
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnderstanding and tuning magnetism in van der Waals-type metal thiophosphates
Rabindra
Basnet
 *a and
Jin
Hu
*bc
*a and
Jin
Hu
*bc
aDepartment of Chemistry & Physics, University of Arkansas at Pine Bluff, Pine Bluff, AR, 71603 USA. E-mail: basnetr@uapb.edu
bDepartment of Physics, University of Arkansas, Fayetteville, Arkansas 72701, USA
cMaterials Science and Engineering Program, Institute for Nanoscience and Engineering, University of Arkansas, Fayetteville, Arkansas 72701, USA. E-mail: jinhu@uark.edu
First published on 5th August 2024
Abstract
Over the past two decades, significant progress in two-dimensional (2D) materials has invigorated research in condensed matter and material physics in low dimensions. While traditionally studied in three-dimensional systems, magnetism has now been extended to the 2D realm. Recent breakthroughs in 2D magnetism have attracted substantial interest from the scientific community, owing to the stable magnetic order achievable in atomically thin layers of the van der Waals (vdW)-type layered magnetic materials. These advances offer an exciting platform for investigating related phenomena in low dimensions and hold promise for spintronic applications. Consequently, vdW magnetic materials with tunable magnetism have attracted significant attention. Specifically, antiferromagnetic metal thiophosphates MPX3 (M = transition metal, P = phosphorus, X = chalcogen) have been investigated extensively. These materials exhibit long-range magnetic order spanning from bulk to the 2D limit. The magnetism in MPX3 arises from localized moments associated with transition metal ions, making it tunable via substitutions and intercalations. In this review, we focus on such tuning by providing a comprehensive summary of various metal- and chalcogen-substitution and intercalation studies, along with the mechanism of magnetism modulation, and a perspective on the development of this emergent material family.
 Rabindra Basnet | Dr Rabindra Basnet is currently a postdoctoral scholar in the Department of Chemistry and Physics at the University of Arkansas at Pine Bluff (UAPB). His research focuses on the synthesis and characterization (structural, electronic, magnetic, and thermodynamic properties) of a wide range of quantum materials including magnetic, topological, strongly correlated materials, and superconductors. He received a MS degree in Physics from Tribhuvan University, Nepal in 2016 and a Ph.D. in Physics from the University of Arkansas in 2023 before starting his current postdoc position at UAPB. |
 Jin Hu | Dr Jin Hu is currently an associate professor in the Department of Physics at the University of Arkansas. His research focuses on discovering novel materials with exotic properties or advanced functionalities, and understanding the physics behind the properties. He received a BS degree in Applied Physics from the University of Science and Technology of China in 2008, and a Ph.D. in Physics from Tulane University in 2013. He established the Quantum Material Research group at the University of Arkansas in 2017, and has been awarded the DOE Early Career Award and the NSF Career award. |
1. Introduction
Since the discovery of graphene,1 numerous two-dimensional (2D) materials have been theoretically predicted and experimentally realized. For a long time, 2D material research was mainly centralized around graphene and transition metal dichalcogenides (TMDs) in terms of their electronic properties because they display almost all functionalities of solid-state systems such as metal, semimetal, semiconductor, and insulator as well as exotic quantum phases and phenomena such as superconductivity, charge density wave, and topological quantum phases. In addition to these properties, magnetism in 2D materials has also attracted intensive attention due to fundamental scientific interest in low dimensional orders and technological demands such as spintronics. Graphene2 and TMDs3–5 have been found to exhibit magnetism. However, owing to the non/weak-magnetic nature of the constituted atoms/ions, magnetism in these materials mainly arises from extrinsic factors such as defects or doping of guest atoms. Therefore, there has been an intensive search for 2D materials with intrinsic magnetism. Recently, there have been breakthroughs in discovering intrinsic magnetic orders in atomically thin layers of FePS3,6 CrI3,7 CrGeTe3,8 CrBr3,9 VI3,10 Fe3GeTe2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 11 and Fe3GaTe2,12 which greatly expand the realm of the 2D magnetism and provide material platforms not only for gaining deep insight into low dimensional magnetism but also for spintronic applications.13–25 These developments have motivated the search for additional novel magnetic van der Waals (vdW) materials, resulting in a blossoming of magnetic vdW materials that can be broadly categorized into two groups: (1) transition metal halides (including both dihalides and trihalides), and (2) transition metal chalcogenides (both binary and ternary) involving transition metals from V to Ni in the periodic table (i.e., V, Cr, Mn, Fe, Co, and Ni). Magnetism in those vdW magnets arises from the magnetic moment of the 3d electrons of the transition metal cations. The metal ions in these compounds are mostly arranged in honeycomb lattices, except for a few compounds such as transition metal dihalides that is characterized by triangular networks of metal ions.26
11 and Fe3GaTe2,12 which greatly expand the realm of the 2D magnetism and provide material platforms not only for gaining deep insight into low dimensional magnetism but also for spintronic applications.13–25 These developments have motivated the search for additional novel magnetic van der Waals (vdW) materials, resulting in a blossoming of magnetic vdW materials that can be broadly categorized into two groups: (1) transition metal halides (including both dihalides and trihalides), and (2) transition metal chalcogenides (both binary and ternary) involving transition metals from V to Ni in the periodic table (i.e., V, Cr, Mn, Fe, Co, and Ni). Magnetism in those vdW magnets arises from the magnetic moment of the 3d electrons of the transition metal cations. The metal ions in these compounds are mostly arranged in honeycomb lattices, except for a few compounds such as transition metal dihalides that is characterized by triangular networks of metal ions.26
Metal thiophosphates are some of the model systems among various 2D magnetic materials. Those materials were discovered in the late 1800s,27,28 usually represented by a chemical formula of MPX3 or M2P2X6, where M, P, and X denote transition metal, phosphorus, and chalcogen elements, respectively. Since their discovery, MPX3 compounds have been studied in various fields including magnetism,6,29–43 magneto-optics,44–50 optoelectronics,27,28,51–53 Li-ion batteries,27,28,54,55 and ferroelectricity.27,28,56–59Their layered structures allow for mechanical exfoliations down to atomically thin layers,6,29,41–43,60 creating pathways to study 2D physics. The magnetic members of the MPX3 (M = V, Cr, Mn, Fe, Co, and Ni) family are antiferromagnets in which the M ions carry localized magnetic moments in a layered honeycomb lattice. Antiferromagnetic (AFM) orders have been found to persist down to atomically thin layers and even to the monolayer limit in some MPX3,6,42,43 thus these materials have been established as important candidates for 2D magnets.6,29,41–43,60 The MPX3 compounds exhibit semiconducting61 or insulating62,63 behavior with band gaps ranging from 1.3 to 3.5 eV. Interestingly, applying high pressure can induce insulator-to-metal transitions64–71 and even superconductivity72 in some MPX3 compounds. Furthermore, these compounds also offer platforms to study correlated electrons in 2D magnetic materials.45,64,73 Recent studies have revealed a strong coupling between optically active excitons and magnetism in MPX3.44–49 For example, in NiPS3, a novel coherent spin–orbit entangled excitonic state stabilized by AFM order44 as well as the correlation between emitted photon and spins45 have been reported. Such coupling can lead to an all-optical control of magnetic anisotropy.50 In addition, a strong correlation of magnons and phonons that generates phonon spin through the transfer of spin angular momentum has been demonstrated in FePS3.42,74 Such intertwined degrees of freedom provide a unique opportunity to explore the interplay between magnetism, strong correlation, and light–matter interactions in 2D magnets.
In addition to fundamental research, this material family also offers a large material pool for potential technological applications. The feasibility of obtaining atomically thin layers enables nanodevices and heterostructures fabrication and integration, opening opportunities for utilization in electronics and spintronics. Moreover, in MPX3, the band gaps ranging from 1.3 to 3.5 eV cover near-infrared to the UV region, enabling the optoelectronic applications in a broad wavelength horizon.27,28 Additionally, the unusual intercalation–reduction behavior and higher ionic conductivity of these materials facilitate potential applications in Li-ion batteries.27,28,54,55 Furthermore, their unusual ferroelectricity could lead to new energy and data storage devices and sensors.27,28
Given such versatility, MPX3 materials have recently gained intensive attention. In particular, magnetism in those materials has been established as an important research topic.75 The magnetic properties in MPX3 strongly depend on the choice of the transition metal M and chalcogen X elements. Not only the AFM transition temperature (Néel temperature, TN) that changes upon substituting M and X,30–39 but also various AFM structures can be obtained through M76–93 and X30–32,94,95 substitutions. Such tuning of magnetism by altering M and X has led to the study of a series of polymetallic76–93 and polychalcogenide30–32,94,95 “mixed” MPX3 compounds. Tunable magnetism has been observed in these mixed compounds, providing promising candidates to explore novel phenomena originating from 2D magnetism. In addition to substitution, the layered structure of MPX3 allows for another doping strategy i.e., the inter-layer intercalation of guest ions. Intercalating various guest species has been reported to cause AFM to ferrimagnetic (FIM)96–100 and ferromagnetic (FM)101 transitions in MPX3. This suggests that the intercalation could be a promising route to achieve additional magnetic phases in MPX3 that may not be accessible by substitution.
Thus far, a wide range of magnetic, electronic, vibrational, optical, and ferroelectric properties of pristine MPX3 compounds have already been summarized by a few excellent review articles.27,28,75 Therefore, the focus of this review will be put on engineering magnetic properties through various doping strategies: metal substitution, chalcogen substitution, and inter-layer intercalation, aiming to provide a comprehensive summary with sufficient depth and width for deeper understanding of magnetism in MPX3 based on magnetic exchanges and anisotropies. This review is organized as follows: summarizing the crystal growth methods and lattice structures, an overall theoretical framework for understanding magnetism in MPX3, experimental efforts in engineering the magnetism, and the perspective of the development of this field. We hope this review can provide the community with comprehensive information on the existing discoveries in doping studies and an in-depth understanding of magnetism, which serves as an important guideline for further exploring the exciting materials physics in MPX3 as well as other 2D magnets and their property tuning. We wish to cover all relevant studies in this review. However, given the rapid development of MPX3 materials, the emerging new discoveries may not be included but can be introduced by future review articles.
2. Crystal growth
The studies on functional materials, their properties, and applications, greatly rely on the development of convenient and reliable synthesis methods to produce high-quality crystals. For solid state systems, single crystals are highly desired because a single crystal possesses well-defined crystalline axes so that the intrinsic anisotropic magnetic, electronic transport and optical properties can be experimentally determined, which are critical to understanding the fundamental physics principles that govern the material properties. In addition, well-defined surfaces and edges of a single crystal enables the surface-sensitive measurements such as angle resolved photoemission spectroscopy (ARPES), scanning tunneling microscopy/spectroscopy (STM/S), etc. which provide valuable information. Conventional single-crystal synthesis techniques include chemical vapor transport (CVT), flux, Bridgman, floating zone (FZ), and Czocharalski methods. The synthesis of MPX3 compounds goes back to the late 19th century when Friedel obtained FePS3 by heating a weighed amount of phosphorus pentasulfide with iron.27,28 This technique was later used by Ferrand27,28 to synthesize other sibling compounds ZnPS3, CdPS3, and NiPS3. Nowadays, CVT and flux have become the main techniques to grow the bulk single crystals of MPX3, which have proven to be effective methods to yield large single crystals for physical characterizations.A CVT method has been widely used to synthesize pristine, metal-, and chalcogen-substituted MPX3 compounds as summarized in Table 1. As shown in Fig. 1(a), this method is based on the vaporization of source materials at the hot end of the growth chamber (usually a quartz ampoule), transportation of the vapor along some temperature gradient, and condensation of vapor at the cold end. For MPX3, a mixture of elementary powders of M, P, and X, or pre-synthesized polycrystal precursors prepared by solid-state reaction, can be used as source materials for CVT. For CVT growth, a transport agent such as I2, TeCl4, SeCl4, etc., is usually added to the source material to aid the vapor transportation process. For MPX3, CVT growth using these transport agents has been reported.31,40,71,87–91,102–106 On the other hand, owing to the volatile elements P and S or Se, CVT growth without a transport agent33,41,107–112 has also been successfully used to synthesize the single crystals of MPX3. Experimentally, the source materials (elementary powders with proper molar ratio or pre-reacted polycrystal precursors) along with the transport agent (if used) are sealed in an evacuated quartz ampoule. The ampoule should be sufficiently long (usually ∼10–15 cm) to create a proper temperature gradient between the source and sink to facilitate the vapor transport. Then, the ampoule is placed in a two-zone furnace [Fig. 1(a)] which provides the needed temperatures and temperature gradient and heated at a certain duration required for the formation of the desired MPX3 phase. The source end is kept at a higher temperature (Thot) than the sink end (Tcold) to establish the thermal gradient, as summarized in Table 1. The heating process must be slow to avoid excessive pressure due to volatile elements P and X. Upon the completion of the growth, the furnace either slowly cools at a controlled slow cooling rate or naturally cools to room temperature, and single crystals can be found at the cold end. Single crystals harvested by quenching in ice water (i.e., very rapid cooling) have also been reported.113 The optical images of single crystals of some pristine and substituted (both metal- and chalcogen-substituted) MPX3 compounds synthesized using the CVT technique are shown in Fig. 1(b–d).
 | ||
| Fig. 1 Crystal growth of MPX3. (a) Schematic of chemical vapor transport (CVT) growth. Optical microscope images of (b) pristine, (c) metal (M)-, (d) chalcogen (X)-substituted MPX3 single crystals synthesized by CVT methods. (e) Optical images of single crystals of MPX3 compounds synthesized by using the reactive P2S5 flux method. Reprinted (adapted) with permission.115 Copyright 2021, American Chemical Society. | ||
| Materials | Growth method | Temperature (°C) | Duration (days) | Ref. | |
|---|---|---|---|---|---|
| T hot | T cold | ||||
| VPS3 | CVT | 600 | 500–350 | 7–30 | 71 and 106 |
| MnPS3 | CVT | 780–650 | 720–600 | 7 | 31, 41, 87, 104 and 238 |
| P2S5 flux | 650 | — | 2 | 115 | |
| FePS3 | CVT | 750–700 | 730–600 | 9–21 | 6, 72, 88, 91, 104, 107 and 239 |
| P2S5 flux | 650 | — | 3 | 114–116 | |
| CoPS3 | CVT | 600 | 550–500 | 7–8 | 108 and 240 |
| P2S5 flux | 580 | — | 2 | 114–116 | |
| NiPS3 | CVT | 760–670 | 690–550 | 7–21 | 31, 88, 89, 91 and 123 |
| P2S5 flux | 650 | — | 2 | 115 | |
| ZnPS3 | CVT | 600 | 550 | 7 | 151 |
| P2S5 flux | 650 | — | 2 | 115 | |
| CdPS3 | CVT | 630 | 600 | 5 | 104 |
| P2S5 flux | 650 | — | 2 | 115 | |
| MgPS3 | CVT | 670 | 550–800 | 90 | 124 |
| P2S5 flux | 650 | — | 2 | 115 | |
| PdPS3 | 500 | 450–500 | 7 | 124 | |
| SnPS3 | 630 | 600 | 3–5 | 104 and 105 | |
| HgPS3 | CVT | 350 | 320–500 | 3 | 124 |
| P2S5 flux | 400 | — | 2 | 118 | |
| PbPS3 | 650 | 620 | 3–5 | 105 | |
| CrPSe3 | 800 | 700 | 10 | 103 | |
| MnPSe3 | 650 | 600 | 7 | 31 and 241 | |
| FePSe3 | 720–700 | 700–600 | 7–10 | 72 and 242 | |
| NiPSe3 | 530 | 500–560 | 90 | 124 | |
| ZnPSe3 | 370 | — | 10 | 243 | |
| CdPSe3 | 610 | 650–550 | 30 | 124 | |
| MgPSe3 | 620 | 480–690 | 90 | 124 | |
| HgPSe3 | 400 | 300 | 5 | 244 | |
| PbPSe3 | 750 | 710 | 3–5 | 105 | |
| CuCrP2S6 | 750–720 | 700–680 | 5–8 | 153 and 154 | |
| CuInP2S6 | CVT | 800 | 700 | 7 | 245 |
| P2S5 flux | 650 | — | 4 | 115 | |
| AgCrP2S6 | 750 | 690 | 5 | 156 | |
| AgInP2S6 | 750–680 | 610–600 | 7–9 | 246 and 247 | |
| Mn1−xFexPS3 | 730 | 700–630 | 5 | 81 | |
| Fe1−xNixPS3 | 760–700 | 690–600 | 9 | 88 and 91 | |
| Ni1−xMnxPS3 | 750–720 | 670–550 | 7–12 | 87, 89 and 152 | |
| Mn1−xCoxPS3 | P2S5 flux | 540 | — | 3 | 117 |
| Fe1−xCoxPS3 | P2S5 flux | 580 | — | 4 | 114–116 |
| Ni1−xCoxPS3 | 625 | 550 | 21 | 90 | |
| Ni1−xCrxPS3 | 750 | 550 | 7 | 177 | |
| Mn1−xZnxPS3 | 700–650 | 680–600 | 7 | 77, 78, 85 and 151 | |
| Fe1−xZnxPS3 | 700 | 650 | 7 | 93 | |
| Fe1−xCuxPS3 | 597 | 577 | 15 | 187 | |
| Fe1−xCdxPS3 | |||||
| Mn1−xFexPSe3 | 750 | 550 | 7 | 122 | |
| (Mn,Fe,Ni,Co)PS3 | 650 | - | 18 | 113 | |
| (V,Mn,Fe,Ni,Co)PS3 | 610 | - | 3 | 113 | |
| (Zn,Mn,Fe,Ni,Co)PS3 | 610 | - | 3 | 113 | |
| (Mg,Mn,Fe,Ni,Co)PS3 | 610 | - | 3 | 113 | |
| (Fe,Mn,Cd,In)PSe3 | 627 | 377 | 7 | 121 | |
| CuxNi2(1−x)CrxP2S6 | 750 | 550 | 7 | 181 | |
| MnPS3−xSex | 650 | 600 | 7 | 31 and 32 | |
| NiPS3−xSex | 750 | 550 | 7 | 31 | |
| FePS3−xSex | 750 | 550 | 7 | 122 | |
In addition to CVT, the reactive flux technique has been recently reported to synthesize pristine and metal-substituted MPX3 single crystals. This method adopts P2X5 (X = S114–118 or Se119) flux and metal precursors, as summarized in Table 1. First, the metal and P2X5 powders in a ratio of 2:3 for monometallic or 1:1:3 for bimetallic compounds is sealed in an evacuated silica tube inside a glove box. The mixture is then heated and maintained at a certain temperature with different heating profiles depending on the target compounds.115 The excess P2S5 (melting point ∼285 °C)114–118 and P2Se5 (melting point ∼214 °C)119 act as a reactive flux that oxidizes the metal element and enhances solubility to reduce reaction times and increase crystallite sizes. Upon the completion of the growth, the excess P2S5 flux is then removed with a 1:1 mixture of ethanol and water followed by a subsequent treatment using deionized water and acetone.114–118 On the other hand, the excess P2Se5 flux is removed by the distillation process at a temperature gradient between 400 °C and room temperature.119 This method can result in sizable single crystals in a relatively shorter synthesis time as compared to a traditional CVT technique, as shown in Fig. 1(e).
