-d) δ: 7.99–7.82 (m, 4H), 7.69 (d, J = 8.5 Hz, 2H), 7.57–7.42 (m, 3H), 7.36 (s, 1H), 3.04–2.87 (m, 1H), 2.83–2.57 (m, 2H), 2.53–2.37 (m, 2H), 2.05–1.86 (m, 1H), 1.84–1.62 (m, 5H), 1.58–1.46 (m, 2H), 1.46–1.20 (m, 2H). 13C NMR (100 MHz, chloroform-d) δ: 171.20, 152.80, 140.52, 130.92, 129.22, 124.18, 122.88, 119.80, 42.95, 39.46, 38.91, 37.78, 26.79, 25.16, 22.45, 21.91.Synthesis of AzoAA33
4-Aminoazobenzene (1 g, 5 mmol) was dissolved in acetic anhydride (12 mL) and stirred for 24 hours. The precipitated solid was filtered and was dissolved in chloroform (20 mL). This solution was washed with 1M HCl (2 × 30 mL), 1 M NaOH (2 × 30 mL), and brine (30 mL), dried over Na2SO4, and filtered and evaporated under reduced pressure. The product was obtained as an orange solid (976 mg, 4 mmol, 81%).1 H NMR (600 MHz, chloroform-d) δ: 7.95–7.87 (m, 4H), 7.68 (d, J = 8.3 Hz, 2H), 7.53–7.48 (m, 2H), 7.48–7.44 (m, 1H), 7.32 (s, 1H), 2.23 (s, 3H).
General method for the synthesis of CIS@n_Azo batches
To a solution of CIS-QDs in chloroform (1 mL, 0.115 μM), a 0.1 M solution of AzoDHLA in chloroform was added. The operation was repeated for 6 different samples, increasing the volume added (see Table S1†) for each. The resulting solutions were stirred and kept under an inert atmosphere at 50 °C for 16 h. The reaction mixtures were concentrated and purified by size exclusion chromatography using chloroform as the eluent. The purified solutions of CIS@n_Azo were then heated in the dark at 40 °C for 48 h to ensure that any potentially formed (Z)-AzoDHLA grafted onto the surfaces of QDs returned to the thermodynamically stable (E)-AzoDHLA isomer.Results and discussion
Synthetic procedures
Copper indium sulfide quantum dots (CuInS2, CIS-QDs) were synthesized according to a heating-up procedure adapted from the literature.3CIS-QDs were capped with 1-octanethiol to ensure good stability of the colloidal suspension in organic solvent (CHCl3). AzoDHLA was obtained by amide coupling and reduction from p-aminoazobenzene (AzoNH2), as depicted in Scheme 1 (see also the Experimental section). AzoAA was synthesized as a model compound according to a slightly modified procedure reported in the literature.33 Hybrids of azobenzene/CIS-QDs with different loadings of photochromic units on the surface (CIS@n_Azo) were prepared by ligand exchange of the native octanethiol ligands with AzoDHLA (see also the Experimental section).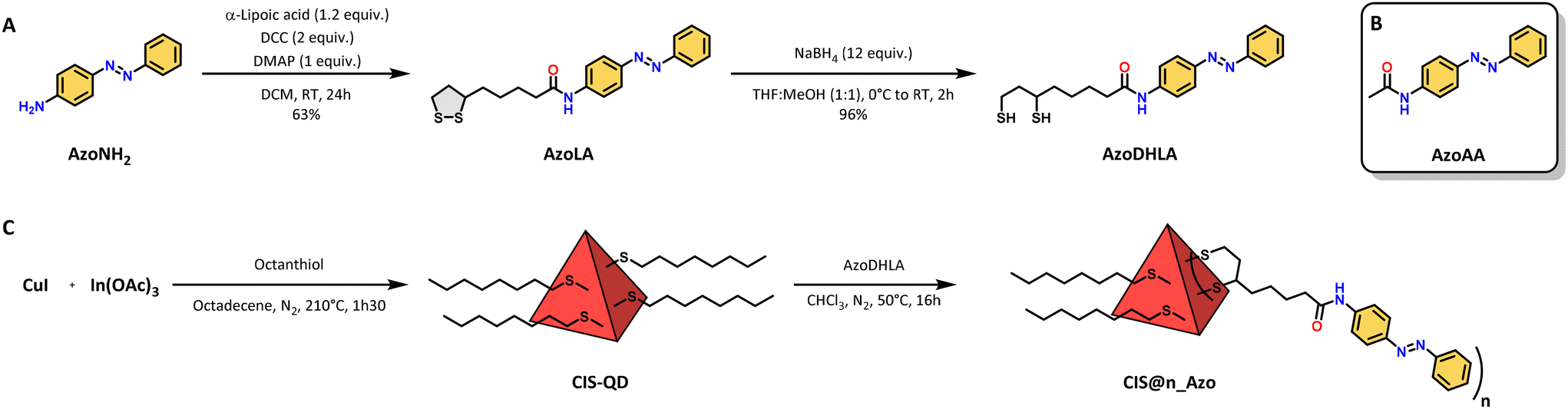 | ||
| Scheme 1 Synthetic representation of the adopted methodology for preparation of hybrids of CIS-QDs/azobenzene. (A) Synthetic procedure for the preparation of AzoLA and AzoDHLA. (B) Representation of the chemical structure of the model compound AzoAA. (C) Heating-up synthesis for the preparation of CIS-QDs capped with 1-octanethiol ligands and ligand-exchange procedure for production of CIS@n_Azo hybrids. | ||
Structural and morphological behaviour
HR-TEM and STEM analyses of CIS-QDs were carried out in order to determine the size and structure of the crystalline core. HRTEM depicted in Fig. S9† displays the morphology of the CIS nanoparticles. The crystalline character of the material is compatible with that of a chalcopyrite phase, observed with greater ease through fast Fourier transform (every FFT refers to a single particle, highlighted in the image by the square dotted in yellow). XRD spectroscopy confirms the chalcopyrite phase of CuInS2, as reported in the literature (Fig. S11†).34–36 In Fig. S10 (a and b)† HAADF–STEM micrographs at different magnifications confirm the observations in the TEM micrographs and enhance the contrast with respect to the carbon thin film due to the difference in atomic weight. By doing this, it is possible to emphasize the triangular shape of the particles, which is characteristic of the chalcopyrite phase, and define the average size of the particles (4 nm). The composition of the quantum dots has been determined by atomic emission spectrometry (MP AES) of digested samples (see the Experimental section). As predicted, the Cu![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :In ratio is 1:1.
