
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Morphological and structural design through hard-templating of PGM-free electrocatalysts for AEMFC applications†
Hilah C.
Honig‡
a,
Silvia
Mostoni‡
 b,
Yan
Presman
a,
Rifael Z.
Snitkoff-Sol
a,
Paolo
Valagussa
b,
Massimiliano
D'Arienzo
b,
Roberto
Scotti
bc,
Carlo
Santoro
b,
Mohsin
Muhyuddin
*b and
Lior
Elbaz
*a
b,
Yan
Presman
a,
Rifael Z.
Snitkoff-Sol
a,
Paolo
Valagussa
b,
Massimiliano
D'Arienzo
b,
Roberto
Scotti
bc,
Carlo
Santoro
b,
Mohsin
Muhyuddin
*b and
Lior
Elbaz
*a
aChemistry Department, Bar-Ilan Center for Nanotechnology and Advanced Materials, Bar-Ilan University, Ramat-Gan 5290002, Israel. E-mail: lior.elbaz@biu.ac.il
bDepartment of Materials Science, University of Milano-Bicocca U5, Via Roberto Cozzi 55, 20125, Milano, Italy. E-mail: m.muhyuddin@campus.unimib.it
cInstitute for Photonics and Nanotechnologies-CNR, Via alla Cascata 56/C, 38123 Povo, TN, Italy
First published on 7th May 2024
Abstract
This study delves into the critical role of customized materials design and synthesis methods in influencing the performance of electrocatalysts for the oxygen reduction reaction (ORR) in anion exchange membrane fuel cells (AEMFCs). It introduces a novel approach to obtain platinum-free (PGM-free) electrocatalysts based on the controlled integration of iron active sites onto the surface of silica nanoparticles (NPs) by using nitrogen-based surface ligands. These NPs are used as hard templates to form tailored nanostructured electrocatalysts with an improved iron dispersion into the carbon matrix. By utilizing a wide array of analytical techniques including infrared and X-ray photoelectron spectroscopy techniques, X-ray diffraction and surface area measurements, this work provides insight into the physical parameters that are critical for ORR electrocatalysis with PGM-free electrocatalysts. The new catalysts showed a hierarchical structure containing a large portion of graphitic zones which contribute to the catalyst stability. They also had a high electrochemically active site density reaching 1.47 × 1019 sites g−1 for SAFe_M_P1AP2 and 1.14 × 1019 sites g−1 for SEFe_M_P1AP2, explaining the difference in performance in fuel cell measurements. These findings underscore the potential impact of a controlled materials design for advancing green energy applications.
Introduction
Green hydrogen is expected to be used in many hard-to-abate sectors, such as the metal industries, fertilizers, automotives, and residential purposes,1 where fuel cell technologies will play a pivotal role in producing electricity directly from hydrogen.The most developed low-temperature fuel cell technology today is the proton exchange membrane fuel cell (PEMFC). These fuel cells rely on platinum group metals (PGMs) supported on carbon (Pt/C) to catalyze both the anodic hydrogen oxidation reaction (HOR) and the cathodic oxygen reduction reaction (ORR). The ORR is considered the bottleneck reaction due to its sluggish kinetics and complex pathways; therefore, high loadings of Pt/C are required. This, in turn, translates into a high cost and large dependence on PGMs that are also considered as critical raw materials (CRMs) and hinder the PEMFC's wide commercialization.2 One way to mitigate this issue is to develop anion exchange membrane fuel cells (AEMFCs), recently proposed as a cost-effective and practical alternative to PEMFCs.3,4 Their main advantage is that operation in an alkaline environment allows the utilization of Earth-abundant transition metals such as iron for the electrocatalysis of the ORR and can completely remove the dependence on scarce and overpriced PGMs.5–9 Particularly, atomically dispersed transition metals coordinated with nitrogen embedded in a carbonaceous support (M–Nx–Cs) are the most promising active sites for the ORR in AEMFCs.10 One drawback of using PGM-free ORR electrocatalysts is their relatively low turnover frequency (TOF) when compared with PGMs.11,12 Therefore, a plethora of attempts have been undertaken to increase the number of active sites in the materials to compensate for their low reaction kinetics.9,13–16 Different synthetic routes have been pursued over the years, mainly based on the mechanical mixing of various precursor species, followed by pyrolytic processes in which the temperature and atmosphere are finely controlled.17 The influence of temperature increments during pyrolysis has been elucidated, shedding light on the temperature's impact on the metal coordination, as well as the activity and durability of the derived electrocatalysts.18–20 Inert atmospheres are more often used during pyrolysis; however, reducing atmospheres containing hydrogen or ammonia have also been reported to have a positive impact on the electrocatalyst performance.21–23
To deal with their intrinsically low TOF, which is approximately two orders of magnitude lower than that of PGMs,24,25 soft and hard templating strategies have been adopted to increase the porosity of electrocatalyst materials, and thus the active sites’ utilization26–28 as well as the electrochemically active site density (EASD).11,29
Soft templating involves a templating agent that disappears during pyrolysis, forming the porosity of the material.30 For tuning the porosity, for example, Zn or ZnO can be used, and later decomposed to create porous structures.31 Another soft templating agent can be urea which decomposes at a relatively low temperature (350 °C), increasing the porosity32,33 and can be used as a source of nitrogen for M–Nx–C.34 Even hard templating methods have been widely used. For example, silica nanoparticles (NPs) that are generally removed through etching methods have been used to create PGM-free electrocatalysts with well-defined porosity.35–40
So far, several different precursors have been used for making active M–NX–C electrocatalysts: porphyrins and phthalocyanines,41–45 metal–organic frameworks (MOFs), particularly ZIF-8,46–48 covalent frameworks,14,49 and various combinations of nitrogen-rich organic precursors and transition metal salts.50–52
The choice of precursors and the synthetic parameters has shown to affect the coordination chemistry of the metal centers53,54 their bond lengths,55 ligand structure,56,57 graphitization of the carbonaceous matrix,58 and surface defects,59 which were manifested in the overall catalytic performance, durability, and EASD.28,60–63 The only commercially available PGM-free ORR electrocatalysts are commercialized by Pajarito Powder64 Precious-Metal-Free™ (PMF) electrocatalysts. These electrocatalysts are produced via the silica hard templating method and have achieved high electrocatalytic activity, and nowadays they are considered as the benchmark for ORR electrocatalysis. However, the intrinsically low TOF of PGM-free ORR electrocatalysts can be resolved by engineering the active site structures while ensuring enough accessibility. This ultimately can improve the EASD, thereby enhancing ORR kinetics. For better control of the iron coordination and its distribution within the electrocatalyst, herein we propose a new synthetic method for synthesizing ORR Fe-based electrocatalysts to substitute the more conventional mechanical mixing approach.
Amino-based functionalizing agents were tethered to silica NPs and used to coordinate iron ions onto the surface of the NPs, which were used as a hard templating sacrificial support to create porous carbonaceous materials through a combined pyrolytic and etching method. Such functionalized silica NPs at the same instance acted as the template to induce porosity and the Fe source that can reduce the undesired coalescence of Fe into the evolution of metallic NPs and ensure the homogeneous distribution of active sites. Later, the effects of synthesis conditions on the physical properties and eventually the performance were investigated. The remarkable performance in AEMFCs is discussed in terms of the synthesis protocol itself and the pyrolysis conditions, which resulted in an increase in the EASD.
Experimental
Materials
All the chemicals and reagents used in the study were of high-purity analytical grade and used as it is without any further processing. For the synthesis of silica templates functionalized with amino-based ligands and iron, tetraethylorthosilicate (TEOS), ammonium hydroxide 25% (NH4OH), and iron sulfate hydrate Fe2(SO4)3·7H2O were purchased from Sigma Aldrich. (3-Aminopropyl)triethoxysilane (APTES) and N-aminoethyl-aminopropyltriethoxysilane (EDTMS) were purchased from abcr Gute Chemie. Ethanol (EtOH) 99.8% was obtained from Exacta Optech LabCenter.Fabrication and functionalization of silica templates
SiO2 NPs (70 ± 5 nm average diameter) were synthesized through a classical Stöber method.65 The procedure is based on the quick addition of TEOS (0.24 M) to a basic ethanol solution (NH4OH 0.36 M) at room temperature (RT) to form colloidal silica NPs. After 24 h, the powder was recovered through centrifugation (9000 rpm, 30 min), washed twice with fresh EtOH and with distilled water and finally dried at 80 °C overnight.SiO2 NPs were then functionalized with APTES or EDTMS to obtain surface amino groups attached to the SiO2 surface suitable for iron coordination. The procedure was adapted from previous publication66 by employing Fe2(SO4)3·7H2O as a suitable iron precursor. The functionalization step was carried out in EtOH at 60 °C for 24 h by adding NH4OH (0.0042 M) to favor the hydrolysis and condensation of the silane over the SiO2 surface. The amounts of APTES or EDTMS used were equal to a molar ratio between the silane and surface OH groups of SiO2 of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (calculated according to the thermogravimetric analysis, TGA in the ESI†). The powder was again recovered through centrifugation, washed twice with EtOH, and dried at 80 °C overnight. The samples are called SA and SE, respectively, where A and E mean APTES or EDTMS.
