
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Membrane-localized magnetic hyperthermia promotes intracellular delivery of cell-impermeant probes†
Javier
Idiago-López‡
ab,
Daniela
Ferreira‡
cd,
Laura
Asín
ab,
María
Moros
 ab,
Ilaria
Armenia
a,
Valeria
Grazú
ab,
Alexandra R.
Fernandes
cd,
Jesús M.
de la Fuente
ab,
Pedro V.
Baptista§
*cd and
Raluca M.
Fratila§
*abe
ab,
Ilaria
Armenia
a,
Valeria
Grazú
ab,
Alexandra R.
Fernandes
cd,
Jesús M.
de la Fuente
ab,
Pedro V.
Baptista§
*cd and
Raluca M.
Fratila§
*abe
aInstituto de Nanociencia y Materiales de Aragón, INMA (CSIC-Universidad de Zaragoza), C/Pedro Cerbuna 12, 50009, Zaragoza, Spain. E-mail: raluca.fratila@csic.es
bCentro de Investigación Biomédica en Red de Bioingeniería, Biomateriales y Nanomedicina (CIBER-BBN), Spain
cAssociate Laboratory i4HB – Institute for Health and Bioeconomy, NOVA School of Science and Technology, NOVA University Lisbon, 2819-516 Caparica, Portugal. E-mail: pmvb@fct.unl.pt
dUCIBIO – Applied Molecular Biosciences Unit, Department of Life Sciences, NOVA School of Science and Technology, NOVA University Lisbon, 2819-516 Caparica, Portugal
eDepartamento de Química Orgánica, Facultad de Ciencias, C/Pedro Cerbuna 12, 50009, Zaragoza, Spain
First published on 25th July 2024
Abstract
In this work, we report the disruptive use of membrane-localized magnetic hyperthermia to promote the internalization of cell-impermeant probes. Under an alternating magnetic field, magnetic nanoparticles (MNPs) immobilized on the cell membrane via bioorthogonal click chemistry act as nanoheaters and lead to the thermal disruption of the plasma membrane, which can be used for internalization of different types of molecules, such as small fluorescent probes and nucleic acids. Noteworthily, no cell death, oxidative stress and alterations of the cell cycle are detected after the thermal stimulus, although cells are able to sense and respond to the thermal stimulus through the expression of different types of heat shock proteins (HSPs). Finally, we demonstrate the utility of this approach for the transfection of cells with a small interference RNA (siRNA), revealing a similar efficacy to a standard transfection method based on the use of cationic lipid-based reagents (such as Lipofectamine), but with lower cell toxicity. These results open the possibility of developing new procedures for “opening and closing” cellular membranes with minimal disturbance of cellular integrity. This on-demand modification of cell membrane permeability could allow the direct intracellular delivery of biologically relevant (bio)molecules, drugs and nanomaterials, thus overcoming traditional endocytosis pathways and avoiding endosomal entrapment.
1. Introduction
Magnetic nanoparticles (MNPs) have been widely exploited for diverse biomedical applications, including imaging, drug delivery, theranostics, separation, and therapeutic hyperthermia.1–5 Magnetic hyperthermia (MH) is based on the ability of certain magnetic materials to generate heat when exposed to an alternating magnetic field (AMF) and has been extensively used mostly in cancer therapy,6 but also for controlled drug release7 and remote control of cellular functions.8–10 MNPs mediate the conversion of the electromagnetic energy from the AMF to thermal energy via different mechanisms such as hysteresis losses, Néel and Brownian relaxation. The heat dissipated by MNPs depends highly on their intrinsic physico-chemical properties (size, shape, composition, anisotropy), as well as on their environment (medium viscosity, aggregation effects) and on the amplitude and frequency of the AMF.11,12In the frame of traditional MH, the purpose is to raise the temperature of the targeted tissue to a level that can induce therapeutic effects, such as MH-induced cell death, modulation of the tumor microenvironment, or activation of the immune response.12,13 However, beyond classical MH, the localized heat generated in the vicinity of MNPs (known also as the “hot-spot effect”14) has also been used for triggered release of drugs from thermoresponsive matrices7,14 or the remote activation of thermosensitive ion channels8 and enzymes,15 without an increase in the global temperature. We are particularly interested in the use of MNPs immobilized on living cell membranes as nanoscale heaters able to induce localized heating areas on the cell membrane and alter the fluidity of the lipid bilayer. Such an effect could serve as a valuable tool for exploring the fundamental aspects of membrane biophysics or for developing new strategies for intracellular delivery of biomolecules, drugs and nanoparticles. Using MNPs coated with oleic acid and deposited on simplified model cell membranes based on 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) and cholesterol, we demonstrated the increase in the fluidity of the membrane structure upon the application of an AMF, which resulted in the embedding of the MNPs into the phospholipidic layer.16 However, studying this effect on living cell membranes is not trivial, as in this case the immobilization of MNPs is a much more challenging task due to the dynamic nature of the cell plasma membrane. A common approach for binding nanoparticles to the cell surface relies on ligand–receptor binding using nanoparticles decorated with targeting elements such as antibodies, peptides, aptamers, etc.17 Nevertheless, this approach can lead to the premature internalization of the nanoparticles, which for our purposes would be counter-productive since the application of the magnetic field and the study of localized heating would require the MNPs to remain on the cell membrane for prolonged times.
We have recently described the covalent immobilization of MNPs on living cell membranes using strain-promoted click azide–alkyne cycloaddition (SPAAC), one of the most powerful reactions in the bioorthogonal chemistry arsenal.18 In this case, azide artificial chemical reporters are introduced on the cell membranes through metabolic glycoengineering and can react with MNPs functionalized with cyclooctyne probes to yield stable triazole adducts (Fig. 1). Metabolic glycoengineering offers the advantage of a dose-dependent expression of chemical reporters (by carefully tuning the concentration of the azide metabolic precursor and the incubation times) and can be applied to virtually any cell line.19
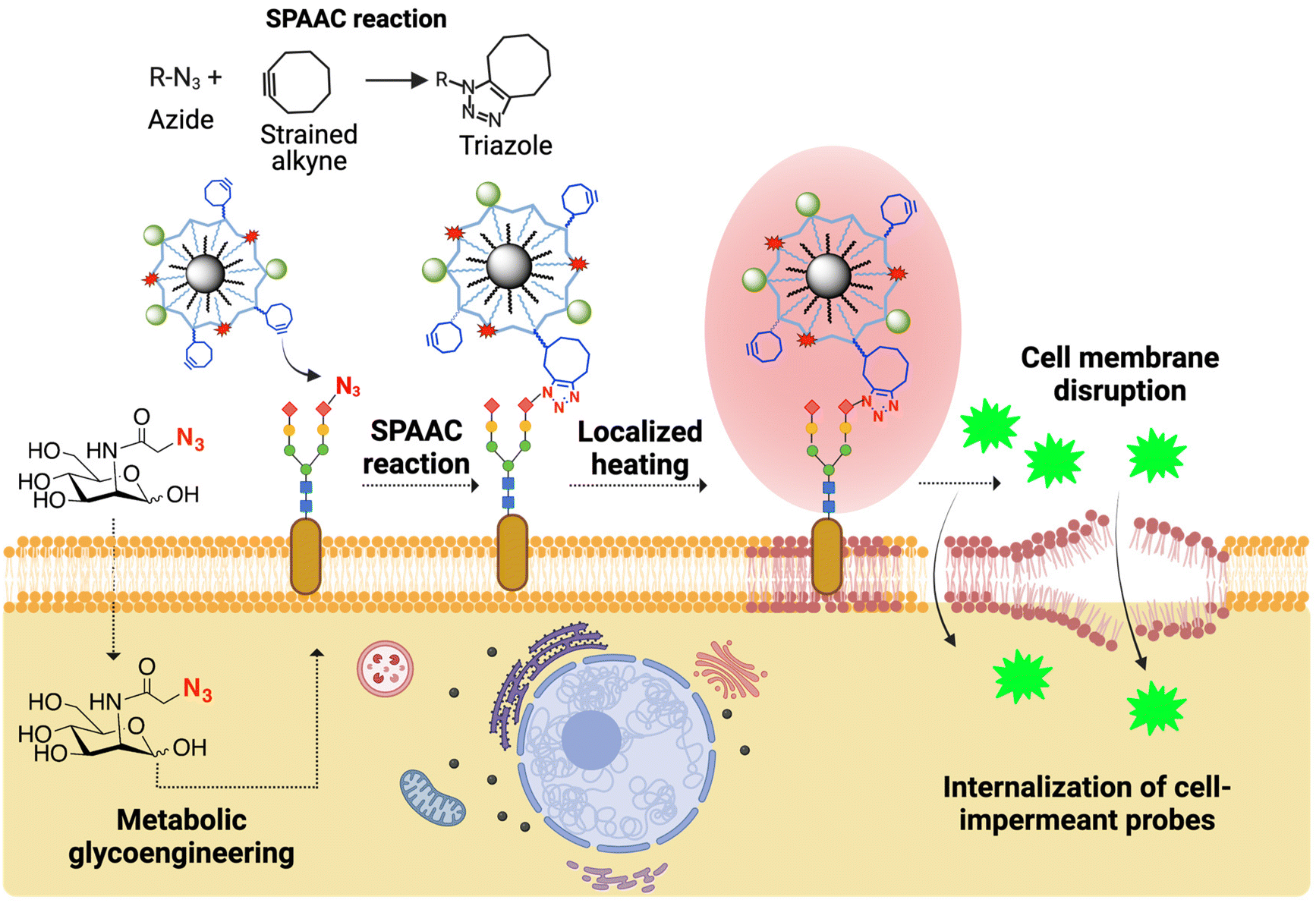 | ||
| Fig. 1 Overview of the general concept of MH-mediated intracellular delivery using MNPs immobilized on the cell membrane via SPAAC bioorthogonal chemistry. The MNPs are functionalized with strained alkynes (in blue) and attached to the membrane of cells previously subjected to metabolic glycoengineering to express unnatural azide bioorthogonal reporters (in red). Created with BioRender.com. | ||
In this work, we report the use of MNPs immobilized on the cell membrane as localized heating sources to promote the internalization of cell-impermeant probes (Fig. 1). As proof of concept, we use YO-PRO®-1, a small fluorescent, cell-impermeant molecule. We show that our approach for the thermal disruption of the plasma membrane does not trigger cell death, oxidative stress and alterations of the cell cycle, although cells can sense and respond to the thermal stimulus through the expression of different types of heat shock proteins (HSPs). Finally, we demonstrate the utility of this approach for biomedical applications, specifically cell transfection, by using a small interference RNA (siRNA) capable of silencing the expression of a green fluorescent protein (GFP). The localized magnetic heating-promoted transfection has a similar efficacy to a standard transfection method based on the use of Lipofectamine reagents, but with lower cell toxicity.
2. Experimental
2.1. Magnetic nanoparticle synthesis, functionalization and characterization
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–50000 g mol−1), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC·HCl), Nα,Nα-bis(carboxymethyl)-L-lysine hydrate, cobalt chloride and 1,2-dihidroxybenzen-3,5-disulfonic acid (Tiron) were purchased from Sigma-Aldrich (St Louis, USA). Tetramethylrhodamine-5-carboxamide (TAMRA) cadaverine was purchased from AnaSpec (Fremont, USA) or Biotium (Fremont, USA). α-Methoxy-ω-amino poly(ethylene glycol) (PEG-NH2, MW = 750 g mol−1) was purchased from Rapp Polymere (Tuebingen, Germany). Dibenzylcyclooctyne-PEG4-NH2 (DBCO-NH2) was purchased from Jena Bioscience (Jena, Germany). Iron standard solutions were purchased from Acros Organics (Geel, Belgium). Amicon® 50 and 100 kDa cut-off and 4 mL centrifuge filters were purchased from Merck Millipore® (Madrid, Spain). Carbon-coated copper grids for TEM were purchased from Electron Microscopy Sciences (Hatfield, USA). Ultrapure water (Milli-Q, a resistivity of 18.2 MΩ cm−1 at 25 °C) was obtained using a Milli-Q® Advantage A10 system (Merck Millipore®, Madrid, Spain).
000–50000 g mol−1), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC·HCl), Nα,Nα-bis(carboxymethyl)-L-lysine hydrate, cobalt chloride and 1,2-dihidroxybenzen-3,5-disulfonic acid (Tiron) were purchased from Sigma-Aldrich (St Louis, USA). Tetramethylrhodamine-5-carboxamide (TAMRA) cadaverine was purchased from AnaSpec (Fremont, USA) or Biotium (Fremont, USA). α-Methoxy-ω-amino poly(ethylene glycol) (PEG-NH2, MW = 750 g mol−1) was purchased from Rapp Polymere (Tuebingen, Germany). Dibenzylcyclooctyne-PEG4-NH2 (DBCO-NH2) was purchased from Jena Bioscience (Jena, Germany). Iron standard solutions were purchased from Acros Organics (Geel, Belgium). Amicon® 50 and 100 kDa cut-off and 4 mL centrifuge filters were purchased from Merck Millipore® (Madrid, Spain). Carbon-coated copper grids for TEM were purchased from Electron Microscopy Sciences (Hatfield, USA). Ultrapure water (Milli-Q, a resistivity of 18.2 MΩ cm−1 at 25 °C) was obtained using a Milli-Q® Advantage A10 system (Merck Millipore®, Madrid, Spain).
Formation of the Lys-NTA–Co2+ complex. A mixture of Nα,Nα-bis(carboxymethyl)-L-lysine hydrate (Lys-NTA, 25 mM) and CoCl2 (30 mM) was prepared in 50 mL of BBS buffer (borate buffered saline, 50 mM, pH 8.0). The pH was then adjusted to 10.5 for precipitation of excess Co2+ ions, the complex was centrifuged (15 min at 4000g) and the final pH of the supernatant was adjusted to 9.0.