3. Crystal structure
The crystal lattice structures and symmetries are critical in determining magnetic properties, as has been theoretically predicted for MPX3.120 Varying lattice parameters have been found to strongly affect the magnetic properties.120 So, understanding crystal structure is essential for gaining deeper insights into magnetism. At first glance, all MPX3 materials exhibit very similar layered structures [Fig. 2(a)]. Careful structure characterizations have revealed monoclinic, rhombohedral, and triclinic structures for various MPX3, as summarized in Table 2. Despite three different lattice structures, MPX3 materials exhibit common structural characteristics. As shown in Fig. 2(b), metal atoms M are arranged in a honeycomb lattice. Each P displays a tetrahedral coordination, which is covalently bonded with one P to form P–P (P2) dimers perpendicular to the hexagonal plane and three X featuring a (P2X6)4− bipyramid structure unit. Such P–P dimer forming (P2X6)4− bipyramids fill the center of the M honeycomb lattice. Therefore, these compounds have also been referred to as M2P2X6 in the literature. For chalcogen atoms X that sandwich the metal honeycomb layer, they form almost close-packed surfaces of the layer with an array of octahedral coordinated sites [Fig. 2(c)]. The monolayer MPX3 can be considered as the monolayer MX2 where the one-third of M sites are substituted by P–P dimers. Thus, in MPX3, two-thirds of the octahedral centers are filled by M2+ cations while the P–P dimers occupy the remaining one-third [Fig. 2(c), P–P dimer in the center is not shown].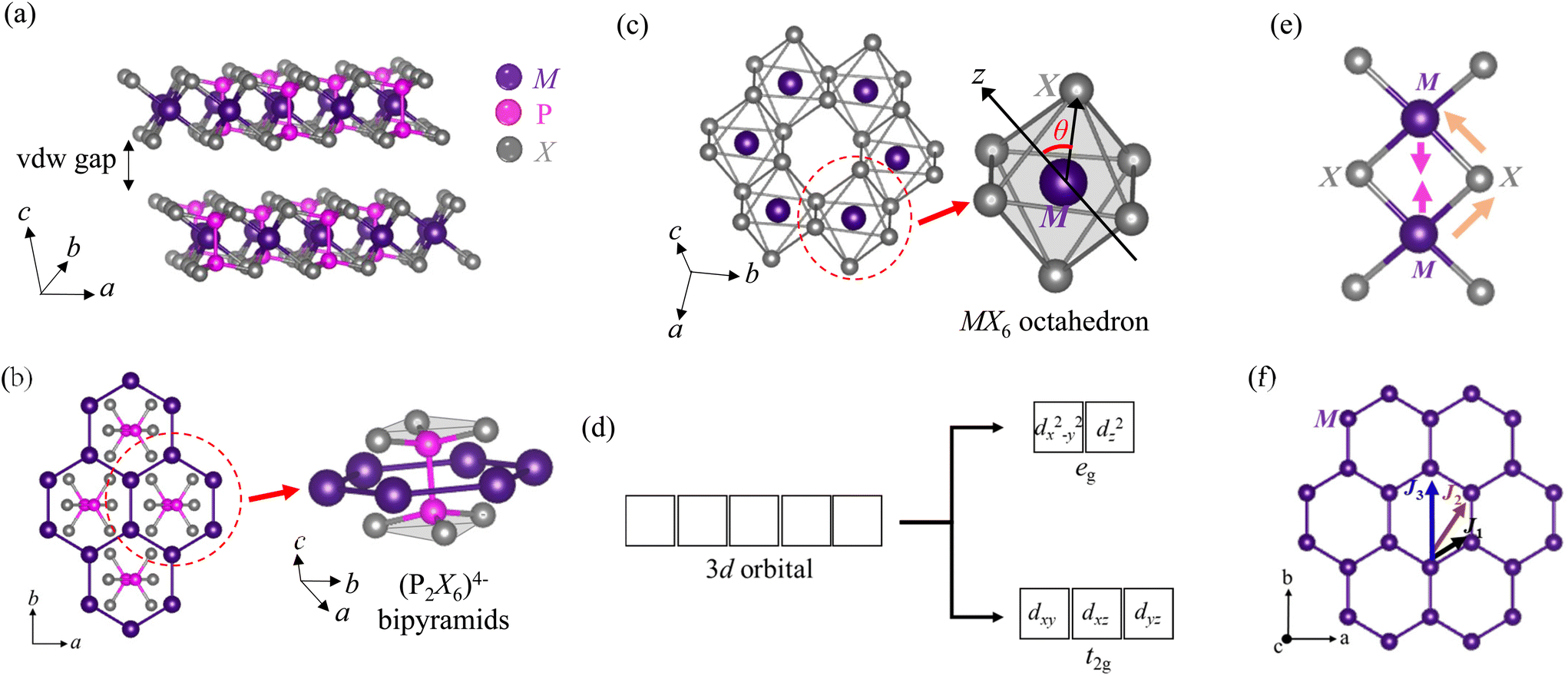 | ||
| Fig. 2 Crystal structures of MPX3. (a) The layered structure of van der Waals material MPX3. (b) The (P2X6)4− (X = S or Se) anion sublattice in each layer. Metal M is arranged in a honeycomb lattice around the (P2X6)4− bipyramids. (c) MX6 octahedron of MPX3. Such MX6 octahedra possesses a trigonal distortion that is characterized by the angle θ between the trigonal axis (perpendicular to the ab-plane) and the M–X bond. (d) Splitting of five 3d orbitals of transition metal cations into three t2g and two eg levels under an octahedral crystal field. (e) Schematic of direct exchange interaction between magnetic ions, and superexchange interactions between two cations through anion. (f) Schematic of nearest-neighbor (J1), second nearest-neighbor (J2), and third nearest-neighbor (J3) interactions in MPX3. | ||
| Materials | Crystal structure | Space group | Ref. |
|---|---|---|---|
| VPS3 | Monoclinic | C2/m | 71 and 106 |
| MnPS3 | Monoclinic | C2/m | 31, 41, 87, 104 and 238 |
| FePS3 | Monoclinic | C2/m | 6, 72, 88, 91, 104, 107 and 239 |
| CoPS3 | Monoclinic | C2/m | 108, 240 |
| NiPS3 | Monoclinic | C2/m | 31, 88, 89, 91 and 123 |
| ZnPS3 | Monoclinic | C2/m | 151 |
| CdPS3 | Monoclinic | C2/m | 104 |
| MgPS3 | Monoclinic | C2/m | 124 |
| PdPS3 | Monoclinic | C2/m | 124 |
| SnPS3 | Monoclinic | C2/m | 104 and 105 |
| HgPS3 | Triclinic |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
124 |
| PbPS3 | Monoclinic | P21/c | 105 |
| CrPSe3 | Monoclinic | C2/m | 103 |
| MnPSe3 | Rhombohedral |
R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) |
31 and 241 |
| FePSe3 | Rhombohedral |
R |
72 and 242 |
| NiPSe3 | Monoclinic | C2/m | 124 |
| ZnPSe3 | Rhombohedral |
R |
243 |
| CdPSe3 | Rhombohedral |
R |
124 |
| MgPSe3 | Rhombohedral |
R |
124 |
| HgPSe3 | Monoclinic | C2/m | 244 |
| PbPSe3 | Monoclinic | P21/c | 105 |
| CuCrP2S6 | Monoclinic | C2/c | 153 and 154 |
| CuInP2S6 | Trigonal |
P1c |
245 |
| AgCrP2S6 | Monoclinic | P2/a | 156 |
| AgInP2S6 | Trigonal | P31c | 246 and 247 |
| Mn1−xFexPS3 | Monoclinic | C2/m | 81 |
| Fe1−xNixPS3 | Monoclinic | C2/m | 88 |
| Ni1−xMnxPS3 | Monoclinic | C2/m | 87, 89 and 152 |
| Mn1−xCoxPS3 | Monoclinic | C2/m | 117 |
| Fe1−xCoxPS3 | Monoclinic | C2/m | 114–116 |
| Ni1−xCoxPS3 | Monoclinic | C2/m | 90 |
| Ni1−xCrxPS3 | Monoclinic | C2/m | 177 |
| Mn1−xZnxPS3 | Monoclinic | C2/m | 151 |
| Fe1−xZnxPS3 | Monoclinic | C2/m | 93 |
| Mn1−xFexPSe3 | Rhombohedral |
R |
86 |
| (Mn,Fe,Ni,Co)PS3 | Monoclinic | C2/m | 113 |
| (V,Mn,Fe,Ni,Co)PS3 | Monoclinic | C2/m | 113 |
| (Zn,Mn,Fe,Ni,Co)PS3 | Monoclinic | C2/m | 113 |
| (Mg,Mn,Fe,Ni,Co)PS3 | Monoclinic | C2/m | 113 |
| (Fe,Mn,Cd,In)PSe3 | Rhombohedral |
R |
121 |
| CuxNi2(1−x)CrxP2S6 | Monoclinic | C2/m | 181 |
| MnPS3−xSex | Mono → Rhom |
C2/m → R |
32 |
| NiPS3−xSex | Monoclinic | C2/m | 31 |
| FePS3−xSex | Mono → Rhom |
C2/m → R |
122 |
As seen in Fig. 2(a), a finite vdW gap of about 3.22–3.24 Å is present in MPX3.27,28,75 Various metal ions M2+ with different ionic radii slightly modify the slab size. The two flat pyramids formed by bonding between a P and three X atoms at the top and bottom of the (P2X6)4− structural unit [shown in Fig. 2(b)] remain invariable, but the P–P distance is slightly adjusted to accommodate the changes in metal cations. For example, the P–P distance elongates from 2.148 Å in NiPS3 to 2.222 Å in CdPS3,27 which is accompanied by increases of the layer thickness.
Although the individual lamella is similar for all MPX3 compounds, their symmetry, and layer stacking to form a bulk lattice structure can vary depending on M and X, as summarized in Table 2. In particular, the symmetries and crystal structures for sulfides and selenides of MPX3 materials are distinct. Generally, almost all the known sulfides MPS3 exhibit monoclinic crystal structures with a C2/m space group, except for PbPS3105 which has been reported to crystalize in a P21/c monoclinic structure as its selenide counterpart, and for HgPS3118 which is triclinic with a P space group symmetry. In sulfides, the monoclinic angle β depends on the metal cation, ranging from 106.97° for MgPS3 to 107.35° for MnPS3. On the other hand, as summarized in Table 2, selenides MPSe3 process larger P–Se bond lengths and Se–P–Se bond angles as compared to their sulfide counterparts, and mostly crystallize in rhombohedral structures with a R space group. The exceptions are NiPSe3, CrPSe3, HgPSe3, and PbPSe3 which exhibit monoclinic crystal structures.
Therefore, the lattice structure of MPX3 appears to be more sensitive to chalcogen rather than metal. This is further confirmed by substitution studies. As summarized in Table 2, substituting metal atoms M does not change the lattice structure in MPX3.76–92,114,116,117 Most known metal-substituted sulfide compounds exhibit monoclinic structure with a C2/m space group in the entire substitutional range. For selenides, previous works on a bimetallic compound Mn1−xFexPSe386 and a high entropy compound Fe1−x−y−zMnxCdyInzSe3121 also found unchanged lattice with rhombohedral R structures. On the other hand, chalcogen S–Se substitution can induce structure transition from monoclinic (C2/m) to rhombohedral (R) in MnPS3−xSex32 and FePS3−xSex,122 which is consistent with the different lattice structures for their sulfide and selenide end compounds. One exception is NiPS3−xSex which displays unchanged monoclinic C2/m structure, which is also in line with the identical C2/m space group for NiPS331,88,89,91,123 and NiPSe3124.
4. Magnetism
4.1. Theoretical background
Owing to the insulating nature of MPX3 compounds, their magnetism can be described by local moment pictures. Magnetism in vdW magnetic materials arises from the magnetic moment of the spin and orbital momenta of the 3d electrons of the transition metal cations. Thus, the choice of transition metal cations plays a vital role in determining magnetism. In the presence of a crystal electric field from the octahedral coordination of the transition metal cations [Fig. 2(c)], the dx2−y2 and dz2 orbitals among the five d-orbitals of the transition metal point in the direction of the ligands, thereby the electrons in these orbitals experience greater repulsion with ligands and increase the energy of these orbitals. Thus, these two d-orbitals are pushed to higher energy levels in the band structure and referred to as two-fold degenerate high-energy eg orbitals. On the other hand, the remaining three d-orbitals dxz, dyz, and dxy do not directly align towards ligands leading to a weaker repulsion and stabilized to a lower energy level than two eg orbitals. Therefore, these three orbitals lie below eg orbitals (dx2−y2 and dz2) in term of energy and constitute three-fold degenerate low-energy t2g orbitals.125,126 The splitting of five 3d orbitals of transition metal ions under an octahedral crystal field is depicted in the schematic in Fig. 2(d). Then, the d electrons of various transition metal cations fill these orbitals following Hund's rule, as presented in Table 3.127 The occupancy of these orbitals determines the total magnetic moment of the 3d transition metal cations, with the total magnetic moment given by gJ[j(j + 1)]1/2μB where |j|, gJ, and μB are the total angular momentum quantum number (= spin + orbital angular momentum), Landé g-factor, and Bohr magneton, respectively. When orbital angular momentum is quenched (i.e., L = 0), only the spin magnetic moment contributes to the total magnetic moment, resulting in a total magnetic moment of 2[s(s + 1)]1/2μB where s is the total spin quantum number, which is generally the case for transition metals.125| Transition metal ions | 3d orbital occupancy |
|---|---|
| V2+ (3d3) |

|
| V3+ (3d2) |

|
| Cr2+ (3d4) |

|
| Cr3+ (3d3) |

|
| Mn2+(3d5) |

|
| Fe2+ (3d6) |

|
| Fe3+ (3d5) |

|
| Co2+ (3d7) |

|
| Ni2+ (3d8) |

|
In the classic Heisenberg model, the magnetism can be explained within the framework of isotropic Heisenberg Hamiltonian: H = −2Σ(JSi·Sj), where J is the exchange coupling, which is considered to be isotropic, the summation is over all pairs of magnetic ions in the lattice and the spins are treated as three-component vectors where Si and Sj are spin magnetic moments of the atomic site i and j respectively. The Heisenberg model provides an accurate account of the interactions between the localized magnetic moments that govern the magnetic properties in insulators (such as MPX3). In vdW materials, magnetic interactions can be anisotropic and are described by a Heisenberg Hamiltonian with anisotropic exchange: H = −2Σ(JxSiSj + JySiSj + JzSiSj), where Jx, Jy, and Jz are exchange couplings along the crystallographic axes. In layered MPX3, magnetic exchanges within the layer are much stronger than inter-layer interactions,128–130 hence the long-range magnetic ordering is believed to be mainly governed by intralayer exchange interactions. The interactions between localized moments in insulating MPX3 compounds are mainly mediated through direct and superexchange interactions, as shown in Fig. 2(e). The direct exchanges between neighboring magnetic moments, which result from the overlapping wavefunctions, mainly depend on their spatial separation, and contribute to the nearest neighbor interaction J1. Despite the fact that the distances between magnetic cations are large (in the order of 3–4 Å), such direct exchange has been found to be important or even dominate the magnetic interactions in some MPX3 compounds such as MnPS3.128 On the other hand, in many other MPX3 materials such as NiPS3, the long-range magnetic coupling between moments is dominated by metal–chalcogen–metal (M–X–M) superexchange interactions mediated through non-magnetic chalcogen X.128 The superexchange couplings contribute to both the nearest-neighbor interaction (J1) and the interactions between further neighbors such as second nearest-neighbor (J2), and third nearest-neighbor (J3) interactions.128 The magnetic interactions J1, J2, and J3 are depicted in Fig. 2(f). The nature and strength of superexchange interactions are determined by M–X–M bonding angle, d-orbital occupancy of M2+ ions, and orbital overlap between M2+ cation and X2− anion:40,128 According to Goodenough–Kanamori rules,131,132 the superexchange interaction is ferromagnetic (FM) if M–X–M bond angle is close to 90° and AFM when M–X–M bond angle is 180°. The occupancy of d orbitals is important. For example, CoPS3 and NiPS3 share the same magnetic structure but possess different strengths for superexchange due to their distinct occupancy of d orbitals for Co2+ (d7) and Ni2+ (d8) ions.129 Such differences could be important in designing different strategies to tune magnetism in CoPS3 and NiPS3. The overlap of atomic orbitals also affects the superexchange interaction. Usually, stronger orbital overlap strengthens the superexchange, therefore substituting Se for S in MPX3 leads to enhanced superexchange because of the extended orbital overlap due to more extended atom orbitals of Se than S.31
Furthermore, structural distortions also affect the magnetism in MPX3.40,128,129 As illustrated in Fig. 2(c) and mentioned above, every metal atom M in MPX3 is located at the center of an octahedron formed by six X atoms. Such MX6 octahedra, however, possess a trigonal distortion that is characterized by the angle θ between the trigonal axis (perpendicular to the ab-plane) and the M–X bond [Fig. 2(c)]. The experimental θ values for various MPX3 compounds are presented in Table 4. An ideal octahedral possesses the ideal octahedral angle θ ≈ 54.75°. The deviation of θ angle from this ideal value characterizes the trigonal distortion. The crystal field arising from trigonal distortion of MX6 octahedra together with spin–orbit coupling (SOC) of the M2+ cations introduce anisotropy in an otherwise isotropic system. Now, with an additional anisotropy term, the full Hamiltonian becomes H = −2 ∑(JijSi·Sj) − A∑(Siz)2, where Jij is exchange interaction between neighboring ith and jth moments and A is single-ion anisotropy parameter that defines the magnetic anisotropy of the system. The strengths of various J's and A determine the magnetism in MPX3. Table 4 summarizes the experimental values of J and A parameters obtained by neutron scattering experiments for various MPX3 materials,35,94,107,128,129 together with theoretical calculated values for VPS3.133 These distinct J and A values for different MPX3 compounds result in diverse AFM structures for this material family, as shown in Fig. 3. When J ≫ A (for very small A), the magnetism is weakly anisotropic and can be explained by the 3D isotropic Heisenberg Hamiltonian, such as MnPS3.35,40 For large A values, on the other hand, the magnetism becomes rather anisotropic and corresponds to XY (such as NiPS340,123 and CoPS3129) or Ising-type (such as FePS340 and FePSe330) magnetism depending on the sign and magnitudes of A. In Mn and Ni systems, the orbital angular momentum (L) is quenched for Mn2+ (d5) and Ni2+ (d8) ions because of their half and fully-filled t2g orbitals respectively, leading to negligible SOC for magnetic ions.40 In MnPS3 which possesses relative weak trigonal distortion (octahedral angle θ ≈ 51.67°), the effect of both spin–orbit splitting and trigonal distortion is found to be negligible for the high spin ground state of Mn2+,40 so the magnetism in MnPS3 is governed by the dipolar anisotropy35,134,135 which results in an out-of-plane moment orientation with a small tilt towards the a-axis (Fig. 3). On the other hand, NiPS3 exhibits greater trigonal distortion characterized by octahedral angle, θ ≈ 51.05° from the undistorted octahedral angle of 54.75°. This leads to remarkable single-ion anisotropy A ≈ 0.3 meV in NiPS3, which is much stronger than that of 0.0086 meV in MnPS3 (Table 4).35,128,129 Consequently, Ni moments are aligned within the basal plane perpendicular to the trigonal axis (Fig. 3),36,40,135 and this system can be modeled by the anisotropic Heisenberg Hamiltonian with XY-type anisotropy. The situation is different for FePS3, which exhibits a substantial magnetic anisotropy with a much higher A (≈2.66 meV) in comparison to MnPS3 and NiPS3.107,128 Thus, FePS3 is best described by the Ising Hamiltonian corresponding to the highly anisotropic Ising-type magnetic ordering with magnetic moments aligned along the out-of-plane direction.40,107 In FePS3, the degenerate t2g orbitals for the high-spin Fe2+ (d6) state lead to a stronger trigonal distortion of the FeS6 octahedra (θ ≈ 51.28° (ref. 107 and 129)) that causes a significant splitting of t2g orbitals to lift the degeneracy while the eg orbitals remain doubly degenerate.136 In addition, the lack of half- or fully-filled t2g orbitals for high-spin Fe2+ ions also contribute to the net orbital moment and consequently enhances the SOC. Although, a recent X-ray photoemission electron microscopy study has demonstrated that the spin–orbit entanglement for Fe2+ under trigonal elongation plays a key role behind the significant magnetic anisotropy in FePS3,137 the SOC alone may not be a dominant factor behind strong magnetic anisotropy in FePS3 because the SOC for Ni2+ (d8) ion is larger than Fe2+ (d6) ion but the resulting compound NiPS3 is less anisotropic than FePS3.40 The large crystal electric field arising from the strong trigonal distortion of the FeS6 octahedra might also be attributed to a substantial magnetic anisotropy in FePS3, which has been confirmed by a recent X-ray absorption spectroscopy experiment.136 In fact, the strong anisotropy due to crystal electrical field i.e., crystal-field anisotropy has been ascribed to Fe2+ (d6) ion in different Fe-based compounds.138,139
 | ||
| Fig. 3 Experimentally determined magnetic structures of various MPX3 materials. For CuCrP2S6, the low-temperature structure is shown. Magnetic structures for (V,Co,Ni)PSe3 and CuCrP2Se6 have not been experimentally determined. | ||
| Parameters | VPS3a133 |
MnPS335 |
FePS3107 |
CoPS3129 |
NiPS3248 |
MnPSe394 |
|---|---|---|---|---|---|---|
| a Theoretical study. | ||||||
| S | 3/2 | 5/2 | 2 | 3/2 | 1 | 5/2 |
| T N (K) | 56 (ref. 106) | 78 | 118 | 120 | 155 | 74 |
| J 1 (meV) | −7.387 | −0.77(9) | 1.46(1) | 2.04 | 1.9(1) | −0.45 |
| J 2 (meV) | −0.068 | −0.07(7) | −0.04(4) | 0.26 | −0.1(1) | −0.03 |
| J 3 (meV) | −0.223 | −0.18(1) | −0.96(5) | −4.21 | −6.90(5) | −0.19 |
| J c (meV) | — | 0.0019(2) | −0.0073(3) | — | 0.32(3) (ref. 130) | −0.031(5) |
| A (meV) | — | 0.0086(9) | 2.66(8) | 2.06 | 0.3(1) | — |
| θ (°) | — | 51.67 (ref. 129) | 51.28 (ref. 129) | 51.38 (ref. 129) | 51.05 (ref. 129) | — |
4.2. Engineering magnetism in MPX3
Engineering magnetism in layered magnetic materials not only optimizes properties for technological applications (such as high transition temperature, wider hysteresis loop, and various types of FM and AFM structures) but also provides an effective approach to clarify the mechanism for the magnetism by identifying the parameters that govern magnetism. Several strategies such as doping,30–32,76–91,94,95,97–99,140–146 high pressure,147–150 and electrostatic gating14,16 have been adopted to tune the magnetic properties in layered magnets. These techniques have been successful in controlling the spin orientation in vdW magnetic materials, which leads to novel magnetic phenomenon arising from 2D magnetism. Tuning magnetic properties has been one major focus and studied for a long time in the MPX3 family.77,78,85 Doping, including metal M and chalcogen X substitutions, and inter-layer intercalation, has been demonstrated as a powerful technique to modulate magnetism in MPX3, which will be the focus of this review as discussed below.76–91,93,114,116,117,151,152 where two different metal atoms are mixed in a honeycomb lattice as shown in Fig. 4(a). In addition to these bimetallic substitutions, the metal substitution in MPX3 has recently been extended to mixing of more than two divalent metal M2+ ions creating medium121 and high entropy113 vdW materials. In addition to these bi- and poly-metallic compounds based on isovalent M2+ ion substitutions, the non-isovalent substitution of monovalent and trivalent ions that leads to a series of quaternary compounds (M1)1+(M2)3+P2X6 (M1 = Li, Cu, Ag, etc.; M2 = V, Cr, In, Bi, etc.; X = S and Se)119,153–161 have also been reported in this material family. Although the focus of this review is the divalent metal substitutions, we have also briefly summarized the magnetic properties of a few magnetic quaternary compounds (Cu,Ag)1+(V,Cr)3+P2(S,Se)6 that have been formed by mixing trivalent ions such as Cr3+ or V3+ with monovalent metal ions such as Cu1+ or Ag1+.153–157 The magnetic exchange J and single-ion anisotropy A parameters are found to be effectively modulated with metal substitutions that lead to efficient control of magnetic properties. Tunable magnetism arising from competing magnetic interactions, magnetic anisotropies, and spin fluctuations has been observed in metal-substituted MPX3 compounds, which will be discussed in the following sections.