:2 (calculated according to the thermogravimetric analysis, TGA in the ESI†). The powder was again recovered through centrifugation, washed twice with EtOH, and dried at 80 °C overnight. The samples are called SA and SE, respectively, where A and E mean APTES or EDTMS.
In the last step, iron was dispersed over SiO2 NPs by exploiting the amino groups of APTES or EDTMS. The synthesis was carried out in EtOH at reflux by using a slight iron excess (Fe:silane molar ratio equal to 1.5:1, as determined by TGA analysis, Fig. S1b†). The final material was recovered through centrifugation, washed with fresh EtOH, dried at 80 °C overnight and finally labeled as SAFe and SEFe, respectively.
Electrocatalyst development
Using iron-functionalized silica templates i.e. SAFe and SEFe, atomically dispersed Fe–N–Cs were developed. This part of the synthesis utilizes the simultaneous opening of the materials’ pores and incorporation of Fe using the hard templating method. In brief, the functionalized silica NPs (SAFe or SEFe), simultaneously acting as the Fe source and templating agent, were mixed with 1-methylimidazole (M, an organic precursor containing both nitrogen and carbon) in a 3:7 weight ratio in deionized water for 24 h under continuous stirring, followed by overnight drying at 80 °C. The obtained dried slurry was ground into a fine powder using a mortar grinder and subsequently subjected to a first pyrolysis (P1) at 900 °C for 1 h under N2 at a heating and cooling rate of 300 °C h−1. The obtained black powders were named SAFe_M_P1 and SEFe_M_P1, respectively. Next, the materials were treated with an acidic solution (2:1 mixture of 25 wt% HF and 35 wt% HNO3) for three days at room temperature to remove the silica templates. The materials obtained after the acid washing were labeled as SAFe_M_P1A and SEFe_M_P1A. Finally, a second pyrolysis treatment (P2), like the previous one with the only difference in the atmosphere (a slightly reducing atmosphere of N2 balanced with 5 wt% H2), was conducted. The samples obtained in the final stage were labeled as SAFe_M_P1AP2 and SEFe_M_P1AP2. The electrocatalysts obtained at different levels of experimental design are summarized in Table S1.†
Characterization
The structural features were investigated using X-Ray Powder Diffraction (XRPD), with a Rigaku MiniFlex 600 diffractometer with 0.154 nm Cu Kα radiation (Rigaku Corporation, Akishima-Shi, Tokyo, Japan). The measurements were performed in the 2θ range of 5–80° (2θ step 0.02°, 1° min−1 scan rate).
Thermogravimetric analysis (TGA) was performed using a Mettler Toledo StarE System TGA/DSC1 (scan range 30–1000 °C, heat rate 10 °C min−1, airflow 50 mL min−1). TGA curves were recorded for the as-prepared SAFe and SEFE NPs to confirm the silica functionalization with APTES and EDTMS, as well as after the acid treatment (P1AP2) to verify the effective silica removal.
Inductively coupled plasma-optical emission spectroscopy (ICP-OES) was used to measure the Fe amount during the preparation steps using an ICP-OES Optima 7000 DV PerkinElmer instrument. The acid digestion was carried out in a microwave Milestone Ethos mineralizer instrument.
X-ray photoelectron spectroscopy (XPS) analysis was conducted using a Nexsa spectrometer equipped with a monochromatic, microfocused, and lower Al Kα X-ray source (photon energy 1486.6 eV).
BET measurements were conducted using a Quantachrome Autosorb iQ instrument with nitrogen as the adsorbate at 77 K. The degassing step included heating the sample to 130 °C (heating rate 5 °C min−1) until the apparatus detected no gas emission from the sample. The pore size distribution was calculated using the DFT method for slit pores.
Further investigation of the morphological structure of the catalysts was done by using a high-resolution transmission electron microscope (HRTEM), JEOL JEM 2100, LaB6 filament, 200 kV.
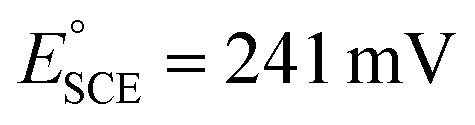 ).69 The measurements were carried out in both O2-saturated acidic and alkaline media of 0.5 M H2SO4 and 0.1 M KOH, respectively. The measured potentials were then converted to reversible hydrogen potential (RHE) using eqn (1), while the potential window was maintained in the potential window of 1.2 to 0.0 V vs. RHE.
).69 The measurements were carried out in both O2-saturated acidic and alkaline media of 0.5 M H2SO4 and 0.1 M KOH, respectively. The measured potentials were then converted to reversible hydrogen potential (RHE) using eqn (1), while the potential window was maintained in the potential window of 1.2 to 0.0 V vs. RHE. | (1) |
By measuring the disk and ring currents (Idisk and Iring, respectively), the peroxide yield (%) and the number of electrons transferred during the ORR (n) were calculated using eqn (2) and (3), respectively.
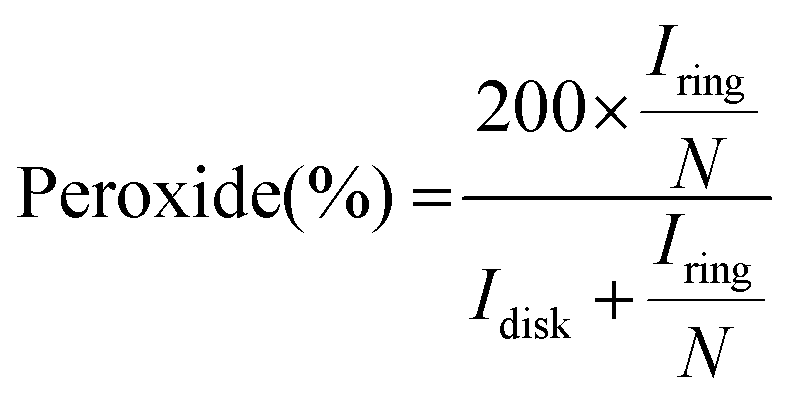 | (2) |
 | (3) |
An accelerated stability test was performed using 2000 voltage cycles to evaluate the stability profiles of the best-performing electrocatalysts. Briefly, 0.6 mg cm−2 loading of the electrocatalyst was drop-cast on a glassy carbon disk of RRDE electrode and cycled 2000 times at the scan rate of 50 mV s−1 between 1.20 and 0.32 V vs. RHE in O2-saturated 0.1 M KOH at a rotation speed of 1600 rpm. For comparison, LSVs in the beginning and at every subsequent 500th cycle were recorded at a slow scan rate of 5 mV s−1.
Fuel cell testing
:H2O solution (3:1 w/w) was added and the ink was sonicated for 1 h. The ink was sprayed using a Sono-Tek ultrasonic spray on a 5 cm2 gas diffusion layer (GDL) (BC-29) until the desired loading was reached. The anode was prepared in the same way using 40 wt% Pt/C (HiSPEC 4000) as an electrocatalyst (0.6 mgPt cm−2). Before the assembly, the membrane (AF1-HNN8-25) and GDEs were activated by soaking in 0.1 M KOH solution for 1 h. The KOH solution was replaced every 20 min. Fuel cell measurements were conducted using the Scribner 850e station. The anode and cathode gases were H2 and O2, respectively. The flow rate was 0.3 L min−1 at the anode and 0.5 L min−1 at the cathode with 100% RH and running at 60/60/60 °C at the anode gas inlet, cell, and cathode gas inlet, respectively. The backpressure was 150 kPa.
