
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Modulating efficacy and cytotoxicity of lipoamino fatty acid nucleic acid carriers using disulfide or hydrophobic spacers†
Ricarda C.
Steffens
ab,
Sophie
Thalmayr
 ac,
Eric
Weidinger
a,
Johanna
Seidl
ac,
Paul
Folda
a,
Miriam
Höhn
a and
Ernst
Wagner
*abc
ac,
Eric
Weidinger
a,
Johanna
Seidl
ac,
Paul
Folda
a,
Miriam
Höhn
a and
Ernst
Wagner
*abc
aPharmaceutical Biotechnology, Department of Pharmacy, LMU Munich, Butenandtstr. 5-13, 81377 Munich, Germany. E-mail: ernst.wagner@lmu.de
bCenter for NanoScience (CeNS), LMU Munich, 80799 Munich, Germany
cCNATM – Cluster for Nucleic Acid Therapeutics Munich, Germany
First published on 5th July 2024
Abstract
Double pH-responsive xenopeptides comprising polar ionizable succinoyl tetraethylene pentamine (Stp) motifs and lipophilic ionizable lipoamino fatty acids (LAFs) were recently found to efficiently transfect mRNA and pDNA at low doses. However, potency was often accompanied with cytotoxicity at higher doses. Insertion of bioreducible disulfide building blocks (ssbb) or non-reducible hydrophobic spacers between polar and apolar ionizable domains of LAF–Stp carriers should mitigate toxicity of xenopeptides. Carriers showed stable nucleic acid complexation and endosomal pH-dependent lytic activities, both of which were abolished after reductive cleavage of ssbb-containing carriers. For pDNA, U-shaped carriers with one Stp and two LAF units or bundle carriers with two Stps and four LAFs displayed highest potency. For mRNA, best transfection was achieved with bundle carriers with one Stp and four LAFs. Both the ssbb and hydrophobic spacer containing analogs displayed improved metabolic activity, reduced membrane damage, and improved cell growth. The ssbb carriers were most beneficial regarding living cell count and low apoptosis rates. Mechanistically, inserted spacers decelerated the transfection kinetics and altered the requirement of endosomal protonation. Overall, mRNA and pDNA carriers with improved biocompatibility have been designed, with their high potency illustrated in transfection of various cell lines including low passage number colon carcinoma cells.
1 Introduction
Nucleic acid therapeutics present a novel frontier in medical science, offering immense potential for treating a wide range of diseases. Beyond their proven efficacy in vaccine development, nucleic acid delivery is a promising approach to cancer immunotherapy, gene and/or protein displacement therapies. Synthetic formulations including polyplexes, lipoplexes and lipid nanoparticles (LNPs) hold great potential for delivery of nucleic acids like DNA, mRNA, siRNA, and Cas9/sgRNA systems.1–4 The involved carriers offer advantages such as relatively facile synthetic accessibility, the potential for precise modulation of nanoparticle features and minimal immunogenicity. However, limitations do exist, such as moderate efficacy requesting administration at very high nanoparticle per cell doses, associated with significant cytotoxicity, and challenges involve careful balancing between particle stability and dynamic nucleic acid release at the right cellular compartment.5 To improve safety and efficacy of polyplexes for the various nucleic acid cargos, researchers have addressed these issues by continuous optimization of formulations and carrier molecules, e.g. by incorporating biocompatible building blocks.6–24Using a chemical evolution approach25 based on solid-phase assisted xenopeptide synthesis,26–29 our group recently developed novel nucleic acid carriers that have proven to be highly effective for delivering a variety of nucleic acid cargos including pDNA and mRNA.30 These carriers are sequence-defined, double pH-responsive xenopeptides and comprise one or several hydrophilic units of cationizable artificial amino acid Stp (succinoyl tetraethylene pentamine)26 and lipophilic, cationizable lipoamino fatty acids (LAFs).30 The Stp units function as polar cationic counterparts to negatively charged nucleic acids, leading to polyplex formation through nucleic acid condensation via electrostatic interaction. The cationizable LAF residues combine advantageous qualities of lipophilic domains, permitting hydrophobic particle stabilization at physiological neutral pH, which is reversible upon protonation at mild acidic endosomal pH, resulting in a strongly enhanced endosomal escape and exceptionally high transfection efficacy. For certain LAF–Stp topologies, mRNA transfection was successful even at ultra-low doses of 3 picograms of mRNA, providing a nanoparticle potency comparable to viral vectors. Additionally, these carriers facilitate remarkable protein expression also in vivo, particularly in the spleen, tumor, lungs, and liver upon intravenous injection of mRNA polyplexes30 or LNPs31 in mice.