 | ||
| Fig. 4 Conceptual schematic of (a) metal (M) substitution, (b) chalcogen (X) substitution, and (c) Li intercalation in MPX3. | ||
4.2.1.1. Isovalent substitution.
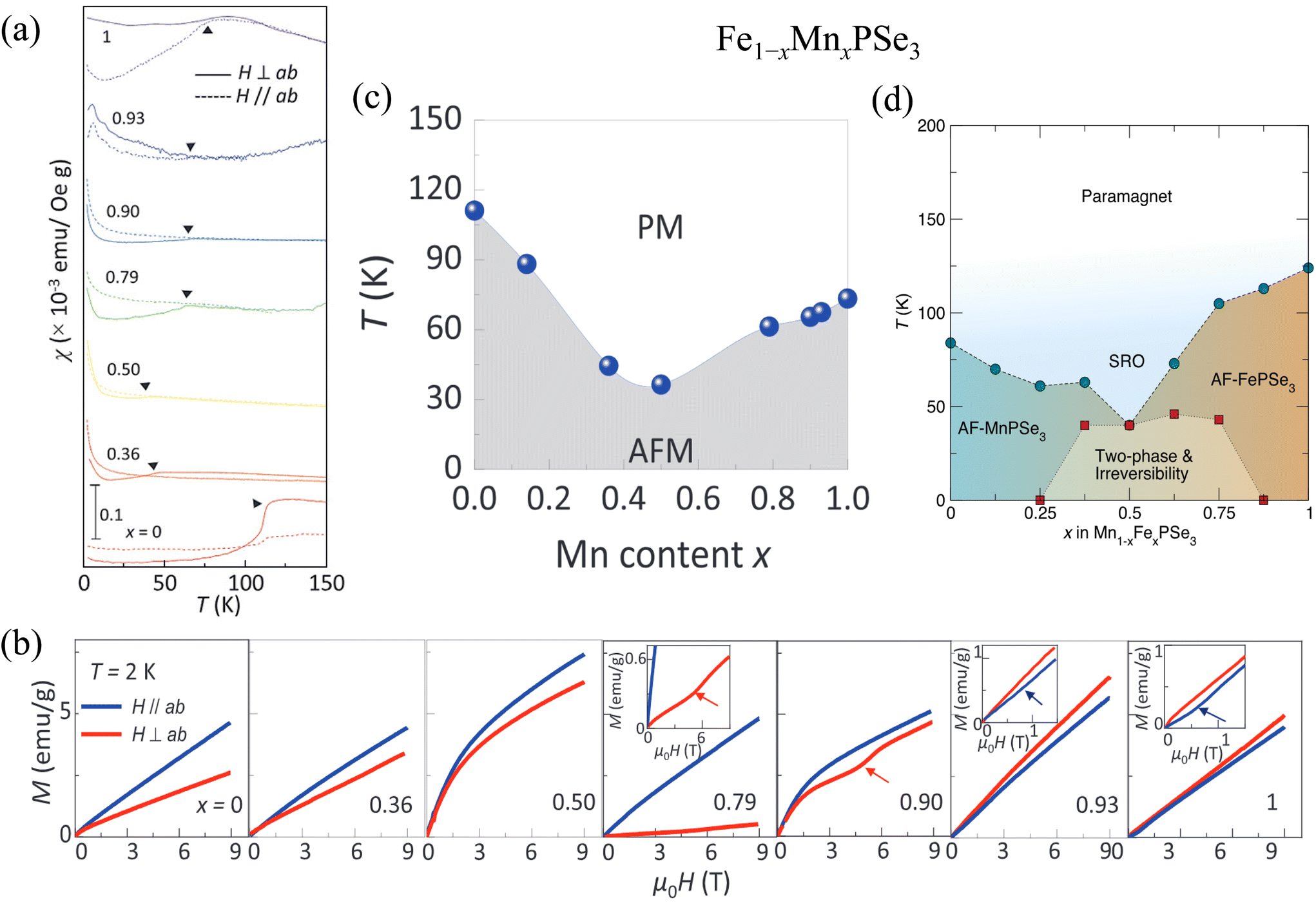
4.2.1.1.1. Mn1−xFexPS3. As shown in Fig. 3, MnPS3 and FePS3 exhibit out-of-plane magnetic moment but different Néel134 and zig-zag107 type AFM structures for each layer, respectively. Therefore, the tuning of magnetic properties arising from the competition between two different AFM interactions is expected when Mn2+ and Fe2+ ions are mixed, which has been studied by Masubuchi et al.81 Single crystals of Mn1−xFexPS3 (0 ≤ x ≤ 1) were synthesized using a CVT technique as detailed in ref. 81.
As shown in Fig. 5, the magnetic properties were characterized by the temperature dependence of magnetization under various magnetic fields along the easy axis (the c* axis in Fig. 5 is perpendicular to the ab-plane. i.e., the out-of-plane direction).81 A few magnetic phases have been identified: AFM order for 0 ≤ x ≤ 0.2 and 0.8 ≤ x ≤ 1 composition, which is characterized by a clear AFM transition in susceptibility without remarkable difference between zero-field-cooling (ZFC) and field-cooling (FC). A broad hump in susceptibility occurs above the AFM ordering temperature TN in end compounds [Fig. 5(a)]. This is a generic feature in many MPX3 compounds, which has been attributed to a short-range 2D or quasi-2D magnetic correlation owing to the layered structures. The broad hump is suppressed by substitution, leading to a sharper peak at TN [Fig. 5(a)], which has been ascribed to the weakening of a short-range magnetic correlation due to randomness introduced by Fe substitution.
 | ||
| Fig. 5 (a and b) Temperature dependence of zero-field cooled (ZFC) susceptibility with the magnetic field applied parallel to the magnetic easy axis (c*-axis) of Mn1−xFexPS3 (x = 0.0, 0.1, 0.2, 0.8, 0.9, 1.0). (c) Magnetic phase diagram of the Mn1−xFexPS3. SG, RSG, AF, and Para indicate a spin glass phase, a reentrant spin glass phase, an antiferromagnetic ordered phase, and a paramagnetic phase, respectively. Reprinted (adapted) with permission.81 Copyright 2008, Elsevier. | ||
Samples in 0.5 ≤ x ≤ 0.6 compositions display significant irreversibility [Fig. 5(b)] and are ascribed to a spin glass phase, which might be attributed to the competition between dominant exchange interactions in MnPS3 and FePS3 phases, namely AFM Mn2+–Mn2+J1 interaction and FM Fe2+–Fe2+J1 interaction, respectively35,107 (Table 4). This is further confirmed by AC susceptibility.81 The glass temperature, defined as the temperature where susceptibility displays irreversibility between ZFC and FC measurement, is found to enhance with the frequency in AC susceptibility measurements. The remaining composition regions, i.e., 0.3 ≤ x ≤ 0.45 and 0.6 < x ≤ 0.7, are characterized by an AFM-like transition without irreversibility at TN. However, irreversibility occurs at a lower temperature. This has been ascribed by a reentrant spin glass transition [RSG in the phase diagram in Fig. 5(c)]. Considering equal Mn–Fe substitutions in both end compounds, the reentrant spin glass phase is found to be stronger towards Fe-rich compositions as compared to Mn-rich compositions, which is manifested by the higher reentrant spin glass transition temperature for x = 0.7 than x = 0.3.81 Such behavior could stem from much larger single-ion anisotropy A for FePS3 (Table 4) as compared to MnPS3, as has been proposed in dilute alloys.162
Overall, Mn1−xFexPS3 displays a non-monotonic doping dependence of TN with a minimum of around x ≈ 0.5. Such behavior is widely seen in other metal-substituted MPX3 and is usually interpreted by spin frustration due to the mixing of two types of magnetic ions compounds (MnPS3 and FePS3) with significantly distinct magnetic exchange interactions J1, J2, and J335,107 as summarized in Table 4.
4.2.1.1.2. Fe1−xNixPS3. For Fe1−xNixPS3, the distinct XY- and Ising-like antiferromagnetism for the end members NiPS3 and FePS3 respectively makes it rather interesting to investigate the evolution between those two magnetic orders with distinct magnetic anisotropies.
Fe1−xNixPS3 single crystals were grown using CVT with a temperature gradient from (760–700) to (690–600) °C for 200 hours.88,91 The magnetic properties of Fe1−xNixPS3 were characterized using temperature and field dependence of magnetization. Fig. 6(a) presents the temperature dependence of susceptibility (χ = M/H) with the magnetic field of 1 T along a, b, and c*. Here, c* represents the direction that is perpendicular to the crystallographic ab-plane, which is slightly deviated from the crystallographic c axis in a monoclinic unit cell. For end compounds FePS3 (x = 0) and NiPS3 (x = 1), susceptibility first increases upon cooling, forming a broad maximum at around temperature Tmax, followed with the AFM transition temperature TN below Tmax. On increasing Ni content x, TN is found to slightly reduce up to x = 0.3, followed by systematic increase with further increasing Ni content [Fig. 6(b)]. Judging from magnetic exchange interaction, such TN increase can be explained by stronger magnetic exchange interactions J1 and J3 for NiPS3128 than FePS3107 (Table 4). Composition dependence for Tmax follows a similar non-monotonic trend, but Tmax increases more remarkably with Ni content in Ni-rich compositions [Fig. 6(b)]. As mentioned in the previous section on Mn1−xFexPS3, the broad maximum exists in pristine MPX3 and has been ascribed to the strong low-dimensional dynamic magnetism pertaining to short-range correlated spins. Similarly, in Fe1−xNixPS3 this broad maximum is also suppressed by substitution, leading to sharper susceptibility transition at TN in substituted samples. This suggests the suppression of the short-range magnetic correlation by substitution in this system. However, the Raman spectroscopy results based on two-magnon Raman scattering reveal that the short-range dynamic magnetism does not quickly quench but rather persists to compositions much below x = 0.9.91 Therefore, more efforts are needed to clarify the nature of such a broad maximum above TN.
 | ||
| Fig. 6 (a) Temperature dependencies of magnetization divided by magnetic field MH−1 for Fe2P2S6, (Fe0.7Ni0.3)2P2S6, (Fe0.5Ni0.5)2P2S6, (Fe0.3Ni0.7)2P2S6, (Fe0.1Ni0.9)2P2S6, and Ni2P2S6, measured with a field of 1 T applied along three different crystallographic directions. TN and Tmax denote Néel temperature and the maximum magnetization temperature. (b) Evolution of TN and Tmax with composition for (Fe1−xNix)2P2S6. Reprinted (adapted) with permission.88 Copyright 2021, American Physical Society. | ||
The rapid suppression of a broad maximum with Fe substitution in NiPS3 also suggests the modification of XY-type magnetism. In fact, the evolution from XY- to Ising-type anisotropy below x < 0.9 is clearly manifested in susceptibility. For pristine NiPS3 (x = 1), susceptibility measured with field along a (χa), b (χb), and c* (χc*) axes overlaps above TN but display significant anisotropy below TN [Fig. 6(a)], implying a moment orientation along the a-axis and consistent with the reported magnetic structure.123 However, by adding Fe, the x ≤ 0.9 (i.e., Fe content ≥ 10%) samples display anisotropy even above TN, and χc* exhibits much stronger temperature dependence. Those observations are characters for highly anisotropic Ising-type magnetism with out-of-plane moment orientation of the end compound FePS3 (x = 0).107 Therefore, a transition from XY- to Ising-type anisotropy may start with only 10% Fe substitution in NiPS3. Such sensitive tuning of magnetic anisotropy in NiPS3 by just 10% Fe substitution might be attributed to a much stronger (≈9 times) single-ion anisotropy (A) for FePS3107 than NiPS3128 (Table 4).
4.2.1.1.3. Ni1−xMnxPS3. Ni1−xMnxPS3 provides a platform to study spin-flop transitions under relatively low magnetic fields. In AFM systems, a magnetic field along the easy axis exceeding a critical spin-flop field HSF drives the magnetic moments to rotate to a canted configuration with a component perpendicular to the field direction, resulting in a net moment along the easy axis.163,164 Such field-driven moment reorientation from AFM to SF state is known as the spin-flop (SF) transition, which manifests into a super-linear behavior in field-dependent magnetization measurements (i.e., magnetization upturn). This is different from the spin–flip transition in which the moments are polarized to the magnetic field direction under a strong field and is characterized by a magnetization saturation-like behavior. For MnPS3 with magnetic moment along the out-of-plane direction (Fig. 3),78,134 a SF transition in the isothermal magnetization has been observed under out-of-plane magnetic field (H⊥ab) but is absent under in-plane field (H∥ab) [Fig. 7(a)–(c)].10,28,65,119,146 During this SF transition, the magnetic moments undergo continuous rotation towards the ab-plane.134 Although the SF transition was discovered a long time ago165 and widely studied from bulk to atomically thin MnPS3,29 it has been reported only recently in NiPS3.89 NiPS3 displays an SF transition under the in-plane field of ∼6 T at 2 K [Fig. 7(c)], which is consistent with the almost in-plane moment orientation of this compound (Fig. 3),33,111,166 as shown by a black arrow in Fig. 7(c). Also, the zig-zag AFM structure with moments oriented along a-axis leads to an anisotropic SF transition that is sensitive to the in-plane field orientations.89
 | ||
| Fig. 7 (a) Schematics of spin-flop transition for MnPS3 and NiPS3.89 (b and c) Magnetic field dependence of magnetization for Ni1−xMnxPS3 under in-plane (H∥ab) and out-of-plane (H⊥ab) magnetic fields.89 Reprinted (adapted) with permission.89 Copyright 2021, American Physical Society. (d) Temperature dependence of magnetization for (Mn1−xNix)2P2S6 under in-plane (H∥ab) and out-of-plane (H∥c) magnetic fields.87 (e) Evolution of TN and Tmax with composition for (Mn1−xNix)2P2S6.87 (f) Doping dependence of Néel temperature (TN), spin-flop field (HSF), and the effective magnetic anisotropy field (HA) of Ni1−xCrxPS3.177 | ||
The distinct SF behavior in NiPS3 and MnPS3 indicates an evolution of SF transition with composition in “mixed” compounds Ni1−xMnxPS3. Indeed, magnetism is found to be highly tunable with composition. Fig. 7(b) and (c) show that the in-plane and out-of-plane magnetizations exhibit a systematic enhancement upon increasing Mn content x, consistent with the greater magnetic moment of Mn2+ than Ni2+. Interestingly, the SF transitions are also extremely sensitive to composition variation.89 The SF transition in NiPS3 under an in-plane field disappears up to 5% Mn substitution (i.e., x = 0.05 in Ni1−xMnxPS3, Fig. 7(c)). Similarly, the HSF for the out-of-plane field in MnPS3 is reduced by half with 5% Ni substitution (i.e., x = 0.95) and disappears upon 10% substitution (x = 0.9) [Fig. 7(b)]. Such highly sensitive nature of SF transition to light substitution of magnetic atoms is very different from that of non-magnetic substitution. It has been found that 20% substitution is needed to reduce HSF by half in Zn-substituted MnPS3.78,85 In Ni-substituted MnPS3, the substantially different single-ion anisotropy (Table 4) may explain the efficient suppression of SF transition. A similar mechanism has also been proposed for the suppression of HSF in MnPS3 under pressure.135 In addition, Ni–Mn substitution can also be viewed as inducing magnetic impurities, especially in low-substitution levels. Hence, it is necessary to consider the magnetic interaction to understand the observed sensitive doping dependence in Ni1−xMnxPS3. The magnetic ordering temperature TN characterizes the strengths of magnetic interactions. For lightly substituted samples with x close to 0 or 1, the TN is found to only change slightly as compared to the parent compounds NiPS3 and MnPS3, which is interesting given that the light Ni–Mn substitution drastically suppresses the SF transitions. This suggests that the efficient suppression of the SF transition with light magnetic substitution in Ni1−xMnxPS3 can be attributed to the tuning of single ion isotropy rather than exchange interaction.
The modification of magnetic anisotropy with Ni–Mn substitution in Mn1−xNixPS387 is further demonstrated by the temperature dependence of susceptibility (χ = M/H) [Fig. 7(d)]. For compositions 0 ≤ x ≤ 0.5, the out-of-plane susceptibility (χc, measured under an out-of-plane field) is significantly reduced below TN in comparison to in-plane susceptibility (χab, measured under an in-plane field), implying moment orientation along the c axis, consistent with the reported magnetic structure of pristine MnPS3.78,134 This susceptibility trend is reversed on further increasing Ni content above x > 0.5 in which χab < χc below TN,87 suggesting switching of magnetic easy axis from out-of-plane to in-plane direction above x > 0.5. These results agree well with a theoretical study that predicts the magnetic transition from Néel-type (where all nearest-neighbor spins are aligned antiparallel as shown in Fig. 3) in pristine MnPS3 to zigzag-type in pristine NiPS3 (Fig. 3) at around x = 0.25–0.5.152
In addition, the TN and Tmax (broad maximum temperature) in susceptibility also provide useful information. As shown in Fig. 7(e), both TN and Tmax undergo non-monotonic dependence on Ni–Mn substitution with a minimum around x = 0.5 (ref. 87, 89 and 152) that can be ascribed to the attenuation of the magnetic interaction due to magnetic disorder in such a magnetically substituted system. Tmax is much larger than TN for both MnPS3 and NiPS3, but is comparable with increasing substitution levels [Fig. 7(e)], reminiscent to the observation for Fe1−xNixPS388,91 described above. Furthermore, for x = 0.5 composition, the PM to AFM transition at TN = 42 K is followed by the emergence of a reentrant spin glass state at low temperature,152 which is manifested by the irreversibility between ZFC and FC below T = 30 K similar to the case of Mn1−xFexPS3 described above.81
4.2.1.1.4. Ni1−xCrxPS3. The Cr-based layered compounds, such as CrI3, Cr2Ge2Te6, CrCl3, and CrPS4, have recently gained intensive attention because of their robust magnetic order persisting in the 2D limit.7,8,167–169 Surprisingly, the study of Cr-based MPX3 compounds has been very limited. So far, only CrPSe3
170 and Cr2/3PS3171 have been reported while the stoichiometric CrPS3 has not been experimentally realized. This has been attributed to the weakened P–P dimerization that favors the formation of Cr3+ cation instead of the +2 metal valence required to stabilize the MPX3 lattice.172 The direct substitution of Cr3+ for M2+ ion in MPX3 is challenging from a chemical valence balance perspective, which may eventually lead to vacancies at metal sites. A similar issue also occurs for VxPS3 (x = 0.78 (ref. 173 and 174) and 0.9 (ref. 71)) in which V2+ and V3+ ions coexist and lead to V vacancies. Indeed, Cr substitution in MPX3 has been found to be challenging in comparison to other divalent metal ion substitutions.87–89,91,175,176
So far, light substitution of up to 9% without inducing a clear signature of vacancies in NiPS3 has been recently reported.177 The lack of vacancies from composition analyses implies an isovalent substitution of +2 valence for Cr ions. Interestingly, a low Cr amount already can efficiently modulate the magnetism in NiPS3. As shown in Fig. 7(f), first, the magnetic transition temperature TN is found to reduce significantly from 155 K in NiPS3 to 34 K in the x = 0.09 sample. Second, the SF transition under an in-plane magnetic field in NiPS3 is significantly suppressed [Fig. 7(f)]. Also, the SF transition starts to occur even under a weak out-of-plane magnetic field of <1 T in the x = 0.09 sample, which strongly implies moment reorientation upon Cr substitution. Evaluating the characteristic field for spin flop, magnetic exchange, and magnetic anisotropy also reveals that the magnetic anisotropy is significantly suppressed by Cr substitution. Furthermore, in addition to SF transition, a moment polarization-like behavior has been observed under both in-plane and out-of-plane fields, with a similar polarization field of ∼8 T and a small saturation moment of 0.24μB per formula unit.
Suppressed SF transition and more isotropic magnetization are consistent with a suppression of magnetic anisotropy upon Cr substitution, which gives rise to more controllable moment orientations and eventually the moment polarization in the x = 0.09 sample. Indeed, similar field-induced moment polarization at relatively low magnetic fields have also been seen in quite a few Cr-based AFM compounds such as CuCrP2S6,153,154 CrPS4,178 CrCl3,179 and CrSBr.180 However, owing to the 3+ valence of Cr ions instead of the 2+ metal valence expected in MPX3, substituting Cr is limited to only 9% in NiPS3, as discussed above. This makes it difficult to explore the potential of this route.
Therefore, recently, an alternative approach that balances the valence by co-substituting mono-valent metal ions together with the tri-valent Cr ions has been reported, through which all M2+ ions can be replaced to tune the magnetism more efficiently.181 This work provides a new doping strategy in the MPX3 family, which successfully substitutes an equal Cu and Cr for Ni leading to a wide range of medium-entropy compositions CuxNi2(1−x)CrxP2S6 (0 ≤ x ≤ 1).