Results and discussion
Herein, we report the development of Fe–N–C ORR electrocatalysts using the hard templating method, as described in the Experimental section (Scheme 1). In contrast to previous studies, where mixtures of all precursors and templating agents are prepared for the pyrolysis step,71–73 in this work, iron was integrated through the ligand-tethered silica templates prior to the pyrolysis process to achieve high EASD. To study this new approach, two different types of surface functionalizing agents (3-aminopropyl)triethoxysilane (APTES) or N-aminoethyl-aminopropyltriethoxysilane (EDTMS) tethered to SiO2-based templating agents were used. | ||
| Scheme 1 Preparation steps of the SAFe_M and SEFe_M type electrocatalysts. | ||
First, SAFe and SEFe (Fe coordinated with SiO2-tethered APTES or EDTMS, respectively) samples were characterized to confirm the effective functionalization of silica NPs with both the silane and iron. FTIR and TGA measurements confirm the presence of surface organic ligands on the SiO2 surface (Fig. S1†). The amounts of APTES and EDTMS were calculated according to the previous publication,66 as described in the ESI.† The amount of iron anchored on SiO2 NPs was measured with ICP-OES and was equal to 1.4 wt% ± 0.1 for SAFe and 1.5 wt% ± 0.1 for SEFe, validating the efficient addition of iron to both samples. SAFe and SEFe were later used as hard templating agents for the electrocatalyst by mixing with 1-methyimidazole (M). FTIR spectra were recorded after each preparation step to follow the material evolution (Fig. 1, SEFe sample reported as an example).
 | ||
| Fig. 1 FTIR spectra of the SEFe sample mixed with 1-methyimidazole and samples acquired after each preparation step. | ||
The FTIR spectrum of SEFe_M is dominated by the typical peaks of M. However, in SEFe_M_P1, the M pattern disappears due to its decomposition during the first pyrolysis; besides, the peaks associated with SiO2 NPs appear with the main broad peak at 1100 cm−1 associated with Si–O–Si stretching. The spectra of both SEFe_M_P1A and SEFe_NC_P1AP2 do not show SiO2 peaks, confirming the effective removal of SiO2 NPs after the acid treatment. Similar results were obtained in the presence of SAFe, as shown in Fig. S2.† Further confirmation of silica removal was obtained by TGA analysis performed before and after the acid treatment (Fig. S3†). The TGA results indicate that after the acid treatment (P1A) the samples do not show any inorganic residues (<5 wt%), whereas the inorganic part after the first pyrolysis was equal to about the 70 wt% of the sample weight. This highlights the good performance of the employed etching procedure.
The XRD patterns of the two samples after the entire preparation procedure are presented in Fig. 2a. These results show one broad peak at approximately 25° and one minor peak at about 44°, which corresponds to the 002-oriented and 101-oriented diffraction peaks of graphite, respectively.74
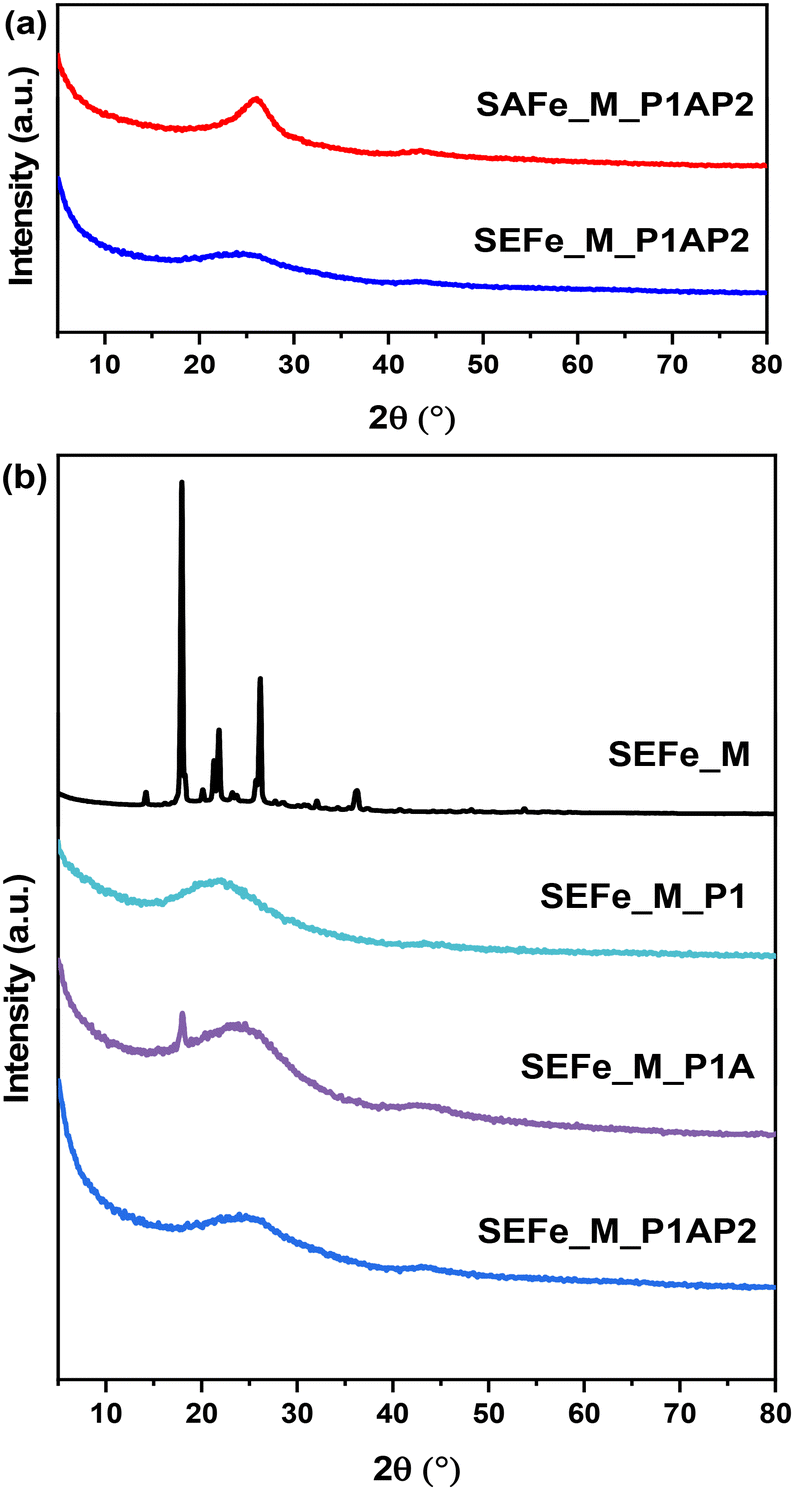 | ||
| Fig. 2 (a) XRD diffractograms of each sample after the complete preparation procedure. (b) XRD diffractograms of the SEFe_M sample after each preparation step. | ||
The comparison with the previous preparation steps (Fig. 2b, SEFe_M series reported as an exemplary sample) underlines the shift of the main broad peak at 22° in the SEFe_M_P1 sample probably due to SiO2 NPs, which become imperceptible after silica removal (SEFe_M_P1A). Besides, the crystalline phase due to methylimidazole (SEFe_M) completely disappears after the first pyrolysis (P1) and no crystalline phases were detected in any of the subsequent preparation steps. This demonstrates that by using this synthetic approach, the formation of the oxide phases is restricted, whereas additional oxide phases were obtained by using other published methods increasing the electrocatalyst speciation.32,33
In addition, there is a slight difference between the graphitic structures of SEFe and SAFe. The broader Lc peak of SEFe suggests smaller crystallite sizes and a possible lower crystallinity or the presence of polycrystalline structures. This distinction in the peak width may provide insight into the structural differences between the two materials, which can impact their ORR catalytic activity.