:In ratio is 1:1.
Photophysical properties
The absorption spectra of pristine CIS-QDs and their hybrids display an ill-defined absorption tail extending up to 700 nm, characteristic of semiconductor nanoparticles (Fig. 1A and B).37CIS@n_(E)-Azo exhibits additional features in the 270–480 nm range in comparison with an azobenzene model compound, which are ascribed to the presence of azobenzene units. In particular, AzoLA was selected as the model azobenzene compound because the (E)-isomer has the same absorption spectrum as (E)-AzoDHLA (see Fig. S10†) and it is stable in air-equilibrated solution, as opposed to AzoDHLA that undergoes oxidation of the dithiol moiety.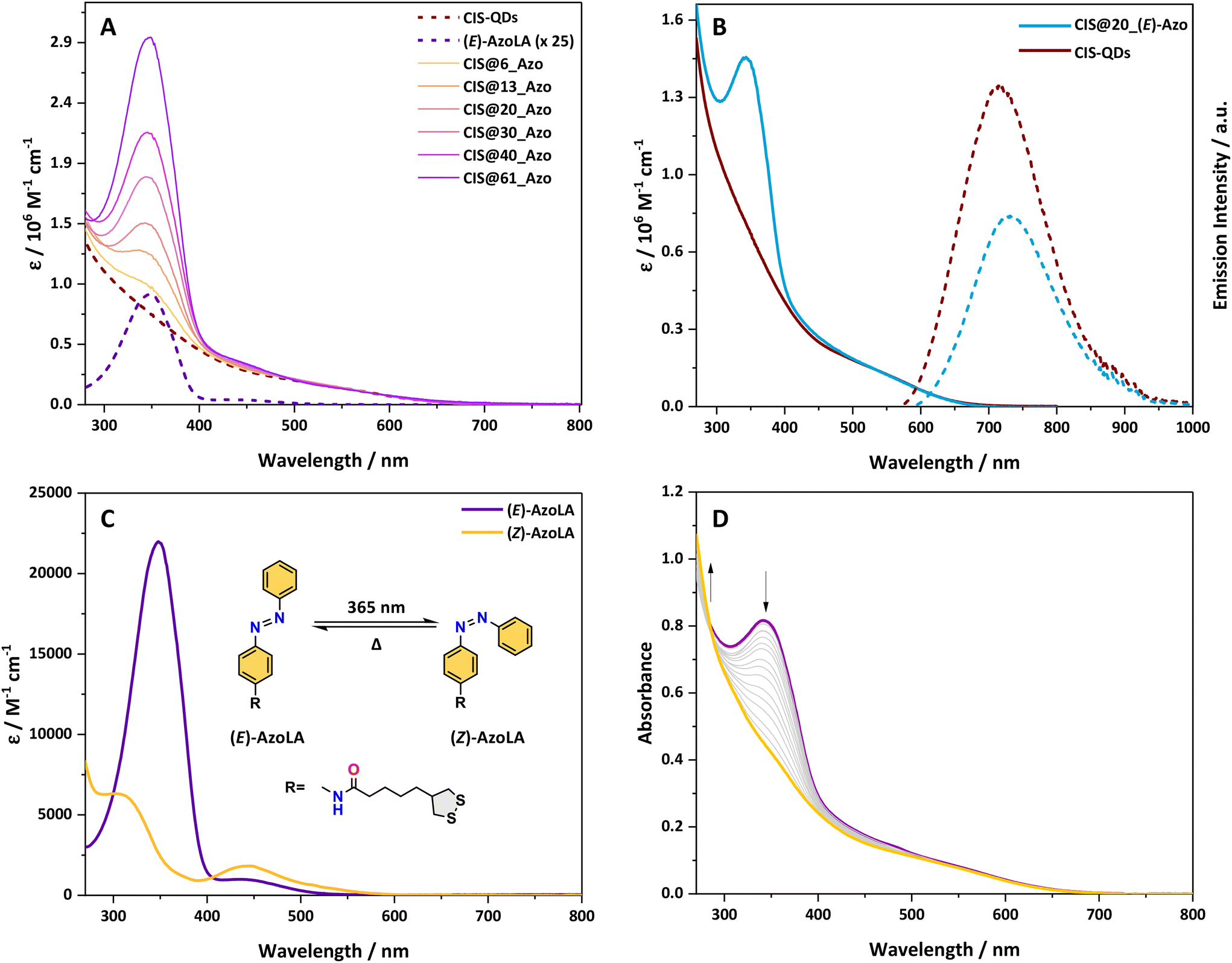 | ||
| Fig. 1 (A) Absorption spectra of CIS-QDs, (E)-AzoLA (ε magnified by 25 times) and CIS@n_(E)-Azo hybrids in chloroform. (B) Absorption (solid lines) and emission spectra (dashed lines) of CIS-QDs (red) and CIS@20_(E)-Azo (blue); λex = 548 nm. (C) Absorption spectra of (E)-AzoLA (purple) and (Z)-AzoLA (yellow) in chloroform. The isomerisation process of AzoLA is depicted in the inset. (D) Absorption spectral evolution of a solution of CIS@20_(E)-Azo in chloroform (7.3 × 10−7 M) during irradiation at λex = 365 nm. | ||
The absorption spectrum of (E)-AzoLA displays a π → π* band at 348 nm and an n → π* band at 442 nm, which matches well with the absorption peaks observed in CIS@n_(E)-Azo (Fig. 1A). The bathochromic shift of the π → π* band and the increased overlap between the two transitions observed in the (E)-AzoLA absorption spectrum compared to normal azobenzene are in accordance with previously reported results for amide-substituted azobenzene.38,39
The absorption spectrum of CIS@n_(E)-Azo matches well with the sum of the absorption spectra of CIS-QDs and (E)-AzoLA. The molar absorption coefficient of CIS-QDs (εCIS) was determined following the procedure reported in the literature,30 which correlates the photophysical properties of the QDs to their dimensions (for more details, see the ESI†). Knowing the molar absorption coefficients of CIS-QDs and (E)-AzoLA, the average number of azobenzenes per QD was evaluated (see the ESI†). For example, the absorption spectrum reported in Fig. 1B corresponds to a sample with an average of 20 azobenzene units per QD, also named CIS@20_(E)-Azo.