MNPs@PMAO functionalization with the Lys-NTA–Co2+ complex. 0.5 mg Fe of MNPs@PMAO were mixed with a mixture of the stock solution of Lys-NTA–Co2+ (32 μmol per mg Fe) and PEG750 (18.5 μmol per mg Fe), adjusting the final reaction volume to 1.5 mL. For activation of the carboxyl groups of the MNPs@PMAO, EDC·HCl (33.2 μmol per mg Fe) was dissolved in 20 μL of BBS buffer (50 mM, pH 9) and two additions of 10 μL each were carried out at time 0 and after 30 min; the reaction mixture was further incubated at 37 °C under gentle stirring for 3 h. Finally, excess reagents were removed by 4 washes with Milli-Q water (10 min at 4800g) carried using centrifugation filters (Amicon, Millipore, 50 kDa).
Transmission electron microscopy (TEM). TEM imaging and size analysis of the MNPs were performed on an FEI Tecnai T20 instrument operating at an accelerating voltage of 80–200 kV (Laboratorio de Microscopias Avanzadas LMA, University of Zaragoza). Samples were prepared on carbon-coated copper grids by depositing a drop of a diluted MNP suspension; TEM grids were allowed to dry at room temperature for 24 h before visualization. The MNP size and size distribution were determined using the ImageJ software by measuring the diameter of at least 200 MNPs per image.
Determination of the iron concentration of MNP suspensions. The iron concentrations of the nanoparticle suspensions were determined using a standard colorimetric method, as previously described.18
Agarose gel electrophoresis. A solution of 1% agar in 0.5× Tris-borate-EDTA (TBE) buffer was prepared. MNP samples mixed with 20% glycerol: 0.5× TBE were loaded in the gel (a final volume of 8 µL per well) and an electric field of 90 V was applied for 45 min.
Dynamic light scattering (DLS) and ζ-potential measurements. Hydrodynamic diameters and ζ-potential values of the different MNPs in suspension were determined using a Malvern Zetasizer Nano considering a refractive index of 2.0 and an absorption index of 1.0 for Fe3O4. Samples were prepared at a concentration of 0.05 mg Fe mL−1 in Milli-Q water. Each sample was measured five times, combining 10 runs per measurement. Results were treated using the Malvern software Zetasizer Nano 7.13.
Characterization of the heating properties of MNPs. SLP measurements were carried out using a D5 Series AMF applicator (nanoScale Biomagnetics, Zaragoza, Spain) with a coil (CAL2) connected to a vacuum pump to achieve thermal isolation of the sample and using a capacitor (120 nF) to evaluate different frequency ranges. MNP samples were prepared at a concentration of 1 mg Fe mL−1 in water and/or cell culture supplemented medium (complete Dulbecco's modified Eagle's medium, cDMEM). For each measurement, the temperature variation was recorded for 5 min and the SLP was calculated by linear curve fitting at the initial times (30–60 s) using the ZAR application provided by the equipment software (MANIAC) and indicating the value of the sample concentration (mg Fe per mL).
For the samples subjected to an AMF to study the effect of local heating, the same protocol was followed with the application of the AMF for 5 min at different frequencies (137, 275, 366, and 445 kHz) at a field amplitude of 16.9 kA m−1 using the D5 series device equipped with the CAL-2 coil-set as an AMF applicator (nanoScale Biomagnetics). The calibration curve obtained using a dry block incubator was used to infer the local temperature of the NPs subjected to the AMF.
2.2. Cell culture and in vitro experiments
Reagents were used as purchased and all reagent solutions and nanoparticle suspensions were sterilized by filtration with 0.22 μm MilliPore® sterile filters before addition to cell cultures.
000 cells per well in Nunc 8-well chamber slides (Lab-Tek™). After 24 h of incubation, the medium was replaced by medium containing Ac4ManNAz (50 μM) for 48 h. Subsequently, the cells were incubated with MNPs@PMAO@PEG@DBCO (100 μgFe mL−1) for 1 h in DMEM for binding to the cell membrane by SPAAC chemistry. Unbound MNPs were then removed by washing twice with DPBS and cells were either fixed immediately or after further incubation for 24 h in cDMEM to promote complete cellular internalization of MNPs. Fixation was carried out by the addition of 300 μL of glutaraldehyde (2%) per well in cacodylate buffer (0.1 M) for 10 min at 37 °C. The fixing agent was replaced by another 300 μL of the same solution and the samples were incubated for 2 h at room temperature. The already fixed cells were washed with phosphate buffer (0.1 M) five times for 2 min each wash and stored at 4 °C. Finally, the cell samples were sectioned and mounted on carbon-coated copper grids for TEM by the Electron Microscopy Service of the Centro de Investigación Príncipe Felipe (CIPF, Valencia, Spain). For this purpose, the samples were treated with OsO4 (2%) for 1 h at room temperature and stained with uranyl acetate for 2 h at 4 °C in the dark. The samples were then washed with distilled water, dehydrated with ethanol, and infiltrated in Durcupan resin (Sigma®) for 24 h. After polymerization, the cells were detached from each well and adhered to Durcupan blocks. Finally, ultrathin sections (0.08 μm) were cut with an Ultracut UC-6 microtome (Leica microsystems®), stained with lead citrate, and analyzed using an FEI TecnaiT20 transmission electron microscope (ThermoFisher Scientific Company, Oregon, USA).
Magnetic hyperthermia equipment and experimental conditions. The study of the effect of localized magnetic hyperthermia was carried out using a D5 Series device (nanoScale Biomagnetics, Zaragoza, Spain) equipped with a planar coil (PC90) and using a capacitor (120 nF) to reach the maximum frequency values available. To maintain the overall temperature of cells constant during the experiment, a thermostat-controlled support was used, connected to a 37 °C water bath (see the ESI† for more details). The frequency and magnetic field amplitude values used were 425 kHz and 16.9 kA m−1, respectively, corresponding to the maximum values available with the planar coil.
Study of the MNP localization change by fluorescence microscopy. HCT116 cells were seeded at a density of 9000 cells per well in polymer bottom 8-well chamber slides (ibidi, Gräfelfing, Germany). After 24 h of incubation, the medium was replaced with medium containing Ac4ManNAz (50 μM) for 48 h. Subsequently, the cells were incubated with MNPs@PMAO@PEG@DBCO (100 μgFe mL−1) for 1 h in DMEM (without phenol red) for binding to the cell membrane by SPAAC chemistry. Unbound MNPs were then removed by washing twice with DPBS and an AMF (425 kHz, 16.9 kA m−1) was applied for 30 min with cell temperature control at 37 °C. In parallel, a control experiment was carried out under the same incubation conditions, but without the application of the AMF. After 30 min (for the slide subjected to the AMF) or 1 h (for the control slide without AMF application) of incubation under cell culture conditions, both slides were observed under a fluorescence microscope (Nikon Eclipse Ti-E inverted microscope, Nikon Europe B.V., Amstelveen, the Netherlands) and images were captured at 40× magnification using the TRITC channel (100 ms exposure time). The cells were imaged without fixing to avoid possible effects of the fixation process in the localization of the MNPs.
YO-PRO®-1 internalization. HCT116 cells were seeded at a density of 10
000 cells per well in polymer bottom 8-well chamber slides (ibidi, Gräfelfing, Germany). After 24 h of incubation, the medium was replaced with medium containing Ac4ManNAz (50 μM) for 48 h. Then, the cells were incubated with MNPs@PMAO@PEG@DBCO (100 μgFe mL−1; MNPs not bearing the TAMRA fluorophore were used) for 1 h in DMEM (without phenol red) for binding to the cell membrane by SPAAC chemistry. Unbound MNPs were removed by washing twice with DPBS, YO-PRO®-1 (0.1 μM) was added in 300 μL of supplemented DMEM (without phenol red) and the AMF (425 kHz, 16.9 kA m−1) was applied for 30 min with cell temperature control at 37 °C or room temperature. After this time, the cells were washed twice with PBS and detached by incubation with 100 μL of Trypsin-EDTA for 5 min. The trypsin was neutralized with 300 μL of supplemented DMEM and the cells were collected in microtubes (Eppendorf®) and centrifuged at 12100g for 15 s. The pellets were resuspended in 300 μL of PBS and the samples were analyzed on a CytoFlex cytometer (Beckman Coulter, Indianapolis, USA), exciting at 488 nm and collecting the FITC channel signal (525/40 nm). Ten thousand events per sample were recorded and data were analyzed using Kaluza 1.5a software (Beckman Coulter, Indianapolis, USA). All samples were prepared and analyzed in duplicate.
000 cells per well in polymer bottom 8-well chamber slides (ibidi, Gräfelfing, Germany). After 24 h of incubation, the medium was replaced by medium containing Ac4ManNAz (50 μM) for 48 h and the cells were incubated with MNPs@PMAO@PEG@DBCO (100 μgFe mL−1) (MNPs without TAMRA) for 1 h in DMEM for binding to the cell membrane by SPAAC chemistry. Unbound MNPs were removed by washing twice with DPBS and the cells were subjected to an alternating magnetic field application (425 kHz, 16.9 kA m−1, 30 min) or further incubated for 24 h to promote MNP internalization prior to AMF application. Cell viability analysis was performed in the same manner for the two MNP localizations (on the cell membrane or internalized).
Analysis of the cell morphology. To evaluate possible morphological changes after the application of the alternating magnetic field, the cells were imaged at 0 h and 24 h after the AMF application using an optical microscope (Nikon Eclipse TE2000-S, Nikon Europe B.V., Amstelveen, the Netherlands).
Analysis of cell viability by flow cytometry. After the AMF application, the culture medium was recovered from each well using microtubes (Eppendorf®), and the cells were washed with PBS and detached with 100 μL of trypsin-EDTA (5 min of incubation at 37 °C). Trypsin was neutralized with 300 μL of supplemented DMEM and the detached cells were transferred to the corresponding microtube containing the cell culture medium previously recovered. Cells were centrifuged at 12
100g for 15 s and the pellet was resuspended in 100 μL of 1× Annexin binding buffer. Then, 5 μL of FITC-Annexin V and 1 μL of propidium iodide (100 μg mL−1) were added. After 15 min incubation at room temperature and in the dark, 100 μL of 1× Annexin binding buffer was added to each microtube, shaken briefly in a vortex and the samples were analyzed on a CytoFlex cytometer (Beckman Coulter, Indianapolis, USA); excitation at 488 nm and signal detection at 585/42 nm (PE channel) and 525/40 nm (FITC channel), respectively. Both a positive cell death control sample (cells incubated at 60 °C for 30 min) and a negative control sample (live cells) were analyzed to adjust the gains of the PE and FITC channels. A total of 20000 events per sample were recorded and the data were analyzed using Kaluza 1.5a software (Beckman Coulter, Indianapolis, USA). All samples were prepared and analyzed in duplicate.
Analysis of the generation of reactive oxygen species (ROS). Before the application of the alternating magnetic field, the cell culture medium was replaced by a solution of 2 μM CM-H2DCFDA in supplemented DMEM. The alternating magnetic field (425 kHz, 16.9 kA m−1, 30 min) was applied and cells were incubated for an additional time of 30 min under standard cell culture conditions. The cells were washed with PBS and detached with trypsin-EDTA (5 min of incubation at 37 °C). Trypsin was neutralized with 300 μL of supplemented DMEM and the detached cells were centrifuged at 12
100g for 15 s, the pellet was resuspended in 100 μL of PBS and samples were analyzed on a CytoFlex cytometer (Beckman Coulter, Indianapolis, USA) using the FITC channel (525/40 nm).
Additionally, ROS generation was assessed by incubating the cells at different global temperatures (37, 39, 42, and 45 °C) for 30 min. For this purpose, 30000 cells per well were seeded in 24-well plates, incubated for 24 h and detached by incubation with trypsin-EDTA (5 min at 37 °C). Trypsin was neutralized with 300 μL of supplemented DMEM and the detached cells were centrifuged at 12100g for 15 s. The pellet was resuspended in 200 μL of 2 μM CM-H2DCFDA in supplemented DMEM and the cells were incubated at different temperatures in a dry block incubator (ThermoMixer F2.0, Eppendorf®) for 30 min. After an additional 30 min of incubation under standard culture conditions, the cells were centrifuged, resuspended, and analyzed by flow cytometry as previously described.
As a positive control for ROS generation, the cells were incubated with 200 μM H2O2 for 30 min and labeled with CM-H2DCFDA under the same conditions. 30000 events were recorded per sample and data were analyzed with Kaluza 1.5a software (Beckman Coulter, Indianapolis, USA). All samples were prepared and analyzed in duplicate.
Cell cycle analysis. After the application of an alternating magnetic field, the cells were incubated for 24 h under standard culture conditions, then washed with PBS and detached by incubation with trypsin-EDTA for 5 min at 37 °C. Trypsin was neutralized with 300 μL of supplemented DMEM and the cells were centrifuged at 12
100g for 15 s. The pellet was carefully resuspended by dropwise addition of ethanol (70%). The cells were fixed for 30 min at 4 °C in ethanol and subsequently washed twice with PBS by centrifugation at 3500g for 2 min. The pellet was resuspended in 100 μL of PBS, and then 1 μL of RNAase (10 mg mL−1) was added and the mixture was incubated for 10 min at 37 °C. Finally, 10 μL of propidium iodide (1 mg mL−1) was added and the cells were analyzed on a CytoFlex cytometer (Beckman Coulter, Indianapolis, USA), exciting at 488 nm and collecting the PE channel signal (585/42 nm). 20000 events per sample were recorded and the data were analyzed using Kaluza 1.5a software (Beckman Coulter, Indianapolis, USA). The cells seeded in parallel, under standard culture conditions and without MNPs and AFM application were used as a control. All samples were prepared and analyzed in duplicate.