The structures of the new materials were also studied using HRTEM (Fig. 3). The images show slight differences in the sample morphology. In agreement with the XRD results, both SAFe_M_P1AP2 and SEFe_M_P1AP2 display similar graphitic structures constituted by NP aggregates. However, while in SAFe_M_P1AP2, the particles seem organized in larger pseudo-spherical aggregates, in SEFe_M_P1AP2, the random orientation of the crystallographic planes may suggest the formation of more elongated aggregates. These features are expected to manifest themselves in the overall performance of these materials in AEMFCs. Fewer active edge groups on ordered materials can lead to lower catalytic ORR activity, as edge sites often play a pivotal role in electrocatalysis.75,76 Conversely, the inclusion of a graphitic structure in electrocatalysts is also important, as it offers a highly conductive framework, facilitating very good electronic conductivity, and increased durability.77,78
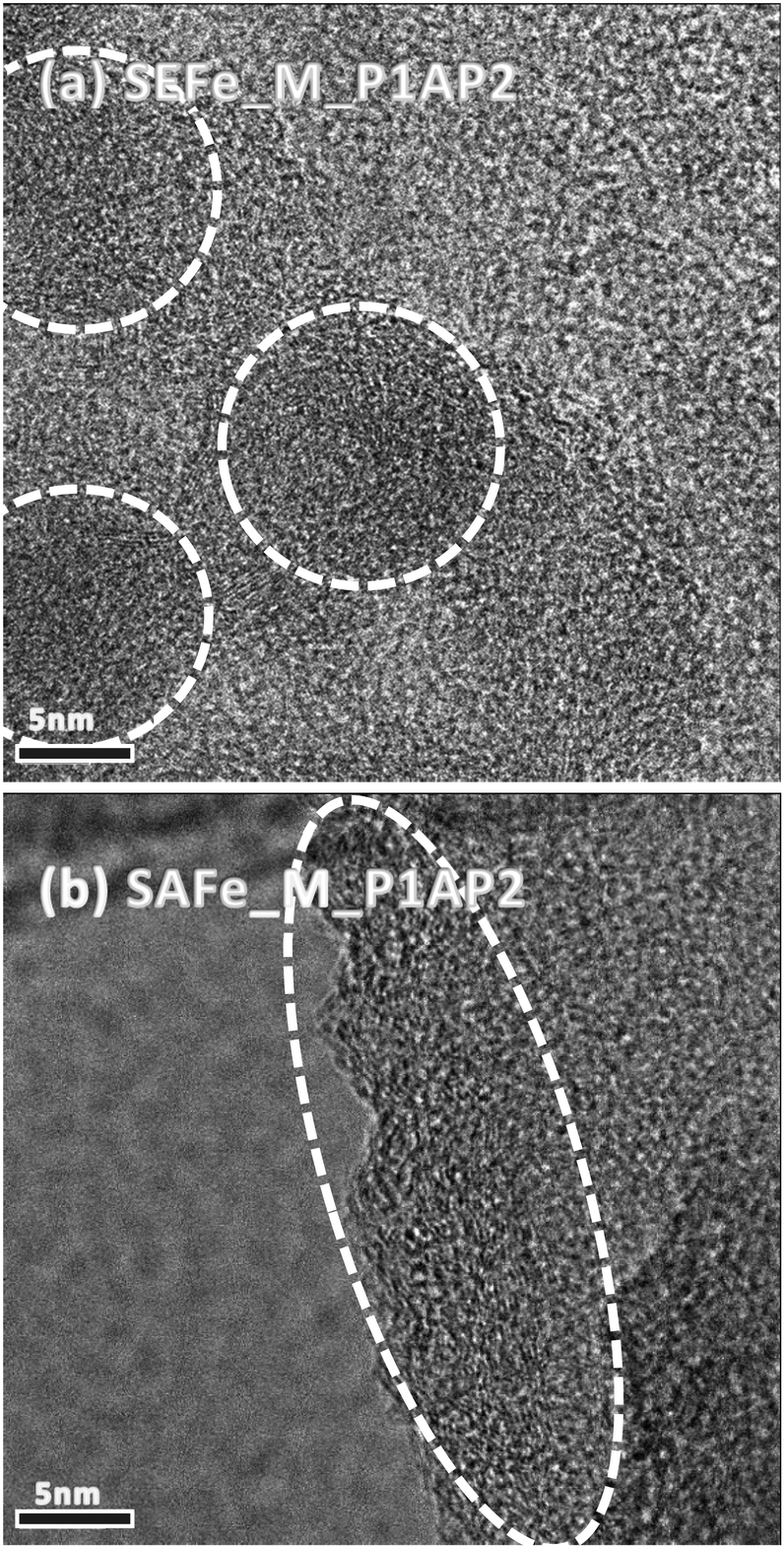 | ||
| Fig. 3 HRTEM images of (a) SEFe_M_ P1AP2 and (b) SAFe_M_P1AP2. | ||
The BET surface area and pore-size distribution were determined from the nitrogen adsorption isotherm for both SEFe_M_P1AP2 and SAFe_M_P1AP2. The SEFe_M_P1AP2 sample has a surface area of 521 m2 g−1, while SAFe_M_P1AP2 has a surface area of 319 m2 g−1. Examination of the cumulative surface area (Fig. 4) revealed a very wide pore-size distribution for the SEFe_M_P1AP2 sample with a pore diameter spanning from 5 to 35 nm, whereas the SAFe_M_P1AP2 sample has a much narrower pore-size distribution, where most of the pores are of 10–15 nm in diameter.
 | ||
| Fig. 4 Pore size distribution calculated using the DFT method for nitrogen adsorption in the slit pores of (a) SEFe_M_P1AP2; (b) SAFe_M_P1AP2. | ||
The analysis of the adsorption data, utilizing the DFT method for pore size distribution calculation, indicates that SEFe displayed a less ordered structure. Yet, both exhibit distinct average pore sizes, ranging from micro- to meso-pores. Such morphologies may favour a high distribution of the active sites and have been found to be instrumental in facilitating the good mass transport of oxygen to the active sites in fuel cells with PGM-free electrocatalysts with potential benefits to their performances.13
XPS was used to study the surface composition and coordination chemistry of the newly developed electrocatalysts. All samples were analyzed after each preparation step to track the structural composition changes during the synthesis. The extensive elemental analysis, depicted in Table S2,† emphasizes the predominance of carbon, nitrogen, and oxygen as principal components in both materials, while silicon is initially detected in the first synthetic steps and decreases in quantity after the etching procedure. High-resolution Fe2p and N1s spectra were recorded to study the coordination chemistry of the metal active site in the materials. Fig. S4† shows that both materials have low iron content manifested in their weak Fe2p spectra, consistent with ICP-OES measurements (<2 wt% iron, Table S3†). Early pyrolysis (P1) alters the iron coordination with Fe 2p3/2 signals at ∼711 eV, indicating the formation of more Fe2+ sites. SAFe_M_P1 and SAFe_M_P1AP2 have similar Fe2+ and Fe3+ species, while SEFe_M_P1 retains its original coordination chemistry confirmed by the N1s spectra (Fig. 5). It shows the existence of diverse nitrogen compounds and their changes throughout each stage of the synthesis. The deconvolution of the N1s peak of both SAFe_M and SEFe_M revealed imine-like N, N–Fe, and pyrrole-like N in both samples. Additionally, a secondary amine is present in the SEFe_M sample, which most probably originates from the pristine SEFe. A noticeable generation of different species was detected during the early pyrolysis stage (P1). The deconvolution analysis revealed the presence of several nitrogen compounds including imine, pyridinic N, Nx–Fe, hydrogenated N, graphitic N, and NOx. The SAFe_M_P1 sample remarkably exhibited a composition closely mirroring that found in its ultimate form (SAFe_M_P1AP2); at this stage, the main nitrogen species originates from hydrogenated N (401 eV), while the secondary major species is pyridinic N (399 eV). The atomic ratio between them is 1.5:1 in relation to the hydrogenated N and it remains consistent after the second pyrolysis step (P1AP2). This was in contrast with the SEFe_M_P1 sample, which exhibited different compositional ratio characteristics of its final product (SEFe_M_P1AP2). The composition of SEFe_P1 initially consisted of equal amounts of hydrogenated nitrogen and pyridinic nitrogen with a ratio of 1:1. However, this ratio changed to 1.5:1 in favor of the hydrogenated nitrogen species during the pyrolysis and acid wash stages (P1AP2). This disparity suggests a more consistent and direct formation of active sites in the SAFe_M_P1AP2 sample as opposed to the SEFe_M_P1AP2 sample. Subsequently, focusing on the final product (SAFe_P1AP2 and SEFe_P1AP2), it becomes apparent that the composition and ratios of the nitrogen species mentioned earlier are similar in SAFe_MP1AP2 and SEFe_MP1AP2. This indicates that the employed synthesis method and the use of the two different silanes do not significantly alter their composition and active site environment.