A larger number of photochromic units grafted onto the surface of CIS-QDs was obtained by increasing the concentrations of AzoDHLA used in the ligand exchange (see Scheme 1 and Table S1†): the absorbance of CIS@n_(E)-Azo at 348 nm (Fig. 1A) shows an increase in the band at 348 nm for samples containing from 6 up to 61 azobenzene units.
The band gap of pristine CIS-QDs as well as that of CIS@n_(E)-Azo was determined through the second derivative method.30 We estimated a similar band gap (2.3 eV) for the QDs before and after ligand exchange, indicating that the photoswitchable moiety does not affect the band position of the nanocrystal core (see Fig. S9†).
The emission of pristine CIS-QDs displays a wide peak centred at 717 nm (FWHM = 134 nm) upon excitation with visible light (i.e., 548 nm; see Fig. 1B). The photoluminescence quantum yield (PLQY) is 12%. A biexponential function was used to fit the emission decay of CIS-QDs (see Fig. S28†), yielding two components: τ1 = 87 ns and τ2 = 352 ns (τave = 219 ns), similar to the values previously reported in the literature.3
CIS@n_(E)-Azo shows the typical emission of CIS-QDs, although the emission peak is red-shifted around 730 nm for all batches (Table 1). Additionally, CIS@n_(E)-Azo batches exhibit reduced PLQY and lifetime values compared to those of pristine CIS-QDs (Table 1). The same effect is observed for CIS-QDs fully capped with dihydrolipoic acid (DHLA), which show a PLQY of 0.1% (see Fig. S27†).
| λ em/nm | FWHM/nm | PLQYa/% | τ ave /ns | |
|---|---|---|---|---|
| a Excitation at 548 nm. Determined by actinometry. b Weighted average lifetime, excitation at 405 nm. Emission detection at 730 nm. c No emission detected. | ||||
| CIS-QDs | 717 | 134 | 12 | 219 |
| (E)-AzoLA | —c | —c | —c | —c |
| CIS@6_(E)Azo | 730 | 128 | 6 | 165 |
| CIS@13_(E)Azo | 733 | 128 | 7 | 130 |
| CI@20_(E)Azo | 731 | 127 | 6 | 154 |
| CIS@30_(E)Azo | 733 | 123 | 4 | 110 |
| CIS@40_(E)Azo | 737 | 123 | 3 | 127 |
| CIS@61_(E)Azo | 732 | 124 | 2 | 77 |
The ligand-exchange procedure alters the local environment of the emitting cores and introduces additional surface defects on them. Therefore, the drop in PLQY and lifetime is most likely due to these effects, which is consistent with earlier studies on functionalized QDs.3,25,40 In QD–azobenzene systems (such as CdS, CdTe, and Si), there is a significant quenching of the QD emission, by up to 16 times, while for CIS-QDs there is no quenching due to energy transfer or electron transfer. This phenomenon likely arises from the unique nature of the emission in CIS-QDs, which is related to Cu trap states rather than the band-gap transitions observed in other QDs.22–25
The photophysical properties of CIS@n_(Z)-Azo were also studied: both PLQY values and the emission lifetimes of all the hybrids do not change significantly according to the isomerisation state of the photoswitch (see Table 1 and Table S2†).
Photochromic behaviour
Due to low stability in solution of samples substituted with a large number of azobenzene units (CIS@30_Azo, CIS@40_Azo and CIS@61_Azo), our photochemical study was limited to QDs substituted with 6, 13 and 20 photoswitchable units.The possibility of photoisomerization of (E)-AzoLA to its corresponding (Z)-AzoLA isomer was demonstrated by absorption spectroscopy in chloroform solution. Upon irradiation at 365 nm, the absorption spectra (Fig. 1C) show a decrease in the π → π* band at 348 nm and the concomitant increase in a new π → π* band at higher energy (λ = 307 nm) attributed to the photoinduced formation of (Z)-AzoLA. At the same time, we observed an increase in the n → π* band at 438 nm.
The photoisomerization reaction was also confirmed by NMR spectroscopy: when a solution of (E)-AzoLA in deuterated chloroform (∼10−3 M) was irradiated at 365 nm, NMR signals of the starting compound were progressively replaced by a new set of peaks at lower field, in accordance with the (E) → (Z) isomerisation of the NN bond (see Fig. S7†). The photostationary state (PSS) at λex = 365 nm was estimated to be 98% of (Z)-isomer by integration of the residual 1H NMR peaks of the (E)-AzoLA.