4.2.1.1.5. Mn1−xCoxPS3. Compared to other metal substitutions, Co substitution has been fairly elusive in MPX3 with only a few recent studies.114,116,117 This might be due to the challenges in crystal growth for CoPS3117 which causes this compound and its derivatives to be less-explored.36,108,129,182 A recent breakthrough in reactive flux technique using P2S5 and metal precursors offers an opportunity to study Co-substituted MPX3 compounds,114–118 as described in section 2. The MnxCo2−xP2S6 single crystals can be obtained by annealing the stoichiometric mixture of Mn, Co, and P2S5 powders at 540 °C for 72 hours.117
The two parent compounds Mn2P2S6 (Néel-type AFM with out-of-plane easy axis) and Co2P2S6 (zig-zag type AFM with in-plane easy axis) exhibit distinct AFM ordering, so magnetism is expected to be tunable with substituting. As shown in Fig. 8(a), the magnetic susceptibility of MnxCo2−xP2S6 lacks significant irreversibility between ZFC and FC in the entire temperature range, suggesting AFM ordering below TN without spin-glass state that is also seen in Fe-81 and Ni-substituted152 Mn2P2S6. Similar to many other metal substituted MPX3,81,85,87,89,152TN also exhibits a non-monotonic composition dependence, showing maximum values for the two end compounds (x = 0 and 1) and reduces upon mixing the metal atoms. In addition, the broad maximum temperature Tmax observed just above TN also displays a similar variation with substitution, except that the broad maximum in susceptibility is completely suppressed for the x = 1 (50% Mn and 50% Co) sample, as shown in Fig. 8(a). Such suppression is also observed in thin flakes of Mn2P2S6, which has been ascribed to the reorientation of Mn2+ moments that gives rise to a weak FM state.183 Similar weak ferromagnetism may also be responsible to the rapid rise in the susceptibility at low temperature below T < 30 K [Fig. 8(a)] and non-linear isothermal magnetization at T = 2 K [Fig. 8(b)] for this x = 1 sample. Nevertheless, one should always be caution about the presence of trace FM impurities in doped materials.
 | ||
| Fig. 8 (a) Field-cooled (FC) magnetic susceptibility of MnxCo2−xP2S6 measured with H = 1000 Oe.117 (b) Magnetization for MnCoP2S6 at 2 K and 150 K.117 Reprinted (adapted) with permission.117 Copyright 2022, American Chemical Society. (c) SEM images of nanosheet (NS) of NiPS3, Ni0.88Co0.12PS3, Ni0.68Co0.32PS3, and Ni0.60Co0.40PS3.90 (d) Magnetization at 5 K for Ni1−xCoxPS3 NS and single crystal samples.90 | ||
The evolution of Curie–Weiss temperature θcw extracted from the linear fit of the high temperature (T > 200 K) paramagnetic (PM) region of the inverse susceptibility also provides useful information.117θcw is negative for AFM MnxCo2−xP2S6, which undergoes a non-monotonic variation with Mn–Co substitution and reaches the maximal (i.e., less negative) at x = 1, indicating the attenuation of AFM interaction around this composition. This is probably due to magnetic frustration owing to the competition between distinct AFM exchange interactions J1, J2, and J3 for Mn2P2S635 and Co2P2S6.129
4.2.1.1.6. Ni1−xCoxPS3. Magnetic properties of Co-substituted NiPS3 (Ni1−xCoxPS3) have been investigated in both single crystals and nanosheet (NS) samples.90 Ni1−xCoxPS3 single crystals were grown using a CVT technique as shown in Table 1. For NS samples, Ni1−xCoxPS3 (0 ≤ x < 0.5) NS of average thickness of 8 nm (around 10 layers) were grown by a chemical vapor conversion method via a two-step process: first, the metal hydroxide NS precursor containing various Ni
:Co ratios were synthesized on carbon cloth through a wet chemical method. Second, these metal hydroxide precursors were loaded downstream of an evacuated custom-designed fused silica socket tube with a mixture of P and S positioned on the other side. Ultrathin Ni1−xCoxPS3 NS can be evenly grown on carbon cloth over a reaction time of 90 min at 480° to 490 °C with an argon gas flow [Fig. 8(c)].90
Magnetization measurements with the background from carbon cloth removed are shown in Fig. 8(d). TN ≈ 155 K for pristine NiPS3 is suppressed by Co substitution for both single crystals and NS, which is in line with the general trend of metal substitution in MPX3 as described above. In contrast to bulk single crystals, susceptibility for all Ni1−xCoxPS3 (0 ≤ x ≤ 0.4) NS samples display low-temperature upturns, suggesting the rise of weak ferromagnetism. Field dependence of magnetization [Fig. 8(d)] also reveals magnetic hysteresis over a low field region (|H| < 300 Oe), which becomes more obvious with increasing Co content.
Providing similar lattice parameters and zig-zag type AFM structures for pristine NiPS3 and CoPS3, it is not surprising that Co–Ni substitution does not cause significant modification in magnetic properties. The low-temperature magnetization upturn and magnetic hysteresis occur in NS samples, which might be attributed to the sulfur vacancy in NSs. Electron spin resonance (ESR) spectroscopy measurements90 have revealed that the sulfur vacancy formation, which is in line with the rise of ferromagnetism with Co substitution. Defect-induced ferromagnetism has been observed in the atomically thin layers of non-magnetic 2D materials such as graphene2,184 and MoS2.4,185 As mentioned earlier in section 4.1, in NiPS3, the magnetic interactions are mainly governed by the Ni–S–Ni superexchange interactions, occurring through the overlapping of half-filled Ni eg and S 3p orbitals that mediates the effective electron hopping between neighboring Ni sites.128 Therefore, tuning of magnetic states due to sulfur vacancy is plausible in Ni1−xCoxPS3 NS. In fact, the theoretical calculations have demonstrated the disruption of anion-mediated superexchange interaction between Ni moments due to sulfur vacancy.90 Furthermore, the calculated electronic structure has revealed unoccupied in-gap-like states that could serve as a carrier trap site and hinder the charge transfer process, further suppressing superexchange interaction and might facilitate other competing magnetic phases in NiPS3.
4.2.1.1.7. Non-magnetic dopant: MnxZn1−xPS3. Magnetic properties of Zn-substituted MnPS3 have been widely studied using magnetization77,78,85 and Raman151 measurements. Up on replacing Mn by non-magnetic Zn, the broad maximum immediately above TN in susceptibility measured with in-plane field starts to vanish with increasing Zn substitution, leading to a sharp susceptibility peak at magnetic transition TN.85 This suggests a breakdown of short-range correlation, as expected for metal substitution in MPX3.81,87,88,91 In addition, for Zn-rich samples (Zn content ≥ 0.5), the low-temperature susceptibility displays an abrupt enhancement, which has been ascribed to a weak PM behavior of the significant number of isolated spins due to magnetic dilution by Zn.85,151
Suppression of transition temperature TN by Zn substitution is seen in both susceptibility85 and Raman151 experiments. Unlike magnetic ion substitutions that always lead to a U-shape non-monotonic composition dependency of TN, non-magnetic Zn substitution gradually suppresses TN to zero temperature up to 50% substitution level in magnetization measurements77,85 [Fig. 9(a)], or even to 70% from extrapolating the results from Raman spectroscopy151 [Fig. 9(b)]. This is surprising given the critical concentration for site dilution in a honeycomb lattice with nearest-neighbor interactions (J1) is x = 0.7.186 Therefore, J1 alone may not explain the behavior of MnxZn1−xPS3, and the second (J2) and the third (J3) nearest-neighbor interactions, though weaker than J1 in MnPS3 (see Table 4), might be ascribed for the magnetic stability below x < 0.7.85 Of course, measurements on the addition of high-Zn samples would provide a better estimate of the critical composition, which was determined by linear extrapolation of a few compositions151 as shown in Fig. 9(b).
 | ||
| Fig. 9 (a and b) Magnetic phase diagrams for MnxZn1−xPS3. Field dependence of phases is also shown in (a).85,151 Reprinted (adapted) with permission.85 Copyright 1998, IOP Publishing. Reprinted (adapted) with permission.151 Copyright 2023, American Physical Society. (c) Temperature dependence of magnetic susceptibilities of Fe1−xZnxPS3 under an in-plane magnetic field of 1000 Oe.93 (d) Temperature dependence of the magnetic susceptibility for Cu0.15Fe0.85PS3 measured under in-plane (H||plane) and out-of-plane (H⊥plane) field of 2000 Oe. (e) Field dependence of magnetization for Cu0.15Fe0.85PS3 at T = 10 K.187 | ||
The impact of Zn substitution in MnPS3 is further manifested as reduced SF transition field HSF.78 Substituting Zn for Mn breaks down the long-range magnetic order, resulting in “weakly bound” Mn moments aligned along the out-of-plane direction. For MnPS3 in which the dipolar anisotropy dominates, the local dipole field along the out-of-plane direction may be attenuated by these weakly bound moments. Furthermore, replacing a Mn with a non-magnetic Zn is accompanied by a vanishing moment at the substitute site, which affects the nearby Mn moments and causes them to be canted.77 On a larger scale, this leads to an average staggered magnetic moment which consequently suppresses the magnetic anisotropy of the system, which eventually suppresses HSF.78
4.2.1.1.8. Non-magnetic dopant: Fe1−xZnxPS3. Magnetic dilution by non-magnetic Zn substitution has also been studied in Ising-type antiferromagnet FePS3.93 Substituting Zn in FePS3 is reported to induce weak ferromagnetism associated with spin/cluster spin glass.93 As shown in Fig. 9(c), substituting Fe with non-magnetic Zn suppresses TN. For Zn content x greater than 0.3, significant irreversibility between ZFC and FC measurements starts to occur at temperatures below TN, which has been ascribed to weak ferromagnetism associated with glassy behavior.93 For high Zn samples (0.7 ≤ x ≤ 0.9), susceptibility upturn appears at low temperatures, which has been attributed to the emergence of FM order with an out-of-plane easy axis.93 This conclusion is also supported by the Curie–Weiss temperature (θCW), which becomes less negative with increasing Zn substitution and even attains positive values for χ⊥ of x = 0.7 and 0.9 samples, suggesting the attenuation of AFM interactions and development of FM correlation with Zn substitution in FePS3.93
The rise of ferromagnetism is further supported by the field dependence of magnetization. The in-plane magnetization displays linear field dependence for all compositions and temperature ranges. However, under H⊥ab magnetic field, the x = 0.9 sample displays magnetic hysteresis and saturation behavior with saturation moment reaching ∼4μB/Fe, which is consistent with the S = 2 Fe2+.93 The theoretical study further reveals the key role played by hole doping to stabilize ferromagnetism in Zn-substituted FePS3.93 In highly Zn-substituted samples x = 0.7 and 0.9,93 the XPS result reveals the presence of Fe3+ ion in addition to Fe2+ ion, leading to hole doping and the subsequent rising of FM interactions mediated by bound magnetic polarons induced impurity-band-exchange in such diluted environment induced by Zn substitution in FePS3.
4.2.1.1.9. Non-magnetic dopant: Fe1−xCuxPS3. In addition to Zn, magnetic dilution in FePS3 has also been realized by Cu substitution.187 Cu substitution up to 15% is found to suppress TN to ≈108 K from that of 120 K in pristine FePS3 [Fig. 9(d)],187 consistent with the result obtained for Zn-substituted FePS3
93 and can be attributed to the suppression of magnetic exchange interactions, as discussed in the previous section. Other than that, the susceptibility for the substituted sample is highly similar to that of the pristine FePS3: the out-of-plane susceptibility measured with a magnetic field perpendicular to the basal plane displays a much stronger temperature dependence below TN than in-plane susceptibility. In addition, in the PM state, susceptibility displays strong anisotropy, with a much higher value for the out-of-plane susceptibility that is similar to undoped FePS3 [Fig. 9(d)]. These observations suggest that magnetic anisotropy is not strongly modified upon Cu substitution up to 15%.
In field-dependent magnetization, the 10% Cu-substituted sample displays a weak non-linearity under an out-of-plane magnetic field [Fig. 9(e)], which was claimed to be a signature of weak ferromagnetism due to the reorientation of canted moments, and has also been observed in Zn-78 and Ni-substituted89 MnPS3. In addition to a moment canting scenario, other mechanisms, such as sulfur vacancy which has been seen in Co-substituted NiPS3 nanoflakes90 (see section 4.2.1.1.5) and the presence of the finite clusters of uncompensated spins187 might also give rise to weak ferromagnetism in Cu0.15Fe0.85PS3.
4.2.1.1.10. Non-magnetic dopant: Fe1−xCdxPS3. Cd as another non-magnetic dopant has been studied a long time ago on a 50% Cd-substituted Fe0.5Cd0.5PS3 sample.188 Magnetic phase transition is not observed in susceptibility measurements down to 57 K. The Curie–Weiss temperature displays positive values, decreasing from 104 K for pristine FePS3 to 40 K for Fe0.5Cd0.5PS3. The positive Curie–Weiss temperature even in pristine FePS3 appears interesting given it well defined AFM ground state. It might be ascribed to the competition between the direct FM J1 and indirect AFM J2 interactions.188 The suppression of Curie–Weiss temperature upon introduction of Cd in FePS3 can be attributed to the attenuation of direct J1 owing to the expanded lattice due to Cd substitution, which increases the nearest neighbor distance of Fe2+ by 1.95%.188 A more concrete understanding may be obtained by future substitution studies covering more Cd compositions.
4.2.1.1.11. Metal substitution in selenides: Fe1−xMnxPSe3. Most of the metal substitutions were performed on sulfides as summarized above, whereas the selenide MPSe3 compounds are relatively less explored. Mn–Fe substitution has been reported in selenide compounds MnPSe3 and FePSe3,86 which are characterized by Néel-type and Ising-type magnetic structures with in-plane and out-of-plane moments, respectively (Fig. 3). Similar to sulfide FePS3 stated above, anisotropy between out-of-plane susceptibility χ⊥ and in-plane susceptibility χ∥above TN is a character of Ising-type magnetism in FePSe3. Such anisotropy is suppressed by Mn substitution for Fe [Fig. 10(a)],122 which is expected since the end compounds FePSe3 and MnPSe3 possess different magnetic structures with different easy axes. Interestingly, the susceptibility anisotropy below TN suggests that the out-of-plane magnetic easy axis for FePSe3 is robust against Mn-substitution up to 90%, which is also supported by the emergence and evolution of the SF transition. SF transition is not observed up to 9 T in FePSe3 due to the strong magnetic anisotropy for Ising-type magnetism. Heavy Mn substitution leads the SF to occur in out-of-plane magnetization only, and further increased Mn content beyond 90% causes the SF transition to appear in in-plane magnetization, implying the switching of easy axis from out-of-plane to in-plane when Mn content x > 0.9 [Fig. 10(b)]. The rotation of the magnetic easy axis occurring only above 90% Mn for Fe substitution suggests a highly anisotropic magnetic order in FePSe3. This can be estimated from the relative magnitudes of single-ion anisotropy (A) for the two end compounds FePSe3 and MnPSe3. Though the experimental value of A is still lacking for FePSe3, in sulfide counterparts FePS3
107 possesses much higher A by >300 times than that of MnPS335 (Table 4). Expecting a similar scenario in selenide samples, the robust magnetic easy axis against up to 90% Mn substitution in FePSe3 can be understood.
 | ||
| Fig. 10 (a) Temperature dependencies susceptibility for Fe1−xMnxPSe3 measured under a magnetic field of 0.1 T.122 (b) Isothermal magnetization at 2 K for Fe1−xMnxPSe3 under out-of-plane (H⊥ab, red) and in-plane (H||ab, blue) magnetic fields. Insets show spin-flop transition at low fields.122 (c and d) Magnetic phase diagrams of Mn1−xFexPSe3 obtained from (c) single crystals122 and (d) polycrystals.86 Reprinted (adapted) with permission.86 Copyright 2020, American Physical Society. | ||
Because both end compounds FePSe342 and MnPSe343 exhibit 2D magnetism down to the monolayer limit, exploring possible 2D magnetism in their mixed compounds (Fe1−xMnxPSe3) is interesting. Having said that, the strong frustration arising from mixing two different magnetic metal ions may destabilize magnetic order in the 2D limit. Indeed, such mixing suppresses TN and leads it to reach a minimum for 50% substitution in studies using both single crystals122 [Fig. 10(c)] and polycrystals86 [Fig. 10(d)]. This appears to be a generic behavior for all polymetallic MPX3.81,85,87,89 In addition, a mixed phase consisting of both Fe2+- and Mn2+-type ordering, forming nano-sized chemically disordered clusters in intermediate compositions (0.25 < x < 0.875) has also been discovered86 [Fig. 10(d)].
4.2.1.1.12. HEPX3 (HE = high entropy; X = S or Se). High entropy (HE) alloy involves the mixing of five or more elements each with a composition of 5–35 atomic percentage. The composition of HE compounds can be greatly varied by adjusting the ratios of constituent elements. Such flexible composition stoichiometry allows for property tuning with a wide compositions range. Incorporating multiple elements also offers various valence states and spin states in a system. Depending on the choices of elements, the distribution of charges and spin interactions may vary significantly. Furthermore, the presence of multiple elements results in a complex and disordered atomic structure. Hence, with the added complexity, HE compounds provides an opportunity to investigate the interplay between charge, spin, and composition degrees of freedom. Recently, the bimetallic substitution in MPX3 has been extended to the realm of HE compounds.113,121,189 Providing various anisotropy of ions and diverse magnetic structures for different MPX3 compounds, such a strategy is expected to lead to rich magnetic properties and phases.
For Mn0.25Fe0.25Co0.25Ni0.27P1.04S3113,189 with equal molar ratio of metal atoms, a well-defined PM to AFM transition at TN = 70 K is found in susceptibility measurements113 [Fig. 11(a)]. Deviation from this equal metal concentration results in broader or less-obvious AFM transition, as observed in Mn0.2Fe0.3Co0.25Ni0.25PS3.189 In Mn0.25Fe0.25Co0.25Ni0.27P1.04S3, the magnetic moments are likely to be aligned perpendicular to the basal plan, providing a weaker temperature dependence for susceptibility under an in-plane field. A spin-glass state is also proposed due to the emergence of irreversibility between ZFC and FC susceptibility at a temperature below TN [Fig. 11(b)]. Similarly, the spin-glass phase has also been observed in a few other HEPS3 compounds at different glass temperatures,113,189 as summarized in Table 5. In addition to sulfides, selenide-based HE compounds have also been studied in Fe–Mn–Cd–In-based HE MPSe3.121 The comparison study on FePSe3, Fe0.8Mn0.1Cd0.05In0.05PSe3, Fe0.7(MnCd)0.1In0.1PSe3, and (FeMnCd)0.25In0.17PSe3 has revealed decreased TN with reducing Fe amount [Fig. 11(c)]. The AFM transition disappears in (FeMnCd)0.25In0.17PSe3, leaving a substantial susceptibility irreversibility around 15 K [Fig. 11(c)] that implies a glass state, which is further confirmed by the frequency-dependent peak in AC susceptibility measurements.121
 | ||
| Fig. 11 (a and b) Magnetization of (Mn,Fe,Co,Ni)PS3 under the external magnetic field of 500 Oe along different directions. The antiferromagnetic transition (TN) and spin glass transition temperature (Tg) are indicated.113 Reprinted (adapted) with permission.113 Copyright 2021, American Chemical Society. (c) Susceptibility (χ) of pristine FePSe3 and (Fe,Mn,Cd,In)PS3.121 Reprinted (adapted) with permission.121 Copyright 2022, American Physical Society. | ||
| HEPX3 compounds | Magnetic properties | Ref. |
|---|---|---|
| Mn0.25Fe0.25Co0.25Ni0.27P1.04S3 | AFM (TN = 70 K) and spin-glass (Tg1 = 35 K, Tg2 = 56 K) | 113 |
| Zn0.29Mn0.14Fe0.17Co0.18Ni0.24PS2.61 | Spin-glass (Tg = 30 K), Tkink = 120 K | 113 |
| Mg0.19Mn0.18Fe0.19Co0.26Ni0.28P1.08S3 | Multikinks in MT at 8 K, 42 K, 60, and 120 K | 113 |
| V0.16Mn0.18Fe0.21Co0.23Ni0.24PS2.62 | Spin-glass (Tg = 37 K), Tkink = 150 K | 113 |
| (FeMnCd)0.25In0.17PSe3 | Spin-glass (Tg = 15 K) | 121 |
4.2.1.2. Non-isovalent substitution.
4.2.1.2.1. CuCrP2X6 (X = S or Se). The metal substitution in M2P2X6 has been extended to the mixing of monovalent M11+ (M1 = Cu or Ag) and trivalent M23+ (M2 = V or Cr) ions to from (M11+)(M23+)P2X6.153–157,190–192 Chemical valence is balanced by maintaining an equal amount of monovalent and trivalent ions. While these compounds have been discovered a long time ago,155,157,190,193 there have been only a few studies on magnetic properties.154,155,157,190,193,194 CuCrP2S6
153,154 has recently gained increased attention. In contrast to the random distribution of dissimilar metal ions in isovalent metal-substituted M2P2X6,76–91 weak repulsive Coulomb interactions and substantial size difference between Cu1+ and Cr3+ cations156 favor an alternating arrangements of them in CuCrP2S6:153,154 as shown in Fig. 12(a), Cr3+ ions are located almost at the center in the vdW layer, whereas Cu+ ions are slightly displaced from the center at the alternative positions above and below the Cr3+ layers.154 This further leads to broken inversion symmetry and ferroelectricity up to room temperature.58,59,195
 | ||
| Fig. 12 (a) Crystal structure of CuCrP2S6. (b and c) Temperature and magnetic field dependencies of magnetic susceptibility of CuCrP2S6. (d) Spin-flop transition when the field is applied along the a-axis.154 | ||
Regarding the magnetic properties which is the focus of this review, CuCrP2S6 displays an AFM order below TN = 32 K (ref. 154) [Fig. 12(b)]. On the other hand, CuCrP2Se6 has a slightly higher TN (= 40 K) than that of CuCrP2S6, which is likely attributed to a stronger Cr–Se–Se–Cr superexchange interaction due to enhanced covalency arising from greater atomic orbitals for Se.196 The much sharper drop in susceptibility measured with the field along the a-axis in comparison to other field directions suggests the magnetic moment orientation along the a-axis. The linear fit of inverse susceptibility yielded a positive Curie–Weiss temperature θcw = 24 K [Fig. 12(b)], implying in-plane FM interaction that may be attributed to an A-type AFM order154 (see Fig. 3). The effective moments of μeff = (3.78 ± 0.05)μB f.u.−1 for H∥ab and μeff = (3.89 ± 0.05)μB f.u.−1 for H⊥ab153 match with the theoretical value of 3.87μB for Cr3+. Like many other Cr-based van der Waals magnets such as CrPS4,178 CrCl3,179 and CrSBr,180 as well as Cr-substitute NiPS3 mentioned above (section 4.2.1.1.6), a field-driven AFM to FM transition has been observed in magnetization measurements with a saturation moment of ≈3μB/Cr [Fig. 12(c)]. Consistent with the easy-axis along the a-axis, an SF transition appears in magnetization with low HSF ≈ 0.4 T [Fig. 12(d)].
As mentioned above in section 4.2.1.1.4, the mono- and tri-valent of Cu+ and Cr3+ allows for co-substitute other bi-valent ions such as Ni2+ to form CuxNi2(1−x)CrxP2S6 (0 ≤ x ≤ 1).181 This doping strategy provides further tuning of magnetism in the MPX3 family and leads to a wide range of medium-entropy compositions.