Assessment of cell viability at various temperatures (external global heating). HCT116 cells were seeded at a density of 50
000 cells per well in 24-well plates. After 48 h of incubation and cell growth under standard cell culture conditions, the cells were washed with PBS and detached by incubation with 100 μL per well of trypsin-EDTA for 5 min. The trypsin was neutralized with 300 μL of supplemented DMEM and the cells corresponding to three wells were collected in microtubes (Eppendorf®). The cells were centrifuged at 12100g for 15 seconds and the pellet was resuspended in 300 μL of supplemented DMEM. The cells were then subjected to different global temperatures (37, 39, 41, 43, 46, 49, and 60 °C) for 30 minutes using a dry block incubator (ThermoMixer® F2.0, Eppendorf, Hamburg, Germany). Finally, 30 μL of cell suspension from each of the conditions were labeled with 30 μL of 0.4% trypan blue and the number of live and dead cells was quantified using a Neubauer chamber. All samples were prepared and analyzed in duplicate.
Quantification of HSP expression by real-time quantitative polymerase chain reaction (RT-qPCR). The expression of heat shock proteins was analyzed in the presence of two different types of thermal stimuli: external global heating and local heating induced by MNPs after the application of an alternating magnetic field. In both cases, RNA was extracted immediately after the end of the thermal stimulus and the same RT-qPCR protocol was followed for both types of samples.
The sequences of the primers corresponding to the HSP genes analyzed are provided in the ESI.† Primers were purchased in the lyophilized format and reconstituted in PCR-grade water following the manufacturer's instructions.
External global heating. Cells were seeded and treated as previously described for the cell viability studies at different temperatures. After 30 min of incubation at 37, 39, 41, and 43 °C, total RNA extraction was performed.
Localized heating. HCT116 cells were seeded at a density of 25
000 cells per well in polymer bottom 8-well chamber slides (ibidi, Gräfelfing, Germany). After 24 h of incubation, the medium was replaced with medium containing Ac4ManNAz (50 μM) for 48 h. Then, the cells were incubated with MNPs@PMAO@PEG@DBCO (100 μgFe mL−1) for 1 h in DMEM for binding to the cell membrane by SPAAC chemistry. Unbound MNPs were removed by washing twice with DPBS and the alternating magnetic field (425 kHz, 16.9 kA m−1) was applied for 30 min at a controlled cell temperature (37 °C). In parallel, 24 h before the application of the magnetic field, the cells were incubated with MNPs@PMAO@PEG@DBCO (100 μg Fe per mL) for 1 h in DMEM; unbound MNPs were removed by washing twice with DPBS and the cells were incubated for 24 h to promote the complete internalization of the MNPs. After 24 h, the alternating magnetic field was applied under the same conditions as described above. After the AMF application, the total RNA was extracted using a commercial kit, following the manufacturer's instructions and eliminating the possible traces of genomic DNA by treatment with DNase I. RNA extraction was carried out in an RNase-free sterility hood previously decontaminated using RNaseZap and using a sterile, RNase-free material. RNA concentration and purity were analyzed by UV spectrophotometry (Thermo Scientific Multiskan GO UV/vis) at 260 and 280 nm and by agarose gel electrophoresis (1%, m/v) stained with Gel Red in 0.5× TBE. The cDNA was synthesized from 1.5 μg of RNA using the SuperScript VILO cDNA Synthesis kit using PCR-grade water and following the manufacturer's instructions.
RT-qPCR was performed using iTaq Universal SYBR Green Supermix (×) in a CFX-96 Real Time PCR System (Bio-Rad, Hercules, USA). The following conditions were used: initial denaturation at 95 °C for 30 s; 40 extension cycles (3 s at 95 °C and 30 s at 60 °C); dissociation curve (to evaluate if unwanted amplification products have been formed): 0.5 °C increments from 59 to 90 °C. The expression of the different genes was evaluated according to the 2−ΔΔCt method, using an 18S ribosomal gene as a reference. Each RT-qPCR reaction was performed in triplicate and at least two biological samples were evaluated.
Magnetic hyperthermia mediated transfection of MCF7/copGFP cells. MCF7 cells were plated at a density of 25
000 cells per well and allowed to adhere for 24 h. Subsequently, the cells were exposed to 100 μM Ac4ManNAz for 48 h before proceeding to magnetic hyperthermia experiments. After 48 h of incubation with Ac4ManNAz, the cells were washed twice with PBS and were incubated for 10 minutes at 37 °C with 10 μgFe mL−1 of MNPs@PMAO@PEG@DBCO and DMEM without FBS. Cells were subjected to two additional washes with PBS, detached using Versene solution, centrifuged, and resuspended in fresh medium.
The magnetic hyperthermia procedure is the same as that used in MCF7 cells transiently transfected with GFP (for full details, see the ESI†). Following transfection, the cells were counted and seeded in a 96-well plate for the cell viability assay and in a 24-well plate for fluorescence microscopy analysis and RNA extraction, under standard cell culture conditions.
copGFP expression analysis. copGFP expression was verified by fluorescence microscopy following 24 h and 48 h of the gene silencing experiment and the images of cells were acquired and analyzed as mentioned previously. Following 48 h post-transfection, total RNA was extracted from samples and reverse transcribed to cDNA as described previously. RT-qPCR was performed using the following conditions: initial denaturation at 95 °C for 5 min; 30 cycles at 95 °C for 30 s, Tm 53 °C for 30 s, 72 °C for 30 s; and a final extension step at 72 °C for 5 min. copGFP gene expression was evaluated according to the 2−ΔΔCt method, using the 18S ribosomal gene as a reference.
Cell viability post-transfection. Cells were plated at approximately 25
000 cells per well in a 96-well plate and incubated for 24 h at 37 °C, under standard cell culture conditions. The MTS assay was performed to determine cytotoxicity following siRNA transfection with magnetic hyperthermia and with Lipofectamine RNAiMax using the MTS assay as described above. At 24 h post-transfection the cell culture medium was removed, MTS solution was added to the cells and incubated for 1 h. The absorbance was measured at 490 nm using a microplate reader. Cell viability was normalized to control cells (cells with culture medium).
2.3 Statistical analysis
Data were analyzed using GraphPad Prism 8.0 (GraphPad Software, San Diego, USA). The results are presented as average ± the standard error mean of at least three independent experiments. One-way ANOVA with Tukey's or Dunnett's multiple comparisons test and unpaired parametric t-test with Welch's correction was used to evaluate differences between groups and were considered statistically significant at p-value < 0.05.3. Results and discussion
3.1 Expression of azide bioorthogonal reporters on cell membranes
As mentioned in the Introduction, for the immobilization of MNPs on living cell membranes using the SPAAC reaction, cells must be engineered to express artificial azide reporters on their surface, while MNPs should be decorated with strained alkynes.For the labeling of cell membranes with azides, we used the sialic acid metabolic pathway to incorporate these chemical reporters into the glycocalyx. Metabolic glycoengineering relies on the use of a biosynthetic precursor containing an azide functional group, typically tetraacetylated N-azidoacetylmannosamine (Ac4ManNAz). This synthetic molecule is taken up by cells and efficiently hydrolyzed to N-α-azidoacetylmannosamine (ManNAz) by cytosolic esterases. ManNAz is then converted to sialic acid via five enzymatic steps and conjugated to the end of the sugar chains, displaying end azide reporters.21 It is worth mentioning that the introduction of azide reporters on cell membranes can be achieved in a controlled manner, by adjusting the concentration of the azide precursor and/or the incubation time.22,23 Indeed, in our previous work we demonstrated this dose-dependent expression of azide groups on the membrane of HCT116 (human colorectal carcinoma) and MCF7 (human breast adenocarcinoma) cells, and established the optimal conditions for each cell line (48 h of incubation with Ac4ManNAz at a concentration of 100 μM for MCF7 and of 50 μM for HCT116 cells).18 The labeling strategy and representative fluorescence microscopy images for both cell lines under optimal metabolic glycoengineering conditions are shown in Fig. 2.
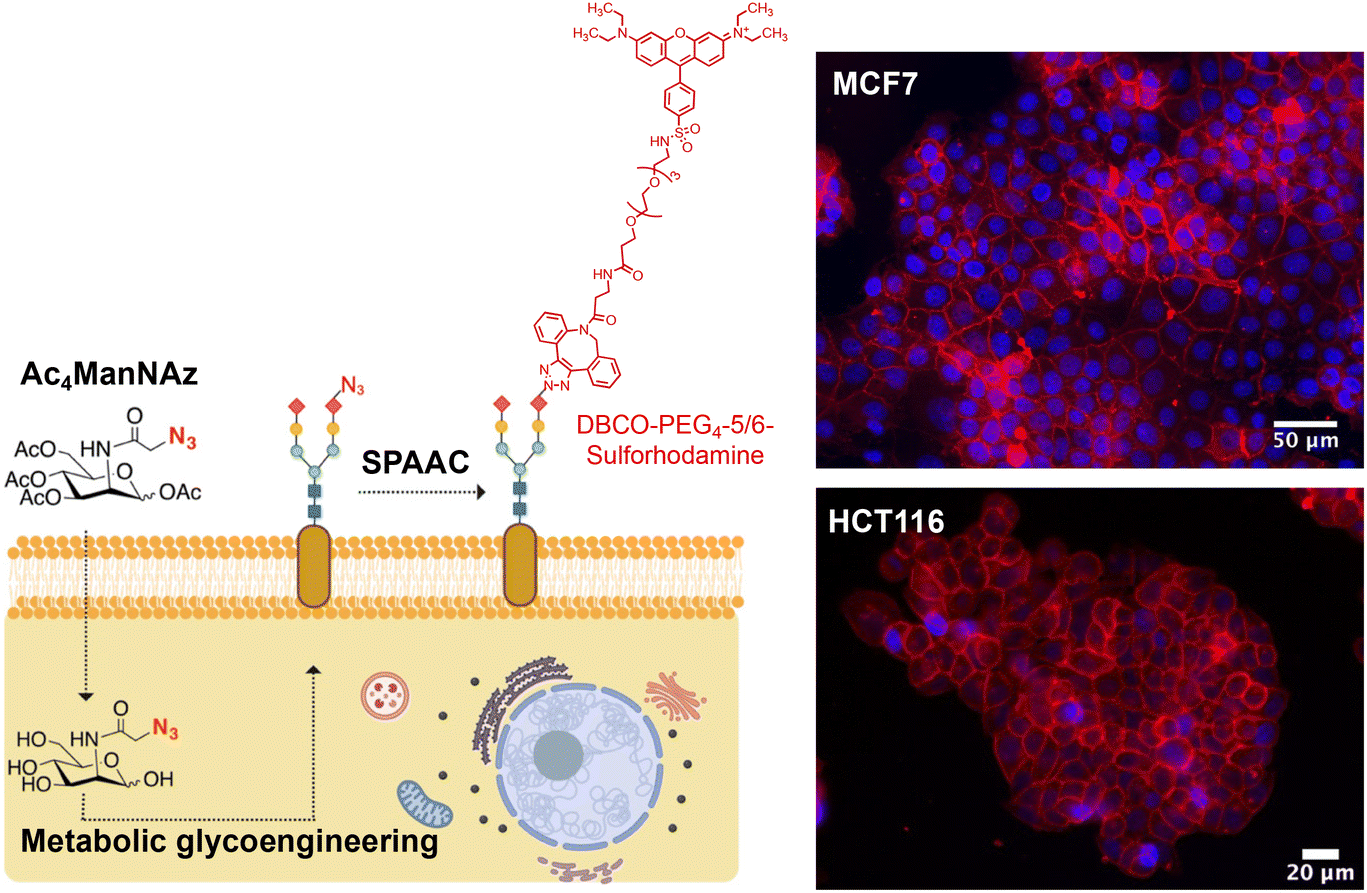 | ||
| Fig. 2 Scheme of the metabolic engineering approach for the expression of azide bioorthogonal reporters on cell surfaces (left) and confocal fluorescence microscopy images of MCF7 and HCT116 cells treated with Ac4ManNAz for 48 h, followed by 30 min of incubation with 20 μM DBCO-sulforhodamine B. Red: DBCO-sulforhodamine B. Blue: DAPI (nuclei staining). Scale bar: 50 μm for MCF7 and 20 μm for HCT116. | ||
3.2 MNP functionalization with strained alkynes
The MNPs used in this work were 13 nm spherical iron oxide MNPs with a carboxylic acid surface functionality, obtained following the methodology previously described and optimized by our group.24,25 The polymer coating the MNPs – poly(maleic anhydride-alt-1-octadecene), PMAO – was modified using a fluorescent dye, tetramethylrhodamine 5-(6)-carboxamide (TAMRA) cadaverine, to allow MNP tracking in vitro by fluorescence microscopy.18,26 The PMAO-coated MNPs, having carboxyl acid groups on their surface, were then functionalized with strained alkynes in a two-step process, by firstly introducing an amino-poly(ethylene glycol) (PEG) derivative to improve the colloidal stability of the MNPs, followed by the incorporation in a second step of a dibenzocyclooctynylamine (DBCO) derivative bearing a short ethylene glycol chain, to yield MNPs@PMAO@PEG@DBCO, as described in our previous work.18,25 This strategy was chosen due to the hydrophobic character of the DBCO derivative, which leads to a loss of the colloidal stability of the MNPs if the functionalization is attempted directly on the MNPs@PMAO.25The preparation and physico-chemical characterization of these different MNP types are reported in detail in our recent work;18 however, relevant characterization data are provided in the ESI for the specific batches employed in the current work (Fig. S1†). The heating efficiency of the MNPs@PMAO@PEG@DBCO, expressed in terms of Specific Loss Power (SLP, defined as the power generated per gram of magnetic material)27 was measured at different AMF amplitudes and frequencies (see Fig. S2†), yielding a maximum value of approximately 100 W gFe−1 at a frequency (f) of 445 kHz and a field amplitude (H) of 16.9 kA m−1 (AMF conditions within the H × f safety limit of 5 × 109 A m−1 s−1 established by Hergt and Dutz28), determined in an aqueous suspension at a concentration of 1 mg Fe mL−1. Noteworthily, the heating efficiency of these MNPs determined in a suspension prepared in cell culture medium (Dulbecco's Modified Eagle's Medium – DMEM, supplemented with 10% of fetal bovine serum, see Fig. S2†) was very similar to the one obtained in water, suggesting that the slightly higher viscosity of the supplemented cell culture medium had no impact on the SLP values.