The photochemical reversibility was then evaluated: upon visible irradiation (λex = 436 or 533 nm) of a (Z)-AzoLA solution in chloroform, the absorption spectrum of the (E)-AzoLA was partially recovered, demonstrating a non-quantitative photoconversion. The PSS was estimated to contain 50% and 92% of (E)-isomer when irradiated at λex = 436 and 533 nm, respectively.
The corresponding photoisomerization quantum yields (Table 2) were determined by fitting the results with the photo-kinetic model reported in the literature:31,32Φ(E)→(Z) = 35.4% and Φ(E)→(Z) = 97% were found upon irradiation at 365 and 436 nm, respectively. The much larger photoisomerization quantum yield obtained upon excitation of the n → π* transition compared to the excitation of the π → π* transition has already been documented, and theoretical calculations have been used to explain it.41–43 The quantum yield of the (Z) → (E) photoisomerization was found to be Φ(Z)→(E) = 39% at λex = 436 nm.
Upon irradiation of chloroform solutions of CIS@n_(E)-Azo at 365 nm, where n = 6, 13 or 20, a decrease in the π → π* band was observed, but unlike the parent AzoLA, no increase in the n → π* band was observed (Fig. 1D). This behaviour suggests that the presence of CIS-QDs interferes with the n → π* transition of (Z)-AzoLA. It is worth noting that this effect has been reported with other hybrids based on azobenzene and various nanoparticles (AuNPs and AgNPs), but no conclusive explanations were reported.24,44–47
The photoconversion from (E) to (Z) was estimated to be quantitative when CIS@n_(E)-Azo hybrids were exposed to UV light (λex = 365 nm) and the isomerization quantum yields of Φ(E)→(Z) ≈ 19% were measured (see Table 2). Such a decrease in Φ(E)→(Z) between CIS@n_Azo and AzoLA (−86%) can be attributed to the different environments of the azobenzene molecules when they are in solution or in proximity to the QDs. In fact, the isomerisation quantum yields of azobenzene photoswitches are highly dependent on their environments (polarity, viscosity, solvent, etc.).43,48,49
Photoinduced back isomerization was investigated on CIS@n_(Z)-Azo. Upon excitation with visible light at 436 or 533 nm, no change in the absorption spectra was observed. It should be noted that this result is notably different from that for (Z)-AzoLA when irradiated at λex = 436 and 533 nm (PSS436 nm = 50% and PSS533 nm = 92% of (E)-isomer). It is also worth noting that any sensitisation process from CIS-QDs to the triplet state of AzoLA has been excluded, since irradiation of CIS@n_(E)-Azo or CIS@n_(Z)-Azo at 650 nm (where only the inorganic core absorbs) did not cause any changes in the absorption spectra of the hybrids. The lack of back photoconversion under visible light could be explained by two main hypotheses: (i) the excited state of grafted (Z)-AzoDHLA is efficiently quenched by the QDs (unlikely, due to the sub-nanosecond excited-state lifetime)13 or (ii) (Z)-AzoDHLA on the surface of the QDs does not absorb light in the visible region.
As a consequence of the previously mentioned effect, excitation of CIS@n_(E)-Azo hybrids by visible light (i.e., λex = 436 or 533 nm) led to a quantitative (E) to (Z) photoisomerization with Φ(E)→(Z) ≈ 37% at λex = 436 nm (see Table 2). Note that the PSS values reached at λex = 436 nm and 533 nm were 50% and 8% of (Z)-isomer, respectively, for AzoLA. The presence of CIS-QDs drastically affects the photoisomerization, changing the photostationary state to a complete conversion to the (Z)-AzoLA isomer. These findings suggest that the photoisomerization is moved to a new photostationary state by the presence of CIS-QDs. The quantitative PSS for (E) to (Z) photoisomerization in the visible range reported here is a notable result. The few previously reported examples of (E) to (Z) photoisomerization by visible light irradiation employ an external photosensitizer or involve the presence of substituents on the azobenzene core.50,51
Heating the CIS@n_(Z)-Azo solution led to the thermal recovery of the CIS@n_(E)-Azo isomer despite photo-triggered back isomerization not being achieved. Kinetic constants of kiso ≈ 1.1 × 10−3 s−1 and kiso = 4.9 × 10−4 s−1 were measured for CIS@n_(Z)-Azo and the free ligand (Z)-AzoLA, respectively (at 70 °C in DMF, Fig. S44†). It is important to note that the amount of azobenzene on the surface of the CIS-QDs does not have any significant effects on kiso. Thermal back isomerization of CIS@n_(Z)-Azo is about twice as fast compared to that of (Z)-AzoLA. This phenomenon has been already reported for hybrids of AuNPs/azobenzene and SiNPs/azobenzene.25,52,53
The remarkable differences in the photochromic behaviour between CIS@n_Azo and the free ligand AzoLA suggest that CIS-QDs are non-innocent in the photochemical processes of the grafted photoswitchable unit.
It is worth noting that previously reported QD–azobenzene systems (such as CdS, CdTe, and Si) focused on the significant quenching of the QD emission upon functionalisation with the chromophore.22,23,25 Only a few cases have observed a change in behaviour of the photochromic units, mainly related to energy or electron transfer favouring the back thermal isomerisation.25,52,53 On the other hand, our system demonstrated a remarkable change in the photoisomerisation behaviour of the dye not related to sensitisation processes, which has never been reported for other systems.