 | ||
| Fig. 5 XPS high-resolution N1s spectra of (a) SEFe_M and (b) SAFe_M after each preparation step. | ||
The electrocatalytic activity of all samples obtained at various stages of the fabrication process in the ORR was first analyzed using a RRDE in an alkaline environment (0.1 M KOH). A summary of the ORR activity, selectivity, and kinetics parameters of the electrocatalysts is given in ESI Fig. S5, S7 and summarized in Table S4.†
The electrocatalytic activity of the newly developed materials was positively affected by the synthetic procedure, resulting in very active electrocatalysts, exhibiting remarkable ORR performance (Fig. 6). This high activity can be attributed to the high active site density obtained after the removal of the templating agent during the acid wash that could promote the good distribution of atomically dispersed active moieties in the micro–mesoporous architecture during the second pyrolysis. Interestingly, after the final synthesis step, i.e. after the second pyrolysis step, both samples showed very high onset potentials and limiting current densities, hinting on the 4-electron selectivity, as can be seen in Fig. 6a (top). The onset potential, here determined at a current density of −0.1 mA cm−2, is 950 and 965 mV vs. RHE for SAFe_M_P1AP2 and SEFe_M_P1AP2, respectively. Both SAFe_M_P1AP2 and SEFe_M_P1AP2 show comparable ORR activity when compared to the state-of-the-art Fe–N–C electrocatalyst (Pajarito Powder®), highlighting SXFe_P1AP2 potential as a practical and effective Fe–N–Cs electrocatalyst for ORR. As can be seen in Fig. 6b (bottom), the peroxide anion yield remained below 22%, in both cases, it ramps up from 0.6 V vs. RHE and lower potentials. This can be explained by the catalysis of the ORR by quinone moieties formed on the carbonaceous material, a well-documented phenomenon under alkaline conditions.79,80 Interestingly, it is more pronounced with the SAFe_M_P1AP2 sample, which could hint at a more oxidized surface and thus higher concentration of quinone moieties there. The calculated number of electrons in the ORR is almost a perfect 4 for both electrocatalysts at the high potential’ region 0.6–0.8 V vs. RHE and slightly decreases below that (Fig. 6b), in full agreement with the peroxide anion yield. Based on the RRDE measurements, Tafel slopes were used to examine the electrocatalyst’ kinetics (Fig. S7†). The final development step provided the lowest value for both electrocatalysts (59 mV dec−1 for SEFe_M_P1AP2 and 62 mV dec−1 for SA_M_P1AP2). The decreases of the Tafel slope's values through the development process can be correlated with the modifications in the active sites, as evidenced in XPS results. Therefore, the electrocatalytic improvement can be linked with the evolution of suitable active site structure during the followed synthesis route. Furthermore, the stability of both electrocatalysts was measured under alkaline conditions (Fig. S8 and S9†). The findings indicate that SEFe_M_P1AP2 had a limiting current reduction of 0.6 mA cm−2 after 2000 cycles, whereas SAFe_M_P1AP2 had a decrease of 1.5 mA cm−2. These results are consistent with SEFe_M_P1AP2 having more graphitic regions, which improve structural robustness and conductivity.
 | ||
| Fig. 6 ORR measurements in 0.1 M KOH demonstrated by electrocatalysts evolved at the final stage of the research design, i.e. after the second pyrolysis (P1AP2), compared to Pajarito Powder®: (a) ring current densities (top) and LSVs obtained at 5 mV s−1 (bottom) and (b) number of electrons transferred (top) and peroxide anion yield (bottom) during the ORR while keeping the electrocatalyst loading at 0.6 mg cm−2 and rotation of the RRDE at 1600 rpm in O2-rich 0.1 M KOH. | ||
The ORR performance of the developed electrocatalysts were also analyzed in an acidic medium (0.5 M H2SO4) and the recorded trends are presented in Fig. S6.† The activity patterns, on the whole, remained consistent with those observed in the alkaline medium where the samples obtained at the final stage of the second pyrolysis demonstrated optimum performance with an aptitude to carry out the ORR following the direct 4-electron route. Notably, the performance in an acidic environment was lower than in an alkaline environment, exhibiting onset potentials of 772 mV and 808 mV for the SEFe_M_P1AP2 and SAFe_M_P1AP samples, respectively.
Durability measurements were conducted for the SEFe_M_P1AP2 and SAFe_M_P1AP2 samples as discussed in the Experimental section and are presented in Fig. S8 and S9.† Interestingly, even after 2000 cycles, only a negligible decay in the Eonset and E1/2 values was observed; however, the limiting current densities were slightly reduced over the course of the stability test, where the major decrease was observed after the first 500 cycles. This is also manifested in a slight rise in the peroxide anion yield by app. 2%. However, afterward, the further increase remained insignificant. Nevertheless, the reaction mechanism was still predominantly a 4-electron reduction to hydroxide, even after 2000 cycles, justifying the efficacy of the developed electrocatalysts. It is noteworthy that the electrocatalyst degradation phenomenon is complex and may involve multiple paths. Their elucidation somehow remains out of the scope of the current study and will be pursued independently in detail to exactly reveal the origin of degradation.
Given the impressive performance in RRDE measurements, both catalysts were studied in AEM (Fig. 7 and S10†) and PEM (Fig. S11†) fuel cells. The AEMFC measurements were conducted with 100% humidified O2 feed to the cathode (0.5 L min−1) and hydrogen to the anode (0.3 L min−1) and operated at 60 °C. The measured open circuit voltage (OCV) in the AEMFC was 0.825 V for SAFe_M_P1AP2 and 0.845 for SEFe_M_P1AP2, respectively. Moreover, SAFe_M_P1AP2 reached a peak power density of 219 mW cm−2 at a current density of 500 mA cm−2, whereas SEFe_M_P1AP2 only reached 103 mW cm−2 at a current density of around 230 mA cm−2 (Fig. 7).
 | ||
| Fig. 7 Polarization curves (circles) and power density curves (triangles) of SAFe (red) and SEFe (blue) in the AEMFC. Temperature (°C) 60/60/60, 0.3 min−1L H2/0.5 min−1L O2, ∼2 mg cm−2 catalyst loading at the cathode, and 0.6 mgPt cm−2 at the anode. | ||
The observed discrepancy in the fuel cell performance, as depicted in Fig. 7 and S12,† evidences that SAFe_M_P1AP2 performs much better under fuel cell conditions in comparison with SEFe_M_P1AP2. To explain this difference, we evaluated the electrochemically active site density in both materials using the FTacV method.28,62,63 Since no peaks associated with the Fe(II)/Fe(III) transition were observed in the CV in the AEMFC, we examined the active site in the PEMFC. After measuring the I–V and CV polarizations, FTacV measurements were conducted in situ in a fuel cell. The potential of the redox peaks is E°′ = 0.76 V vs. RHE, measured by the CV and the FTacV measurements. Fig. S12† demonstrates the 5th, 6th and 7th FTacV harmonics and their fitting simulations. The EASD was calculated from the 5th harmonic peak current for both electrocatalysts. The EASD observed for SAFe_M_P1AP2 is 1.47 × 1019 sites per g, while that observed for SEFe_M_P1AP2 EASD is 1.14 × 1019 sites per g. These results strongly support the higher fuel cell performance observed with SAFe_M_P1AP2. Besides, a defined and uniform pore size distribution, as was demonstrated by SAFe_M_P1AP2, ensures good accessibility and exposure of the active sites, facilitating improved mass transport of reactants to the active sites, ultimately enhancing the performance in the fuel cell. Finally, both the XRD and HRTEM results of SAFe_M_P1AP2 showed a higher graphitization that provides a conductive and stable framework for the active sites, facilitating efficient electron transfer and catalytic reaction kinetics during the ORR.
Thus, both the specific pore size distribution and increased graphitic surface area lead to a greater EASD, as was evaluated by FTacV, offering more reactive areas for the ORR to occur. These morphological differences significantly increase the availability of overall catalytic activity by SAFe_M_P1AP2, ensuring effective and efficient electrochemical conversion of oxygen in the fuel cell environment.
Conclusions
This study highlights the crucial role of tailored materials design, structural characteristics, and synthesis methods in shaping the electrocatalytic performance for ORR in AEMFCs. Through a range of analytical techniques, the investigation elucidated the impact of controlled integration of silica and metals, removal of templating agents, and the use of nitrogen-rich organic precursors on the electrocatalyst morphology, porosity, and chemical composition. The results indicate a substantial improvement in the ORR performance as fabrication stages progress, illustrating the potential of PGM-free electrocatalysts in facilitating the ORR in AEMFCs. The enhanced ORR activity was notably observed in the final stage (P1AP2) of the development process. The analysis emphasized the significance of the specific pore size distribution and increased the graphitic surface area in enhancing the EASD and fuel cell performance in AEMFCs. This study also highlights the successful synthesis methods using combined pyrolytic processes and templating strategies to enhance the electrocatalytic activity. Overall, this work offers insights into the controlled materials design and tailored synthesis methods necessary to optimize electrocatalysts for improved ORR performance and their potential for green energy applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
H. C. H. would like to thank The Israeli Smart Transportation Research Center (ISTRC), the Israeli Ministry of Energy and the Katz’ family fellowship for funding her research. This work was supported by funding from the Israeli Ministry of Science. C. S. and M. D. A. would like to acknowledge the Ministry of Foreign Affairs and International Cooperation – Directorate General for Cultural and Economic Promotion and Innovation (Italian Republic) within the bilateral project Italy-Israel (WE-CAT).References
- IRENA (2022), Green hydrogen for industry: A guide to policy making, International Renewable Energy Agency, Abu Dhabi, 2022 Search PubMed.
- S. T. Thompson, B. D. James, J. M. Huya-Kouadio, C. Houchins, D. A. DeSantis, R. Ahluwalia, A. R. Wilson, G. Kleen and D. Papageorgopoulos, Direct hydrogen fuel cell electric vehicle cost analysis: System and high-volume manufacturing description, validation, and outlook, J. Power Sources, 2018, 399, 304–313 CrossRef CAS.