Alongside with favorable efficacy of these novel LAF-carriers, in several cases high potency was observed to correlate with increased cytotoxicity. This can be compensated by application of low dosages,30 nevertheless raises safety concerns regarding future therapeutic administration. While the superior ability of these nanoparticles to interact with cell membranes is highly desirable for nanoparticle uptake by endocytosis and endosomal escape, prolonged intracellular presence of membrane-active carriers may lead to reduced cell viability. Cytotoxicity induced by cationic polymers or lipids can be attributed to several causes. Carrier molecules and their metabolites may have the potential to cause cellular membrane disruption or nuclear damage, mitochondrial dysfunction, generation of reactive oxygen species (ROS), among other factors. For example, a two-stage cytotoxicity32,33 was reported for polyplexes consisting of polyethylenimine (PEI),12–14,34 a most frequently used gold-standard for nucleic acid delivery. An early phase I toxicity resulting from compromised cellular membrane integrity, and a later phase II cytotoxicity due to a mitochondrial apoptotic program was reported.35 Additionally, PEI-based transfection efficiency and cytotoxicity was found to be associated with polymer size and type,14,16,20 and with permeabilization of nuclear membranes.36,37 Similar observations were made for a series of other cationic carriers; characteristics that favorably enhance intracellular transfer of nucleic acids across membrane barriers are likely to be also associated with increased cytotoxicity due to side effects of membrane disruption. The question has been raised whether the observed correlation of efficacy and cytotoxicity can be uncoupled. In fact, for polymeric cationic carriers it was already demonstrated that efficiency/cytotoxicity ratios can be modulated by either optimizing the size of the polymer14,16,38 or its cationic subunits.7,39 For example, separating the efficient but also toxic multi-cationic structure of PEI into several small cationic (oligo)aminoethylene units resulted into highly potent but far less toxic polymers of similar sizes.7 Molecular reasons are at least twofold; defined small subunits may have inherent favorable efficacy/toxicity properties39 based on amphiphilicity or pKa profile, as observed and defined in the even-odd rule of oligoethylenimines.40 Secondly, biodegradable linkages between the oligocationic subunits can be designed, reducing the persistence of polycationic molecules and this leading to favorable cytotoxicity profiles. Esters,17,18,21,41 acetals,22,42 or disulfides15,19,23 were integrated into synthetic PEI carriers, ‘breaking up the correlation between efficacy and toxicity’.19 Disulfide linkages have been applied to various other cationic carriers including ionizable lipids.43–52 Disulfide bonds are stable in the extracellular environment but are subsequently reduced within cells after endosomal release by high concentrations of cytosolic glutathione (GSH), leading to fragmentation of the carriers.53 Hereby, both release of the nucleic acid cargo from the carrier is facilitated and cytotoxic effect associated with highly lytic properties of the carrier can be minimized.
In our previous work on sequence-defined T-shaped lipo-xenopeptides the introduction of a bioreducible disulfide bond between the cationic polar backbone and the standard, non-ionizable fatty acid domain was advantageous for mRNA and siRNA delivery,51,52 reducing cytotoxic effects and enhancing efficacy by solving the balancing act between extracellular polyplex stability and cytosolic nucleic acid release.52,54 The precise positioning of the disulfide cleavage site at the desired location was managed by design of a protected cystamine-based disulfide building block (ssbb) for use in solid-phase synthesis.51
In the current work, novel LAF carriers with U-shape or bundle topology were modified with bioreducible or non-reducible spacers that are integrated in the oligomer sequence at precise positions based on the rationale of separating cationic and lipophilic domains of the carrier oligomers. By introducing disulfide spacers, we aimed to maintain favorable characteristics of the polyplexes while consequently improving biocompatibility of the carriers after endosomal release. As the introduction of reducible linkers changes the geometry and distances between lipophilic and hydrophilic subdomains of carriers, we also designed analogous carriers with isosteric hydrophobic nonreducible spacers. In the following we demonstrate the dynamic nucleic acid binding ability of the disulfide-modified carriers and polyplex destabilization as a result of the reductive cytosolic environment. Consequently, bioreducible carriers showed beneficial impact on cell viability while maintaining high nucleic acid transfer activity promoted by LAF carriers. Interestingly, introduction of hydrophobic non-reducible linkers also modulated the transfection/cytotoxicity profile. Altogether this highlights a beneficial influence of altered chemical topology plus optional biodegrading bonds on the biological carrier characteristics.
2 Experimental
2.1 Erythrocyte leakage assay with and without GSH treatment
The erythrocyte leakage assay was performed following a protocol published by Krhac Levacic et al.52 Erythrocytes were isolated by repeated washing of fresh, citrate buffered (25 mM citrate) human blood with a solution of phosphate buffered saline (PBS) supplemented with 25 mM citrate and subsequent centrifugation (800 rcf, 10–15 min at 4 °C). Washing was considered as completed when the supernatant was clear after centrifugation. The cell pellet was then diluted to 5 × 107 erythrocytes per mL with PBS at different pH values (pH 5.5, 6.5 and 7.4) and stored on ice. The oligomers were diluted with PBS at the indicated pH value to a concentration of 2.5 μM and pipetted into a V-bottom 96-well plate as quadruplicate (75 μL per well). For evaluation of lytic potential after incubation under reductive conditions, the oligomers were incubated with 10 mM GSH (final concentration) in HEPES (20 mM, pH 7.4) at the indicated pH value for 90 minutes at 37 °C while shaking at 500 rpm, followed by pipetting into the V-bottom 96-well plate. 75 μL of the erythrocyte suspension at the same pH value was added to each well.Then, the V-bottom 96-well plates were incubated under slow constant shaking for 60 min at 37 °C. Afterwards, the samples were centrifuged (800 rcf, 4 °C, 10 min), and 100 μL of the supernatant were transferred to a TPP-ft 96-well plate. To determine the lytic activity, the samples were analyzed for hemoglobin release at wavelength λ = 405 nm using a microplate reader (Spark, Tecan, Männedorf, Switzerland). PBS at the indicated pH values was used as negative control (0% value) and samples treated with 1% Triton X-100 at the indicated pH value served as positive control (100% value). Data are presented as mean value (±SD) out of quadruplicates.