Photochemical behaviour of ungrafted azobenzene chromophores in the presence of CIS-QDs
Based on the previously discussed results, we decided to investigate the interaction of CIS-QDs with free azobenzene derivatives in solution. For this purpose, chloroform solutions of (E)-AzoLA (∼10−5 M) and CIS-QDs (∼10−6–10−7 M) were placed in two separate compartments of a two-chamber Yankeelov cuvette. The latter was irradiated at λex = 533 nm from the QD side. When the PSS was reached, the solutions were mixed, and then irradiated again at λex = 533 nm. The absorption spectrum revealed that the initial PSS containing 8% of (Z)-isomer shifted to a full conversion (100% of (Z)-isomer) after mixing (see Fig. 2A). These results indicate that pre-grafting on the surface of the QDs is not necessary. To rule out possible light-induced grafting of AzoLA on the surfaces of the QDs, the same experiment was repeated with the model azobenzene AzoAA (Scheme 1B), which does not possess 1,2-dithiolane units. We measured a PSS of 28% of (Z)-isomer at 533 nm before mixing the two compartments and total photoconversion to the (Z)-isomer after shaking (see Fig. S21†). Surface grafting of CIS-QDs with a dithiol linker is not required and the same singular photoisomerization properties are observed.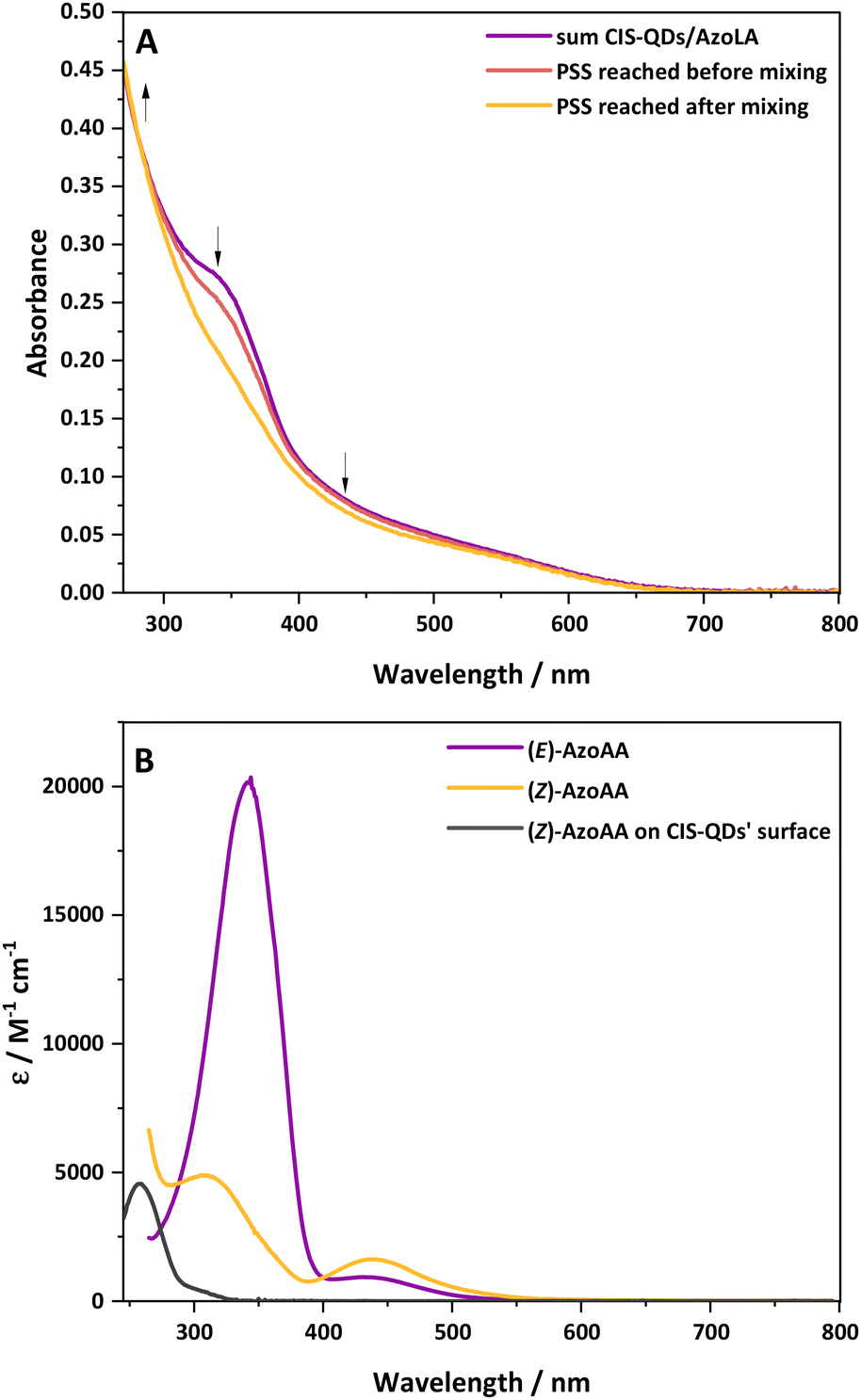 | ||
| Fig. 2 (A) Absorption spectra of solutions of CIS-QDs (2.26 × 10−7 M) and AzoLA (1.25 × 10−5 M) in chloroform, recorded using a two-chamber Yankeelov cuvette. Before mixing and irradiation at 533 nm (purple), before mixing and after irradiation at 533 nm to reach PSS (red) and after mixing and further irradiation at 533 nm to achieve PSS (yellow). (B) Absorption spectra of solutions of (E)-AzoAA (purple), (Z)-AzoAA (yellow) and spectrum of (Z)-AzoAA on the surfaces of CIS-QDs (black, obtained by subtraction of the absorption of CIS-QDs from the absorption spectrum of CIS ⊂ (Z)-AzoAA). | ||
These findings, together with the lack of an n → π* band arising during the photoisomerization of CIS@n_(E)-Azo, led us to consider the possibility of a direct interaction between the azobenzene core and CIS-QDs.