- M. Hren, M. Božič, D. Fakin, K. S. Kleinschek and S. Gorgieva, Alkaline membrane fuel cells: anion exchange membranes and fuels, Sustainable Energy Fuels, 2021, 5(3), 604–637 RSC.
- H. Chen, R. Tao, K.-T. Bang, M. Shao and Y. Kim, Anion Exchange Membranes for Fuel Cells: State-of-the-Art and Perspectives, Adv. Energy Mater., 2022, 12(28), 2200934 CrossRef CAS.
- N. Levy and L. Elbaz, Design of PGM -free ORR Catalysts: From Molecular to the State of the Art, in Electrocatalysis for Membrane Fuel Cells, 2023, pp. 175–203 Search PubMed.
- N. Zion, A. Friedman, N. Levy and L. Elbaz, Bioinspired Electrocatalysis of Oxygen Reduction Reaction in Fuel Cells Using Molecular Catalysts, Adv. Mater., 2018, 30(41), e1800406 CrossRef PubMed.
- L. Elbaz, G. Wu and P. Zelenay, Heat-Treated Non-precious-Metal-Based Catalysts for Oxygen Reduction, in Electrocatalysis in Fuel Cells, ed. M. Shao, Springer London, 2013, vol. 9, pp. 213–246 Search PubMed.
- C. Santoro, A. Lavacchi, P. Mustarelli, V. Di Noto, L. Elbaz, D. R. Dekel and F. Jaouen, What is Next in Anion-Exchange Membrane Water Electrolyzers? Bottlenecks, Benefits, and Future, ChemSusChem, 2022, 15(8), e202200027 CrossRef CAS PubMed.
- N. Zion, J. C. Douglin, D. A. Cullen, P. Zelenay, D. R. Dekel and L. Elbaz, Porphyrin Aerogel Catalysts for Oxygen Reduction Reaction in Anion-Exchange Membrane Fuel Cells, Adv. Funct. Mater., 2021, 31, 2100963 CrossRef CAS.
- M. M. Hossen, M. S. Hasan, M. R. I. Sardar, J. b. Haider, Mottakin, K. Tammeveski and P. Atanassov, State-of-the-art and developmental trends in platinum group metal-free cathode catalyst for anion exchange membrane fuel cell (AEMFC), Appl. Catal., B, 2023, 325, 121733 CrossRef CAS.
- M. Primbs, Y. Sun, A. Roy, D. Malko, A. Mehmood, M.-T. Sougrati, P.-Y. Blanchard, G. Granozzi, T. Kosmala, G. Daniel, P. Atanassov, J. Sharman, C. Durante, A. Kucernak, D. Jones, F. Jaouen and P. Strasser, Establishing reactivity descriptors for platinum group metal (PGM)-free Fe–N–C catalysts for PEM fuel cells, Energy Environ. Sci., 2020, 13(8), 2480–2500 RSC.
- J. S. Shpilman, A. Friedman, N. Zion, N. Levy, D. T. Major and L. Elbaz, Combined Experimental and Theoretical Study of Cobalt Corroles as Catalysts for Oxygen Reduction Reaction, J. Phys. Chem. C, 2019, 123(50), 30129–30136 CrossRef CAS.
- Y. Persky, Y. Yurko, R. Z. Snitkoff-Sol, N. Zion and L. Elbaz, Tuning the performance of Fe–porphyrin aerogel-based PGM-free oxygen reduction reaction catalysts in proton exchange membrane fuel cells, Nanoscale, 2024, 16(1), 438–446 RSC.
- Y. Persky, Ł Kielesiński, S. N. Reddy, N. Zion, A. Friedman, H. C. Honig, B. Koszarna, M. J. Zachman, I. Grinberg, D. T. Gryko and L. Elbaz, Biomimetic Fe–Cu Porphyrrole Aerogel Electrocatalyst for Oxygen Reduction Reaction, ACS Catal., 2023, 13(16), 11012–11022 CrossRef CAS.
- L. Peles-Strahl, Y. Persky and L. Elbaz, Design of advanced aerogel structures for oxygen reduction reaction electrocatalysis, SusMat, 2023, 3(1), 44–57 CrossRef CAS.
- N. Zion, D. A. Cullen, P. Zelenay and L. Elbaz, Heat-Treated Aerogel as a Catalyst for the Oxygen Reduction Reaction, Angew. Chem., Int. Ed., 2020, 59(6), 2483–2489 CrossRef CAS PubMed.
- M. Muhyuddin, E. Berretti, S. A. Mirshokraee, J. Orsilli, R. Lorenzi, L. Capozzoli, F. D'Acapito, E. Murphy, S. Guo, P. Atanassov, A. Lavacchi and C. Santoro, Formation of the active site structures during pyrolysis transformation of Fe-phthalocyanine into Fe-Nx-C electrocatalysts for the oxygen reduction reaction, Appl. Catal., B, 2024, 343, 123515 CrossRef CAS.
- F. Lorandi, K. Vezzù, A. Nale, G. Pagot, Y. H. Bang, E. Negro and V. Di Noto, Tuning synthesis parameters and support composition for high-performing and durable core-shell Pt–Ni carbon nitride electrocatalysts for the oxygen reduction reaction, J. Power Sources, 2023, 555, 232390 CrossRef CAS.
- S. A. Mirshokraee, M. Muhyuddin, J. Orsilli, E. Berretti, L. Capozzoli, A. Lavacchi, C. Lo Vecchio, V. Baglio, A. Galli, A. Zaffora, F. Di Franco, M. Santamaria, L. Olivi, S. Pollastri and C. Santoro, Mono-, bi- and tri-metallic platinum group metal-free electrocatalysts for hydrogen evolution reaction following a facile synthetic route, Ind. Chem. Mater., 2023, 1(3), 343–359 RSC.
- J. Li, L. Jiao, E. Wegener, L. L. Richard, E. Liu, A. Zitolo, M. T. Sougrati, S. Mukerjee, Z. Zhao, Y. Huang, F. Yang, S. Zhong, H. Xu, A. J. Kropf, F. Jaouen, D. J. Myers and Q. Jia, Evolution Pathway from Iron Compounds to Fe1(II)–N4 Sites through Gas-Phase Iron during Pyrolysis, J. Am. Chem. Soc., 2020, 142(3), 1417–1423 CrossRef CAS PubMed.
- F. Luo, A. Roy, M. T. Sougrati, A. Khan, D. A. Cullen, X. Wang, M. Primbs, A. Zitolo, F. Jaouen and P. Strasser, Structural and Reactivity Effects of Secondary Metal Doping into Iron-Nitrogen-Carbon Catalysts for Oxygen Electroreduction, J. Am. Chem. Soc., 2023, 145(27), 14737–14747 CrossRef CAS PubMed.
- U. I. Kramm and L. Elbaz, Modifying Fe–N interaction to boost catalytic performance, Nat. Catal., 2023, 6(12), 1111–1112 CrossRef CAS.
- I. Herrmann, U. I. Kramm, J. Radnik, S. Fiechter and P. Bogdanoff, Influence of sulfur on the pyrolysis of CoTMPP as electrocatalyst for the oxygen reduction reaction, J. Electrochem. Soc., 2009, 156(10), B1283–B1292 CrossRef CAS.
- F. Luo, C. H. Choi, M. J. M. Primbs, W. Ju, S. Li, N. D. Leonard, A. Thomas, F. Jaouen and P. Strasser, Accurate Evaluation of Active-Site Density (SD) and Turnover Frequency (TOF) of PGM-Free Metal–Nitrogen-Doped Carbon (MNC) Electrocatalysts using CO Cryo Adsorption, ACS Catal., 2019, 9(6), 4841–4852 CrossRef CAS.
- V. C. A. Ficca, C. Santoro, E. Placidi, F. Arciprete, A. Serov, P. Atanassov and B. Mecheri, Exchange Current Density as an Effective Descriptor of Poisoning of Active Sites in Platinum Group Metal-free Electrocatalysts for Oxygen Reduction Reaction, ACS Catal., 2023, 13(4), 2162–2175 CrossRef CAS.
- X. Yang, C. Priest, Y. Hou and G. Wu, Atomically dispersed dual-metal-site PGM-free electrocatalysts for oxygen reduction reaction: Opportunities and challenges, SusMat, 2022, 2(5), 569–590 CrossRef CAS.
- L. Osmieri, L. Pezzolato and S. Specchia, Recent trends on the application of PGM-free catalysts at the cathode of anion exchange membrane fuel cells, Curr. Opin. Electrochem., 2018, 9, 240–256 CrossRef CAS.
- R. Z. Snitkoff-Sol and L. Elbaz, Assessing and measuring the active site density of PGM-free ORR catalysts, J. Solid State Electrochem., 2022, 26(9), 1839–1850 CrossRef CAS.