2.2 Polyplex formation
Polyplex formation was performed as follows: the respective nucleic acid was diluted in HBG (HEPES buffered glucose, 20 mM HEPES, 5% glucose (w/v), pH 7.4) and the calculated amount of LAF carrier at the indicated N/P (nitrogen/phosphate) ratio was diluted in Millipore water from a LAF carrier stock solution (10 mg mL−1 in EtOH/H2O at carrier-specific solvent ratios, for details see Table S1, ESI†). For calculation of the N/P ratio, secondary amines of Stp units (3 amines per Stp), terminal primary amines and tertiary amines of the LAF residues were considered. Polyplexes were formed upon addition of equal volumes of nucleic acid solution (in HBG) to the LAF carrier solution (in water), followed by rapid pipetting and incubation for 40 min at room temperature (RT) in a closed Eppendorf tube. In case of control polyplexes, i.e., LPEI (N/P 6) for pDNA polyplexes and succPEI (weight/weight (w/w) ratio of 4) for mRNA polyplexes, the respective carrier was diluted in HBG before mixing with the nucleic acid solution as described above. Final concentrations of nucleic acid were 12.5 μg mL−1 for mRNA and 10 μg mL−1 for pDNA.2.3 Experiments under reductive conditions
For experiments mimicking cytosolic reductive environment, the polyplexes were prepared at the indicated N/P ratio in the required volume with a pDNA concentration of 12.5 μg mL−1 and a mRNA concentration of 15.6 μg mL−1. After polyplex formation, GSH solution (50 mM GSH in HBG, pH adjusted to 7.4 by addition of 1 M NaOHaq) was added to the to the polyplex solution to obtain a final GSH concentration of 10 mM. For negative controls, the respective volume of HBG was added to the polyplex solutions. The samples were incubated at 37 °C for 90 min while being placed on a mechanical shaker at 500 rpm before further evaluation.2.4 Agarose gel shift assay
1% (w/v) agarose gel was prepared by microwave-assisted heating of agarose in TBE buffer (18.0 g of tris(hydroxymethyl)aminomethane, 5.5 g of boric acid, 2 mM EDTA at pH 8 in 1 L of water). After cooling down to about 50 °C, 1000× GelRed (0.001% (v/v)) was added to the solution. Afterwards, the solution was casted into an electrophoresis unit and cooled down for gelation. Polyplexes were formulated as described in section 2.2 with a pDNA concentration of 10 μg mL−1 and mRNA concentration of 12.5 μg mL−1, respectively. After 40 min of incubation, loading buffer (6×; prepared from 6 mL of glycerol, 1.2 mL of 0.5 M EDTA, 2.8 mL of H2O, 0.02 g of bromophenol blue) was added to the polyplex solution to obtain a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 dilution. 20 μL (pDNA polyplexes) or 18 μL (mRNA polyplexes) of the samples were loaded to the gel and electrophoresis was performed at 120 mV for 70 min in 1× TBE buffer. Free pDNA (c = 10 μg mL−1) or mRNA (c = 12.5 μg mL−1) diluted in HBG was used as control.
:6 dilution. 20 μL (pDNA polyplexes) or 18 μL (mRNA polyplexes) of the samples were loaded to the gel and electrophoresis was performed at 120 mV for 70 min in 1× TBE buffer. Free pDNA (c = 10 μg mL−1) or mRNA (c = 12.5 μg mL−1) diluted in HBG was used as control.
2.5 Ethidium bromide exclusion assay
Quantification of EtBr fluorescence was carried out using a microplate reader (Spark, Tecan, Männedorf, Switzerland) with an excitation wavelength λex = 535 nm and an emission wavelength λem = 590 nm. Polyplexes were prepared as described in section 2.2 with a total volume of 50 μL at a final pDNA concentration of 10 μg mL−1. After the indicated incubation time, 250 μL of aqueous EtBr solution (c = 0.5 μg mL−1) were added to the polyplexes. After additional incubation for 10 min at room temperature, 260 μL of each sample were transferred into a TPP-ft 96-well-plate and fluorescence intensity was measured. A calibration curve with free pDNA (linear concentration range from 0 to 10 μg mL−1) diluted in HBG was used for quantification. The amount of free, i.e., non-compacted pDNA was calculated based on the calibration of free pDNA and displayed as percentage of EtBr fluorescence in relation to 100% of free pDNA.2.6 Complexation efficiency of mRNA polyplexes with and without reductive conditions