4.2.1.2.2. AgMP2X6 (M = V or Cr; X = S or Se). AgVP2S6 and AgCrP2S6 have been studied a long time ago.191,192 Structurally, these materials form a zig-zag chain of V3+ (S = 1) and Cr3+ (S = 3/2) ions respectively along the a-axis [Fig. 13(a)], which is distinct from other MPX3 compounds. Long-range AFM order is absent in AgVP2S6 from 5 to 200 K in susceptibility measurements191 [Fig. 13(b)]. In contrast, AgCrP2S6 is reported to be AFM below TN ≈ 7 K (ref. 192) [Fig. 13(c)]. Such TN from susceptibility is lower than TN ≈ 20 K obtained from powder neutron diffraction experiment.155 Both compositions display enhanced susceptibility at low temperatures, but AgVP2S6
191 is characterized by isotropic susceptibility for magnetic field directions along and perpendicular to the zig-zag chain direction [Fig. 13(a)], while AgCrP2S6 features a small anisotropy at low temperatures192 [Fig. 13(b)].
 | ||
| Fig. 13 (a) Crystal structure of AgVP2S6 and AgCrP2S6. (b and c) Temperature dependence of the susceptibility for AgVP2S6191 and AgCrP2S6.192 Reprinted (adapted) with permission.191 Copyright 1994, Elsevier. Reprinted (adapted) with permission.192 Copyright 1990, Elsevier. (d) Crystal structure (top and side views) of AgVP2Se6. (e) Temperature (left) and magnetic field (right) dependencies of magnetization for AgVP2Se6.197 | ||
A more recent study has expanded to selenide counterpart AgVP2Se6197 and AgCrP2Se6.198 These compounds are structurally slightly different from the above AgVP2S6 and AgCrP2S6 [Fig. 13(d)]. Interestingly, susceptibility irreversibility and magnetization saturation of AgVP2S6197 [Fig. 13(e)] implies ferromagnetism with a Tc ∼ 18.5 K, which is distinct from the AFM ground state for other MPX3. On the other hand, AgCrP2Se6198 exhibits AFM ground state below TN ∼ 42 K, which features field-driven moment polarization under the high field of B ∼ 3–6 T. Furthermore, polar-reflective magnetic circular dichroism (RMCD) experiments on AgVP2Se6197 found a growing hysteresis loop with reducing thickness in thin flakes, which persist down to 6.7 nm flake thickness with unchanged Tc.
4.2.2.1. MnPS3−xSex. In MnPS3−xSex, the successful substitution can be readily seen by the colors of the crystal. Upon increasing the Se content, these relatively transparent crystals gradually change color from green to wine red [Fig. 14(a)], indicating the variation of the optical gap.31 Upon Se substitution, TN displays a monotonic, slight reduction from 78.5 K for MnPS3 to 74 K for MnPSe3
31,32 [Fig. 14(b)], which is drastically different from non-monotonic composition dependence seen in metal-substituted MnPS3.81,85,87,89 This is found to be caused by the suppressed exchange interaction.31 Magnetism in MnPS3 is governed by the nearest-neighbor exchange interaction (J1) from the Mn–Mn direct exchange,39,128,129 and the third nearest-neighbor interaction (J3) is also considerable (Table 5). Se substitution for S expands in-plane lattice202,203 and elongates Mn–Mn distance, which attenuates the direct exchange and consequently reduces TN.31,32
 | ||
| Fig. 14 (a) Images of MnPS3−xSex single crystals. Doping dependence of (b) Néel temperature (TN) and (c) spin-flop (SF) field for MnPS3−xSex. (d) Images of NiPS3−xSex single crystals (0 ≤ x ≤ 1.3). (e) Doping dependence of TN for NiPS3−xSex. (f) Field dependence of magnetization of NiPS3−xSex at T = 2 K.31 Reprinted (adapted) with permission.31 Copyright 2022, American Physical Society. | ||
In addition to magnetic exchange interactions, chalcogen S–Se substitution also modifies magnetic anisotropy.31 This is expected because the two end compounds MnPS3 and MnPSe3, though both possess Néel-type AFM structure within each vdW layer where all nearest-neighbor spins are aligned antiparallel, feature different magnetic out-of-plane78,134 and almost-in-plane30,86,94,95 easy axis, respectively30,78,86,94,95,134 (Fig. 3). The modification of magnetic anisotropy is reflected by the evolution of SF transition. SF transition in MnPS3 occurs under an in-plane field. The SF field HSF is gradually suppressed to zero upon increasing Se content near x = 1 (MnPSSe2). On the other hand, the SF transition re-appears but under an out-of-plane field for x > 1 samples. Since SF transition in AFM material is driven by magnetic field component along the magnetic moment direction (i.e., easy axis), this implies the rotation of easy axis from out-of-plane towards the in-plane direction with Se substitution, which can be attributed to the enhanced single-ion anisotropy upon Se substitution that usually favors the in-plane moment orientation in MPX3. The switching of the easy axis is found to happen closer to the MnPS3 side i.e., between x = 0.7 and 1.2 [Fig. 14(c)], implying the magnetism in MnPS3 is softer than MnPSe3, which is also consistent with the results obtained from the theoretical calculations that reveal much smaller anisotropy between the in-plane and out-of-plane directions of the spins in MnPS3 than MnPSe3.31
4.2.2.2. NiPS3−xSex. The study on Ni-based NiPS3−xSex faces difficulty in synthesizing single crystals for Se-rich compositions. Indeed, the studies on NiPSe3 are very limited39 as compared to the sulfide counterpart NiPS3. A recent investigation on NiPS3−xSex
31 combining single crystal with Se content up to x = 1.3 [Fig. 14(d)] and polycrystals up to x = 3 have revealed monotonic enhancement in TN with increasing Se content, from ∼155 K for NiPS3 to ∼210 K for NiPSe3 [Fig. 14(e)]. This observation is distinct from the reduced TN due to metal substitution in NiPS3.87,89 Interestingly, compared to MnPS3−xSex in which Se substitution slightly reduces TN,31,32 in NiPS3 the similar Se substitution causes strong TN enhancement.31 Such results have been ascribed to different governing exchanges in Mn- and Ni-based systems.31 Magnetism of both systems are determined by the nearest J1 and third nearest J3 while the second nearest-neighbor interaction J2 is negligible (Table 4). Unlike MnPS3 in which the direct exchange between the neighboring metal ions contributes to J as described in the previous section, neutron scattering experiments39,128 have demonstrated that the magnetic interactions in NiPS3 are superexchange in nature. The direct exchange between Ni2+, however, does not exist due to the filled t2g orbitals for Ni2+.39,128 Therefore, compared to S, Se with extended 4p-orbitals favors orbital overlap and enhances superexchange between Ni2+ through chalcogen, giving rise to enhanced TN.
Similar to the case of MnPS3, Se substitution also tunes SF transitions in NiPS3.31 The SF transition under an in-plane field of ∼6 T for NiPS3 (x = 0) occurs at a higher in-plane field of ∼8 T for x = 0.25 [Fig. 14(f)]. Further increasing Se content up to 1.3 results in essentially linear magnetization under both in-plane (H∥ab) and out-of-plane (H⊥ab) fields up to 9 T. This is consistent with enhanced exchange interactions with Se substitution, based on evaluating the SF field,  where HE and HA are effective exchange and magnetic anisotropy fields, respectively.204 SF transition also provides a measure of magnetic anisotropy. When SF transition switches from one magnetic field direction to another, it suggests rotation of magnetic easy axis as seen in aforementioned MnPS3−xSex. For NiPS3−xSex, however, unavailable single crystals for Se-rich compositions, and the lack of experimentally determined magnetic structure for end compound NiPSe3, make it difficult to judge the evolution of magnetic anisotropy based on magnetization study.
where HE and HA are effective exchange and magnetic anisotropy fields, respectively.204 SF transition also provides a measure of magnetic anisotropy. When SF transition switches from one magnetic field direction to another, it suggests rotation of magnetic easy axis as seen in aforementioned MnPS3−xSex. For NiPS3−xSex, however, unavailable single crystals for Se-rich compositions, and the lack of experimentally determined magnetic structure for end compound NiPSe3, make it difficult to judge the evolution of magnetic anisotropy based on magnetization study.
4.2.2.3. FeP(S1−xSex)3. In the MPX3 family, FePS3 has been identified as a representative material that displays Ising-type magnetism characterized by out-of-plane magnetic moments (Fig. 3).6,40,107 Such Ising-type magnetism in FePS3 has been proposed to originate from the combination of the strong spin–orbit coupling (SOC) of the high-spin Fe2+ (d6) state and the trigonal distortion of FeS6 octahedra.40 Another Fe-based MPX3 compound FePSe3 which was discovered a few decades ago30,100 has so far received surprisingly less attention than FePS3 despite the fact that both FePS3
6,40,107 and FePSe330,42 exhibit similar Ising-type zig-zag AFM ordering from bulk to the monolayer limit. This is in stark contrast to the distinct magnetic structures (experimentally or theoretically determined) for sulfide and selenide compounds in some other known MPX3 such as MnP(S,Se)3 and NiP(S,Se)3.31,205 Such lack of change in magnetic structure in FeP(S,Se)3 naturally raises a question of whether chalcogen substitution may play any role in modifying magnetic properties, which has been investigated in ref. 122. For the entire composition range, magnetic susceptibility displays strong anisotropy both below and above TN, with the in-plane susceptibility measured under the in-plane field (H∥ab) showing much weaker temperature dependence as compared to the out-of-plane susceptibility [Fig. 15(a)], implying the persistence of the Ising-type magnetism up on Se substitution. In Mn- and Ni-based MPX3, the magnetic anisotropy mainly originate from anisotropic superexchange interaction that arises from SOC of non-magnetic ligands.206,207 However, in FeP(Se1−xSx)3, magnetic anisotropy predominantly stems from the strong crystal electric field on Fe2+ ion (discussed in section 4.1.),138,139 and thus S–Se substitution does not substantially change magnetic anisotropy and consequently maintains the robust Ising-type magnetic ordering in FeP(Se1−xSx)3.122
 | ||
| Fig. 15 (a) Temperature dependencies of susceptibility of FeP(Se1−xSx)3 measured under a magnetic field of 0.1 T. Doping dependence of (b) TN and (c) Fe–Fe distance for FeP(Se1−xSx)3.122 | ||
The evolution of TN with Se substitution is also interesting. Unlike Mn- and Ni-based MPX3 in which chalcogen substitution leads to monotonic decrease or increase in TN, respectively (see sections 4.2.2.1 and 4.2.2.2), in Fe-based system, a non-monotonic dependent is observed122 [Fig. 15(b)]. This has been attributed to the evolution of magnetic exchange:122 previous neutron scattering measurement has revealed dominant J1 in FePS3107 (Table 4), which is FM in nature and sensitive to Fe–Fe distance due to direct exchange interaction of the nearest-neighbor Fe moments.128,188 A similar direct exchange scenario occurs for MnPS3−xSex except J1 is AFM in nature (see sections 4.2.2.1). With the consideration that the competition between FM and AFM correlations determines the magnetic ordering type and transition temperature, the evolution of TN can be understood in terms of the nearest-neighbor Fe–Fe distance that governs the FM J1 in FeP(Se1−xSx)3. As shown in Fig. 15(c), the nearest-neighbor Fe–Fe distance in FeP(Se1−xSx)3 first reduces with S substitution up to x = 0.5. Further increasing S content slightly modifies the lattice structure and leads to elongated Fe–Fe distance. This causes a non-monotonic composition dependence for J1 and eventually results in non-monotonic evolution for TN.
4.2.3.1. Lithium intercalation. Lithium (Li) intercalation in MPX3 has been reported a long time ago.54,100,208,209 Earlier work mainly focused on the electrical, optical, and electrochemical properties of Li-intercalated MPX3. Regarding magnetic properties, the effects of Li intercalation on NiPS3, FePS3, FePSe3, and MnPSe3 compounds were first studied four decades ago.100 The intercalation was achieved by reacting MPX3-powered samples and n-butyllithium solution in hexane at room temperature in a dry, oxygen-free environment for 10–15 days. The duration of the reaction and concentration of n-butyllithium was found to affect the amount of intercalated Li. In this work,100 resistivity reduction in those powered samples implies successful intercalation. However, magnetic properties including transition temperature, effective moment, and Weiss temperature extracted from temperature-dependent magnetic susceptibility measurements do not display notable changes [Fig. 16(a)].
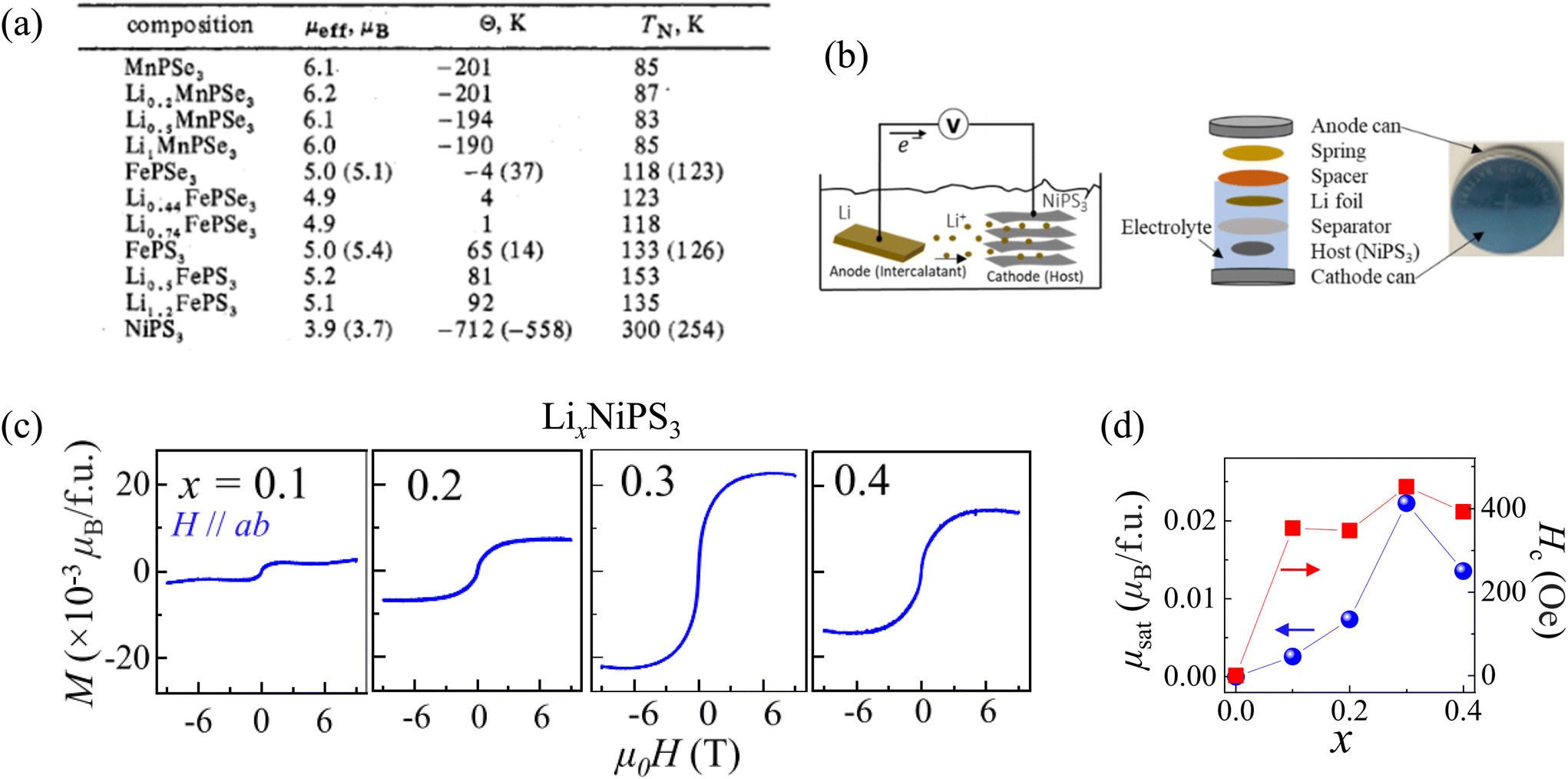 | ||
| Fig. 16 (a) Magnetic properties for LixMnPSe3, LixFePSe3, LixFePS3, and LixNiPS3.100 Reprinted (adapted) with permission.100 Copyright 1979, American Chemical Society. (b) Conceptual schematic of electrochemical intercalation process and the coin cell setup for intercalation.98 (c) Non-linear field dependence of magnetization of LixNiPS3 at T = 2 K after removing the linear magnetization background.98 (d) Evolution of saturation moment (μsat) and coercive fields (Hc) with Li content.98 | ||
Recent studies adopting electrochemical interaction to intercalate Li in single crystalline samples reveal modified magnetic properties in LixNiPS398 and LixFePS3.210 The intercalation in both studies was performed using a battery setup [Fig. 16(b)] and the expanded c-axis implies successful intercalation. For NiPS3, samples with Li content up to x = 0.4 are air-stable and maintain good crystallinity based on X-ray diffraction and scanning electron microscope imaging, though non-magnetic Li3PS4 impurity starts to occur for x ≥ 0.3. It is worth noting that the Li amount is the nominal content calculated based on the intercalation duration and electrical current because neither X-ray diffraction nor energy dispersive X-ray spectroscopy can directly probe Li. Similar to the earlier study using n-butyllithium solution for Li intercalation,100 the electrochemically intercalated LixNiPS3 (x = 0–0.4) samples do not display clear changes in TN. However, the development of non-linear field dependence for magnetization, and its saturation-like behavior after removing the linear background [Fig. 16(c)], and the rise of the hysteresis loop and tiny magnetic moment [Fig. 16(d)] have been ascribed to ferrimagnetism due to uncompensated antiparallel magnetic moments.98 For FePS3,210 in contrast, TN is found to be reduced with the expansion of the c-axis by Li-intercalation, suggesting a possible role of inter-layer magnetic exchange. The Ising-type magnetism characterized by strong anisotropy in magnetic susceptibility in the high-temperature paramagnetic state remains robust. At low temperatures, non-linear field dependence for magnetization is also observed, similar to Li-intercalated NiPS3.
4.2.3.2. Organic-ion intercalation. The vdW gap allows for not only the intercalation of small Li atoms or ions but also the much larger organic molecules or ions.96,97,99,211,212 Organic ion intercalation has been mainly performed on NiPS3.96,97,99 Intercalating tetraheptyl ammonium (THA+),97 tetrabutylammonium (TBA+),99 cobaltocenium ions [Co(Cp)2+, where Cp is a cyclopentadienyl ring C5H5−],99 and 1,10-phenanthroline96 using electrochemical97,99 and solution reaction96 have been performed. Compared to Li-intercalation, intercalating large organic ions results in a much more significant elongation of inter-layer spacing.96,97,99 It has also been found that the intralayer lattice structure does not display remarkable change upon intercalation.96
Organic-ions intercalation in NiPS3 is found to drastically modify the magnetic properties,96,97,99 mainly giving rise to a low-temperature FIM state97,99 that is also reported in Li-intercalated NiPS398 described in the previous section. The conclusion has been drawn based on the transition-like sharp susceptibility upturn around and below 100 K [Fig. 17(a) and (c)], as well as magnetic hysteresis loops in THA+-,97 TBA+-,99 and Co(Cp)2+ (ref. 99)-intercalated NiPS3 [Fig. 17(b) and (d)]. It has been proposed99 that the cation intercalation causes the reduction of certain Ni from the Ni2+ to the Ni0 state, together with its displacement from the Oh to the Td lattice site. Such transition from high spin Ni2+–Oh atoms ([Ar]3d8s0) to zero-spin Ni0–Td ([Ar]3d10s0) leads to uncompensated antiferromagnetism, i.e. a FIM state. An alternative mechanism based on the Stoner effect has also been proposed:97 the charge doping due to organic-ion intercalation enhances the density of states (DOS) at the Fermi level and eventually triggers the inter-chain Stoner splitting of the itinerant electrons, resulting in the higher electron concentration in one Ni chain to create net magnetic moment which gives rise to ferrimagnetism.
 | ||
| Fig. 17 (a) Temperature and (b) field dependencies of magnetization of intercalated THA-NiPS3.97 (c) Field-cooled temperature-dependent magnetization and (d) magnetic hysteresis for NiPS3, TBA0.25NiPS3, and [Co(Cp)2]0.25NiPS3.99 | ||
Recently, a new method using iron dopant as reaction active sites to intercalating organic-ion in NiPS3 has been reported.96 In this method, light Fe-substituted NiPS3 (i.e., Fe0.02Ni0.98PS3) single crystals are synthesized, which is followed by intercalation of a complexing agent 1,10-phenanthroline by solution reaction with aniline chloride.96 In this process, 1,10-phenthroline removes Fe2+ ions into the solution, creating metal-ion vacancies. The aniline chloride provides a proton to 1,10-phenthroline, and such protonated 1,10-phenanthroline is attracted towards the vacant metal sites, forcing intercalation of 1,10-phenanthroline into inter-layer spacing. The intercalated samples also display the signature of ferrimagnetism.
5. Engineering magnetism through pressure
In addition to chemical doping, pressure has also been established as an efficient external stimulus to tune magnetic properties. In MPX3, high pressure has been found to effectively modulate electronic properties.64–72 Recently, pressure has also been used to control magnetism in this material family.213–216 A recent review article has extensively summarized the theoretical and experimental progress of the structural, electronic, magnetic, and optical properties tuning of MPX3 under pressure.217 In this review, we focus on engineering magnetism in MPX3 materials. Since pressure generally tunes lattice structures and possibly induces structure transitions, it also sheds light on tuning magnetism through changing magnetic exchange and anisotropy. Therefore, here we briefly describe the pressure-driven modification of magnetism in MPX3. The magnetic properties of MPX3 are found to be sensitive to high pressure. TN systematically increases with the pressure for MnPS3213,216 and FePS3,214 which can be explained by the enhanced magnetic interactions under pressure. In particular, compressing the crystal planes closer strengthens the inter-layer interaction.214,216 For MnPS3, the TN also enhances with applying pressure, with different reported rates of 6.7 K GPa−1 (ref. 213) and 13 K GPa−1 (ref. 216) depending on the type of samples (polycrystal or single crystal). In addition, SF transition is tunable by pressure. In MnPS3,216 the SF field decreases at a rate of 4.0 × 103 Oe GPa−1, which is likely due to the reduction of magnetic anisotropy under pressure. Furthermore, the magnetic structure for MnPS3 remains unchanged up to ∼3.6 GPa (ref. 213) but changes at ∼2 GPa for FePS3214 and ∼4 GPa for NiPSe3.215 Such changes in magnetic structures are accompanied by the insulator-to-metal transition and even superconductivity in FePSe372 and NiPSe3.215 Therefore, high pressure, as well as strain, are excellent tools that can be combined with chemical tuning (substitution and intercalation) to achieve additional properties, such as the combination of the desired magnetic structure and electrical conductivity.