- A. Friedman, I. Saltsman, Z. Gross and L. Elbaz, Electropolymerization of PGM-free molecular catalyst for formation of 3D structures with high density of catalytic sites, Electrochim. Acta, 2019, 310, 13–19 CrossRef CAS.
- X. Yin, H. T. Chung, U. Martinez, L. Lin, K. Artyushkova and P. Zelenay, PGM-Free ORR Catalysts Designed by Templating PANI-Type Polymers Containing Functional Groups with High Affinity to Iron, J. Electrochem. Soc., 2019, 166(7), F3240 CrossRef CAS.
- M. Kim, K. L. Firestein, J. F. S. Fernando, X. Xu, H. Lim, D. V. Golberg, J. Na, J. Kim, H. Nara, J. Tang and Y. Yamauchi, Strategic design of Fe and N co-doped hierarchically porous carbon as superior ORR catalyst: from the perspective of nanoarchitectonics, Chem. Sci., 2022, 13(36), 10836–10845 RSC.
- L. Du, G. Zhang, X. Liu, A. Hassanpour, M. Dubois, A. C. Tavares and S. Sun, Biomass-derived nonprecious metal catalysts for oxygen reduction reaction: The demand-oriented engineering of active sites and structures, Carbon Energy, 2020, 2(4), 561–581 CrossRef CAS.
- L. Álvarez-Manuel, C. Alegre, D. Sebastián, A. Eizaguerri, P. F. Napal and M. J. Lázaro, N-doped carbon xerogels from urea-resorcinol-formaldehyde as carbon matrix for Fe-N-C catalysts for oxygen reduction in fuel cells, Catal. Today, 2023, 418, 114067 CrossRef.
- K. Veske, A. Sarapuu, M. Käärik, A. Kikas, V. Kisand, H.-M. Piirsoo, A. Treshchalov, J. Leis, A. Tamm and K. Tammeveski, Cobalt-Containing Nitrogen-Doped Carbon Materials Derived from Saccharides as Efficient Electrocatalysts for Oxygen Reduction Reaction, Catalysts, 2022, 12(5), 568 CrossRef CAS.
- A. Serov, K. Artyushkova, N. I. Andersen, S. Stariha and P. Atanassov, Original Mechanochemical Synthesis of Non-Platinum Group Metals Oxygen Reduction Reaction Catalysts Assisted by Sacrificial Support Method, Electrochim. Acta, 2015, 179, 154–160 CrossRef CAS.
- S. Stariha, K. Artyushkova, M. J. Workman, A. Serov, S. McKinney, B. Halevi and P. Atanassov, PGM-free Fe-N-C catalysts for oxygen reduction reaction: Catalyst layer design, J. Power Sources, 2016, 326, 43–49 CrossRef CAS.
- A. Serov, A. D. Shum, X. Xiao, V. De Andrade, K. Artyushkova, I. V. Zenyuk and P. Atanassov, Nano-structured platinum group metal-free catalysts and their integration in fuel cell electrode architectures, Appl. Catal., B, 2018, 237, 1139–1147 CrossRef CAS.
- A. Cosenza, L. Delafontaine, A. Ly, H. Wang, E. Murphy, Y. Liu, S. Specchia and P. Atanassov, Novel acid-free process intensification for the synthesis of non-precious metal-nitrogen-carbon electrocatalysts for oxygen reduction reaction, J. Power Sources, 2023, 556, 232382 CrossRef CAS.
- K. Artyushkova, S. Rojas-Carbonell, C. Santoro, E. Weiler, A. Serov, R. Awais, R. R. Gokhale and P. Atanassov, Correlations between Synthesis and Performance of Fe-Based PGM-Free Catalysts in Acidic and Alkaline Media: Evolution of Surface Chemistry and Morphology, ACS Appl. Energy Mater., 2019, 2(8), 5406–5418 CrossRef CAS.
- K. Kisand, A. Sarapuu, J. C. Douglin, A. Kikas, A. Treshchalov, M. Käärik, H.-M. Piirsoo, P. Paiste, J. Aruväli, J. Leis, V. Kisand, A. Tamm, D. R. Dekel and K. Tammeveski, Templated Nitrogen-, Iron-, and Cobalt-Doped Mesoporous Nanocarbon Derived from an Alkylresorcinol Mixture for Anion-Exchange Membrane Fuel Cell Application, ACS Catal., 2022, 12(22), 14050–14061 CrossRef CAS.
- Z. Zhang, M. Dou, J. Ji and F. Wang, Phthalocyanine tethered iron phthalocyanine on graphitized carbon black as superior electrocatalyst for oxygen reduction reaction, Nano Energy, 2017, 34, 338–343 CrossRef CAS.
- K. Chen, K. Liu, P. An, H. Li, Y. Lin, J. Hu, C. Jia, J. Fu, H. Li, H. Liu, Z. Lin, W. Li, J. Li, Y.-R. Lu, T.-S. Chan, N. Zhang and M. Liu, Iron phthalocyanine with coordination induced electronic localization to boost oxygen reduction reaction, Nat. Commun., 2020, 11(1), 4173 CrossRef CAS PubMed.
- Y. Liu, Y.-Y. Wu, G.-J. Lv, T. Pu, X.-Q. He and L.-L. Cui, Iron(II) phthalocyanine covalently functionalized graphene as a highly efficient non-precious-metal catalyst for the oxygen reduction reaction in alkaline media, Electrochim. Acta, 2013, 112, 269–278 CrossRef CAS.
- M. Muhyuddin, A. Friedman, F. Poli, E. Petri, H. Honig, F. Basile, A. Fasolini, R. Lorenzi, E. Berretti, M. Bellini, A. Lavacchi, L. Elbaz, C. Santoro and F. Soavi, Lignin-derived bimetallic platinum group metal-free oxygen reduction reaction electrocatalysts for acid and alkaline fuel cells, J. Power Sources, 2023, 556, 232416 CrossRef CAS.
- H. C. Honig and L. Elbaz, Degradation Mechanisms of Platinum Group Metal-Free Oxygen Reduction Reaction Catalyst based on Iron Phthalocyanine, ChemElectroChem, 2023, 10(7), e202300042 CrossRef CAS.
- Q. Ren, H. Wang, X.-F. Lu, Y.-X. Tong and G.-R. Li, Recent Progress on MOF-Derived Heteroatom-Doped Carbon-Based Electrocatalysts for Oxygen Reduction Reaction, Adv. Sci., 2018, 5(3), 1700515 CrossRef PubMed.
- L. Tang, Q. Xu, Y. Zhang, W. Chen and M. Wu, Correction to: MOF/PCP-based Electrocatalysts for the Oxygen Reduction Reaction, Electrochem. Energy Rev., 2022, 5(4), 10 CrossRef.
- J. Tian, A. Morozan, M. T. Sougrati, M. Lefèvre, R. Chenitz, J.-P. Dodelet, D. Jones and F. Jaouen, Optimized Synthesis of Fe/N/C Cathode Catalysts for PEM Fuel Cells: A Matter of Iron–Ligand Coordination Strength, Angew. Chem., Int. Ed., 2013, 52(27), 6867–6870 CrossRef CAS PubMed.
- N. Zion, L. Peles-Strahl, A. Friedman, D. A. Cullen and L. Elbaz, Electrocatalysis of Oxygen Reduction Reaction in a Polymer Electrolyte Fuel Cell with a Covalent Framework of Iron Phthalocyanine Aerogel, ACS Appl. Energy Mater., 2022, 5(7), 7997–8003 CrossRef CAS.
- L. Peles-Strahl, N. Zion, O. Lori, N. Levy, G. Bar, A. Dahan and L. Elbaz, Bipyridine Modified Conjugated Carbon Aerogels as a Platform for the Electrocatalysis of Oxygen Reduction Reaction, Adv. Funct. Mater., 2021, 31(26), 2100163 CrossRef CAS.
- A. Friedman, M. Mizrahi, N. Levy, N. Zion, M. Zachman and L. Elbaz, Application of Molecular Catalysts for the Oxygen Reduction Reaction in Alkaline Fuel Cells, ACS Appl. Mater. Interfaces, 2021, 13(49), 58532–58538 CrossRef CAS PubMed.
- A. Sarapuu, J. Lilloja, S. Akula, J. H. Zagal, S. Specchia and K. Tammeveski, Recent Advances in Non-Precious Metal Single-Atom Electrocatalysts for Oxygen Reduction Reaction in Low-Temperature Polymer-Electrolyte Fuel Cells, ChemCatChem, 2023, 15(22), e202300849 CrossRef CAS.