Quant-iT™ RiboGreen RNA Assay-Kit (Thermo Fisher Scientific) was used to determine the encapsulation efficiency [ee (%)] of mRNA polyplexes. Polyplexes were formulated as described in section 2.2 at a concentration of 15.6 μg mL−1. For stress testing, HBG or GSH solution (50 mM, section 2.3) were added to a final mRNA concentration of 12.5 μg mL−1 and a GSH concentration of 10 mM. The samples were incubated for 90 min at 37 °C under constant shaking at 400 rpm. Unstressed control samples were prepared as described above with addition of HBG and without further incubation. All samples were diluted with 1× TE (10 mM Tris–HCl, 1 mM EDTA, pH 7.5 in RNase-free water) to an mRNA concentration of 2 μg mL−1. 50 μL of the diluted samples were mixed with 50 μL of 1× TE as ‘untreated samples’. For complete release of mRNA, unstressed samples were diluted with 50 μL of 1× TE containing 2% (v/v) Triton X-100 and 250 IU mL−1 heparin to a final mRNA concentration of 2 μg mL−1 (i.e., ‘Triton sample’). All samples were incubated for 10 min at 37 °C under constant shaking at 150 rpm. After cooling down at RT for 5 min, 100 μL of RiboGreen reagent diluted 200-fold in 1× TE were added to each sample. After 5 min under light protection, the fluorescence intensity was measured in a Tecan microplate reader (Spark, Tecan, Männedorf, Switzerland) at excitation/emission wavelength of 485/535 nm in duplicates. Background was measured for the respective conditions (i.e. HBG or HBG with GSH 10 mM) in 1× TE, or in 1× TE supplemented with Triton X-100 and heparin, treated in the same manner as the respective polyplex samples. The following formula was used to calculate encapsulation efficiency after background subtraction of each sample: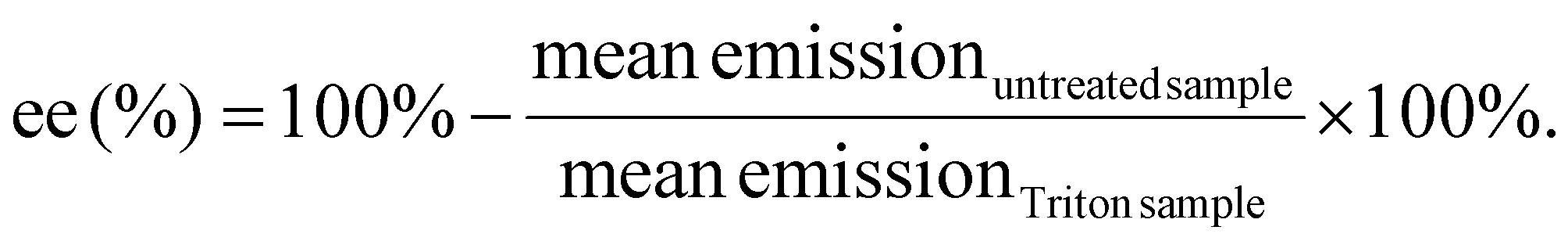
2.7 Dynamic light scattering (DLS)
Measurements were performed using a Zetasizer Nano ZS (Malvern Instruments, Malvern, Worcestershire, United Kingdom) using a folded capillary cell (DTS1070) by dynamic and electrophoretic laser light scattering (DLS, ELS). For determination of particle size and polydispersity index (PDI), 100 μL of pDNA polyplex solutions or 80 μL of mRNA polyplex solutions were formed as described above at a pDNA concentration of 10 μg mL−1 or an mRNA concentration of 12.5 μg mL−1, respectively, and measured with the settings as follows: 30 s of equilibration time, temperature of 25 °C, refractive index 1.330, viscosity 0.8872 mPa s. Each sample was measured three times with six sub-runs per measurement. For determination of zeta potential, the polyplex solution was diluted with HBG to a final volume of 800 μL immediately prior to measurement. Measurement parameters were identical to size determination, with an increase of equilibration time to 60 s. Each sample was measured with 15 sub-runs (10 s each) and zeta potential were calculated by Smoluchowski equation. All values (size, PDI and zeta potential) were displayed as mean ± SD of these measurements.2.8 Cell culture
HeLa (human adherent cervix carcinoma cell line), HeLa-Gal8-mRuby stably expressing a galectin8-mRuby3 fusion protein55–57 and N2a (murine neuroblastoma cell line Neuro2a) cells were cultured in Dulbecco's Modified Eagle Medium (DMEM) low glucose (1 g L−1 glucose). Huh7 wt cells (human adherent hepatic carcinoma cell line) were cultured in DMEM Ham's F12 medium. Cell culture media were supplemented with 10% fetal calf serum (FCS), 4 mM of stable glutamine, 100 U mL−1 penicillin and 100 μg mL−1 streptomycin. Cells were cultured at 37 °C with 5% CO2 and a relative humidity of 95% in an incubator. Human colon carcinoma COGA-2 and COGA-12 low passage cell lines,58 generated in a previous clinical study approved by local ethical committee with written, informed consent from all patients and molecularly characterized in great detail, were cultured in RPMI 1640 medium containing L-glutamine and sodium bicarbonate, supplemented with 10% FCS, 100 U mL−1 penicillin and 100 μg mL−1 streptomycin). COGA-2 cells were cultured in flasks manually pre-coated with collagen A (1:10 (v/v) in PBS).
2.9 Transfection screening
5000 HeLa, 10000 N2a, 8000 Huh7 or 10000 COGA-2 or COGA-12 cells were seeded in a 96-well plate about 24 hours prior to transfection. For COGA-2 cells, plates were coated with collagen A (1:10 (v/v) in PBS) before seeding. Polyplexes were prepared as described above. Prior to treatment, cell culture medium was replaced by suitable amounts of fresh medium to obtain a final volume of 100 μL per well after transfection. Different doses of nucleic acid per well were evaluated. Each polyplex was added to the wells as triplicate. For pDNA, indicated doses of pDNA polyplex solution containing 10 μg mL−1 pCMVLuc (Plasmid Factory GmbH, Bielefeld, Germany) were prepared as described in section 2.2. From this polyplex solution, required volumes (in the range of 5–20 μL) were added to the wells; for detailed information see indicated doses in the figures. LPEI (MW = 22 kDa) polyplex solution (N/P 6) was used as positive control at indicated pDNA doses. For mRNA, indicated doses of mRNA polyplexes containing 12.5 μg mL−1 CleanCap® FLuc mRNA (5moU) (Trilink Biotechnologies, San Diego, CA, USA) (i.e. suitable volumes in a range of 1.25–20 μL) of polyplex solution were transferred to the corresponding wells. As positive control, succinylated branched PEI (succPEI) at a w/w ratio of 4 was used at indicated doses. Each well was filled up with the corresponding volume of HBG to reach a final volume of 100 μL per well.