The interaction between the azobenzene core and surface of CIS-QDs was demonstrated by comparison of the absorption spectrum of (Z)-AzoAA with or without CIS-QDs (Fig. 2B). The former (black line in Fig. 2B) was obtained by subtraction of the absorbance of QDs from the absorption spectrum of a solution of CIS-QDs and (Z)-AzoAA, assuming that the absorbance of the QDs does not change significantly in the presence of the azobenzene model compound. (Z)-AzoAA on the surface of CIS-QDs displays a narrow band centred at 257 nm (ε = ∼4600 L mol−1 cm−1) and no absorption features at wavelengths higher than 300 nm. Therefore, the optical properties of (Z)-AzoAA are strongly modified on the surfaces of the QDs. These results confirm the hypothesis of a direct interaction between the azobenzene moiety and the core of the QDs: a plausible explanation is an interaction of the nitrogen lone pairs with the metal ions at the surface of the QD, a hypothesis corroborated by the decrease in the n → π* transition. A new species (CIS ⊂ (Z)-AzoAA, Scheme 2) is therefore formed, which can account for the peculiar properties observed.
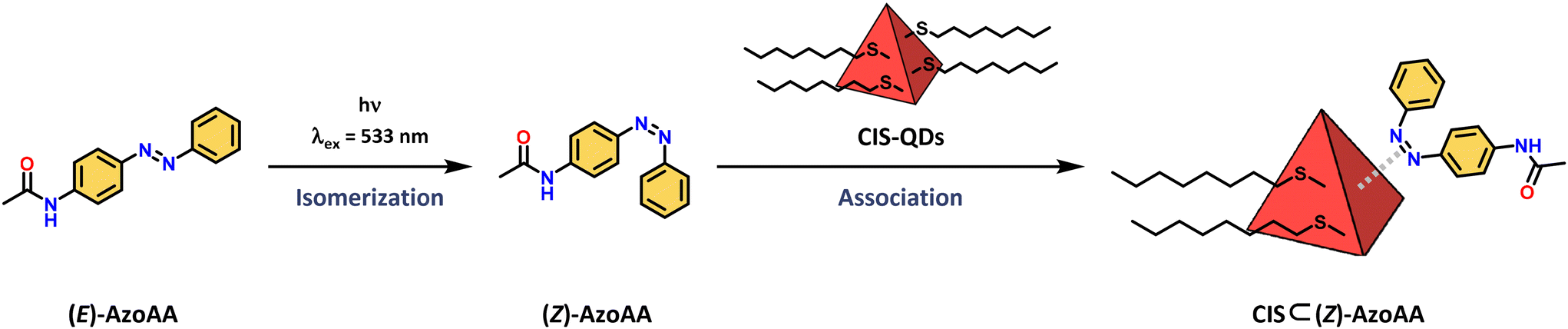 | ||
| Scheme 2 Schematic representation of isomerization of (E)-AzoAA to (Z)-AzoAA followed by the association to CIS-QDs yielding quantitatively CIS ⊂ (Z)-AzoAA. | ||
The formation of CIS ⊂ (Z)-AzoAA was also demonstrated through NMR spectroscopy. Upon irradiation at λexc = 365 nm of (E)-AzoAA solution (1 × 10−4 M), the characteristic signature of (Z)-AzoAA at 6.86 ppm was observed according to what was explained in the previous sections (Fig. S46†). After 10 minutes of irradiation, 0.1 equivalent of CIS-QDs was added to the solution, and a new set of peaks arose around 7.2 ppm, while the peaks of (Z)-AzoAA decreased. The new signals are ascribed to the formation of CIS ⊂ (Z)-AzoAA (Fig. S46†). After equilibration of the solution, further irradiation led to the conversion of (E)-AzoAA to (Z)-AzoAA, confirming that the presence of CIS-QDs is affecting the PSS.
Therefore, NMR results confirm that (Z)-AzoAA interacts with CIS-QDs. No such effect has been observed with (E)-AzoAA, demonstrating that CIS-QDs interact exclusively with the (Z)-isomer (Fig. S47†).
The absence of any absorption features in the visible spectrum of (Z)-AzoAA on the surfaces of the QDs suggests that (Z) → (E) photoisomerisation is prevented upon irradiation at λex ≥ 300 nm. It should be noted that excitation at λex = 254 nm led to photodegradation of the complex without any evidence of (E)-AzoAA restoration. No interaction has been observed between CIS-QDs and (E)-AzoAA because no differences have been observed before and after mixing solutions of the previously mentioned compounds (see Fig. S22†). Therefore, (E)-AzoAA can isomerise to its (Z)-isomer under visible light irradiation even in the presence of CIS-QDs, while the reverse process is not possible photochemically. Indeed, the (E)-isomer configuration does not favour a direct interaction on the QD surface.
The interaction between CIS-QDs and the studied azobenzene chromophores can thus account for the quantitative (E) → (Z) photoconversion observed.