The SLP measurements described above were performed at a relatively high MNP concentration, ten times higher than the one used for cell membrane immobilization (100 μgFe mL−1, see below). At low concentrations, MNPs can continue to generate heat locally under the application of an AMF with suitable parameters, although a variation in the temperature of the medium in which they are dispersed is no longer detected. For our work using MNPs immobilized on the cell membrane, the estimation of the local temperature (TLOC) on the surface of the MNPs is crucial, as the alteration of cell membrane fluidity depends directly on the local temperature increase. To determine the temperature on the MNP surface, different types of thermal transducers can be used, including nucleic acid probes, organic fluorescent dyes, polymers, lanthanide-based nanoparticles, etc.8,29–32 Here, we used the green fluorescent protein (GFP), based on a recent report from our group in which we demonstrated the usefulness of fluorescent proteins as thermal probes of the local temperature in the vicinity of the MNP surface.20 When subjected to thermal denaturation, this protein follows an irreversible unfolding of the β-barrel around the fluorophore, leading to a linear fluorescence loss.33 To simplify the MNP functionalization with the GFP (which would be difficult to incorporate on the surface of the MNPs already modified with PEG and DBCO), we adapted the strategy described by Ovejero et al.20 MNPs@PMAO were functionalized first with a nitriloacetic acid-Co2+ (NTA-Co2+) complex and PEG, and then with a GFP genetically modified to include a hexahistidine tag (6His-tag) at its N-terminus (Fig. 3a). The 6His-tag forms a coordination bond with the Co2+ ions, resulting in an oriented binding of the protein on the MNP surface.20,34
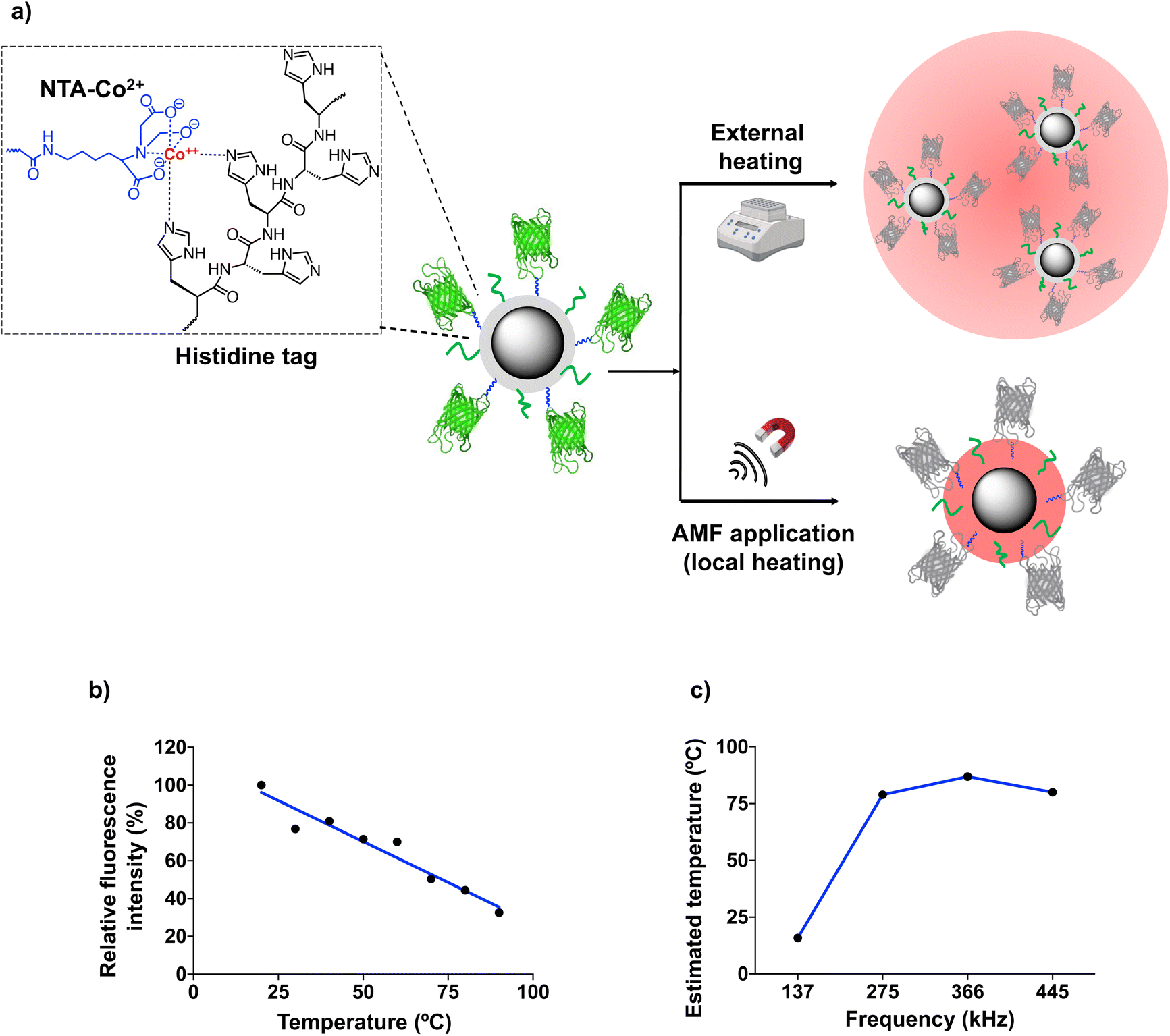 | ||
| Fig. 3 (a) Schematic depiction of the strategy used to assess the effect of global and local heating on the fluorescence of GFP linked to the MNPs through metal affinity binding. (b) Effect of temperature on the fluorescence of the GFP immobilized on MNPs and subjected to macroscopic heating. The fluorescence intensity at 20 °C was fixed at 100%. (c) Temperature estimation based on the fluorescence intensity measured for the MNP-GFP complexes exposed to AMFs (16.9 kA m−1 and increasing frequencies). | ||
To determine if the GFP immobilization on the MNP surface causes a non-linear fluorescence loss (due to changes in the protein structure or its rigidification upon immobilization), MNP@PMAO@GFP conjugates were first exposed to external, macroscopic heating using a hot bath. The fluorescence vs. temperature curve depicted in Fig. 3b confirmed that the GFP fluorescence intensity decreased linearly with the temperature. Fig. 3c shows the TLOC values determined from the fluorescence intensity of GFP after subjecting a diluted suspension (10 μgFe mL−1) of MNP@PMAO@GFP conjugates to an AMF at different frequencies and an amplitude of 16.9 kA m−1 for 5 min. The temperature of the MNP suspension remained constant at 18 ± 0.5 °C throughout the AMF application. This observation indicates that the heating was confined to the immediate environment of the MNPs, due to the low concentration of the suspension, and in line with the previous finding from our group.20 As can be inferred from Fig. 3c, at the lowest AMF frequency of 137 kHz, the increase in temperature on the MNP surface was negligible (TLOC of approximately 12 °C); however, at higher frequencies, TLOC in the range of 79 to 87 °C were recorded. The plateau effect observed (the frequency change from 275 to 445 kHz did not lead to a relevant change in the maximum temperature reached on the MNP surface) suggested that the denaturation of GFP reached a maximum at the local temperature produced by the application of an AMF with a frequency of 275 kHz. In summary, the determination of TLOC using the GFP as a local thermal probe revealed that indeed very high temperatures can be reached close to the MNP surface, even under diluted conditions as the ones used for cell work.
Finally, to ensure the accuracy of the temperature measured on the MNP surface, we verified that the MNPs@PMAO and the MNPs@PMAO@PEG@DBCO had the same heating efficiency. The SLP values measured for these two particles were practically identical under the AMF conditions employed (104 W gFe−1 for the MNPs@PMAO and 100 W gFe−1 for the MNPs@PMAO@PEG@DBCO at a frequency of 445 kHz and a field amplitude of 16.9 kA m−1). This means that the TLOC estimated for the MNP@PMAO@GFP can be extrapolated to the MNPs@PMAO@PEG@DBCO.
3.3 Effect of the localized heating on cell membrane permeability and cell viability
The application of the AMF in cell cultures was carried out using a circular planar coil of 90 mm diameter with a radial field homogeneity of up to 90% in a diameter of 35 mm (typical size of a cell culture dish) and fitted with a water jacket to ensure that the cells were maintained at 37 °C during the experiments (Fig. S3†). Due to the thickness of the water jacket, the cells are positioned 6 mm from the surface of the coil, which means an intrinsic decrease of the field amplitude of about 25% with respect to the center of the coil (see the ESI†). For this reason, we chose to use the maximum field amplitude and frequency combination permitted for this planar coil (16.9 kA m−1 and 425 kHz, respectively).HCT116 cells engineered to express azide groups were incubated with MNPs@PMAO@PEG@DBCO for 1 h at an MNP concentration of 100 μgFe mL−1, as optimized in our previous work.18 After MNP immobilization on the cell surface by SPAAC, the cells were exposed for 30 minutes to an AMF with the parameters mentioned above. The first indication of a possible thermal disruption of the cell membrane was the change observed in the cellular localization of the MNPs: 30 minutes after the AMF application, a significant portion of the total number of MNPs bound to the cell membrane was found to form intracellular agglomerates (Fig. S4a†). In contrast, in cells not exposed to the AMF, MNPs retained a homogeneous distribution along the entire membrane (Fig. S4b†).
This effect was further demonstrated by the internalization of YO-PRO®-1, a small (629 Da), exogenous fluorescent probe, which is cell-impermeant under normal conditions of membrane fluidity. However, at high enough concentrations, YO-PRO®-1 could cross cell membranes by gradient diffusion, an effect commonly known as non-specific internalization. Therefore, an optimization of the YO-PRO®-1 concentration was initially carried out to reduce non-specific internalization of the probe and to better assess MH-promoted internalization. A concentration of 0.1 μM was determined as the optimal concentration at which non-specific internalization was minimized while ensuring good detection sensitivity by flow cytometry (see ESI and Fig. S5†).
We then assessed YO-PRO®-1 internalization after the AMF application (Fig. 4). Interestingly, when performing the AMF application at room temperature, for all four experimental conditions tested (depicted in Fig. 4a), we observed a very low degree of internalization, which could be attributed to a non-specific uptake of the probe (Fig. 4b, top). In contrast, when the experiment was carried out at physiological temperature, maintaining the cell culture temperature constant at 37 °C, significant differences appeared between the different conditions evaluated (Fig. 4b, bottom). Cells in which MNPs were immobilized on the membrane by click chemistry showed the highest level of YO-PRO®-1 internalization, although non-specific binding of MNPs in cells without Ac4ManNAz treatment also resulted in probe internalization after the application of the AMF, but to a lesser extent due to a lower number of “hotspots” able to alter the membrane permeability. These results highlight the importance of the cell temperature in thermal stimulation experiments. At temperatures below physiological conditions (e.g., room temperature) the local heat generated by the MNPs is invested in restoring normal membrane fluidity, without altering the biophysical properties of the membrane. However, if thermal stimulation starts at a normal state of membrane fluidity, the local temperature increase generated is directly harnessed for increasing it, leading to the internalization of exogenous molecules.
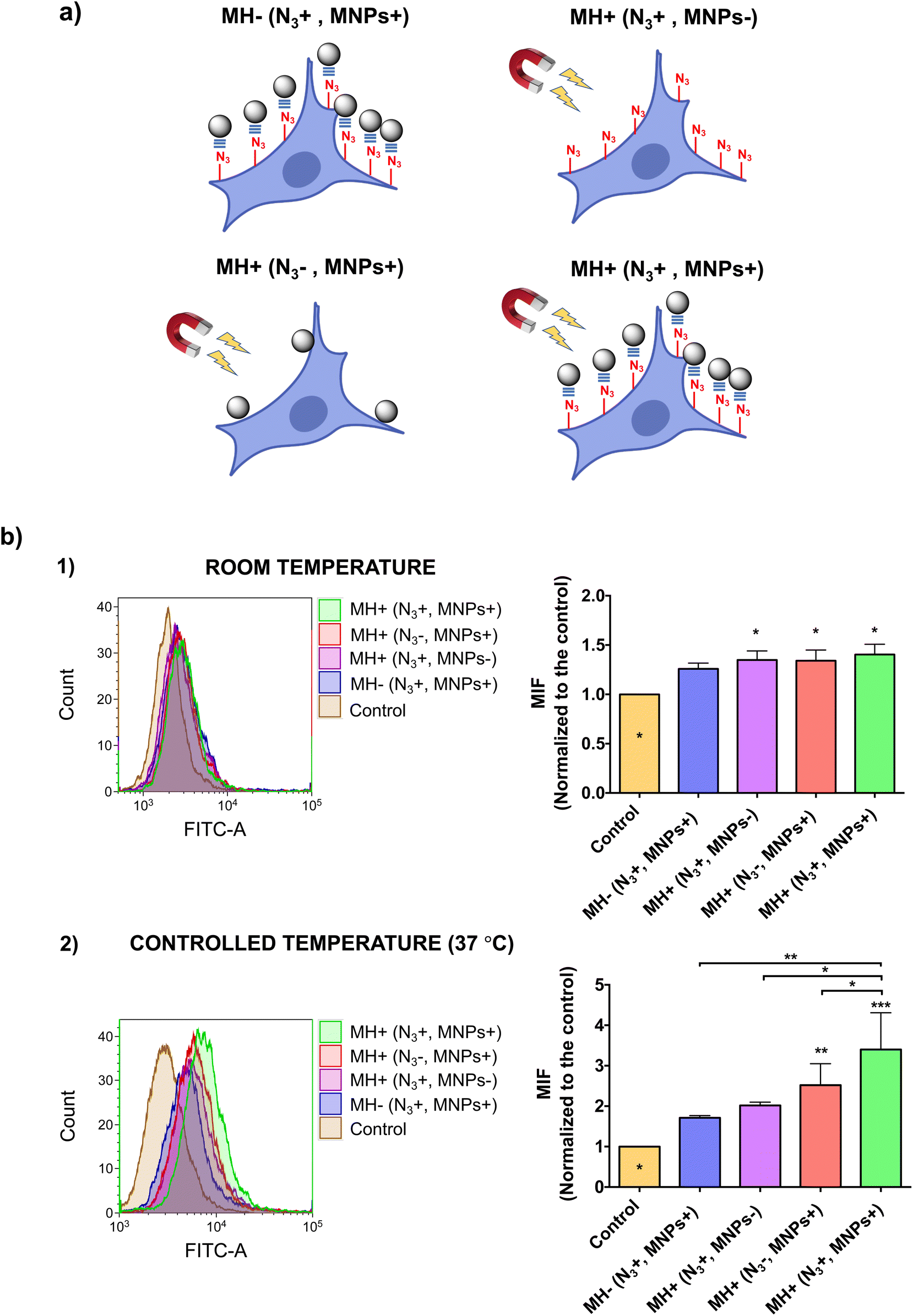 | ||
| Fig. 4 (a) Experimental conditions evaluated for intracellular delivery of YO-PRO®-1. (b) Flow cytometry analysis of YO-PRO®-1internalization with MH. HCT116 cells were treated with Ac4ManNAz for 48 h and then incubated with MNPs@PMAO@PEG@DBCO for 1 h before the AMF application (30 min, 425 kHz, 16.9 kA m−1) at room temperature (1) and 37 °C (2). Black asterisks indicate statistical difference between the normalized fluorescence intensity of the different samples (*p < 0.05; **p < 0.01; one-way ANOVA, Tukey's multiple comparisons test). Data analyses are expressed as mean ± standard deviation of two independent experiments. MFI: mean fluorescence intensity. | ||
For biomedical applications based on cell membrane fluidization, assessing the cell viability at different levels after the cell membrane-localized MH stimulus is essential for establishing the limits of thermal stress. Once the thermal disruption of the plasma membrane ceases, if the cell can cope with the level of thermal stress, it will activate repair processes, such as HSP expression. However, if the thermal stimulation is too severe or prolonged, the cells may not be able to recover and cell death processes could be initiated. The temperature increase induced in the cell may also have effects at the molecular level (e.g., on DNA, proteins, lysosomal membranes, etc.) that eventually could also trigger cell death. With this in mind, we studied the cell viability after MH application at different levels – morphological changes, expression of ROS, apoptosis/necrosis and cell cycle alteration (Fig. 5). We compared the effect of the cell-membrane localized MH and the intracellular MH (see Fig. S6†), to better understand how the localization of the heat source can influence different aspects of cell viability.