2.10 Luciferase expression assay
Transfection efficacy of the polyplexes was evaluated after incubation for 24 hours at 37 °C. The medium was removed, and the cells were lysed with 0.5× lysis buffer (100 μL per well) and stored at −80 °C at least overnight. Before determination of luciferase expression, plates were allowed to come up to RT and incubated for 1 hour. For luciferase activity of mRNA-treated cells, the cell lysate was diluted in PBS at a ratio of 1:100. Luciferase activity was measured using a Centro LB 960 plate reader luminometer (Berthold Technologies GmbH & Co. KG, Bad Wildbad, Germany). Hereto, 35 μL of the cell lysate were measured for 10 s after automatic addition of 100 μL LAR buffer (20 mM glycylglycine; 1 mM MgCl2; 0.1 mM ethylenediaminetetraacetic acid; 3.3 mM dithiothreitol; 0.55 mM adenosine 5′-triphosphate; 0.27 mM coenzyme A, pH 8–8.5) supplemented with 5% (v/v) of a mixture of 10 mM luciferin and 29 mM glycylglycine. Transfection efficiency was calculated for the seeded number of cells and presented as relative light units (RLU) per well. In case of mRNA, data was presented after background subtraction (i.e., HBG-treated control cells). In the case of pDNA polyplexes, the HBG background was not subtracted but depicted in the related graphs.
2.11 Cell metabolic activity by CellTiter-Glo®
Transfections were performed as described in section 2.9. The supernatant was removed at 24 h after transfection, and 25 μL of medium mixed with 25 μL of CellTiter-Glo® Reagent (Promega) were added to each well. After incubation on an orbital shaker for 30 min at RT, luminescence was recorded using a Centro LB 960 plate reader luminometer (Berthold Technologies, Bad Wildbad, Germany). The luminescence signals (in RLU) of the samples were set in relation to the luminescence signal of the negative control (HBG buffer-treated control cells). Experiments were performed as triplicates.2.12 Confocal laser scanning microscopy (CLSM)
15000 HeLa Gal8 mRuby cells55–57 per well were seeded one day prior to transfection in ibidi μ-slide 8-well ibidiTreat chamber slides and cultured at 37 °C, with 5% CO2 and 95% relative humidity. Polyplexes were formed as described above at a Luc-mRNA concentration of 12.5 μg mL−1 with 20% (w/w) Cy5 labelled firefly luciferase mRNA. Medium was changed prior to transfection with adequate volumes to reach a final volume of 300 μL per well after transfection with indicated doses of mRNA polyplexes. After transfection, the cells were incubated for 2 h or 4 h. Medium was removed, cells were washed twice with PBS and fixed with 4% (w/v) paraformaldehyde in PBS for 1 h at RT in the dark. Subsequently, cells were washed twice with PBS and nuclei were stained with DAPI (1 μg mL−1 in PBS) for 30 min at RT in the dark. The staining solution was replaced with fresh PBS and cells were stored under light protection at 4 °C until imaging. Imaging was performed with a Leica-TCS-SP8 CLSM equipped with an HC PL APO 63× 1.4 objective and images were processed with the LAS X software from Leica.
2.13 Bafilomycin A1 assay
5000 HeLa cells were seeded into 96-well plates one day prior to transfection. 30 min before transfection, medium was replaced with either fresh medium or medium containing bafilomycin A1 (BafA1) (0.1 μg mL−1 in DMSO) to reach a final concentration of 200 nM BafA1 after polyplex addition. Polyplexes were formed as described above and indicated doses of pCMVLuc pDNA or Luc-mRNA polyplexes were subjected to the cells. After 4 h of incubation, cells treated with mRNA polyplexes were lysed and evaluated via luciferase expression assay as described in section 2.10. For pDNA, medium was replaced by fresh medium without BafA1 after 4 h and incubated further 20 h before analysis. Transfections were performed in triplicates.2.14 Cell viability by LDH cytotoxicity assay
LDH cytotoxicity assay was performed according to the manufacturers’ protocol (Promega). Briefly, 5000 HeLa cells per well were seeded into a 96-well plate one day prior to transfection. Polyplexes were prepared as described in section 2.2. After replacement of medium with suitable amounts of fresh medium, the test compounds were added to the cells in the indicated concentration (i.e., 100 ng per well for pDNA polyplexes and 63 ng per well for mRNA polyplexes) to reach a final volume of 100 μL per well. As control groups, cells treated with the indicated amount of HBG buffer or 20 μL of either LPEI polyplexes containing 10 μg mL−1 pDNA or succPEI polyplexes containing 12.5 μg mL−1 mRNA were used. Cells without any treatment (neither polyplex nor HBG) served as negative control. After transfection, the cells were incubated at 37 °C for the desired exposure time (i.e., 8 h, 12 h, 16 h, 24 h). 45 min before end of the incubation time and addition of CytoTox® reagent, a triplicate of untreated cells was treated with 10 μL of 10× lysis solution as maximum LDH release control. Then, 50 μL of the supernatant of each well was transferred to a fresh transparent, flat-bottom 96-well plate, followed by addition of 50 μL of CytoTox® reagent and incubation in the dark for 30 min at RT while slowly shaking. After 30 min, 50 μL of stop solution was added to each well and the absorbance was immediately measured at 490 nm (OD490) using a multiplate reader (Spark, Tecan, Männedorf, Switzerland). For calculation of released LDH, the average values of the untreated cells was subtracted from all values of experimental cells. From the corrected experimental values, the following equation was used to calculated the percentage of LDH release:
2.15 Microscope images
The polyplexes were prepared as described in section 2.2 and transfected to the cells as described in section 2.9. Subsequently the cells were incubated for 24 hours after transfection, and then examined microscopically by an Axiovert 200 (Zeiss, Oberkochen, Germany). Photographic images were taken by a INFINITY2-3C (3.3 Megapixel Color CCD Camera USB2.0), Lumenera Corporation/Ottawa (purchased from FRAMOS GmbH, Taufkirchen, Germany) and processed by Infinity Capture Software.2.16 Cell watcher analysis
For the cell watcher analysis, HeLa cells were seeded 24 hours prior to transfection to a 48 well plate (5000 cells per well) and monitored by Cellwatcher M (PHIO Scientific GmbH, Munich, Germany). Confluency was calculated by the corresponding software. 24 hours after seeding, medium was replaced by fresh, pre-warmed medium and the cells were transfected with polyplex solution at indicated doses. Cell growth was monitored for a total incubation time of 48 hours after transfection.2.17 Flow cytometry analysis
40000 HeLa cells per well were seeded 24 hours prior to transfection. Polyplexes were prepared as described in section 2.2 and transfected to the cells with 100 ng per well for pDNA polyplexes (pDNA concentration of 10 μg mL−1) and 63 ng per well for mRNA polyplexes (mRNA concentration of 12.5 μg mL−1). After 24 hours of incubation, the medium was removed, and the cells were gently washed with cold PBS (4 °C) and trypsinized for 5 minutes at 37 °C. The cell suspension was diluted with FACS buffer (10% FBS in PBS, 0.1% (v/v) DAPI) and transferred to a FACS tube. Cells were analyzed by flow cytometry using a CytoFLEX S Flow Cytometer (Beckman Coulter, Brea, CA, USA) over 60 seconds at a flow rate of 170 μL min−1. Flow cytometry data were analyzed using FlowJo™ v10.8 flow cytometric analysis software by FlowJo, LLC (Becton, 20 Dickinson and Company, USA).
2.18 Kinetics of cellular association and expression
To evaluate the cell association and expression kinetics, 18000 HeLa cells per well were seeded in a 24 well-plate 24 h prior to transfection. Polyplexes were prepared at N/P 24 with a w/w mixture of 20% (w/w) EZ Cap Cy5 Firefly Luciferase mRNA (5moUTP) and 80% (w/w) CleanCap® EGFP mRNA (5moU) at a total mRNA concentration of 12.5 μg mL−1. The cells were transfected in duplicates with a dose of 100 ng total mRNA and incubated for 30 min, 2 h, 4 h, 6 h, 24 h and 48 h. After incubation, the cells were prepared for flow cytometer measurement as described in section 2.17, without the addition of DAPI. The measurement was performed using a CytoFLEX S Flow Cytometer (Beckman Coulter, Brea, CA, USA) at suitable flow rates and times of acquisition. However, at least 8000 cells per analysis were recorded. Flow cytometry data were analyzed using FlowJo™ v10.8 flow cytometric analysis software by FlowJo, LLC (Becton, 20 Dickinson and Company, U.S.).
2.19 Annexin V/propidium iodide assay
Polyplex treatments were performed in duplicates in 24-well plates. HeLa cells were seeded 24 h prior to transfection in a 24-well-plate (40000 cells per well). Immediately prior to transfection, the medium was replaced with 475 μL of fresh medium containing 10% (v/v) FBS for pDNA polyplexes and with 487.5 μL for mRNA polyplexes. The nanoparticles were prepared as described in section 2.2. Transfection was performed with 12.5 μL of polyplexes containing 125 ng per well pDNA or 6.3 μL polyplex solution containing 78 ng per well mRNA, respectively. HBG was then added to reach a final volume of 500 μL per well, i.e., 12.5 μL per well for cells transfected with pDNA polyplexes and 6.2 μL for cells treated with mRNA polyplexes. HBG buffer (25 μL per well for pDNA polyplexes and 12.5 μL per well for mRNA polyplexes) was used as negative control. After 24 h of incubation, the cells were collected and incubated with annexin V incubation reagent (prepared according to manufacturer's protocol of Bio-Techne GmbH, Wiesbaden, Germany) for 10 min before flow cytometer analysis using a CytoFLEX S Flow Cytometer (Beckman Coulter, Brea, CA, USA). Cells were analyzed with a flow rate of 170 μL min−1 until 10000 events were counted. Gates were set compared to control measurements with HBG-buffer treated cells and with exclusion of cell debris. Annexin V and propidium iodide negative cells were considered as healthy cells. Cells shifted to the annexin V and propidium iodide positive gate indicated late apoptotic cells, whereas cells shifted to only annexin V-positive gate were considered as early apoptotic, and PI-positive, but annexin-V-negative cells were not viable. Flow cytometry data were analyzed using FlowJo™ v10.8 flow cytometric analysis software by FlowJo, LLC (Becton, 20 Dickinson and Company, U.S.).