Conclusions
We have successfully grafted azobenzene derivatives onto the surface of copper indium sulfide quantum dots. The photophysical properties of the quantum dots are affected by this functionalization: a decrease in lifetime and emission quantum yield is observed, and it is more pronounced with an increase in the average number of azobenzene units grafted onto the QD surface. The interaction between azobenzene and QDs results in an even more pronounced effect on the photochemistry of azobenzene. Classically, (E) → (Z) photoisomerization of azobenzene chromophores is achieved by UV excitation of the π–π* band. Here, we report one of the first examples of complete (E) to (Z) photoisomerization triggered by visible light irradiation of the n–π* band (in the green and blue regions). These results are observed not only for azobenzene grafted onto surfaces of CIS-QDs, but also for ungrafted chromophores, physically mixed with CIS-QDs. We demonstrated that the (Z)-AzoAA isomer in the presence of CIS-QDs leads to a new species CIS ⊂ (Z)-AzoAA with significantly different absorption properties with respect to the (Z)-AzoAA isomer in solution.The reported results are of the utmost importance with respect to tuning the photochemical properties of azobenzene derivatives in solution, forcing a complete isomerization caused by a strong interaction between the dye and the nanoparticle. This effect could be a pillar for new light-induced complexation on nanoparticles and light-triggered ligand exchange.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The University of Bologna and the European Union's Horizon 2020 research and innovation program under grant agreement no. 101006839 (CONDOR) are gratefully acknowledged for financial support. Z. Z. and P. C. acknowledge the National Recovery and Resilience Plan (NRRP), Mission 4 Component 2 Investment 1.3 – Call for tender no. 1561 of 11.10.2022 from the Ministero dell'Università e della Ricerca (MUR), funded by the European Union – NextGenerationEU. A. G acknowledge the Project funded by the European Union – NextGenerationEU under the National Recovery and Resilience Plan project IR0000027, CUP: B33C22000710006 – iENTRANCE@ENL: Infrastructure for Energy TRAnsition and Circular Economy @ EuroNanoLab. A. G acknowledge the Project funded by Horizon Europe Grant agreement ID: 101094299 – IMPRESS | Interoperable electron Microscopy Platform for advanced Research and Services.References
- A. Junk and F. Riess, Am. J. Phys., 2006, 74, 825–830 CrossRef CAS.
- C. B. Murray, D. J. Norris and M. G. Bawendi, J. Am. Chem. Soc., 1993, 115, 8706–8715 CrossRef CAS.
- G. Morselli, A. Gradone, V. Morandi and P. Ceroni, Nanoscale, 2022, 14, 3013–3019 RSC.
- H. Zhong, Z. Bai and B. Zou, J. Phys. Chem. Lett., 2012, 3, 3167–3175 CrossRef CAS PubMed.
- A. D. P. Leach and J. E. Macdonald, J. Phys. Chem. Lett., 2016, 7, 572–583 CrossRef CAS PubMed.
- J. Kolny-Olesiak and H. Weller, ACS Appl. Mater. Interfaces, 2013, 5, 12221–12237 CrossRef CAS PubMed.
- S. Kobatake, S. Takami, H. Muto, T. Ishikawa and M. Irie, Nature, 2007, 446, 778–781 CrossRef CAS PubMed.
- W. Szymański, J. M. Beierle, H. A. V. Kistemaker, W. A. Velema and B. L. Feringa, Chem. Rev., 2013, 113, 6114–6178 CrossRef PubMed.
- Y.-J. Zheng, Y.-F. Shi, C.-B. Tian, H. Lin, L.-M. Wu, X.-T. Wu and Q.-L. Zhu, Chem. Commun., 2019, 55, 79–82 RSC.
- M. Baroncini, S. Silvi and A. Credi, Chem. Rev., 2020, 120, 200–268 CrossRef CAS PubMed.
- Z. Ziani, F. Loiseau, E. Lognon, M. Boggio-Pasqua, C. Philouze, S. Cobo and G. Royal, Chem. Eur. J., 2021, 27, 16642–16653 CrossRef CAS PubMed.
- D. M. Junge, D. V. McGrath and de B. den Berg, J. Chem. Soc., 1996, 97, 4912–4913 Search PubMed.
- H. M. D. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- M. Villa, G. Bergamini, P. Ceroni and M. Baroncini, Chem. Commun., 2019, 55, 11860–11863 RSC.
- M. Baroncini, S. d'Agostino, G. Bergamini, P. Ceroni, A. Comotti, P. Sozzani, I. Bassanetti, F. Grepioni, T. M. Hernandez, S. Silvi, M. Venturi and A. Credi, Nat. Chem., 2015, 7, 634–640 CrossRef CAS PubMed.
- F. Vögtle, M. Gorka, R. Hesse, P. Ceroni, M. Maestri and V. Balzani, Photochem. Photobiol. Sci., 2002, 1, 45–51 CrossRef PubMed.
- Z. Ziani, S. Cobo, F. Loiseau, D. Jouvenot, E. Lognon, M. Boggio-Pasqua and G. Royal, JACS Au, 2023, 3, 131–142 CrossRef CAS PubMed.
- E. Fischer, M. Frankel and R. Wolovsky, J. Chem. Phys., 1955, 23, 1367–1367 CrossRef CAS.
- J. Volarić, J. Buter, A. M. Schulte, K.-O. van den Berg, E. Santamaría-Aranda, W. Szymanski and B. L. Feringa, J. Org. Chem., 2022, 87, 14319–14333 CrossRef PubMed.
- D. Bléger, J. Schwarz, A. M. Brouwer and S. Hecht, J. Am. Chem. Soc., 2012, 134, 20597–20600 CrossRef PubMed.
- W. A. Velema, W. Szymanski and B. L. Feringa, J. Am. Chem. Soc., 2014, 136, 2178–2191 CrossRef CAS PubMed.
- S. Saeed, J. Yin, M. A. Khalid, P. A. Channar, G. Shabir, A. Saeed, M. A. Nadeem, C. Soci and A. Iqbal, J. Photochem. Photobiol., A, 2019, 375, 48–53 CrossRef CAS.
- H. Javed, K. Fatima, Z. Akhter, M. A. Nadeem, M. Siddiq and A. Iqbal, Proc. R. Soc. Lond. A. Math. Phys. Sci., 2016, 472, 20150692 Search PubMed.