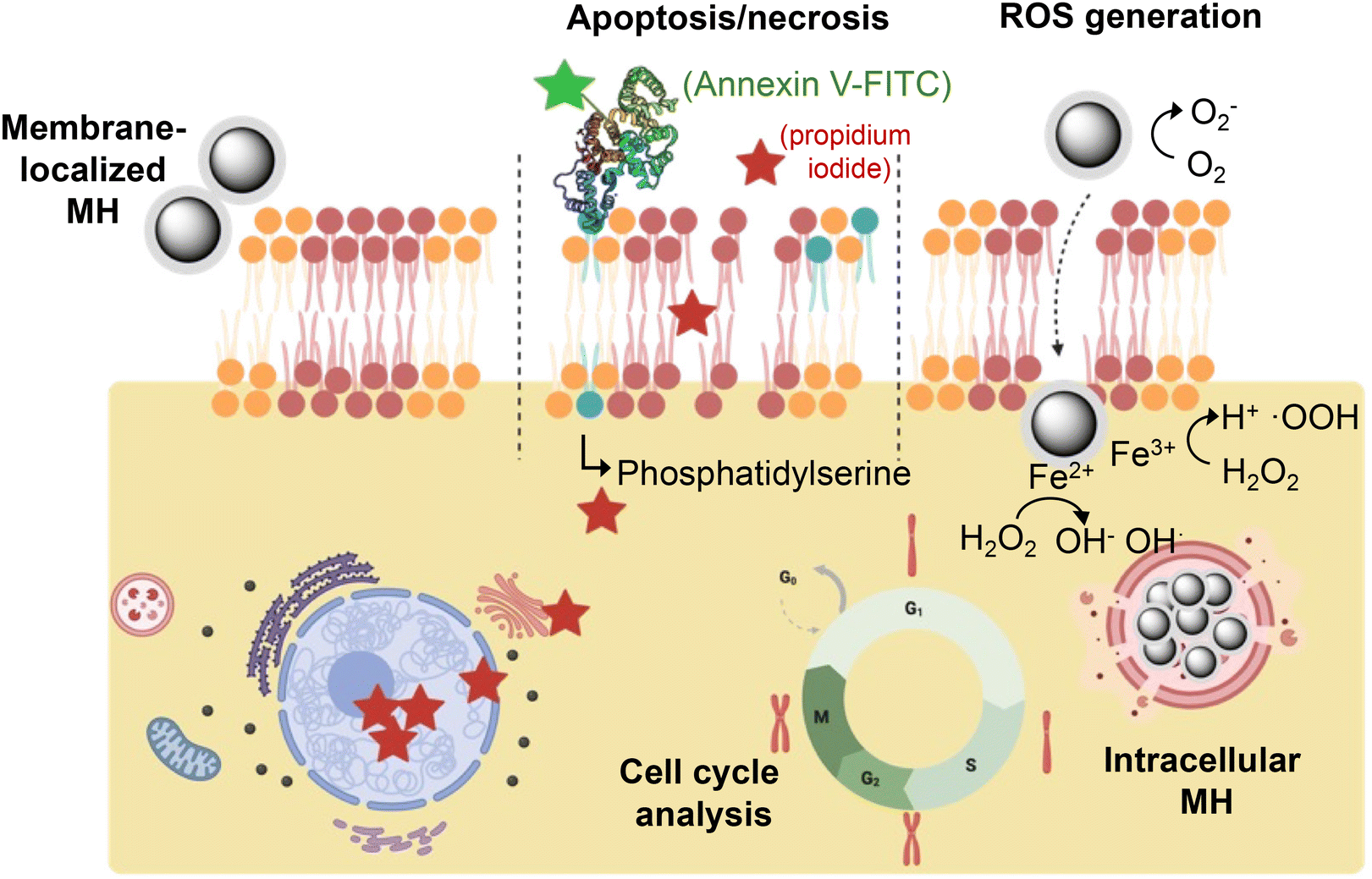 | ||
| Fig. 5 Schematic representation of the different aspects of cell viability post-MH evaluated in this study (apoptosis/necrosis, ROS generation, and cell cycle analysis). The phospholipids in blue correspond to phosphatidylserine and the red stars to propidium iodide. Created with BioRender.com. | ||
Cells subjected to intolerable heat stress undergo easily detectable morphological changes such as an increase in size, stellate morphology, swelling of the nucleus, or even cytoplasmic bubbles as an early indicator of cell death.35 In addition to effects arising from the disruption of the homeostatic equilibrium across the membrane, heat stress can trigger effects at the cytoskeleton level, such as the rearrangement of actin filaments36 or protein denaturation phenomena at the mitotic spindle level. After the application of the thermal stimulus (MH localized at the cell membrane or inside the cell), we observed slight changes in the cell morphology (i.e., slightly rounded morphologies), both for cells in which MNPs were localized on the membrane and for those that had internalized the MNPs. Nevertheless, these changes were transient, and the cells recovered their normal morphology rapidly; after 24 h of incubation, all cells exhibited normal morphology and growth, as evidenced by the increase in cell confluence (Fig. S7†).
Under heat stress, increased cellular metabolism leads to a loss of balance between cellular oxidative production and defense mechanisms (e.g., superoxide dismutase, glutathione peroxidase, ascorbic acid, etc.), resulting in increased oxidative stress.37 Furthermore, it has been shown that despite the high biocompatibility of Fe3O4 MNPs, they can catalyze the generation of ROS both on the surface of the MNP and by degradation and release of Fe3+ ions in the cytosol.38 To quantify the generation of ROS, we used a chloromethyl derivative of dichlorodihydrofluorescein diacetate (CM-H2DCFDA), which can diffuse easily inside the cell. Once in the cytoplasm, this compound is deacetylated by cellular esterases and in the presence of ROS can rapidly oxidize, giving rise to a fluorescent compound that can be detected and quantified by fluorescence microscopy and flow cytometry. We first studied the expression of ROS in response to macroscopic cell heating. For this purpose, cells were heated in an external hot bath for 30 min at different temperatures, ranging from 37 to 45 °C (Fig. S8†). As expected, we observed an increase in ROS production at higher temperatures, with a maximum of 45 °C. For cells incubated at 37 °C, a 24% increase in ROS expression was observed compared to control cells incubated under normal cell culture conditions. Since the macroscopic heating was performed with cells in suspension (due to the configuration of the heating bath), this effect can be attributed to metabolic stress induced by cell detachment, which can trigger the dysregulation of basal ROS levels.39 When analyzing the effect of MH on ROS production, a clear difference was observed depending on the location of the MNPs (Fig. S9†). While for cells containing internalized MNPs the AMF application led to an increase in ROS production of about 53%, in cells with membrane-bound MNPs and under the same MH conditions the increase in ROS production was only 13.5% (this value is in the range obtained for control cells containing internalized or membrane-bound MNPs, but not subjected to an AMF). The difference observed between intracellular and membrane MH can be attributed to effects associated with the localization of the MNPs in intracellular compartments such as lysosomes or the mitochondrial membrane. For example, Domenech et al. observed an increase in lysosomal membrane permeabilization, which correlated with increased levels of cathepsin B (a lysosomal proteolytic enzyme) in the cytoplasm and the production of ROS.40 Likewise, Connord et al. verified in real time an increase in lysosomal membrane permeability and ROS production with only 30 min of application of an AMF.41 Thus, while heating due to internalized MNPs increases cellular ROS production, membrane-localized MH does not cause significant oxidative stress.
If cells cannot cope with heat stress and/or disruption of the cell membrane, they will enter a process of programmed (apoptotic) or necrotic cell death.42,43 We analyzed the effect of MH, both in cells with membrane-bound MNPs and with internalized MNPs, at two different time points: immediately (0 h) and a later time (24 h) after the thermal stimulation (depending on the level of cellular damage, apoptosis may not occur immediately, as indicated in previous reports44). In both cases, we did not observe cell death by apoptosis or necrosis (Fig. S10†). These results confirmed that the localized MH conditions used allow the modification of the fluidity and/or permeability of the cell membrane without affecting cell viability, although the cell can sense the thermal stress and activate molecular response mechanisms, as demonstrated by the HSP expression. Furthermore, the absence of apoptosis supports the internalization of YO-PRO®-1 as a consequence of an increased membrane fluidity through localized MH, as several literature reports are pointing out to YO-PRO®-1 as an indicator of early apoptosis (YO-PRO®-1 can enter the cell through P2X7 receptors, a type of ion channel that is highly active in cells in an apoptotic state45,46).
We next focused our attention on the analysis of the cell cycle. Due to heat stress, the cell cycle can be affected as a consequence of the inhibition of DNA replication or the activation of proteins that act as regulators of cell cycle progression.47 Moreover, numerous studies have shown that intracellular accumulation of MNPs and other types of NPs can affect the cell cycle.48 We used propidium iodide (PI), a fluorescent molecule able to bind the DNA; since the DNA content varies throughout the different stages of the cell cycle, a relationship can be established between the fluorescence intensity detected in each cell and its DNA content.49 We did not observe alterations of the cell cycle under any of the different conditions of thermal stress, or caused by MNP presence on the cell membrane or in intracellular compartments (Fig. S11†). This observation is in line with previous studies realized by our group using similar MNPs; after internalization in B16 cells (mouse melanoma cells) and AMF application, no variations in cell cycle phases were observed, even at higher AMF frequencies and amplitudes compared to those employed in this work (835 kHz and 20.05 kA m−1vs. 425 kHz and 16.9 kA m−1).50
Taken together, our results demonstrate that cell membrane-localized MH can act as a disruptive agent of cell membrane fluidity and permeability, leading to the internalization of cell-impermeant probes without impairing cell viability.