2.20 Statistical analysis
Experimental results are presented as arithmetic mean of triplicates, if not stated otherwise, with error bars displaying the standard deviation (SD). Statistical analysis of the results (mean ± SD) was performed by applying unpaired Student's two-tailed t-test with Welch's correction. Calculations and graphical presentation were performed with Prism 10.1.2 (GraphPad Software Inc.). ns (statistically not significant) p > 0.05; *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001.3 Results and discussion
3.1 Design and synthesis of disulfide and hydrophobic spacer analogs
In recent work, our group has developed a library of novel nucleic acid carriers with versatile sequences and topologies.30 The novel xenopeptides are comprised of protonatable Stp units, derived from aminoethylene units of PEI, enabling nucleic acid compaction and providing endosomal buffer capacity, α,ε-amidated lysines as branching points and lipoamino fatty acids (LAF) with central tertiary amines, which display favorable, pH-dependent properties supporting polyplex stabilization by lipophilic characteristics at extracellular pH and particle disassembly upon protonation in endosomal environment. The LAFs used in the new study were 8Oc (based on reductive bisalkylation of 8-aminooctanoic acid with octanal) and 12Oc (obtained by the reductive amination of dodecanal with 8-aminooctanoic acid), see Scheme 1. Variations of the lipophilic chain length and the position of the tertiary amine have been used to tailor endosomal escape by endosomolytic activity. Each LAF-xenopeptide was assigned to a distinct, consecutive ID number, and an additional nomenclature, providing information about type of LAF, topology (B2 bundle, or U-shape) and the Stp/LAF ratio (e.g.1621: LAF = 8Oc, topology = B2, Stp/LAF = 1:4). Among the xenopeptides of the library, the carriers 1621 (8Oc-B2-1:4) and 1611 (12Oc-U1-1:2) have demonstrated great potencies for the delivery of mRNA and DNA, respectively. Especially for mRNA, both carriers exhibited excellent transfection efficacies, even at ultra-low mRNA doses. For B2 bundle topology, shorter LAFs such as 8Oc in 1621 were favorable for transfection efficiency compared to the longer 12Oc LAF units, although cytotoxicity was observed for 1621 at high doses. In case of pDNA polyplexes, formulations with a higher Stp/LAF ratio (1:2) as present in U-shape carrier 1611 are more effective in terms of gene transfer.30 This is consistent with the hypothesis that the PEI-like Stp units are beneficial for DNA condensation into polyplexes.8,29,38
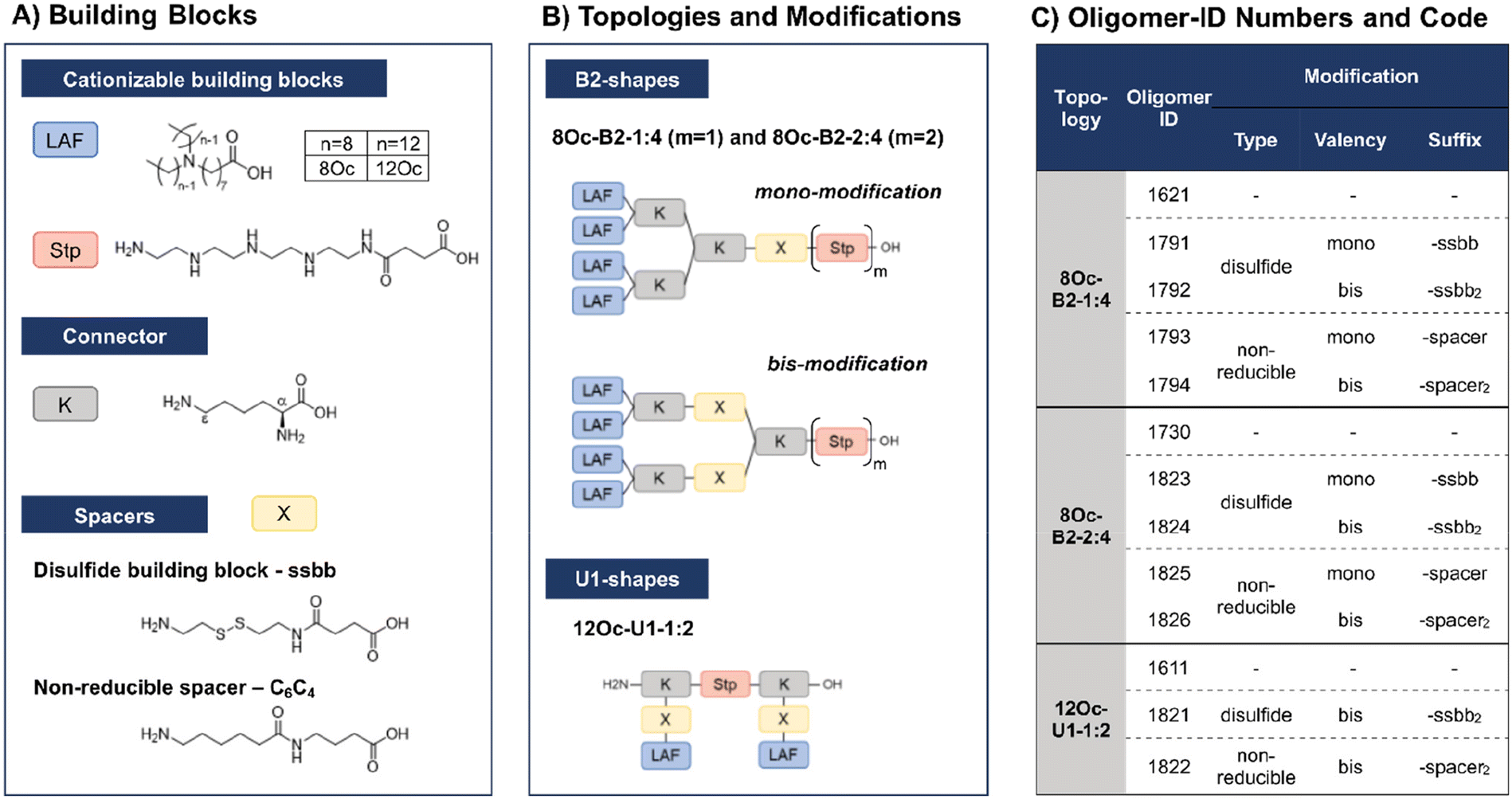 | ||
| Scheme 1 Library design of lipoamino fatty acid containing LAF–Stp carriers containing reducible (ssbb) and non-reducible (C6C4) spacer domains. (A) Building blocks used in solid phase assisted assembly (displayed in unprotected form). (B) Different topologies and positioning of bioreducible or non-reducible building blocks. (C) Summarized overview of the library and its identification (ID) numbers. | ||
Based on these considerations we converted the sequence of B2-1:4 1621 into the B2-2:4 carrier 1730. As demonstrated here, DNA polyplexes formed with the 1730, providing two Stp units, showed superior properties over 1621 (Fig. S1, ESI†). For example, polyplex size was reduced from 220 nm to 80 nm at N/P 12 and transfection efficiency on N2a cells was 108-times increased compared to 1621 (Fig. S1C, ESI†). However, cell viability, determined by a CellTiter Glo® assay, was still reduced compared to LPEI, but improved over 1621 at high N/P ratios (Fig. S1D, ESI†). Altogether, these new findings make 1730 a highly promising LAF carrier for pDNA delivery.