- M. Mazzucato and C. Durante, How determinant is the iron precursor ligand in Fe-N-C single-site formation and activity for oxygen reduction reaction?, Electrochim. Acta, 2021, 394, 139105 CrossRef CAS.
- Y. Zhao, R. Fan, Z. Chen, Q. Zhao, J. Li, L. Yang and J. Xue, Engineering beneficial structures and morphologies of M-N-C oxygen-reduction catalysts derived from different metal-containing precursors, Ionics, 2018, 24(6), 1733–1744 CrossRef CAS.
- Z. Miao, X. Wang, Z. Zhao, W. Zuo, S. Chen, Z. Li, Y. He, J. Liang, F. Ma, H.-L. Wang, G. Lu, Y. Huang, G. Wu and Q. Li, Improving the Stability of Non-Noble-Metal M–N–C Catalysts for Proton-Exchange-Membrane Fuel Cells through M–N Bond Length and Coordination Regulation, Adv. Mater., 2021, 33(39), 2006613 CrossRef CAS PubMed.
- F. Roncaroli, E. S. D. Molin, F. A. Viva, M. M. Bruno and E. B. Halac, Cobalt and Iron Complexes with N-heterocyclic Ligands as Pyrolysis Precursors for Oxygen Reduction Catalysts, Electrochim. Acta, 2015, 174, 66–77 CrossRef CAS.
- L. Wang, L. Zhang and J. Zhang, Improved ORR activity of non-noble metal electrocatalysts by increasing ligand and metal ratio in synthetic complex precursors, Electrochim. Acta, 2011, 56(16), 5488–5492 CrossRef CAS.
- S. Gadipelli, T. Zhao, S. A. Shevlin and Z. Guo, Switching effective oxygen reduction and evolution performance by controlled graphitization of a cobalt–nitrogen–carbon framework system, Energy Environ. Sci., 2016, 9(5), 1661–1667 RSC.
- W. d. S. Freitas, A. D'Epifanio, V. C. A. Ficca, E. Placidi, F. Arciprete and B. Mecheri, Tailoring active sites of iron-nitrogen-carbon catalysts for oxygen reduction in alkaline environment: Effect of nitrogen-based organic precursor and pyrolysis atmosphere, Electrochim. Acta, 2021, 391, 138899 CrossRef CAS.
- S. Specchia, P. Atanassov and J. H. Zagal, Mapping transition metal–nitrogen–carbon catalyst performance on the critical descriptor diagram, Curr. Opin. Electrochem., 2021, 27, 100687 CrossRef CAS.
- J. H. Zagal, S. Specchia and P. Atanassov, Mapping transition metal-MN4 macrocyclic complex catalysts performance for the critical reactivity descriptors, Curr. Opin. Electrochem., 2021, 27, 100683 CrossRef CAS.
- A. Friedman, R. Z. Snitkoff-Sol, H. C. Honig and L. Elbaz, Simplified FTacV model to quantify the electrochemically active site density in PGM-free ORR catalysts, Electrochim. Acta, 2023, 463, 142865 CrossRef CAS.
- R. Z. Snitkoff-Sol, O. Rimon, A. M. Bond and L. Elbaz, Direct measurement of the oxygen reduction reaction kinetics on iron phthalocyanine using advanced transient voltammetry, Nat. Catal., 2024, 7, 139–147 CrossRef CAS.
- https://pajaritopowder.com .
- W. Stöber, A. Fink and E. Bohn, Controlled growth of monodisperse silica spheres in the micron size range, J. Colloid Interface Sci., 1968, 26(1), 62–69 CrossRef.
- S. Mostoni, M. D'Arienzo, B. Di Credico, L. Armelao, M. Rancan, S. Dirè, E. Callone, R. Donetti, A. Susanna and R. Scotti, Design of a Zn single-site curing activator for a more sustainable sulfur cross-link formation in rubber, Ind. Eng. Chem. Res., 2021, 60(28), 10180–10192 CrossRef CAS PubMed.
- M. Muhyuddin, J. Filippi, L. Zoia, S. Bonizzoni, R. Lorenzi, E. Berretti, L. Capozzoli, M. Bellini, C. Ferrara, A. Lavacchi and C. Santoro, Waste Face Surgical Mask Transformation into Crude Oil and Nanostructured Electrocatalysts for Fuel Cells and Electrolyzers, ChemSusChem, 2022, 15(2), e202102351 CrossRef CAS PubMed.
- M. Muhyuddin, N. Zocche, R. Lorenzi, C. Ferrara, F. Poli, F. Soavi and C. Santoro, Valorization of the inedible pistachio shells into nanoscale transition metal and nitrogen codoped carbon-based electrocatalysts for hydrogen evolution reaction and oxygen reduction reaction, Mater. Renew. Sustain. Energy., 2022, 11(2), 131–141 CrossRef.
- M. Muhyuddin, D. Testa, R. Lorenzi, G. M. Vanacore, F. Poli, F. Soavi, S. Specchia, W. Giurlani, M. Innocenti, L. Rosi and C. Santoro, Iron-based electrocatalysts derived from scrap tires for oxygen reduction reaction: Evolution of synthesis-structure-performance relationship in acidic, neutral and alkaline media, Electrochim. Acta, 2022, 433, 141254 CrossRef CAS.
- R. Z. Snitkoff-Sol, A. Friedman, H. C. Honig, Y. Yurko, A. Kozhushner, M. J. Zachman, P. Zelenay, A. M. Bond and L. Elbaz, Quantifying the electrochemical active site density of precious metal-free catalysts in situ in fuel cells, Nat. Catal., 2022, 5(2), 163–170 CrossRef CAS.
- A. Serov, K. Artyushkova, E. Niangar, C. Wang, N. Dale, F. Jaouen, M.-T. Sougrati, Q. Jia, S. Mukerjee and P. Atanassov, Nano-structured non-platinum catalysts for automotive fuel cell application, Nano Energy, 2015, 16, 293–300 CrossRef CAS.
- K. Kumar, T. Asset, X. Li, Y. Liu, X. Yan, Y. Chen, M. Mermoux, X. Pan, P. Atanassov, F. Maillard and L. Dubau, Fe–N–C Electrocatalysts’ Durability: Effects of Single Atoms’ Mobility and Clustering, ACS Catal., 2021, 11(2), 484–494 CrossRef CAS.
- Y. Chen, R. Gokhale, A. Serov, K. Artyushkova and P. Atanassov, Novel highly active and selective Fe-N-C oxygen reduction electrocatalysts derived from in situ polymerization pyrolysis, Nano Energy, 2017, 38, 201–209 CrossRef CAS.
- Y. Xia and R. Mokaya, Synthesis of Ordered Mesoporous Carbon and Nitrogen-Doped Carbon Materials with Graphitic Pore Walls via a Simple Chemical Vapor Deposition Method, Adv. Mater., 2004, 16(17), 1553–1558 CrossRef CAS.
- C. Tang and Q. Zhang, Nanocarbon for Oxygen Reduction Electrocatalysis: Dopants, Edges, and Defects, Adv. Mater., 2017, 29(13), 1604103 CrossRef PubMed.
- Q. Yang, Z. Xiao, D. Kong, T. Zhang, X. Duan, S. Zhou, Y. Niu, Y. Shen, H. Sun, S. Wang and L. Zhi, New insight to the role of edges and heteroatoms in nanocarbons for oxygen reduction reaction, Nano Energy, 2019, 66, 104096 CrossRef CAS.
- D. Yan, Y. Li, J. Huo, R. Chen, L. Dai and S. Wang, Defect Chemistry of Nonprecious-Metal Electrocatalysts for Oxygen Reactions, Adv. Mater., 2017, 29(48), 1606459 CrossRef PubMed.
- Z. Liu, Z. Zhao, Y. Wang, S. Dou, D. Yan, D. Liu, Z. Xia and S. Wang, In Situ Exfoliated, Edge-Rich, Oxygen-Functionalized Graphene from Carbon Fibers for Oxygen Electrocatalysis, Adv. Mater., 2017, 29(18), 1606207 CrossRef PubMed.
- S. Rojas-Carbonell, K. Artyushkova, A. Serov, C. Santoro, I. Matanovic and P. Atanassov, Effect of pH on the Activity of Platinum Group Metal-Free Catalysts in Oxygen Reduction Reaction, ACS Catal., 2018, 8(4), 3041–3053 CrossRef CAS.
- A. Friedman, N. R. Samala, H. C. Honig, M. Tasior, D. T. Gryko, L. Elbaz and I. Grinberg, Control of Molecular Catalysts for Oxygen Reduction by Variation of pH and Functional Groups, ChemSusChem, 2021, 14(8), 1886–1892 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr01779j |
| ‡ The authors have equally contributed to the manuscript. |
| This journal is © The Royal Society of Chemistry 2024 |