- D. Hu, J. Lin, S. Jin, Y. Hu, W. Wang, R. Wang and B. Yang, Mater. Chem. Phys., 2016, 170, 108–112 CrossRef CAS.
- M. Villa, S. Angeloni, A. Bianco, A. Gradone, V. Morandi and P. Ceroni, Nanoscale, 2021, 13, 12460–12465 RSC.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- M. Montalti, A. Credi, L. Prodi and M. T. Gandolfi, Handbook of Photochemistry, CRC Press, 2006 Search PubMed.
- G. A. Crosby and J. N. Demas, J. Phys. Chem., 1971, 75, 991–1024 CrossRef CAS.
- K. Suzuki, A. Kobayashi, S. Kaneko, K. Takehira, T. Yoshihara, H. Ishida, Y. Shiina, S. Oishi and S. Tobita, Phys. Chem. Chem. Phys., 2009, 11, 9850–9860 RSC.
- M. Booth, A. P. Brown, S. D. Evans and K. Critchley, Chem. Mater., 2012, 24, 2064–2070 CrossRef CAS.
- M. Maafi and R. G. Brown, J. Photochem. Photobiol., A, 2007, 187, 319–324 CrossRef CAS.
- M. Maafi, Front. Chem., 2023, 11, 1233151–1233173 CrossRef CAS PubMed.
- G. Leriche, G. Budin, L. Brino and A. Wagner, Eur. J. Org. Chem., 2010, 4360–4364 CrossRef CAS.
- C. Wang, X. Tong, W. Wang, J.-Y. Xu, L. V. Besteiro, A. I. Channa, F. Lin, J. Wu, Q. Wang, A. O. Govorov, A. Vomiero and Z. M. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 36277–36286 CrossRef CAS PubMed.
- G. Wang, H. Wei, J. Shi, Y. Xu, H. Wu, Y. Luo, D. Li and Q. Meng, Nano Energy, 2017, 35, 17–25 CrossRef CAS.
- W. Bi, L. Zhang, H. Jiang, C. Li and Y. Hu, Chem. Eng. J., 2022, 433, 133679 CrossRef CAS.
- C. Xia, P. Tamarat, L. Hou, S. Busatto, J. D. Meeldijk, C. de M. Donega and B. Lounis, ACS Nano, 2021, 15, 17573–17581 CrossRef CAS PubMed.
- A. Mourot, M. A. Kienzler, M. R. Banghart, T. Fehrentz, F. M. E. Huber, M. Stein, R. H. Kramer and D. Trauner, ACS Chem. Neurosci., 2011, 2, 536–543 CrossRef CAS PubMed.
- M. Dong, A. Babalhavaeji, S. Samanta, A. A. Beharry and G. A. Woolley, Acc. Chem. Res., 2015, 48, 2662–2670 CrossRef CAS PubMed.
- N. Tsolekile, S. Parani, M. C. Matoetoe, S. P. Songca and O. S. Oluwafemi, Nano-Struct. Nano-Objects, 2017, 12, 46–56 CrossRef CAS.
- I. Conti, M. Garavelli and G. Orlandi, J. Am. Chem. Soc., 2008, 130, 5216–5230 CrossRef CAS PubMed.
- C. Ciminelli, G. Granucci and M. Persico, Chem. – Eur. J., 2004, 10, 2327–2341 CrossRef CAS PubMed.
- H. Dürr and H. Bouas-Laurent, Photochromism: molecules and systems, Elsevier, Amsterdam Boston, Rev. edn, 2003 Search PubMed.
- J. Li and X. Jia, Nanomaterials, 2023, 13, 2562 CrossRef CAS PubMed.
- D. Manna, T. Udayabhaskararao, H. Zhao and R. Klajn, Angew. Chem., 2015, 127, 12571–12574 CrossRef.
- L. Sun, C. Wang, Y. Pan, T. Chen and Z. Lv, J. Raman Spectrosc., 2020, 51, 756–763 CrossRef CAS.
- G. Angelini, L. Scotti, A. Aceto and C. Gasbarri, J. Mol. Liq., 2019, 284, 592–598 CrossRef.
- K. Stranius and K. Börjesson, Sci. Rep., 2017, 7, 41145 CrossRef CAS PubMed.
- G. Granucci and M. Persico, Theor. Chem. Acc., 2007, 117, 1131–1143 Search PubMed.
- J. Gemen, J. R. Church, T.-P. Ruoko, N. Durandin, M. J. Białek, M. Weißenfels, M. Feller, M. Kazes, M. Odaybat, V. A. Borin, R. Kalepu, Y. Diskin-Posner, D. Oron, M. J. Fuchter, A. Priimagi, I. Schapiro and R. Klajn, Science, 2023, 381, 1357–1363 CrossRef CAS PubMed.
- D. Bléger and S. Hecht, Angew. Chem., Int. Ed., 2015, 54, 11338–11349 CrossRef PubMed.
- E. Titov, L. Lysyakova, N. Lomadze, A. V. Kabashin, P. Saalfrank and S. Santer, J. Phys. Chem. C, 2015, 119, 17369–17377 CrossRef CAS.
- G. L. Hallett-Tapley, C. D'Alfonso, N. L. Pacioni, C. D. McTiernan, M. González-Béjar, O. Lanzalunga, E. I. Alarcon and J. C. Scaiano, Chem. Commun., 2013, 49, 10073 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR spectra, emission lifetimes, emission and photoreaction quantum yields, absorption and emission spectroscopic data. See DOI: https://doi.org/10.1039/d4nr01997k |
| ‡ Authors contributed equally. |
| § Current position: Department of Chemistry, University of Basel, St. Johanns-Ring 19, 4056, Basel, Switzerland. |
| This journal is © The Royal Society of Chemistry 2024 |