3.4 Molecular study of the localized hyperthermia effect – heat shock protein (HSP) activation
Although no direct impact of the localized MH on cell viability was detected, we questioned whether the cells could actively sense the thermal effects. It is widely acknowledged that cells respond to thermal stress by activating different molecular mechanisms to repair or limit the damage caused. Expression of HSPs is one of the predominant molecular mechanisms of response to heat stress; therefore, these proteins have been extensively used to assess the effects of MH at the cellular level.50–52 However, in all these studies, MNPs were internalized; to the best of our knowledge, there is no report on the effect of cell-membrane localized heating on HSP expression. On the other hand, several studies in bacteria, yeast and mammalian cells have indicated a link between the fluidity of the cell membrane and the induction of HSP expression, but all these studies have been performed using chemical agents for membrane fluidization (such as benzyl and phenethyl alcohol).53–56 In this work, we investigated for the first time the expression of different HSPs in response to the thermal stimulus induced by MH localized on the cell membrane and compared it to the one triggered by intracellular MH. The HSP70 family is the most evolutionarily conserved amongst all HSPs; all eukaryotes present more than one gene encoding HSP70 proteins with different preferential intracellular localizations.57 We selected three members of this family, namely HSP70-1A, HSP70–8 and HSP70–9, and analyzed the expression of their genes (HSPA1A, HSPA8, and HSPA9, respectively) by real-time quantitative polymerase chain reaction (RT-qPCR) due to its high sensitivity, which allows detection of subtle expression changes. In both scenarios (membrane-localized MH and intracellular MH), MNPs were attached to HCT116 cell membranes via click chemistry. For membrane-localized MH studies, cells were exposed to the AMF immediately after the click reaction, while for intracellular MH the MNPs were allowed to internalize for 24 h after the click reaction (Fig. 6a). However, as after performing the click reaction the unreacted MNPs were washed, we expect the same overall amount of MNPs in both cases. | ||
| Fig. 6 (a) Schematic representation of the experimental conditions for assessing the effect of MH at different cellular localizations (on the cell membrane and inside the cell). (b) Expression of HSPA1A, HSPA8 and HSPA9 genes after exposure to heat shock at different temperatures (macroscopic heating) or after application of an AMF (30 min, 425 kHz, 16.9 kA m−1) in cells with internalized or membrane immobilized MNPs. Expression levels were normalized with respect to the 18S reference gene and compared to gene expression in control cells incubated at 37 °C. Black asterisks indicate significant differences with respect to the control (*p < 0.05; **p < 0.01; and ***p < 0.001; one-way ANOVA, Dunnett's multiple comparisons test). | ||
The HSP expression under the MH stimulus was compared to the one triggered by macroscopic heating of the cells at different temperatures (39, 41, and 43 °C) using an external hot water bath. The heat shock response is characterized by a rapid increase in HSP expression after exposure to environmental stress; however, as mRNA has a short half-life (less than one hour58), the level of expression can decrease rapidly. Indeed, previous studies carried out in our group revealed a clear descent in the expression of the mouse hsp70 gene after the MH stimulus, with basal expression values 120 min after the AMF application.50 For this reason, the analysis of the expression of the selected genes was carried out on the mRNA extracted immediately after the application of the thermal stimulus (macroscopic heating or MH). We observed that the expression levels of the analyzed genes depended highly on both the type of HSP and the type and localization of the thermal stimulus (Fig. 6b). HSPA1A had the highest expression level in the case of macroscopic heating, probably due to the role of HSP70-1A protein in the repair of heat-induced DNA damage.59 However, cells exposed to membrane-localized or intracellular MH had a relatively low expression of HSP70 mRNA, with only a two-fold increase compared to the control (cells incubated at 37 °C). As the heating sources (MNPs) are very localized (attached to cell membrane glycoproteins or confined inside lysosomes), the thermal effect produced is likely not high enough to alter DNA stability and trigger the activation of HSPA1A. In the case of HSPA8, similar levels of expression were observed under cell-membrane localized and intracellular MH conditions, both being higher than for macroscopic heating. HSP70-8 proteins interact with membrane lipids directly through the C-terminal end and play a key role in the folding of membrane proteins, thus facilitating the translocation of polypeptides across membranes.60 They are also involved in restoring cell membrane fluidity through a cholesterol-dependent mechanism.61 These two functions can account for the expression levels observed for the HSPA8 gene when MH is localized on the cell membrane. On the other hand, HSPA8 proteins have a protecting role against reactive oxygen species (ROS) production,62 which could explain their overexpression when cells are subjected to thermal stress from intracellular MNPs. Finally, for HSPA9 we observed a higher level of expression for intracellular MH and cell membrane-localized than in the case of macroscopic heating, with the higher expression in the case of cells with internalized MNPs and subjected to the AMF. The expression of HSP70-9, a protein primarily localized in mitochondria, is independent of heat stress and is regulated by alterations in the Ca2+ influx, decreasing glucose levels, and disruption of glycolysis.63 We hypothesize that an effect on mitochondrial stability induced by intracellular MH could trigger its expression,64 in line with the results observed for the ROS expression under intracellular MH conditions. Taken together, these results confirmed the molecular response of HCT116 cells to the thermal stimulus delivered by cell membrane-localized and intracellular MNP-based hyperthermia, indicating that the cell is able to respond to the disruption of membrane fluidity through the expression of membrane-stabilizing HSPs, such as HSP70-8. The differences in the HSP expression observed between MH or exposure to macroscopic increases in temperature reflected how the cell can activate the molecular defense response to heat stress differently when subjected to local thermal stimulation by MNPs, as previously described by other authors.50–52
3.5 Transfection based on membrane-localized magnetic hyperthermia
After confirming the potential of cell membrane-localized MH to induce changes in the membrane fluidity and favor the internalization of impermeant exogenous molecules without detrimental effects on cell viability, we sought to extend its use to a specific biomedical application, namely cell transfection. Cell transfection, i.e., the incorporation of exogenous nucleic acids into target cells holds therapeutic promise for many diseases linked to genetic disorders.65–67 Interference RNA (RNAi), genome, and gene editing tools allow the manipulation of DNA and edition of gene sequences, regulate gene expression, and reconfigure chromatin structure, thus modulating specific traits in the target cells’ phenotype.68,69 In particular, small interference RNA (siRNA) has been profusely used to target specific messenger RNA (mRNA) in the cells’ cytoplasm to suppress protein translation.70 The efficacy of siRNA action depends on the capability to penetrate the cell membrane and avoid intracellular enzymatic degradation or lysosomal entrapment. Transfection of nucleic acids may be achieved via many different approaches, including viral vectors,71 physical72 or chemical (e.g., lipids)73 methods, all having disadvantages, such as direct cell toxicity, loading efficiency or cell specificity.74Advances in materials science and nanotechnology in the past decades have put forward a range of nanomaterials as tools for the transport and delivery of (therapeutic) nucleic acids.75 While MNPs have been widely exploited for magnetic-field guided delivery of nucleic acids, a technique known as “magnetofection”,76–79 the localized heating that takes place in the vicinity of the MNP surface when they are exposed to external AMFs has not been used to date for transfection purposes. Herein, we propose the use of MNPs as “hotspots” able to induce changes in the fluidity of cell membranes and promote the transient transfection of antisense siRNA in MCF7 cells to silence the expression of a green fluorescent protein (Fig. 7). The MCF7 cell line is derived from a pleural effusion acquired from metastatic breast cancer and is frequently used as a model of estrogen-positive breast cancer. Besides, MCF7 cells can be addressed as an interesting model for the development of novel therapies against breast cancer (e.g., gene therapy) due to their remarkable genetic characteristics, such as chromosomal instability and clonal heterogeneity.80,81 Several commercial viral and non-viral transfection reagents have been reported as suitable for the transfection of nucleic acids in MCF7 cells.82 However, MCF7 cells typically grow in clumps or clusters, which makes the transfection and the uptake of the therapeutic cargo difficult due to lower exposure of the cell's membrane surface. Therefore, we considered the MCF7 cell line as a relevant model for developing a new transfection method based on localized magnetic hyperthermia and cell surface engineering through bioorthogonal chemistry. An overview of the general concept of cell membrane-localized MH-mediated transfection using MNPs immobilized on the cell membrane via SPAAC bioorthogonal chemistry is presented in Fig. 7.
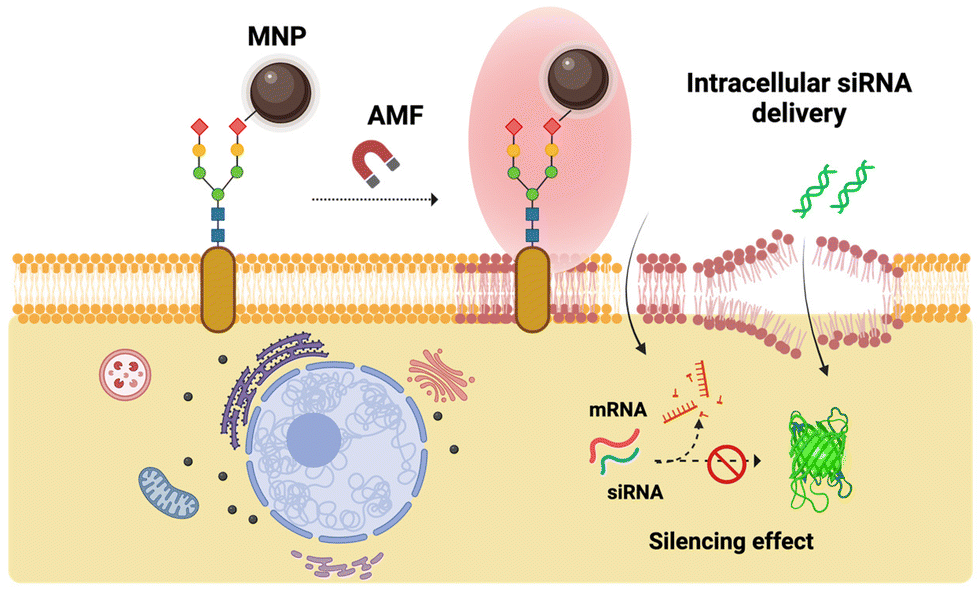 | ||
| Fig. 7 Overview of the general concept of cell membrane-localized MH-mediated transfection. Upon application of the AMF, the thermal disruption of the cell membrane is used for intracellular delivery of siRNA to silence the constitutively expressed copGFP. Created with BioRender.com. | ||
Optimization of the conditions for cell transfection experiments was initially carried out in MCF7 cells co-transfected with the pAcGFP1 Nuc vector; full details on this optimization are provided in the ESI (Fig. S12–S14†). For MH silencing experiments, MCF7 cells constitutively expressing copGFP were incubated with Ac4ManNAz for 48 h before MH to provide them with artificial azide reporters for the click immobilization of MNPs on the membrane, as described earlier (Day 2 and Day 3 in Fig. 8). Hereafter cells were incubated with MNPs@PMAO@PEG@DBCO (at 10 µgFe mL−1 for 10 minutes) and the AMF (23.9 kA m−1 and 418 kHz) was applied for 30 minutes with pulses (each pulse with a duration of five minutes, with a sixty second pause between pulses, Day 4 in Fig. 8). The transfection efficiency was compared with a positive control using the Lipofectamine RNAiMax reagent under the same experimental conditions of MH samples. Moreover, control experiments using cells transfected with siRNA and cells with Ac4ManNAz treatment, and incubated with MNPs, but without MH application (MNPs + N3 + MH−) were used to verify if the presence of MNPs could promote the intracellular siRNA delivery. copGFP silencing was followed for all the conditions tested using fluorescence microscopy, 24 and 48 h after MH (Day 5 and Day 6 in Fig. 8) (Fig. S15a†). After 48 h, RNA was extracted and expression analysis by RT-qPCR confirmed the effectiveness of MH on the silencing of copGFP (Fig. 9a). The results revealed that in cells with MNPs attached to the membrane via click chemistry and exposed to the AMF (MNPs + N3 + MH+), and copGFP expression was 40% lower. This silencing was comparable (no statistical difference) with that obtained with the commercial Lipofectamine RNAiMax (around 40% reduction in AcGFP1 expression; Fig. 9a). Moreover, in the absence of AMF (MNPs + N3 + MH−), no statistically significant reduction of copGFP expression was observed compared to controls (cells with and without siRNA), indicating the potential of localized MH for transfection in MCF7/GFP cells. The quantitative analysis of corrected total cell fluorescence (CTCF) of fluorescence microscopy images from the 48 h post-MH, demonstrated a statistically significant difference between MNPs + N3 + MH+ in the reduction of copGFP protein expression (Fig. S15b†). Together, these results confirmed that the MNPs immobilized on the cell membrane acted as hotspots under an AMF, inducing localized heating and triggering an increase in the fluidity of the membrane, which enhances the transfection of exogenous molecules. The effect observed reached its maximum when the cell membrane glycocalyx was labeled with azides, as the density of hotspots generated on the membrane was the highest among all the different conditions tested.
 | ||
| Fig. 8 Scheme of the experimental design used to evaluate the siRNA transfection via MH in MCF7 cells expressing GFP, labeled with azide groups. Created with BioRender.com. | ||
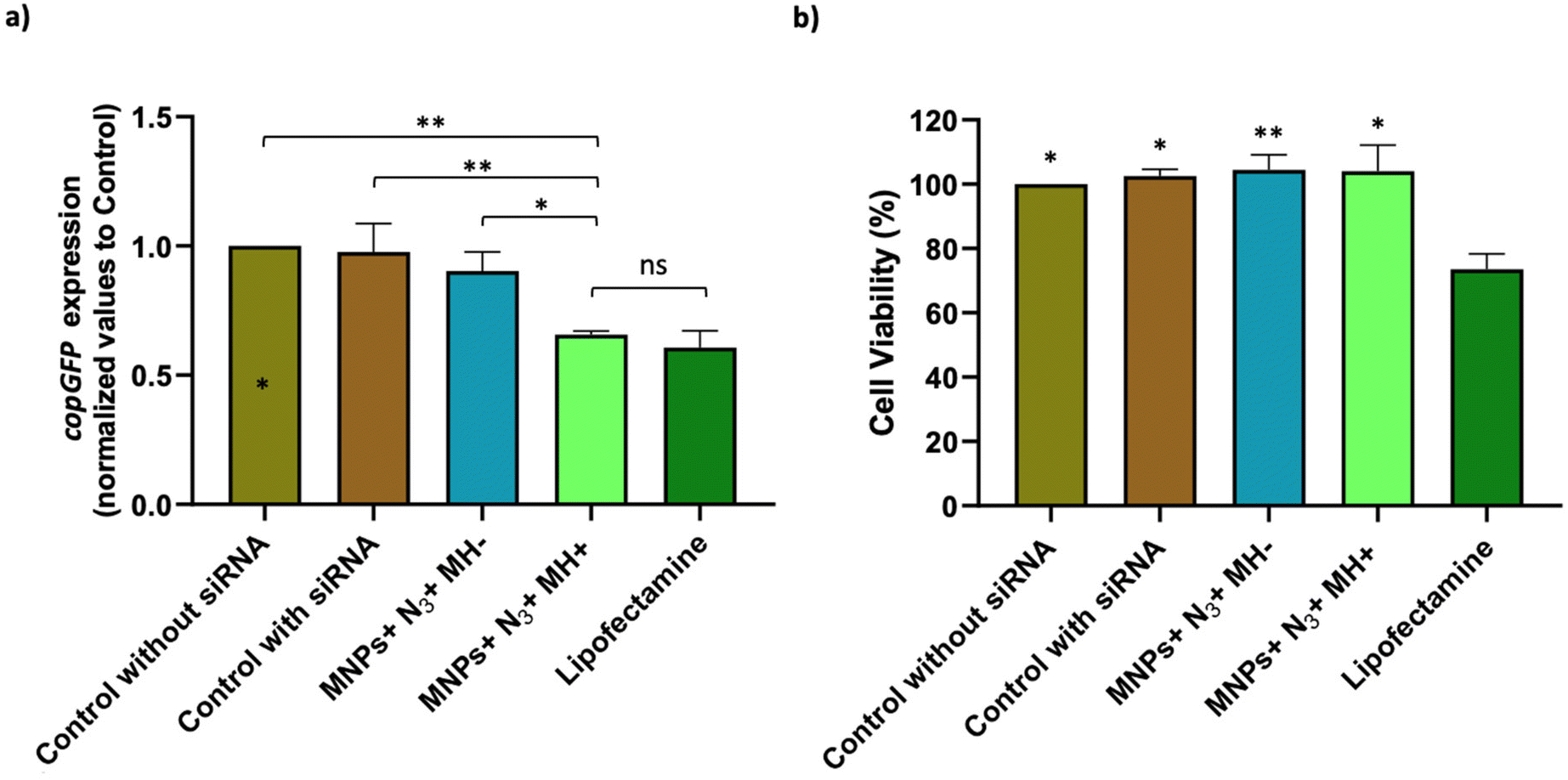 | ||
| Fig. 9 RT-qPCR analysis and cell viability assessment 48 h post-siRNA transfection in MCF7/GFP cells. (a) RT-qPCR to evaluate copGFP gene expression in the different samples tested. The gene expression levels were normalized to the control. Black asterisks indicate statistical differences between MNPs + N3 + MH+ and the controls (cells with and without siRNA) and MNPs + N3 + MH− (**p < 0.01; *p < 0.05, respectively; unpaired parametric t-test with Welch's correction). Data represent the mean value ± the standard error mean of three biologically independent experiments with two technical replicates for each. (b) Cell viability analysis of the different conditions tested after MH via MTS assay. The statistical significance between the cells treated with lipofectamine and the other samples is represented (*p < 0.05; **p < 0.01; unpaired parametric t-test with Welch's correction). | ||
One of the most important limitations of the commercially available transfection methods is their impact on cell viability, with many reports describing the cytotoxicity of cationic lipid reagents82,83 or the impact of electroporation on cell viability.84 For this reason, once the efficiency of the transfection and the silencing of the copGFP gene was demonstrated, the impact on cell viability was assessed. Lipofectamine was found to induce a statistically significant reduction of cell viability of circa 25% not observed for MH-treated cells (MNPs + N3 + MH+) (cell viability of 100%) (Fig. 9b).