6. Summary and perspectives
In this review article, we provide a summary covering the crystal growth, structure, and magnetic properties of the vdW-type AFM MPX3 material family, with a focus on property tuning by various doping scenarios. Metal substitution, chalcogen substitution, and inter-layer intercalation are highly effective in tuning the magnetic properties in MPX3, including magnetic ordering, exchange interactions, magnetic anisotropy, etc. Generally, AFM ground states are robust, and most of those metal and chalcogen substitution tune properties within antiferromagnetism, except for a few cases such as Fe1−xZnxPS3 and AgVP2Se6 that display FM-like behaviors such as irreversibility, hysteresis, and saturation magnetization. However, magnetization is not a bulk measurement as a tiny number of magnetic impurities may give rise to a strong magnetization signal. Also, quite a few FM-like signatures may originate from a spin–flip transition or a canted AFM state, as seen in AFM CuCrP2S6 and Ni1−xCrxPS3. Therefore, other bulk measurements such as neutron diffraction are needed to clarify the magnetic ground state as well as any field-induced phase transitions. Indeed, the rapid development of MPX3 and its derivatives calls for more neutron resource allocations on these interesting vdW magnetic compounds.On the other hand, intercalations of a few guest species have been reported to result in a FIM state at low temperatures. The differences in substitution and intercalation might be attributed to different consequences: substitution within each individual layer plays a role mainly in altering magnetic anisotropy and exchange interactions while intercalation in the inter-layer spacing might lead to carrier doping or reductive reaction for metal ions. Also, intercalation is efficient in tuning inter-layer distance, especially when intercalating large ions such as organic intercalation. This effectively reduces interlayer interaction and may push the magnetism approach to a 2D limit. Therefore, intercalation might be a good tool to engineering magnetism for compounds in which the interlayer exchange Jz is important. So far, there are not many reported intercalation studies, therefore more efforts toward this direction are needed to fully explore this promising material tuning approach. Similarly, a direct probe for magnetic ordering such as neutron diffraction is also highly desired in this direction. Another urgent need is an accurate and convenient approach to determine the amount of Li for Li-intercalated samples.
High-entropy (HE) MPX3 is an emerging field with great opportunities owing to the expanded degrees of freedom. The combination of multiple elements in HE compounds offers greater versatility and tunability of material properties as compared to conventional bi-metallic and bi-chalcogenide alloys by varying the composition of M and X elements. Incorporating multiple components in an HE compound could induce a severe lattice distortion due to the difference in their atomic sizes, which provides opportunities for crystal structure and symmetry engineering for possible novel functional properties. The HE alloys are expected to generate unusual magnetic orderings due to competing magnetic interactions enabled by the interplay between spin, charge, and orbital degrees of freedom.218 On the other side, the randomness introduced by high entropy may also quench the magnetic order and simultaneously facilitate the insulator-to-metal transition and subsequent emergence of superconductivity, which may be extremely challenging but has been observed in FePSe3-based HE compounds under external pressure.121 Further exploring HE MPX3 compounds could provide pathways to realize enhanced conductivity and even superconductivity at an ambient pressure or much lower pressure. Understanding the effect of entropy on the structural stability and material properties of complex HE compounds is critical for their optimal design and utilization in practical applications. However, the controlled synthesis of the alloy composition, and the large parameter space, pose significant challenges for controlled study (e.g., fixing the content of one or more metals) to clarify the mechanism behind the property changes. More effort, especially in synthesis and applying additional experimental tools, would be beneficial.
One driving force of the study of MPX3 is the feasibility of obtaining their atomically thin layers that provide invaluable opportunities for studying new phenomena originating from 2D magnetism and future device applications. The tunable magnetism summarized in this review further demonstrates the great potential of this material platform. Despite their well-established magnetic orders in bulk materials and the fact of being one of a few first demonstrated 2D magnets in the atomically thin layer form, the study of 2D magnetism in MPX3 is still in an early stage with only a few pristine compositions being investigated, as summarized in Table 6. It has been found that the long-range magnetic orders in atomically mono- or bi-layers only exist in a few MPX3 compounds such as MnPS3,29,109 FePS3,6 NiPS3,60 CoPS3,108 MnPSe3,43 and FePSe3.42 Whereas the 2D magnetism in substituted and intercalated MPX3 compounds remains elusive. So far, few challenges to exploring magnetism in 2D have been identified in the MPX3 family. First, because of the absence of net magnetization, direct probe of AFM states in 2D layers of MPX3 is challenging by using nanoscale magnetic characterization techniques such as scanning single-spin magnetometry,219 magneto-optical Kerr effect microscopy,7,8 polar reflective magnetic circular dichroism (RMCD),10 X-ray magnetic dichroism (XMCD),167 and optical linear dichroism (LD).48,220 In addition to developing more experimental techniques for probing AFM state in nanoscale, inducing ferromagnetism in MPX3 can be a shortcut for 2D magnetism study. We hope the various doping techniques summarized in this review can provide some insight into creating FM ground states for field-driven polarized FM states in these materials. Another advantage of inducing ferromagnetism is the breaking of time-reversal symmetry that may bring in other exotic phenomena. Recent studies have predicted the non-trivial topological states in the FM phase of MPX3,221,222 providing a rare platform for investigating the interplay between magnetism and band topology in the 2D limit.
Although this review mainly focuses on magnetism, we wish to take this opportunity to comment on the electronic properties of MPX3. Those materials are well-known wide band gap insulators and electronic transport measurements in bulk materials are almost impossible or only possible in the high-temperature range.71,197 Enabling electronic studies in vdW-type magnetic MPX3 could be an important direction. Exploiting the coupling between magnetic and electronic properties would provide a platform for tunable electrical properties to develop quantum materials and devices with multifunctional magnetic and electronic properties. Therefore, though applying high pressure has been demonstrated to be effective,64–72 further development in approaches or materials that are compatible with 2D magnetism and device integration is critical, which calls for synergistic efforts in material modeling, high-throughput calculations, synthesis, characterization, and device study.
The vdW structure of MPX3 materials is naturally advantageous for fabricating devices for electronic and optoelectronic applications. However, the highly insulating nature of MPX3 materials has somewhat hindered the rapid development of device applications. To the best of our knowledge, so far the field-effect transistors (FET) using atomically thin layers of only a few MPX3 compounds such as NiPS3223 and MnPS353 have been reported. The FET based on NiPS3223 and MnPS353 thin flakes have been found to exhibit n-type and p-type semiconducting behavior with on/off ratios of ∼103–105 and ∼102–103 respectively at room temperature, with low charge carrier mobilities of ∼0.5–1 cm2 V−1 s−1 and 8.3 × 10−3 cm2 V−1 s−1 respectively. On the other hand, recent works have mainly focused on optoelectronic devices such as photodetectors,52,224–226 phototransistors,227,228 and sensors229 based on a few layers of MPX3. High-performance and sensitive photodetectors working in a wide frequency range from near-infrared226 to ultraviolet52 have been successfully fabricated using the thin layers of MnPS3,53 FePS3,52,224 MnPSe3,225 and FePSe3.226 The few layers NiPS3228 and MnPSe3227 have also been integrated into phototransistors for ultrasensitive light detection. Furthermore, the MnPS3 thin flakes229 has been demonstrated as sensors for sensitive and selective moisture sensing. Providing this progress, efficiently tuning the magnetism would possibly lead to new optoelectronic and photonic applications, especially the light polarization-based applications for which magnetism can play a role. Furthermore, metalizing MPX3via combined chemical and strain may pave a way for electronic applications.
Finally, more attention should be given to atomic ordering in these substituted and intercalated compounds. The distribution of the dopants and possibly the associated vacancies may play important roles in mediating properties. The ultimate examples are well-defined atomic orderings such as superlattice or Peierls transition, which can be relatively easily probed by high-resolution structure characterization tools such as High-resolution/Scanning transmission electron microscopy (HRTEM/STEM) and synchrotron XRD. However, chemical short-range order, which is another ultimate example, is difficult to characterize and has not been widely studied in crystalline solids. Such chemical short-range order is distinct from the well-known short-range order in condensed matter physics that is defined from the long-range ordering. Roughly speaking, the long-range order is based on periodic ordering with long-range correlation length, while the short-range order is normally referred to as similar ordering but with much-shortened correlation length, which usually occurs with the disturbance of the long-range order by doping or increasing the temperature to around the phase transition temperature. In contrast, chemical short-range order occurs in a doped system and characterizes the deviation of the atomic distribution from the perfect random distributions. The study started from the III–V semiconductor alloys decades ago which mainly focused on long-range order,230 and has been recently extended to chemical short-range order in other systems such as group-IV semiconductors,231,232 metal alloys,233,234 thermoelectric materials,235,236 and oxides.237 Thus far, chemical short-range order remains to be an extremely less-explored topic in condensed matter physics, probably owing to the lack of efficient experimental tools to probe it. In doped MPX3 systems, chemical short-range order should also occur and may affect the magnetic properties. Providing rich and tunable magnetism, the doped MPX3 compounds provide an ideal platform for studying the nature of the possible various chemical short-range orders in vdW materials and their impacts on magnetism, which is a new field awaiting exploration.
Author contributions
R. B. and J. H. designed, wrote, and edited this manuscript.Data availability
No primary research results, software or code have been included and no new data were generated as part of this review.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
R. B. acknowledges the support from μ-ATOMS, an Energy Frontier Research Center funded by DOE, Office of Science, Basic Energy Sciences, under Award DE-SC0023412. J. H. acknowledges the support from the U.S. Department of Energy, Office of Science, Basic Energy Sciences program under Grant No. DE-SC0022006. The authors are grateful to T. Li at George Washington University for informative discussions.References
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- W. Han, R. K. Kawakami, M. Gmitra and J. Fabian, Nat. Nanotechnol., 2014, 9, 794–807 CrossRef CAS PubMed.
- B. Li, T. Xing, M. Zhong, L. Huang, N. Lei, J. Zhang, J. Li and Z. Wei, Nat. Commun., 2017, 8, 1958 CrossRef PubMed.
- Z. Guguchia, A. Kerelsky, D. Edelberg, S. Banerjee, F. von Rohr, D. Scullion, M. Augustin, M. Scully, D. A. Rhodes, Z. Shermadini, H. Luetkens, A. Shengelaya, C. Baines, E. Morenzoni, A. Amato, J. C. Hone, R. Khasanov, S. J. L. Billinge, E. Santos, A. N. Pasupathy and Y. J. Uemura, Sci. Adv., 2018, 4, eaat3672 CrossRef CAS PubMed.
- K.-T. Ko, K. Kim, S. B. Kim, H.-D. Kim, J.-Y. Kim, B. I. Min, J.-H. Park, F.-H. Chang, H.-J. Lin, A. Tanaka and S.-W. Cheong, Phys. Rev. Lett., 2011, 107, 247201 CrossRef PubMed.
- J.-U. Lee, S. Lee, J. H. Ryoo, S. Kang, T. Y. Kim, P. Kim, C.-H. Park, J.-G. Park and H. Cheong, Nano Lett., 2016, 16, 7433–7438 CrossRef CAS PubMed.
- B. Huang, G. Clark, E. Navarro-Moratalla, D. R. Klein, R. Cheng, K. L. Seyler, D. Zhong, E. Schmidgall, M. A. McGuire, D. H. Cobden, W. Yao, D. Xiao, P. Jarillo-Herrero and X. Xu, Nature, 2017, 546, 270–273 CrossRef CAS PubMed.
- C. Gong, L. Li, Z. Li, H. Ji, A. Stern, Y. Xia, T. Cao, W. Bao, C. Wang, Y. Wang, Z. Q. Qiu, R. J. Cava, S. G. Louie, J. Xia and X. Zhang, Nature, 2017, 546, 265–269 CrossRef CAS PubMed.
- Z. Zhang, J. Shang, C. Jiang, A. Rasmita, W. Gao and T. Yu, Nano Lett., 2019, 19, 3138–3142 CrossRef CAS PubMed.
- Z. Lin, B. Huang, K. Hwangbo, Q. Jiang, Q. Zhang, Z. Liu, Z. Fei, H. Lv, A. Millis, M. McGuire, D. Xiao, J.-H. Chu and X. Xu, Nano Lett., 2021, 21, 9180–9186 CrossRef CAS PubMed.
- Z. Fei, B. Huang, P. Malinowski, W. Wang, T. Song, J. Sanchez, W. Yao, D. Xiao, X. Zhu, A. F. May, W. Wu, D. H. Cobden, J.-H. Chu and X. Xu, Nat. Mater., 2018, 17, 778–782 CrossRef CAS PubMed.
- G. Zhang, F. Guo, H. Wu, X. Wen, L. Yang, W. Jin, W. Zhang and H. Chang, Nat. Commun., 2022, 13, 5067 CrossRef CAS PubMed.
- S. Albarakati, C. Tan, Z.-J. Chen, J. G. Partridge, G. Zheng, L. Farrar, E. L. H. Mayes, M. R. Field, C. Lee, Y. Wang, Y. Xiong, M. Tian, F. Xiang, A. R. Hamilton, O. A. Tretiakov, D. Culcer, Y.-J. Zhao and L. Wang, Sci. Adv., 2019, 5, eaaw0409 CrossRef CAS PubMed.
- S. Jiang, L. Li, Z. Wang, K. F. Mak and J. Shan, Nat. Nanotechnol., 2018, 13, 549–553 CrossRef CAS PubMed.
- Z. Wang, T. Zhang, M. Ding, B. Dong, Y. Li, M. Chen, X. Li, J. Huang, H. Wang, X. Zhao, Y. Li, D. Li, C. Jia, L. Sun, H. Guo, Y. Ye, D. Sun, Y. Chen, T. Yang, J. Zhang, S. Ono, Z. Han and Z. Zhang, Nat. Nanotechnol., 2018, 13, 554–559 CrossRef CAS PubMed.
- B. Huang, G. Clark, D. R. Klein, D. MacNeill, E. Navarro-Moratalla, K. L. Seyler, N. Wilson, M. A. McGuire, D. H. Cobden, D. Xiao, W. Yao, P. Jarillo-Herrero and X. Xu, Nat. Nanotechnol., 2018, 13, 544–548 CrossRef CAS PubMed.
- Y. Deng, Y. Yu, Y. Song, J. Zhang, N. Z. Wang, Z. Sun, Y. Yi, Y. Z. Wu, S. Wu, J. Zhu, J. Wang, X. H. Chen and Y. Zhang, Nature, 2018, 563, 94–99 CrossRef CAS PubMed.
- T. Song, X. Cai, M. W.-Y. Tu, X. Zhang, B. Huang, N. P. Wilson, K. L. Seyler, L. Zhu, T. Taniguchi, K. Watanabe, M. A. McGuire, D. H. Cobden, D. Xiao, W. Yao and X. Xu, Science, 2018, 360, 1214–1218 CrossRef CAS PubMed.
- Y. Wu, S. Zhang, J. Zhang, W. Wang, Y. L. Zhu, J. Hu, G. Yin, K. Wong, C. Fang, C. Wan, X. Han, Q. Shao, T. Taniguchi, K. Watanabe, J. Zang, Z. Mao, X. Zhang and K. L. Wang, Nat. Commun., 2020, 11, 3860 CrossRef PubMed.
- D. R. Klein, D. MacNeill, J. L. Lado, D. Soriano, E. Navarro-Moratalla, K. Watanabe, T. Taniguchi, S. Manni, P. Canfield, J. Fernández-Rossier and P. Jarillo-Herrero, Science, 2018, 360, 1218–1222 CrossRef CAS PubMed.
- J. Shang, X. Tang, X. Tan, A. Du, T. Liao, S. C. Smith, Y. Gu, C. Li and L. Kou, ACS Appl. Nano Mater., 2020, 3, 1282–1288 CrossRef CAS.
- D. Zhong, K. L. Seyler, X. Linpeng, R. Cheng, N. Sivadas, B. Huang, E. Schmidgall, T. Taniguchi, K. Watanabe, M. A. McGuire, W. Yao, D. Xiao, K.-M. C. Fu and X. Xu, Sci. Adv., 2017, 3, e1603113 CrossRef PubMed.
- Z. Wang, I. Gutiérrez-Lezama, N. Ubrig, M. Kroner, M. Gibertini, T. Taniguchi, K. Watanabe, A. Imamoğlu, E. Giannini and A. F. Morpurgo, Nat. Commun., 2018, 9, 2516 CrossRef PubMed.
- T. Song, M. W.-Y. Tu, C. Carnahan, X. Cai, T. Taniguchi, K. Watanabe, M. A. McGuire, D. H. Cobden, D. Xiao, W. Yao and X. Xu, Nano Lett., 2019, 19, 915–920 CrossRef CAS PubMed.
- Y. Wang, J. Balgley, E. Gerber, M. Gray, N. Kumar, X. Lu, J.-Q. Yan, A. Fereidouni, R. Basnet, S. J. Yun, D. Suri, H. Kitadai, T. Taniguchi, K. Watanabe, X. Ling, J. Moodera, Y. H. Lee, H. O. H. Churchill, J. Hu, L. Yang, E.-A. Kim, D. G. Mandrus, E. A. Henriksen and K. S. Burch, Nano Lett., 2020, 20, 8446–8452 CrossRef CAS PubMed.
- K. F. Mak, J. Shan and D. C. Ralph, Nat. Rev. Phys., 2019, 1, 646–661 CrossRef.
- F. Wang, T. A. Shifa, P. Yu, P. He, Y. Liu, F. Wang, Z. Wang, X. Zhan, X. Lou, F. Xia and J. He, Adv. Funct. Mater., 2018, 28, 1802151 CrossRef.
- R. Samal, G. Sanyal, B. Chakraborty and C. S. Rout, J. Mater. Chem. A, 2021, 9, 2560–2591 RSC.
- G. Long, H. Henck, M. Gibertini, D. Dumcenco, Z. Wang, T. Taniguchi, K. Watanabe, E. Giannini and A. F. Morpurgo, Nano Lett., 2020, 20, 2452–2459 CrossRef CAS PubMed.
- A. Wiedenmann, J. Rossat-Mignod, A. Louisy, R. Brec and J. Rouxel, Solid State Commun., 1981, 40, 1067–1072 CrossRef CAS.
- R. Basnet, K. M. Kotur, M. Rybak, C. Stephenson, S. Bishop, C. Autieri, M. Birowska and J. Hu, Phys. Rev. Res., 2022, 4, 023256 CrossRef CAS.
- H. Han, H. Lin, W. Gan, Y. Liu, R. Xiao, L. Zhang, Y. Li, C. Zhang and H. Li, Appl. Phys. Lett., 2023, 122, 033101 CrossRef CAS.
- A. R. Wildes, V. Simonet, E. Ressouche, G. J. Mcintyre, M. Avdeev, E. Suard, S. A. Kimber, D. Lançon, G. Pepe and B. Moubaraki, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 224408 CrossRef.
- A. R. Wildes, H. M. Rønnow, B. Roessli, M. J. Harris and K. W. Godfrey, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 094422 CrossRef.
- A. R. Wildes, B. Roessli, B. Lebech and K. W. Godfrey, J. Phys.: Condens. Matter, 1998, 10, 6417–6428 CrossRef CAS.
- A. R. Wildes, V. Simonet, E. Ressouche, R. Ballou and G. J. McIntyre, J. Phys.: Condens. Matter, 2017, 29, 455801 CrossRef CAS PubMed.
- T. Sekine, M. Jouanne, C. Julien and M. Balkanski, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 42, 8382–8393 CrossRef CAS PubMed.
- Y. Takano, N. Arai, A. Arai, Y. Takahashi, K. Takase and K. Sekizawa, J. Magn. Magn. Mater., 2004, 272–276, E593–E595 CrossRef CAS.
- G. Le Flem, R. Brec, G. Ouvard, A. Louisy and P. Segransan, J. Phys. Chem. Solids, 1982, 43, 455–461 CrossRef CAS.
- P. A. Joy and S. Vasudevan, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 5425–5433 CrossRef CAS PubMed.
- G. Long, T. Zhang, X. Cai, J. Hu, C. Cho, S. Xu, J. Shen, Z. Wu, T. Han, J. Lin, J. Wang, Y. Cai, R. Lortz, Z. Mao and N. Wang, ACS Nano, 2017, 11, 11330–11336 CrossRef CAS PubMed.
- J. Luo, S. Li, Z. Ye, R. Xu, H. Yan, J. Zhang, G. Ye, L. Chen, D. Hu, X. Teng, W. A. Smith, B. I. Yakobson, P. Dai, A. H. Nevidomskyy, R. He and H. Zhu, Nano Lett., 2023, 23, 2023–2030 CrossRef CAS PubMed.
- Z. Ni, A. V. Haglund, H. Wang, B. Xu, C. Bernhard, D. G. Mandrus, X. Qian, E. J. Mele, C. L. Kane and L. Wu, Nat. Nanotechnol., 2021, 16, 782–787 CrossRef CAS PubMed.
- S. Kang, K. Kim, B. H. Kim, J. Kim, K. I. Sim, J.-U. Lee, S. Lee, K. Park, S. Yun, T. Kim, A. Nag, A. Walters, M. Garcia-Fernandez, J. Li, L. Chapon, K.-J. Zhou, Y.-W. Son, J. H. Kim, H. Cheong and J.-G. Park, Nature, 2020, 583, 785–789 CrossRef CAS PubMed.
- X. Wang, J. Cao, Z. Lu, A. Cohen, H. Kitadai, T. Li, Q. Tan, M. Wilson, C. H. Lui, D. Smirnov, S. Sharifzadeh and X. Ling, Nat. Mater., 2021, 20, 964–970 CrossRef CAS PubMed.
- F. Dirnberger, R. Bushati, B. Datta, A. Kumar, A. H. MacDonald, E. Baldini and V. M. Menon, Nat. Nanotechnol., 2022, 17, 1060–1064 CrossRef CAS PubMed.
- K. Hwangbo, Q. Zhang, Q. Jiang, Y. Wang, J. Fonseca, C. Wang, G. M. Diederich, D. R. Gamelin, D. Xiao, J.-H. Chu, W. Yao and X. Xu, Nat. Nanotechnol., 2021, 16, 655–660 CrossRef CAS PubMed.
- H. Zhang, Z. Ni, C. E. Stevens, A. Bai, F. Peiris, J. R. Hendrickson, L. Wu and D. Jariwala, Nat. Photonics, 2022, 16, 311–317 CrossRef CAS.
- C. A. Belvin, E. Baldini, I. O. Ozel, D. Mao, H. C. Po, C. J. Allington, S. Son, B. H. Kim, J. Kim, I. Hwang, J. H. Kim, J.-G. Park, T. Senthil and N. Gedik, Nat. Commun., 2021, 12, 4837 CrossRef CAS PubMed.
- D. Afanasiev, J. R. Hortensius, M. Matthiesen, S. Mañas-Valero, M. Šiškins, M. Lee, E. Lesne, H. S. J. van der Zant, P. G. Steeneken, B. A. Ivanov, E. Coronado and A. D. Caviglia, Sci. Adv., 2021, 7, eabf3096 CrossRef CAS PubMed.
- J. Chu, F. Wang, L. Yin, L. Lei, C. Yan, F. Wang, Y. Wen, Z. Wang, C. Jiang, L. Feng, J. Xiong, Y. Li and J. He, Adv. Funct. Mater., 2017, 27, 1701342 CrossRef.
- Y. Gao, S. Lei, T. Kang, L. Fei, C.-L. Mak, J. Yuan, M. Zhang, S. Li, Q. Bao, Z. Zeng, Z. Wang, H. Gu and K. Zhang, Nanotechnology, 2018, 29, 244001 CrossRef PubMed.
- R. Kumar, R. N. Jenjeti, M. P. Austeria and S. Sampath, J. Mater. Chem. C, 2019, 7, 324–329 RSC.
- A. H. Thompson and M. S. Whittingham, Mater. Res. Bull., 1977, 12, 741–744 CrossRef CAS.
- P. J. S. Foot, T. Katz, S. N. Patel, B. A. Nevett, A. R. Pieecy and A. A. Balchin, Phys. Status Solidi A, 1987, 100, 11–29 CrossRef CAS.
- F. Liu, L. You, K. L. Seyler, X. Li, P. Yu, J. Lin, X. Wang, J. Zhou, H. Wang, H. He, S. T. Pantelides, W. Zhou, P. Sharma, X. Xu, P. M. Ajayan, J. Wang and Z. Liu, Nat. Commun., 2016, 7, 12357 CrossRef CAS PubMed.
- A. Belianinov, Q. He, A. Dziaugys, P. Maksymovych, E. Eliseev, A. Borisevich, A. Morozovska, J. Banys, Y. Vysochanskii and S. V. Kalinin, Nano Lett., 2015, 15, 3808–3814 CrossRef CAS PubMed.
- W. F. Io, S.-Y. Pang, L. W. Wong, Y. Zhao, R. Ding, J. Mao, Y. Zhao, F. Guo, S. Yuan, J. Zhao, J. Yi and J. Hao, Nat. Commun., 2023, 14, 7304 CrossRef CAS PubMed.
- C. B. Park, A. Shahee, K.-T. Kim, D. R. Patil, S. A. Guda, N. Ter-Oganessian and K. H. Kim, Adv. Electron. Mater., 2022, 8, 2101072 CrossRef CAS.
- K. Kim, S. Y. Lim, J.-U. Lee, S. Lee, T. Y. Kim, K. Park, G. S. Jeon, C.-H. Park, J.-G. Park and H. Cheong, Nat. Commun., 2019, 10, 345 CrossRef PubMed.
- K. Du, X. Wang, Y. Liu, P. Hu, M. I. B. Utama, C. K. Gan, Q. Xiong and C. Kloc, ACS Nano, 2016, 10, 1738–1743 CrossRef CAS PubMed.
- Y. Jin, M. Yan, T. Kremer, E. Voloshina and Y. Dedkov, Sci. Rep., 2022, 12, 735 CrossRef CAS PubMed.
- A. Kamata, K. Noguchi, K. Suzuki, H. Tezuka, T. Kashiwakura, Y. Ohno and S. Nakai, J. Phys. Soc. Jpn., 1997, 66, 401–407 CrossRef CAS.
- H.-S. Kim, K. Haule and D. Vanderbilt, Phys. Rev. Lett., 2019, 123, 236401 CrossRef CAS PubMed.
- Y. Wang, Z. Zhou, T. Wen, Y. Zhou, N. Li, F. Han, Y. Xiao, P. Chow, J. Sun, M. Pravica, A. L. Cornelius, W. Yang and Y. Zhao, J. Am. Chem. Soc., 2016, 138, 15751–15757 CrossRef CAS PubMed.
- R. A. Evarestov and A. Kuzmin, J. Comput. Chem., 2020, 41, 1337–1344 CrossRef CAS PubMed.
- M. Tsurubayashi, K. Kodama, M. Kano, K. Ishigaki, Y. Uwatoko, T. Watanabe, K. Takase and Y. Takano, AIP Adv., 2018, 8, 101307 CrossRef.
- C. R. S. Haines, M. J. Coak, A. R. Wildes, G. I. Lampronti, C. Liu, P. Nahai-Williamson, H. Hamidov, D. Daisenberger and S. S. Saxena, Phys. Rev. Lett., 2018, 121, 266801 CrossRef CAS PubMed.
- M. J. Coak, D. M. Jarvis, H. Hamidov, C. R. S. Haines, P. L. Alireza, C. Liu, S. Son, I. Hwang, G. I. Lampronti, D. Daisenberger, P. Nahai-Williamson, A. R. Wildes, S. S. Saxena and J.-G. Park, J. Phys.: Condens. Matter, 2019, 32, 124003 CrossRef PubMed.
- X. Ma, Y. Wang, Y. Yin, B. Yue, J. Dai, J. Cheng, J. Ji, F. Jin, F. Hong, J.-T. Wang, Q. Zhang and X. Yu, Sci. China: Phys., Mech. Astron., 2021, 64, 297011 CAS.
- M. J. Coak, S. Son, D. Daisenberger, H. Hamidov, C. R. S. Haines, P. L. Alireza, A. R. Wildes, C. Liu, S. S. Saxena and J.-G. Park, npj Quantum Mater., 2019, 4, 1–6 CrossRef.
- Y. Wang, J. Ying, Z. Zhou, J. Sun, T. Wen, Y. Zhou, N. Li, Q. Zhang, F. Han, Y. Xiao, P. Chow, W. Yang, V. V. Struzhkin, Y. Zhao and H. Mao, Nat. Commun., 2018, 9, 1914 CrossRef PubMed.
- S. Y. Kim, T. Y. Kim, L. J. Sandilands, S. Sinn, M.-C. Lee, J. Son, S. Lee, K.-Y. Choi, W. Kim, B.-G. Park, C. Jeon, H.-D. Kim, C.-H. Park, J.-G. Park, S. J. Moon and T. W. Noh, Phys. Rev. Lett., 2018, 120, 136402 CrossRef CAS PubMed.
- S. Liu, A. Granados del Águila, D. Bhowmick, C. K. Gan, T. Thu Ha Do, M. A. Prosnikov, D. Sedmidubský, Z. Sofer, P. C. M. Christianen, P. Sengupta and Q. Xiong, Phys. Rev. Lett., 2021, 127, 097401 CrossRef CAS PubMed.
- M. A. Susner, M. Chyasnavichyus, M. A. McGuire, P. Ganesh and P. Maksymovych, Adv. Mater., 2017, 29, 1602852 CrossRef PubMed.
- N. Chandrasekharan and S. Vasudevan, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 14903–14906 CrossRef CAS PubMed.
- D. J. Goossens, A. J. Studer, S. J. Kennedy and T. J. Hicks, J. Phys.: Condens. Matter, 2000, 12, 4233–4242 CrossRef CAS.
- A. M. Mulders, J. C. P. Klaasse, D. J. Goossens, J. Chadwick and T. J. Hicks, J. Phys.: Condens. Matter, 2002, 14, 8697–8705 CrossRef CAS.
- Y. Takano, A. Arai, Y. Takahashi, K. Takase and K. Sekizawa, J. Appl. Phys., 2003, 93, 8197–8199 CrossRef CAS.
- J. N. Graham, M. J. Coak, S. Son, E. Suard, J.-G. Park, L. Clark and A. R. Wildes, Phys. Rev. Mater., 2020, 4, 084401 CrossRef CAS.
- T. Masubuchi, H. Hoya, T. Watanabe, Y. Takahashi, S. Ban, N. Ohkubo, K. Takase and Y. Takano, J. Alloys Compd., 2008, 460, 668–674 CrossRef CAS.
- V. Manríquez, P. Barahona and O. Peña, Mater. Res. Bull., 2000, 35, 1889–1895 CrossRef.
- D. J. Goossens, S. Brazier-Hollins, D. R. James, W. D. Hutchison and J. R. Hester, J. Magn. Magn. Mater., 2013, 334, 82–86 CrossRef CAS.
- Y. He, Y.-D. Dai, H. Huang, J. Lin and Y. Hsia, J. Alloys Compd., 2003, 359, 41–45 CrossRef CAS.
- D. J. Goossens and T. J. Hicks, J. Phys.: Condens. Matter, 1998, 10, 7643–7652 CrossRef CAS.
- A. Bhutani, J. L. Zuo, R. D. McAuliffe, C. R. dela Cruz and D. P. Shoemaker, Phys. Rev. Mater., 2020, 4, 034411 CrossRef CAS.
- Y. Shemerliuk, Y. Zhou, Z. Yang, G. Cao, A. U. B. Wolter, B. Büchner and S. Aswartham, Electron. Mater, 2021, 2, 284–298 CrossRef.
- S. Selter, Y. Shemerliuk, M.-I. Sturza, A. U. B. Wolter, B. Büchner and S. Aswartham, Phys. Rev. Mater., 2021, 5, 073401 CrossRef CAS.
- R. Basnet, A. Wegner, K. Pandey, S. Storment and J. Hu, Phys. Rev. Mater., 2021, 5, 064413 CrossRef CAS.
- F. Wang, N. Mathur, A. N. Janes, H. Sheng, P. He, X. Zheng, P. Yu, A. J. DeRuiter, J. R. Schmidt, J. He and S. Jin, Sci. Adv., 2000, 7, eabj4086 CrossRef PubMed.
- S. Lee, J. Park, Y. Choi, K. Raju, W.-T. Chen, R. Sankar and K.-Y. Choi, Phys. Rev. B, 2021, 104, 174412 CrossRef CAS.
- J. P. Odile, J. J. Steger and A. Wold, Inorg. Chem., 1975, 14, 2400–2402 CrossRef CAS.
- J. Peng, X. Yang, Z. Lu, L. Huang, X. Chen, M. He, J. Shen, Y. Xing, M. Liu, Z. Qu, Z. Wang, L. Li, S. Dong and J.-M. Liu, Adv. Quantum Technol., 2023, 6, 2200105 CrossRef CAS.
- S. Calder, A. V. Haglund, A. I. Kolesnikov and D. Mandrus, Phys. Rev. B, 2021, 103, 024414 CrossRef CAS.
- P. Jeevanandam and S. Vasudevan, J. Phys.: Condens. Matter, 1999, 11, 3563–3570 CrossRef CAS.
- X. Ma, L. Zhang, C. Xu, Q. Dong, R. I. Walton, Z. Li, H. Shi, G. Chen, J. Hu, J. Li and H. Yang, Chem. Commun., 2020, 56, 4603–4606 RSC.
- M. Mi, X. Zheng, S. Wang, Y. Zhou, L. Yu, H. Xiao, H. Song, B. Shen, F. Li, L. Bai, Y. Chen, S. Wang, X. Liu and Y. Wang, Adv. Funct. Mater., 2022, 32, 2112750 CrossRef CAS.
- R. Basnet, D. Ford, K. TenBarge, J. Lochala and J. Hu, J. Phys.: Condens. Matter, 2022, 34, 434002 CrossRef CAS PubMed.
- D. Tezze, J. M. Pereira, Y. Asensio, M. Ipatov, F. Calavalle, F. Casanova, A. M. Bittner, M. Ormaza, B. Martín-García, L. E. Hueso and M. Gobbi, Nanoscale, 2022, 14, 1165–1173 RSC.
- R. Brec, D. M. Schleich, G. Ouvrard, A. Louisy and J. Rouxel, Inorg. Chem., 1979, 18, 1814–1818 CrossRef CAS.
- E. Manova, A. Leaustic, I. Mitov, D. Gonbeau and R. Clement, Mol. Cryst. Liq. Cryst. Sci. Technol., Sect. A, 1998, 311, 155–160 CrossRef.
- J. J. Abraham, Y. Senyk, Y. Shemerliuk, S. Selter, S. Aswartham, B. Büchner, V. Kataev and A. Alfonsov, Phys. Rev. B, 2023, 107, 165141 CrossRef CAS.
- M. Baithi, N. T. Dang, T. A. Tran, J. P. Fix, D. H. Luong, K. P. Dhakal, D. Yoon, A. V. Rutkauskas, S. E. Kichanov, I. Y. Zel, J. Kim, N. J. Borys, D. P. Kozlenko, Y. H. Lee and D. L. Duong, Inorg. Chem., 2023, 62, 12674–12682 CrossRef CAS PubMed.
- R. Nitsche and P. Wild, Mater. Res. Bull., 1970, 5, 419–423 CrossRef CAS.
- C. D. Carpentier and R. Nitsche, Mater. Res. Bull., 1974, 9, 401–410 CrossRef CAS.
- C. Liu, Z. Li, J. Hu, H. Duan, C. Wang, L. Cai, S. Feng, Y. Wang, R. Liu, D. Hou, C. Liu, R. Zhang, L. Zhu, Y. Niu, A. A. Zakharov, Z. Sheng and W. Yan, Adv. Mater., 2023, 35, 2300247 CrossRef CAS PubMed.
- D. Lançon, H. C. Walker, E. Ressouche, B. Ouladdiaf, K. C. Rule, G. J. McIntyre, T. J. Hicks, H. M. Rønnow and A. R. Wildes, Phys. Rev. B, 2016, 94, 214407 CrossRef.
- Q. Liu, L. Wang, Y. Fu, X. Zhang, L. Huang, H. Su, J. Lin, X. Chen, D. Yu, X. Cui, J.-W. Mei and J.-F. Dai, Phys. Rev. B, 2021, 103, 235411 CrossRef CAS.
- K. Kim, S. Y. Lim, J. Kim, J.-U. Lee, S. Lee, P. Kim, K. Park, S. Son, C.-H. Park, J.-G. Park and H. Cheong, 2D Mater., 2019, 6, 041001 CrossRef CAS.
- C.-T. Kuo, M. Neumann, K. Balamurugan, H. J. Park, S. Kang, H. W. Shiu, J. H. Kang, B. H. Hong, M. Han, T. W. Noh and J.-G. Park, Sci. Rep., 2016, 6, 20904 CrossRef CAS PubMed.
- H. Chu, C. J. Roh, J. O. Island, C. Li, S. Lee, J. Chen, J.-G. Park, A. F. Young, J. S. Lee and D. Hsieh, Phys. Rev. Lett., 2020, 124, 027601 CrossRef CAS PubMed.
- S. Lee, K.-Y. Choi, S. Lee, B. H. Park and J.-G. Park, APL Mater., 2016, 4, 086108 CrossRef.
- T. Ying, T. Yu, Y.-S. Shiah, C. Li, J. Li, Y. Qi and H. Hosono, J. Am. Chem. Soc., 2021, 143, 7042–7049 CrossRef CAS PubMed.
- M. Cheng, Y.-S. Lee, A. K. Iyer, D. G. Chica, E. K. Qian, M. A. Shehzad, R. dos Reis, M. G. Kanatzidis and V. P. Dravid, Inorg. Chem., 2021, 60, 17268–17275 CrossRef CAS PubMed.
- D. G. Chica, A. K. Iyer, M. Cheng, K. M. Ryan, P. Krantz, C. Laing, R. dos Reis, V. Chandrasekhar, V. P. Dravid and M. G. Kanatzidis, Inorg. Chem., 2021, 60, 3502–3513 CrossRef CAS PubMed.
- M. Cheng, A. K. Iyer, A. P. Douvalis, S. M. Ribet, M. G. Kanatzidis and V. P. Dravid, Chem. Mater., 2023, 35, 1458–1465 CrossRef CAS.
- M. Cheng, A. K. Iyer, X. Zhou, A. Tyner, Y. Liu, M. A. Shehzad, P. Goswami, D. Y. Chung, M. G. Kanatzidis and V. P. Dravid, Inorg. Chem., 2022, 61, 13719–13727 CrossRef CAS PubMed.
- L.-J. Yao, C.-L. Hu, Z. Fang and J.-G. Mao, J. Solid State Chem., 2022, 315, 123433 CrossRef CAS.
- E. K. Qian, A. K. Iyer, M. Cheng, K. M. Ryan, L. Jirousek, D. G. Chica, P. Krantz, Y.-S. Lee, V. Chandrasekhar, V. P. Dravid and M. G. Kanatzidis, Chem. Mater., 2023, 35, 3671–3685 CrossRef CAS.
- B. L. Chittari, Y. Park, D. Lee, M. Han, A. H. MacDonald, E. Hwang and J. Jung, Phys. Rev. B, 2016, 94, 184428 CrossRef.
- X. Chen, J. Wang, T. Ying, D. Huang, H. Gou, Q. Zhang, Y. Li, H. Hosono, J. Guo and X. Chen, Phys. Rev. B, 2022, 106, 184502 CrossRef CAS.
- R. Basnet, T. Patel, J. Wang, D. Upreti, S. K. Chhetri, G. Acharya, M. R. U. Nabi, J. Sakon and J. Hu, Adv. Electron. Mater., 2024, 10, 2300738 CrossRef CAS.
- A. R. Wildes, V. Simonet, E. Ressouche, G. J. McIntyre, M. Avdeev, E. Suard, S. A. J. Kimber, D. Lançon, G. Pepe, B. Moubaraki and T. J. Hicks, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 224408 CrossRef.
- W. Klingen, R. Ott and H. Hahn, Z. Anorg. Allg. Chem., 1973, 396, 271–278 CrossRef CAS.
- D. Khomskii, Transition Metal Compounds, Cambridge University Press, 2014 Search PubMed.
- M. Dalal, A Textbook of Inorganic Chemistry – Volume 1, Dalal Institute, 2017 Search PubMed.
- R. Basnet, Understanding and Tuning Magnetism in van der Waals Magnetic Compounds, Doctoral dissertation, University of Arkansas, 2023 Search PubMed.
- D. Lançon, R. A. Ewings, T. Guidi, F. Formisano and A. R. Wildes, Phys. Rev. B, 2018, 98, 134414 CrossRef.
- C. Kim, J. Jeong, P. Park, T. Masuda, S. Asai, S. Itoh, H.-S. Kim, A. Wildes and J.-G. Park, Phys. Rev. B, 2020, 102, 184429 CrossRef CAS.
- A. R. Wildes, J. R. Stewart, M. D. Le, R. A. Ewings, K. C. Rule, G. Deng and K. Anand, Phys. Rev. B, 2022, 106, 174422 CrossRef CAS.
- J. B. Goodenough, Phys. Rev., 1955, 100, 564–573 CrossRef CAS.
- J. Kanamori, J. Phys. Chem. Solids, 1959, 10, 87–98 CrossRef CAS.
- N. Bazazzadeh, M. Hamdi, F. Haddadi, A. Khavasi, A. Sadeghi and S. M. Mohseni, Phys. Rev. B, 2021, 103, 014425 CrossRef CAS.
- D. J. Goossens, A. R. Wildes, C. Ritter and T. J. Hicks, J. Phys.: Condens. Matter, 2000, 12, 1845–1854 CrossRef CAS.
- D. J. Goossens, Eur. Phys. J. B, 2010, 78, 305–309 CrossRef CAS.
- A. G. Chang, L.-W. Lan, Y.-J. Chan, C.-N. Kuo, T. Chen, C.-H. Huang, T.-H. Chuang, D.-H. Wei, C.-S. Lue and C.-C. Kuo, Phys. Rev. B, 2022, 106, 125412 CrossRef CAS.
- Y. Lee, S. Son, C. Kim, S. Kang, J. Shen, M. Kenzelmann, B. Delley, T. Savchenko, S. Parchenko, W. Na, K.-Y. Choi, W. Kim, H. Cheong, P. M. Derlet, A. Kleibert and J.-G. Park, Adv. Electron. Mater., 2023, 9, 2200650 CrossRef CAS.
- R. R. Rao and A. K. Raychaudhuri, J. Phys. Chem. Solids, 1992, 53, 577–583 CrossRef CAS.
- L. Bevaart, E. Frikkee and L. J. de Jongh, Phys. Rev. B: Condens. Matter Mater. Phys., 1979, 19, 4741–4756 CrossRef CAS.
- C.-K. Tian, C. Wang, W. Ji, J.-C. Wang, T.-L. Xia, L. Wang, J.-J. Liu, H.-X. Zhang and P. Cheng, Phys. Rev. B, 2019, 99, 184428 CrossRef CAS.
- X. Hu, D.-X. Yao and K. Cao, Phys. Rev. B, 2022, 106, 224423 CrossRef CAS.
- C. Tian, F. Pan, S. Xu, K. Ai, T. Xia and P. Cheng, Appl. Phys. Lett., 2020, 116, 202402 CrossRef CAS.
- G. Drachuck, Z. Salman, M. W. Masters, V. Taufour, T. N. Lamichhane, Q. Lin, W. E. Straszheim, S. L. Bud'ko and P. C. Canfield, Phys. Rev. B, 2018, 98, 144434 CrossRef CAS.
- S. Pan, Y. Bai, J. Tang, P. Wang, Y. You, G. Xu and F. Xu, J. Alloys Compd., 2022, 908, 164573 CrossRef CAS.
- M. Abramchuk, S. Jaszewski, K. R. Metz, G. B. Osterhoudt, Y. Wang, K. S. Burch and F. Tafti, Adv. Mater., 2018, 30, 1801325 CrossRef PubMed.
- T. A. Tartaglia, J. N. Tang, J. L. Lado, F. Bahrami, M. Abramchuk, G. T. McCandless, M. C. Doyle, K. S. Burch, Y. Ran, J. Y. Chan and F. Tafti, Sci. Adv., 2020, 6, eabb9379 CrossRef CAS PubMed.
- T. Masubuchi, X. Jin, K. Koyama, Y. Takahashi, K. Takase, Y. Uwatoko, Y. Takano and K. Sekizawa, AIP Conf. Proc., 2006, 850, 1279–1280 CrossRef CAS.
- T. Li, S. Jiang, N. Sivadas, Z. Wang, Y. Xu, D. Weber, J. E. Goldberger, K. Watanabe, T. Taniguchi, C. J. Fennie, K. Fai Mak and J. Shan, Nat. Mater., 2019, 18, 1303–1308 CrossRef CAS PubMed.
- S. Mondal, M. Kannan, M. Das, L. Govindaraj, R. Singha, B. Satpati, S. Arumugam and P. Mandal, Phys. Rev. B, 2019, 99, 180407 CrossRef CAS.