Finally, it is worth mentioning that the results obtained for siRNA transfection in MCF7 cells with transient expression of GFP (see details in the ESI, Fig. S16 and S17†) were similar to the ones attained in MCF7 cells constitutively expressing copGFP, both in terms of efficacy and cell viability. Altogether, our results indicate that this new transfection method can attain similar efficiencies of transfection compared to the gold standard lipofectamine without cellular cytotoxicity. Consequently, it is crucial to optimize in each situation the amount of silencing oligo (siRNA) to be used to improve gene silencing.
4. Conclusions
In this work, we report the use of cell membrane-localized MH as a disruptive agent of cell membrane permeability. We investigated different aspects related to cell viability following the discontinuation of the thermal stimulus, confirming the absence of apoptotic/necrotic events, the generation of ROS, and alterations of the cell cycle. However, the molecular study of heat stress response revealed that not only were HSPs activated in response to the thermal stimulus, but their expression was different, depending on the nature and location of the heat shock agent, and associated with the role and preferential localization and the site of action of each HSP studied.The alteration of the membrane permeability was first verified by the internalization of YO-PRO®-1, a cell-impermeant fluorescent molecule. Then, as a global proof-of-concept of potential biomedical applications, we validated the localized MH successful transfection of siRNA into MCF7 cells (transiently or stably expressing GFP) and the subsequent GFP silencing. The membrane localized hyperthermia-mediated transfection had similar efficacy to “gold standard” methods based on lipofectamine, but without affecting cell viability. This approach addresses some of the challenges associated with standard transfection methods, providing a safer method for improving the delivery and action of (therapeutic) nucleic acids, with spatio-temporal control through the application of an AMF. Taken together, these results suggest the modulation of the cell membrane permeability and fluidity based on the so-called “hotspot” behavior of the MNPs. These findings are supported by a recent report from our group on the effect of the localized increase in temperature in the immediate environment of individual MNPs deposited on a model cell membrane upon the application of the AMF.16 The use of MH as a stimulus for inducing membrane permeabilization has also been documented by Alvarez-Berríos et al., who proved that Caco-2 cells subjected to magnetic fluid hyperthermia using iron oxide carboxymethyl dextran nanoparticles showed a higher sensitivity to cisplatin therapy than in the case of a similar temperature increase (41 °C) induced by hot water hyperthermia, an effect attributed to an increase of cell membrane fluidity favoring the passive transport of the drug across the membrane.85 However, in this report the MNPs were not immobilized onto the membranes of living cells, but suspended in the cell culture medium. On the other hand, Hulangamuwa et al. found that the use of internalized MNPs in human lymphoma cells (U937) and the application of very short pulses (200 μs) of an inhomogeneous magnetic field at a frequency of 30.9 kHz favored the internalization of a model drug (doxorubicin) due to the change in membrane permeability.86 Nonetheless, this phenomenon did not stem from the “hotspot” effect of the MNPs; rather, it was caused by the transmission of ultrasonic frequency sound waves resulting from the translational motion of MNPs within the applied magnetic field.
While further research into the cell plasma membrane thermal disruption with localized MH and the mechanism of internalization of different probes is needed, we believe that our strategy could lead to the development of a more efficient procedure for the “opening and closing” of cellular membranes with minimal disturbance of cellular integrity. This on-demand modification of cell membrane permeability will allow the MH-mediated intracellular transport of biologically relevant (bio)molecules, drugs and nanomaterials, promoting direct intracellular delivery and thus overcoming traditional endocytosis pathways and avoiding endosomal entrapment. Moreover, cell surface engineering with artificial chemical reporters such as the azide groups and the nanoparticle immobilization via bioorthogonal chemistry could provide a universal tool for intracellular delivery, applicable practically to any cell line and nanoparticle type.
Author contributions
J.I.L.: conceptualization, investigation, data curation, methodology, formal analysis, and writing – original draft; D.F.: conceptualization, investigation, data curation, methodology, formal analysis, and writing – original draft; L.A.: investigation, data curation, and formal analysis; M.M.: investigation, formal analysis, funding acquisition, and writing – review and editing; I.A.: investigation, data curation, formal analysis, and writing – review and editing; V.G.: conceptualization, funding acquisition, project administration, supervision, and writing – review and editing; A.R.F.: conceptualization, funding acquisition, project administration, supervision, and writing – original draft, review and editing; J.M.F.: conceptualization, funding acquisition, project administration, supervision, and writing – review and editing; P.V.B.: conceptualization, funding acquisition, project administration, supervision, and writing – original draft, review and editing; R.M.F.: conceptualization, investigation, funding acquisition, project administration, supervision, and writing – original draft, review and editing.All authors have given approval to the final version of the manuscript.
Data availability
Most data supporting this article have been included as part of the main text and the ESI.† Additional data (data regarding the MNP surface temperature estimation, magnetic measurements, and qPCR data for the HSP expression) can be found in the Zenodo repository (https://zenodo.org/records/12683703). Further data (raw flow cytometry data for YO-PRO internalization, cell viability, generation of reactive oxygen species and cell cycle analysis and raw RT-qPCR data for the transfection experiments) are available from the corresponding authors upon reasonable request.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported by the following grants: AEI, Grant PCIN-2017-060 funded by MICIU/AEI/10.13039/501100011033 and co-funded by the European Union (M-ERA.NET COFUND call 2016), grants PGC2018-096016-B-I00 to R. M. F and BIO 2017-84246-C2-1R to V. G. and J. M. F. funded by MICIU/AEI/10.13039/501100011033 and by “ERDF A way of making Europe”, grant RYC2015-17640 to R. M. F. and RYC2019-026860-I to M. M. funded by MICIU/AEI/10.13039/501100011033 and by “ESF Investing in your future”, and grant CNS2023-144436 funded by MICIU/AEI/10.13039/501100011033 and by “European Union Next Generation EU/PRTR”. This work was also financed by national funds from FCT – Fundação para a Ciência e a Tecnologia, I. P., in the scope of the project UIDP/04378/2020 and UIDB/04378/2020 of the Research Unit on Applied Molecular Biosciences – UCIBIO and the project LA/P/0140/2020 of the Associate Laboratory Institute for Health and Bioeconomy – i4HB, project M-ERA.NET2/0008/2016, DF also acknowledges the FCT/MCTES grant 2020.06599.BD. J. I. L. acknowledges financial support for his predoctoral fellowships from Gobierno de Aragón (DGA 2017-2021 call, co-funded by the Programa Operativo Fondo Social Europeo de Aragón 2014–2020). L. A. acknowledges support from the José Castillejo program (CAS18/00233). The authors also acknowledge support from Gobierno de Aragón and Fondos Feder for funding the Bionanosurf (E15_23R) research group. The authors would like to acknowledge the use of Servicios Cientifícos Técnicos del CIBA (IACS-Universidad de Zaragoza), the Advanced Microscopy Laboratory (Universidad de Zaragoza), for access to their instrumentation and expertise and for the use of Servicio General de Apoyo a la Investigación-SAI, Universidad de Zaragoza, BIOLAB@UCIBIO, NOVA School of Science and Technology for flow cytometry experiments. We also thank Eduardo Moreno-Antolín (Bionanosurf group, INMA, UNIZAR-CSIC) for insightful discussions and help with the preparation of the MNPs.References
- R. Hao, R. Xing, Z. Xu, Y. Hou, S. Gao and S. Sun, Adv. Mater., 2010, 22, 2729–2742 CrossRef CAS PubMed.
- S. Laurent, A. A. Saei, S. Behzadi, A. Panahifar and M. Mahmoudi, Expert Opin. Drug Delivery, 2014, 11, 1449–1470 CrossRef CAS PubMed.
- J. Mosayebi, M. Kiyasatfar and S. Laurent, Adv. Healthcare Mater., 2017, 6, 1700306 CrossRef PubMed.
- S. M. Dadfar, K. Roemhild, N. I. Drude, S. von Stillfried, R. Knüchel, F. Kiessling and T. Lammers, Adv. Drug Delivery Rev., 2019, 138, 302–325 CrossRef CAS PubMed.
- A. Farzin, S. A. Etesami, J. Quint, A. Memic and A. Tamayol, Adv. Healthcare Mater., 2020, 9, 1901058 CrossRef CAS PubMed.
- P. Das, M. Colombo and D. Prosperi, Colloids Surf., B, 2019, 174, 42–55 CrossRef CAS PubMed.
- M. Moros, J. Idiago-López, L. Asín, E. Moreno-Antolín, L. Beola, V. Grazú, R. M. Fratila, L. Gutiérrez and J. M. de la Fuente, Adv. Drug Delivery Rev., 2019, 138, 326–343 CrossRef CAS PubMed.
- H. Huang, S. Delikanli, H. Zeng, D. M. Ferkey and A. Pralle, Nat. Nanotechnol., 2010, 5, 602–606 CrossRef CAS PubMed.
- R. Munshi, S. M. Qadri, Q. Zhang, I. Castellanos Rubio, P. del Pino and A. Pralle, eLife, 2017, 6, e27069 CrossRef PubMed.
- R. Munshi, S. M. Qadri and A. Pralle, Front. Neurosci., 2018, 12, 560 CrossRef PubMed.
- A. Chiu-Lam and C. Rinaldi, Adv. Funct. Mater., 2016, 26, 3933–3941 CrossRef CAS PubMed.
- S. Noh, S. H. Moon, T.-H. Shin, Y. Lim and J. Cheon, Nano Today, 2017, 13, 61–76 CrossRef CAS.
- M. Saeed, W. Ren and A. Wu, Biomater. Sci., 2018, 6, 708–725 RSC.
- E. Cazares-Cortes, S. Cabana, C. Boitard, E. Nehlig, N. Griffete, J. Fresnais, C. Wilhelm, A. Abou-Hassan and C. Ménager, Adv. Drug Delivery Rev., 2019, 138, 233–246 CrossRef CAS PubMed.
- I. Armenia, M. V. Grazú Bonavia, L. De Matteis, P. Ivanchenko, G. Martra, R. Gornati, J. M. de la Fuente and G. Bernardini, J. Colloid Interface Sci., 2019, 537, 615–628 CrossRef CAS PubMed.
- S. Ruiz-Rincón, A. González-Orive, V. Grazú, R. M. Fratila, J. M. de la Fuente and P. Cea, Colloids Surf., B, 2020, 196, 111315 CrossRef PubMed.
- H. Koo, M. S. Huh, I.-C. Sun, S. H. Yuk, K. Choi, K. Kim and I. C. Kwon, Acc. Chem. Res., 2011, 44, 1018–1028 CrossRef CAS PubMed.
- J. Idiago-López, E. Moreno-Antolín, M. Eceiza, J. M. Aizpurua, V. Grazú, J. M. de la Fuente and R. M. Fratila, Bioconjugate Chem., 2022, 33, 1620–1633 CrossRef PubMed.
- H. Y. Yoon, H. Koo, K. Kim and I. C. Kwon, Biomaterials, 2017, 132, 28–36 CrossRef CAS PubMed.
- J. G. Ovejero, I. Armenia, D. Serantes, S. Veintemillas-Verdaguer, N. Zeballos, F. López-Gallego, C. Grüttner, J. M. de la Fuente, M. del Puerto Morales and V. Grazu, Nano Lett., 2021, 21, 7213–7220 CrossRef CAS PubMed.
- J. Du, M. A. Meledeo, Z. Wang, H. S. Khanna, V. D. P. Paruchuri and K. J. Yarema, Glycobiology, 2009, 19, 1382–1401 CrossRef CAS PubMed.