- Y. Peng, Z. Lin, G. Tian, J. Yang, P. Zhang, F. Wang, P. Gu, X. Liu, C.-W. Wang, M. Avdeev, F. Liu, D. Zhou, R. Han, P. Shen, W. Yang, S. Liu, Y. Ye and J. Yang, Adv. Funct. Mater., 2022, 32, 2106592 CrossRef CAS.
- R. Oliva, E. Ritov, F. Horani, I. Etxebarria, A. K. Budniak, Y. Amouyal, E. Lifshitz and M. Guennou, Phys. Rev. B, 2023, 107, 104415 CrossRef CAS.
- Z. Lu, X. Yang, L. Huang, X. Chen, M. Liu, J. Peng, S. Dong and J.-M. Liu, J. Phys.: Condens. Matter, 2022, 34, 354005 CrossRef CAS PubMed.
- S. Selter, K. K. Bestha, P. Bhattacharyya, B. Özer, Y. Shemerliuk, M. Roslova, E. Vinokurova, L. T. Corredor, L. Veyrat, A. U. B. Wolter, L. Hozoi, B. Büchner and S. Aswartham, Phys. Rev. Mater., 2023, 7, 033402 CrossRef CAS.
- X. Wang, Z. Shang, C. Zhang, J. Kang, T. Liu, X. Wang, S. Chen, H. Liu, W. Tang, Y.-J. Zeng, J. Guo, Z. Cheng, L. Liu, D. Pan, S. Tong, B. Wu, Y. Xie, G. Wang, J. Deng, T. Zhai, H.-X. Deng, J. Hong and J. Zhao, Nat. Commun., 2023, 14, 840 CrossRef CAS PubMed.
- C. Payen, H. Mutka, J. L. Soubeyroux, P. Molinié and P. Colombet, J. Magn. Magn. Mater., 1992, 104–107, 797–798 CrossRef CAS.
- S. Selter, Y. Shemerliuk, B. Büchner and S. Aswartham, Crystals, 2021, 11, 500 CrossRef CAS.
- H. Mutka, C. Payen, P. Molinié, J. L. Soubeyroux, P. Colombet and A. D. Taylor, Phys. Rev. Lett., 1991, 67, 497–500 CrossRef CAS PubMed.
- M. A. Gave, D. Bilc, S. D. Mahanti, J. D. Breshears and M. G. Kanatzidis, Inorg. Chem., 2005, 44, 5293–5303 CrossRef CAS PubMed.
- D. Kim, E. K. Qian, D. G. Chica, Y.-H. Chiang, M. G. Kanatzidis and Q. Tu, ACS Appl. Nano Mater., 2023, 6, 8214–8221 CrossRef CAS.
- A. Kargar, H. Hong, J. Tower, P. Guggenheim, E. Qian, C. Brown, M. Breen, J. F. Christian, K. Shah, E. Lukosi, R. Golduber, J. Gallagher, A. Benkechkache, M. Kanatzidis and M. R. Squillante, in Hard X-Ray, Gamma-Ray, and Neutron Detector Physics XXIV, SPIE, 2022, vol. 12241, pp. 59–12268 Search PubMed.
- D. G. Chica, Y. He, K. M. McCall, D. Y. Chung, R. O. Pak, G. Trimarchi, Z. Liu, P. M. De Lurgio, B. W. Wessels and M. G. Kanatzidis, Nature, 2020, 577, 346–349 CrossRef CAS PubMed.
- A. del Moral, J. I. Arnaudas and P. M. Gehring, J. Phys.: Condens. Matter, 1994, 6, 4779 CrossRef CAS.
- F. B. Anderson and H. B. Callen, Phys. Rev., 1964, 136, A1068–A1087 CrossRef.
- L. J. D. Jongh and A. R. Miedema, Adv. Phys., 2001, 50, 947–1170 CrossRef.
- K. Okuda, K. Kurosawa, S. Saito, M. Honda, Z. Yu and M. Date, J. Phys. Soc. Jpn., 1986, 55, 4456–4463 CrossRef CAS.
- A. P. Dioguardi, S. Selter, U. Peeck, S. Aswartham, M.-I. Sturza, R. Murugesan, M. S. Eldeeb, L. Hozoi, B. Büchner and H.-J. Grafe, Phys. Rev. B, 2020, 102, 064429 CrossRef CAS.
- A. Bedoya-Pinto, J.-R. Ji, A. K. Pandeya, P. Gargiani, M. Valvidares, P. Sessi, J. M. Taylor, F. Radu, K. Chang and S. S. P. Parkin, Science, 2021, 374, 616–620 CrossRef CAS PubMed.
- X. Cai, T. Song, N. P. Wilson, G. Clark, M. He, X. Zhang, T. Taniguchi, K. Watanabe, W. Yao, D. Xiao, M. A. McGuire, D. H. Cobden and X. Xu, Nano Lett., 2019, 19, 3993–3998 CrossRef CAS PubMed.
- J. Son, S. Son, P. Park, M. Kim, Z. Tao, J. Oh, T. Lee, S. Lee, J. Kim, K. Zhang, K. Cho, T. Kamiyama, J. H. Lee, K. F. Mak, J. Shan, M. Kim, J.-G. Park and J. Lee, ACS Nano, 2021, 15, 16904–16912 CrossRef CAS PubMed.
- M. Baithi, N. T. Dang, T. A. Tran, J. P. Fix, D. H. Luong, K. P. Dhakal, D. Yoon, A. V. Rutkauskas, S. E. Kichanov, I. Y. Zel, J. Kim, N. J. Borys, D. P. Kozlenko, Y. H. Lee and D. L. Duong, Inorg. Chem., 2023, 62, 12674–12682 CrossRef CAS PubMed.
- Z.-L. Huang, J.-T. Zhao, J.-X. Mi, S.-Y. Mao and L.-S. Zheng, J. Solid State Chem., 1999, 144, 388–391 CrossRef CAS.
- D. Sen and T. Saha-Dasgupta, Phys. Rev. Mater., 2023, 7, 064008 CrossRef CAS.
- G. Ouvrard, R. Fréour, R. Brec and J. Rouxel, Mater. Res. Bull., 1985, 20, 1053–1062 CrossRef CAS.
- K. Ichimura and M. Sano, Synth. Met., 1991, 45, 203–211 CrossRef CAS.
- D. J. Goossens, D. James, J. Dong, R. E. Whitfield, L. Norén and R. L. Withers, J. Phys.: Condens. Matter, 2011, 23, 065401 CrossRef CAS PubMed.
- P. Fuentealba, C. Olea, H. Aguilar-Bolados, N. Audebrand, R. C. de Santana, C. Doerenkamp, H. Eckert, C. J. Magon and E. Spodine, Phys. Chem. Chem. Phys., 2020, 22, 8315–8324 RSC.
- R. Basnet, D. Upreti, T. Patel, S. Karki Chhetri, G. Acharya, M. R. U. Nabi, M. M. Sharma, J. Sakon, M. Mortazavi and J. Hu, Phys. Rev. B, 2024, 109, 184405 CrossRef.
- Y. Peng, S. Ding, M. Cheng, Q. Hu, J. Yang, F. Wang, M. Xue, Z. Liu, Z. Lin, M. Avdeev, Y. Hou, W. Yang, Y. Zheng and J. Yang, Adv. Mater., 2020, 32, 2001200 CrossRef CAS PubMed.
- M. A. McGuire, G. Clark, S. KC, W. M. Chance, G. E. Jellison, V. R. Cooper, X. Xu and B. C. Sales, Phys. Rev. Mater., 2017, 1, 014001 CrossRef.
- E. J. Telford, A. H. Dismukes, K. Lee, M. Cheng, A. Wieteska, A. K. Bartholomew, Y.-S. Chen, X. Xu, A. N. Pasupathy, X. Zhu, C. R. Dean and X. Roy, Adv. Mater., 2020, 32, 2003240 CrossRef CAS PubMed.
- D. Upreti, R. Basnet, M. M. Sharma, S. K. Chhetri, G. Acharya, M. R. U. Nabi, J. Sakon, M. Mortazavi and J. Hu, 2024, arXiv:2408.02858. DOI:10.48550/arXiv.2408.02858.
- T. Matsuoka, R. Rao, M. A. Susner, B. S. Conner, D. Zhang and D. Mandrus, Phys. Rev. B, 2023, 107, 165125 CrossRef CAS.
- W. Bai, Z. Hu, C. Xiao, J. Guo, Z. Li, Y. Zou, X. Liu, J. Zhao, W. Tong, W. Yan, Z. Qu, B. Ye and Y. Xie, J. Am. Chem. Soc., 2020, 142, 10849–10855 CrossRef CAS PubMed.
- O. V. Yazyev and L. Helm, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 125408 CrossRef.
- L. Cai, J. He, Q. Liu, T. Yao, L. Chen, W. Yan, F. Hu, Y. Jiang, Y. Zhao, T. Hu, Z. Sun and S. Wei, J. Am. Chem. Soc., 2015, 137, 2622–2627 CrossRef CAS PubMed.
- V. K. S. Shante and S. Kirkpatrick, Adv. Phys., 1971, 20, 325–357 CrossRef.
- V. Pashchenko, O. Bludov, D. Baltrunas, K. Mazeika, S. Motria, K. Glukhov and Yu. Vysochanskii, Condens. Matter Phys., 2022, 25, 43701 CAS.
- A. Bhowmick, B. Bal, S. Ganguly, M. Bhattacharya and M. L. Kundu, J. Phys. Chem. Solids, 1992, 53, 1279–1284 CrossRef CAS.
- T. Ying, T. Yu, Y. Qi, X. Chen and H. Hosono, Adv. Sci., 2022, 9, 2203219 CrossRef CAS PubMed.
- P. Colombet, A. Leblanc, M. Danot and J. Rouxel, J. Solid State Chem., 1982, 41, 174–184 CrossRef CAS.
- T. Asano, Y. Ajiro, M. Mekata, H. Yamazaki, N. Hosoito, T. Shinjo and H. Kikuchi, Solid State Commun., 1994, 90, 125–128 CrossRef CAS.
- C. Payen, P. Molinie, P. Colombet and G. Fillion, J. Magn. Magn. Mater., 1990, 84, 95–101 CrossRef CAS.
- S. Lee, P. Colombet, G. Ouvrard and R. Brec, Mater. Res. Bull., 1986, 21, 917–928 CrossRef CAS.
- S. Selter, K. K. Bestha, P. Bhattacharyya, B. Özer, Y. Shemerliuk, L. T. Corredor, L. Veyrat, A. U. B. Wolter, L. Hozoi, B. Büchner and S. Aswartham, Phys. Rev. Mater., 2022, 7, 033402 CrossRef.
- Y. Ma, Y. Yan, L. Luo, S. Pazos, C. Zhang, X. Lv, M. Chen, C. Liu, Y. Wang, A. Chen, Y. Li, D. Zheng, R. Lin, H. Algaidi, M. Sun, J. Z. Liu, S. Tu, H. N. Alshareef, C. Gong, M. Lanza, F. Xue and X. Zhang, Nat. Commun., 2023, 14, 7891 CrossRef CAS PubMed.
- X. Bourdon, V. Maisonneuve, V. B. Cajipe, C. Payen and J. E. Fischer, J. Alloys Compd., 1999, 283, 122–127 CrossRef CAS.
- Y. Peng, X. Cheng, P. Gu, F. Wang, J. Yang, M. Xue, W. Yang, C. Wang, S. Liu, K. Watanabe, T. Taniguchi, Y. Ye and J. Yang, Adv. Funct. Mater., 2020, 30, 1910036 CrossRef CAS.
- M. A. Susner, B. S. Conner, E. Rowe, R. Siebenaller, A. Giordano, M. V. McLeod, C. R. Ebbing, T. J. Bullard, R. Selhorst, T. J. Haugan, J. Jiang, R. Pachter and R. Rao, J. Phys. Chem. C, 2024, 128, 4265–4276 CrossRef CAS.
- X. Yan, X. Chen and J. Qin, Mater. Res. Bull., 2011, 46, 235–238 CrossRef CAS.
- G. Kliche, Z. Naturforsch., A: Astrophys., Phys. Phys. Chem., 1983, 38, 1133–1137 CrossRef.
- B. Zapeka, M. Kostyrko, I. Martynyuk-Lototska and R. Vlokh, Philos. Mag., 2015, 95, 382–393 CrossRef CAS.
- B. Hillman, L. Noren and D. J. Goossens, in Proceedings of the 35th Annual Australia/New Zealand Condensed Matter and Materials Meeting, (Australian Institute of Physics, 2011), p. 50.
- Y. Gu, Q. Zhang, C. Le, Y. Li, T. Xiang and J. Hu, Phys. Rev. B, 2019, 100, 165405 CrossRef CAS.
- Y. Yu, G. Deng, Y. Cao, G. J. McIntyre, R. Li, N. Yuan, Z. Feng, J.-Y. Ge, J. Zhang and S. Cao, Ceram. Int., 2019, 45, 1093–1097 CrossRef CAS.
- A. M. Ruiz, D. L. Esteras, A. Rybakov and J. J. Baldoví, Dalton Trans., 2022, 51, 16816–16823 RSC.
- J. L. Lado and J. Fernández-Rossier, 2D Mater., 2017, 4, 035002 CrossRef.
- G. Wang, K. Yang, Y. Ma, L. Liu, D. Lu, Y. Zhou and H. Wu, Chin. Phys. Lett., 2023, 40, 077301 CrossRef.
- A. Le Méhauté, G. Ouvrard, R. Brec and J. Rouxel, Mater. Res. Bull., 1977, 12, 1191–1197 CrossRef.
- P. J. S. Foot and B. A. Nevett, Solid State Ionics, 1983, 8, 169–172 CrossRef CAS.
- D. Upreti, R. Basnet, M. M. Sharma, S. K. Chhetri, G. Acharya, M. R. U. Nabi, J. Sakon, B. Da, M. Mortazavi and J. Hu, 2024, DOI:10.48550/arXiv.2407.12662.
- C. Yang, X. Chen, J. Qin, K. Yakushi, Y. Nakazawa and K. Ichimura, J. Solid State Chem., 2000, 150, 281–285 CrossRef CAS.
- J. S. O. Evans, D. O'Hare, R. Clement, A. Leaustic and P. Thuéry, Adv. Mater., 1995, 7, 735–739 CrossRef CAS.
- D. P. Kozlenko, O. N. Lis, N. T. Dang, M. Coak, J.-G. Park, E. V. Lukin, S. E. Kichanov, N. O. Golosova, I. Yu. Zel and B. N. Savenko, Phys. Rev. Mater., 2024, 8, 024402 CrossRef CAS.
- M. J. Coak, D. M. Jarvis, H. Hamidov, A. R. Wildes, J. A. M. Paddison, C. Liu, C. R. S. Haines, N. T. Dang, S. E. Kichanov, B. N. Savenko, S. Lee, M. Kratochvílová, S. Klotz, T. C. Hansen, D. P. Kozlenko, J.-G. Park and S. S. Saxena, Phys. Rev. X, 2021, 11, 011024 CAS.
- H. Sun, L. Qiu, Y. Han, E. Yi, J. Li, M. Huo, C. Huang, H. Liu, M. Li, W. Wang, D.-X. Yao, B. A. Frandsen, B. Shen, Y. Hou and M. Wang, Mater. Today Phys., 2023, 36, 101188 CrossRef CAS.
- W. Toyoshima, T. Masubuchi, T. Watanabe, K. Takase, K. Matsubayashi, Y. Uwatoko and Y. Takano, J. Phys.: Conf. Ser., 2009, 150, 042215 CrossRef.
- T. Matsuoka, H.-S. Kim, S. Samanta, J. L. Musfeldt and D. G. Mandrus, Front. Mater., 2024, 11, 1362744 CrossRef.
- L. Min, M. Sretenovic, T. W. Heitmann, T. W. Valentine, R. Zu, V. Gopalan, C. M. Rost, X. Ke and Z. Mao, Commun. Phys., 2022, 5, 1–7 CrossRef.
- L. Thiel, Z. Wang, M. A. Tschudin, D. Rohner, I. Gutiérrez-Lezama, N. Ubrig, M. Gibertini, E. Giannini, A. F. Morpurgo and P. Maletinsky, Science, 2019, 364, 973–976 CrossRef CAS PubMed.
- Q. Zhang, K. Hwangbo, C. Wang, Q. Jiang, J.-H. Chu, H. Wen, D. Xiao and X. Xu, Nano Lett., 2021, 21, 6938–6945 CrossRef CAS PubMed.
- J. Yang, Y. Zhou, Y. Dedkov and E. Voloshina, Adv. Theory Simul., 2020, 3, 2000228 CrossRef CAS.
- N. Sheremetyeva, I. Na, A. Saraf, S. M. Griffin and G. Hautier, Phys. Rev. B, 2023, 107, 115104 CrossRef CAS.
- R. N. Jenjeti, R. Kumar, M. P. Austeria and S. Sampath, Sci. Rep., 2018, 8, 8586 CrossRef PubMed.
- J. Duan, P. Chava, M. Ghorbani-Asl, Y. Lu, D. Erb, L. Hu, A. Echresh, L. Rebohle, A. Erbe, A. V. Krasheninnikov, M. Helm, Y.-J. Zeng, S. Zhou and S. Prucnal, ACS Appl. Mater. Interfaces, 2022, 14, 11927–11936 CrossRef CAS PubMed.
- P. Liu and Y. Pu, Results Phys., 2021, 29, 104750 CrossRef.
- T. Xu, M. Luo, N. Shen, Y. Yu, Z. Wang, Z. Cui, J. Qin, F. Liang, Y. Chen, Y. Zhou, F. Zhong, M. Peng, M. Zubair, N. Li, J. Miao, W. Lu, C. Yu and W. Hu, Adv. Electron. Mater., 2021, 7, 2100207 CrossRef CAS.
- X. Han, P. Song, J. Xing, Z. Chen, D. Li, G. Xu, X. Zhao, F. Ma, D. Rong, Y. Shi, Md. R. Islam, K. Liu and Y. Huang, ACS Appl. Mater. Interfaces, 2021, 13, 2836–2844 CrossRef CAS PubMed.
- Y. Liu and X. Sun, Magnetochemistry, 2022, 8, 101 CrossRef CAS.
- R. Kumar, R. N. Jenjeti and S. Sampath, Adv. Mater. Interfaces, 2019, 6, 1900666 CrossRef CAS.
- Spontaneous Ordering in Semiconductor Alloys, ed. A. Mascarenhas, Springer US, Boston, MA, 2002 Search PubMed.
- B. Cao, S. Chen, X. Jin, J. Liu and T. Li, ACS Appl. Mater. Interfaces, 2020, 12, 57245–57253 CrossRef CAS PubMed.
- X. Jin, S. Chen and T. Li, Commun. Mater., 2022, 3, 1–9 CrossRef PubMed.
- R. Zhang, S. Zhao, J. Ding, Y. Chong, T. Jia, C. Ophus, M. Asta, R. O. Ritchie and A. M. Minor, Nature, 2020, 581, 283–287 CrossRef CAS PubMed.
- J. Ding, Q. Yu, M. Asta and R. O. Ritchie, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 8919–8924 CrossRef CAS PubMed.
- B. Jiang, Y. Yu, J. Cui, X. Liu, L. Xie, J. Liao, Q. Zhang, Y. Huang, S. Ning, B. Jia, B. Zhu, S. Bai, L. Chen, S. J. Pennycook and J. He, Science, 2021, 371, 830–834 CrossRef CAS PubMed.
- S. Roychowdhury, T. Ghosh, R. Arora, M. Samanta, L. Xie, N. K. Singh, A. Soni, J. He, U. V. Waghmare and K. Biswas, Science, 2021, 371, 722–727 CrossRef CAS PubMed.
- H. Ji, A. Urban, D. A. Kitchaev, D.-H. Kwon, N. Artrith, C. Ophus, W. Huang, Z. Cai, T. Shi, J. C. Kim, H. Kim and G. Ceder, Nat. Commun., 2019, 10, 592 CrossRef CAS PubMed.
- K. Kim, S. Y. Lim, J. Kim, J.-U. Lee, S. Lee, P. Kim, K. Park, S. Son, C.-H. Park, J.-G. Park and H. Cheong, 2D Mater., 2019, 6, 041001 CrossRef CAS.
- K. C. Rule, G. J. McIntyre, S. J. Kennedy and T. J. Hicks, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 134402 CrossRef.
- Y. Jin, Y. Jin, K. Li, M. Yan, Y. Guo, Y. Zhou, A. Preobrajenski, Y. Dedkov and E. Voloshina, J. Phys. Chem. Lett., 2022, 13, 10486–10493 CrossRef CAS PubMed.
- T. T. Mai, K. F. Garrity, A. McCreary, J. Argo, J. R. Simpson, V. Doan-Nguyen, R. V. Aguilar and A. R. H. Walker, Sci. Adv., 2021, 7, eabj3106 CrossRef CAS PubMed.
- D. Jana, P. Kapuscinski, A. Pawbake, A. Papavasileiou, Z. Sofer, I. Breslavetz, M. Orlita, M. Potemski and C. Faugeras, Phys. Rev. B, 2023, 108, 144415 CrossRef CAS.
- R. Gusmão, Z. Sofer, D. Sedmidubský, Š. Huber and M. Pumera, ACS Catal., 2017, 7, 8159–8170 CrossRef.
- L. Liao, E. Kovalska, V. Mazanek, L. Valdman, L. Dekanovsky, W. Bing, D. Sedmidubský, J. Luxa, Š. Huber, A. P. Herman, R. Kudrawiec and Z. Sofer, J. Mater. Chem. C, 2022, 10, 8834–8844 RSC.
- J. Zhang, W. Xu, L. Wang, Z. Zheng, F. Liu, P. Yu and G. Yang, Cryst. Growth Des., 2023, 23, 1259–1268 CrossRef CAS.
- X. Zhao, X. Yin, D. Liu, C. Huo, G.-Z. Dong, S. Chen, Z.-B. Liu, X.-Q. Yan and J.-G. Tian, J. Phys. Chem. C, 2022, 126, 6837–6846 CrossRef CAS.
- X. Wang, K. Du, W. Liu, P. Hu, X. Lu, W. Xu, C. Kloc and Q. Xiong, Appl. Phys. Lett., 2016, 109, 123103 CrossRef.
- D. Lançon, R. A. Ewings, T. Guidi, F. Formisano and A. R. Wildes, Phys. Rev. B, 2018, 98, 134414 CrossRef.
| This journal is © The Royal Society of Chemistry 2024 |