- H. Koo, S. Lee, J. H. Na, S. H. Kim, S. K. Hahn, K. Choi, I. C. Kwon, S. Y. Jeong and K. Kim, Angew. Chem., Int. Ed., 2012, 51, 11836–11840 CrossRef CAS PubMed.
- S. Lee, H. Koo, J. H. Na, S. J. Han, H. S. Min, S. J. Lee, S. H. Kim, S. H. Yun, S. Y. Jeong, I. C. Kwon, K. Choi and K. Kim, ACS Nano, 2014, 8, 2048–2063 CrossRef CAS PubMed.
- M. Moros, B. Pelaz, P. López-Larrubia, M. L. García-Martin, V. Grazú and J. M. de la Fuente, Nanoscale, 2010, 2, 1746 RSC.
- R. M. Fratila, M. Navascuez, J. Idiago-López, M. Eceiza, J. I. Miranda, J. M. Aizpurua and J. M. de la Fuente, New J. Chem., 2017, 41, 10835–10840 RSC.
- M. Moros, B. Hernáez, E. Garet, J. T. Dias, B. Sáez, V. Grazú, Á. González-Fernández, C. Alonso and J. M. de la Fuente, ACS Nano, 2012, 6, 1565–1577 CrossRef CAS PubMed.
- A. Makridis, S. Curto, G. C. van Rhoon, T. Samaras and M. Angelakeris, J. Phys. D: Appl. Phys., 2019, 52, 255001 CrossRef CAS.
- R. Hergt and S. Dutz, J. Magn. Magn. Mater., 2007, 311, 187–192 CrossRef CAS.
- A. Riedinger, P. Guardia, A. Curcio, M. A. Garcia, R. Cingolani, L. Manna and T. Pellegrino, Nano Lett., 2013, 13, 2399–2406 CrossRef CAS PubMed.
- L. Polo-Corrales and C. Rinaldi, J. Appl. Phys., 2012, 111, 07B334 CrossRef.
- R. Piñol, C. D. S. Brites, R. Bustamante, A. Martínez, N. J. O. Silva, J. L. Murillo, R. Cases, J. Carrey, C. Estepa, C. Sosa, F. Palacio, L. D. Carlos and A. Millán, ACS Nano, 2015, 9, 3134–3142 CrossRef PubMed.
- J. T. Dias, M. Moros, P. Del Pino, S. Rivera, V. Grazú and J. M. de la Fuente, Angew. Chem., Int. Ed., 2013, 52, 11526–11529 CrossRef CAS PubMed.
- T. Melnik, T. Povarnitsyna, H. Solonenko and B. Melnik, Thermochim. Acta, 2011, 512, 71–75 CrossRef CAS.
- C. Castro-Hinojosa, S. Del Sol-Fernández, E. Moreno-Antolín, B. Martín-Gracia, J. G. Ovejero, J. M. de la Fuente, V. Grazú, R. M. Fratila and M. Moros, Bioconjugate Chem., 2023, 34, 2275–2292 CrossRef CAS PubMed.
- F. Wang, Y. Li, L. Chen, D. Chen, X. Wu and H. Wang, Biomicrofluidics, 2012, 6, 014120 CrossRef PubMed.
- R. M. Rivera, K. L. Kelley, G. W. Erdos and P. J. Hansen, Biol. Reprod., 2004, 70, 1852–1862 CrossRef CAS PubMed.
- I. Belhadj Slimen, T. Najar, A. Ghram, H. Dabbebi, M. Ben Mrad and M. Abdrabbah, Int. J. Hyperthermia, 2014, 30, 513–523 CrossRef CAS PubMed.
- T. Mai and J. Z. Hilt, Colloids Surf., A, 2019, 576, 9–14 CrossRef CAS.
- J. A. Eble and F. F. de Rezende, Antioxid. Redox Signal., 2014, 20, 1977–1993 CrossRef CAS PubMed.
- M. Domenech, I. Marrero-Berrios, M. Torres-Lugo and C. Rinaldi, ACS Nano, 2013, 7, 5091–5101 CrossRef CAS PubMed.
- V. Connord, P. Clerc, N. Hallali, D. El Hajj Diab, D. Fourmy, V. Gigoux and J. Carrey, Small, 2015, 11, 2437–2445 CrossRef CAS PubMed.
- B. V. Harmon, A. M. Corder, R. J. Collins, G. C. Gobé, J. Allen, D. J. Allan and J. F. R. Kerr, Int. J. Radiat. Biol., 1990, 58, 845–858 CrossRef CAS PubMed.
- I. Shlomovitz, M. Speir and M. Gerlic, Cell Commun. Signaling, 2019, 17, 139 CrossRef PubMed.
- C. Blanco-Andujar, D. Ortega, P. Southern, S. A. Nesbitt, N. T. K. Thanh and Q. A. Pankhurst, Nanomedicine, 2016, 11, 121–136 CrossRef CAS PubMed.
- T. Idziorek, J. Estaquier, F. De Bels and J.-C. Ameisen, J. Immunol. Methods, 1995, 185, 249–258 CrossRef CAS PubMed.
- F. Rassendren, G. N. Buell, C. Virginio, G. Collo, R. A. North and A. Surprenant, J. Biol. Chem., 1997, 272, 5482–5486 CrossRef CAS PubMed.
- A. K. Velichko, E. N. Markova, N. V. Petrova, S. V. Razin and O. L. Kantidze, Cell. Mol. Life Sci., 2013, 70, 4229–4241 CrossRef CAS PubMed.
- M. Mahmoudi, K. Azadmanesh, M. A. Shokrgozar, W. S. Journeay and S. Laurent, Chem. Rev., 2011, 111, 3407–3432 CrossRef CAS PubMed.
- Y. Shen, P. Vignali and R. Wang, Bio-Protoc., 2017, 7(16), e2517 Search PubMed.
- M. Moros, A. Ambrosone, G. Stepien, F. Fabozzi, V. Marchesano, A. Castaldi, A. Tino, J. M. de la Fuente and C. Tortiglione, Nanomedicine, 2015, 10, 2167–2183 CrossRef CAS PubMed.
- A. Mukherjee, M. Castanares, M. Hedayati, M. Wabler, B. Trock, P. Kulkarni, R. Rodriguez, R. H. Getzenberg, T. L. DeWeese, R. Ivkov and S. E. Lupold, Nanomedicine, 2014, 9, 2729–2743 CrossRef CAS PubMed.
- M. E. De Sousa, A. Carrea, P. Mendoza Zélis, D. Muraca, O. Mykhaylyk, Y. E. Sosa, R. G. Goya, F. H. Sánchez, R. A. Dewey and M. B. Fernández Van Raap, J. Phys. Chem. C, 2016, 120, 7339–7348 CrossRef CAS.
- L. Vigh, B. Maresca and J. L. Harwood, Trends Biochem. Sci., 1998, 23, 369–374 CrossRef CAS PubMed.
- I. Gombos, T. Crul, S. Piotto, B. Güngör, Z. Török, G. Balogh, M. Péter, J. P. Slotte, F. Campana, A.-M. Pilbat, Á. Hunya, N. Tóth, Z. Literati-Nagy, L. Vígh, A. Glatz, M. Brameshuber, G. J. Schütz, A. Hevener, M. A. Febbraio, I. Horváth and L. Vígh, PLoS One, 2011, 6, e28818 CrossRef CAS PubMed.
- B. Csoboz, G. E. Balogh, E. Kusz, I. Gombos, M. Peter, T. Crul, B. Gungor, L. Haracska, G. Bogdanovics, Z. Torok, I. Horvath and L. Vigh, Int. J. Hyperthermia, 2013, 29, 491–499 CrossRef CAS PubMed.
- G. Balogh, I. Horváth, E. Nagy, Z. Hoyk, S. Benkõ, O. Bensaude and L. Vígh, FEBS J., 2005, 272, 6077–6086 CrossRef CAS PubMed.
- M. Daugaard, M. Rohde and M. Jäättelä, FEBS Lett., 2007, 581, 3702–3710 CrossRef CAS PubMed.
- N. G. Theodorakis and R. I. Morimoto, Mol. Cell. Biol., 1987, 7, 4357–4368 CAS.
- S. B. Nadin, F. D. Cuello-Carrión, M. L. Sottile, D. R. Ciocca and L. M. Vargas-Roig, Int. J. Hyperthermia, 2012, 28, 191–201 CrossRef CAS PubMed.
- P. R. Dores-Silva, D. M. Cauvi, A. L. S. Coto, N. S. M. Silva, J. C. Borges and A. De Maio, Cell Stress Chaperones, 2021, 26, 671–684 CrossRef CAS PubMed.
- N. Moein-Vaziri, I. Phillips, S. Smith, C. Almiňana, C. Maside, M. A. Gil, J. Roca, E. A. Martinez, W. V. Holt, A. G. Pockley and A. Fazeli, Reproduction, 2014, 147, 719–732 CAS.
- F. Wang, S. R. Bonam, N. Schall, L. Kuhn, P. Hammann, O. Chaloin, J.-B. Madinier, J.-P. Briand, N. Page and S. Muller, Sci. Rep., 2018, 8, 16820 CrossRef PubMed.
- M. Gagaoua, E. M. C. Terlouw, A. M. Mullen, D. Franco, R. D. Warner, J. M. Lorenzo, P. P. Purslow, D. Gerrard, D. L. Hopkins, D. Troy and B. Picard, Meat Sci., 2021, 172, 108311 CrossRef CAS PubMed.
- J. L. Roti Roti, Int. J. Hyperthermia, 2008, 24, 3–15 CrossRef PubMed.
- S. R. Patil, I. A. Al-Zoubi, P. H. Raghuram, N. Misra, N. Yadav and M. K. Alam, Int. Med. J., 2018, 25, 361–364 CAS.
- H. Moradian, T. Roch, A. Lendlein and M. Gossen, Sci. Rep., 2020, 10, 1–15 CrossRef PubMed.
- Z. S. Guo, Q. Li, D. L. Bartlett, J. Y. Yang and B. Fang, Trends Mol. Med., 2008, 14, 410–418 CrossRef CAS PubMed.
- C. B. Waryah, C. Moses, M. Arooj and P. Blancafort, Zinc Fingers, TALEs, and CRISPR Systems: A Comparison of Tools for Epigenome Editing, In Epigenome Editing. Methods in Molecular Biology, ed. A. Jeltsch and M. Rots, Humana Press, New York, NY, 2018, vol. 1767, DOI:10.1007/978-1-4939-7774-1_2.
- S. K. Gupta and P. Shukla, Crit. Rev. Biotechnol., 2017, 37, 672–684 CrossRef CAS PubMed.
- H. Dana, G. M. Chalbatani, H. Mahmoodzadeh, R. Karimloo, O. Rezaiean, A. Moradzadeh, N. Mehmandoost, F. Moazzen, A. Mazraeh, V. Marmari, M. Ebrahimi, M. M. Rashno, S. J. Abadi and E. Gharagouzlo, Int. J. Biomed. Sci., 2017, 13, 48–57 CrossRef PubMed.
- J. L. Howarth, Y. B. Lee and J. B. Uney, Cell Biol. Toxicol., 2010, 26, 1–20 CrossRef CAS PubMed.
- M. Lee, K. Chea, R. Pyda, M. Chua and I. Dominguez, J. Biomol. Tech., 2017, 28, 67–74 CrossRef PubMed.
- F. Cardarelli, L. Digiacomo, C. Marchini, A. Amici, F. Salomone, G. Fiume, A. Rossetta, E. Gratton, D. Pozzi and G. Caracciolo, Sci. Rep., 2016, 6, 25879 CrossRef CAS PubMed.
- T. K. Kim and J. H. Eberwine, Anal. Bioanal. Chem., 2010, 397, 3173–3178 CrossRef CAS PubMed.
- V. Sokolova and M. Epple, Angew. Chem., Int. Ed., 2008, 47, 1382–1395 CrossRef CAS PubMed.
- C. Plank, U. Schillinger, F. Scherer, C. Bergemann, J.-S. Rémy, F. Krötz, M. Anton, J. Lausier and J. Rosenecker, Biol. Chem., 2003, 384, 737–747 CAS.
- O. Mykhaylyk, O. Zelphati, J. Rosenecker and C. Plank, Curr. Opin. Mol. Ther., 2008, 10, 493–505 CAS.
- C. Plank, O. Zelphati and O. Mykhaylyk, Adv. Drug Delivery Rev., 2011, 63, 1300–1331 CrossRef CAS PubMed.
- E. Delyagina, W. Li, N. Ma and G. Steinhoff, Nanomedicine, 2011, 6, 1593–1604 CrossRef CAS PubMed.
- M. Nugoli, P. Chuchana, J. Vendrell, B. Orsetti, L. Ursule, C. Nguyen, D. Birnbaum, E. J. Douzery, P. Cohen and C. Theillet, BMC Cancer, 2003, 3, 13 CrossRef PubMed.
- G. N. Raval, S. Bharadwaj, E. A. Levine, M. C. Willingham, R. L. Geary, T. Kute and G. L. Prasad, Oncogene, 2003, 22, 6194–6203 CrossRef CAS PubMed.
- Z. X. Chong, S. K. Yeap and W. Y. Ho, PeerJ, 2021, 9, e11165 CrossRef PubMed.
- T. Wang, L. Larcher, L. Ma and R. Veedu, Molecules, 2018, 23, 2564 CrossRef PubMed.
- T. Batista Napotnik, T. Polajžer and D. Miklavčič, Bioelectrochemistry, 2021, 141, 107871 CrossRef CAS PubMed.
- M. P. Alvarez-Berríos, A. Castillo, J. Mendéz, O. Soto, C. Rinaldi and M. Torres-Lugo, Int. J. Nanomed., 2013, 8, 1003–1013 Search PubMed.
- W. Hulangamuwa, B. Acharya, V. Chikan and R. J. Rafferty, ACS Appl. Nano Mater., 2020, 3, 2414–2420 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr01955e |
| ‡ These authors contributed equally and share the first author position. |
| § These authors contributed equally and share the last author position. |
| This journal is © The Royal Society of Chemistry 2024 |