Due to their great potential for nucleic acid delivery, xenopeptides 1611, 1621 and 1730 were selected for further sequence-defined chemical evolution by introduction of spacers between the Stp and LAF units. Previous work demonstrated that topologies between or within the various subunits (‘LAF nitrogen catwalk’) can modulate potency but also cytotoxicity of carriers. Based on established Fmoc solid-phase assisted synthesis with protected artificial amino acids,26–29 we now generated novel spacer-containing xenopeptides as listed in Scheme 1 and Table S1.† As spacers, either Fmoc-protected succinoyl-cystamine as a solid-phase-synthesis compatible disulfide building block (ssbb)51,59 or an analogous non-reducible amino hexanoic acid-amino-butanoic acid dipeptide spacer (C6C4) was used, providing comparable spatial distance (ssbb = 20.6 Å vs. C6C4 = 19.6 Å) and similar hydrophilic/hydrophobic characteristics.
The incorporation of the disulfide building blocks provides carriers with redox-responsive cleavage sites for degradation in the intracellular, reductive cytosolic environment with increased concentration of GSH.53,60,61 Cytosolic carrier cleavage is expected to destabilize the polyplexes and to abolish the amphiphilic and potentially lytic character of the xenopeptides. Both spacer- and disulfide-analogs were set in comparison with their original LAF–Stp structures regarding their nucleic acid compaction ability and resulting physicochemical properties (size, polydispersity, surface charge) as well as their potency to deliver pDNA and mRNA to various cell lines without having a cytotoxic influence on the cells. For carriers of the U1-1:2 topology, the disulfide modification was introduced in the lysine side chain before coupling of 12Oc for maintaining symmetry and generating a system that is cleaved in small fragments of mostly hydrophilic and lipophilic properties upon disulfide reduction (see Scheme 1). For bundle structures, multiple options for the positioning of the disulfide-motifs were possible, leading to various cleavage products. At first, the ssbb was positioned behind the cationic Stp unit. Upon disulfide reduction, fragmentation into a cationic Stp-domain and a large, mostly hydrophobic LAF domain will occur. As a second option, the disulfide was integrated after the first branching lysine. In this case, disulfide reduction would lead to fragmentation into a Stp-lysine fragment and two smaller LAF fragments. As a last option, the ssbb was introduced directly before the LAF, leading to a structure with four cleavage sites. However, synthetic issues regarding disulfide instability occurred during the assembly of tetra-ssbb analogs (data not shown) and prevented the synthesis. The identity of all synthesized carriers was confirmed by MALDI-TOF mass spectrometry and 1H-NMR spectroscopy. The analytical data are summarized in the ESI.†
In order to investigate the biodegradability of the reducible carriers under reductive stress, MALDI-TOF analysis was performed after incubation with the physiological antioxidant GSH (c = 10 mM). To mimic intracellular reducing conditions, the disulfide-containing oligomers were dissolved in HBG and incubated with GSH at a final concentration of 10 mM for 90 min at 37 °C. Oligomer fragments of GSH-induced disulfide reduction were subsequently detected by MALDI analysis, besides small peaks of intact oligomer (Fig. 1 and Table S2, ESI†). In case of the exemplarily presented mass spectrum, mass peaks associated with the Stp backbone after partial and complete disulfide reduction were found, as well as the mass peaks of the hydrophobic LAF arm.
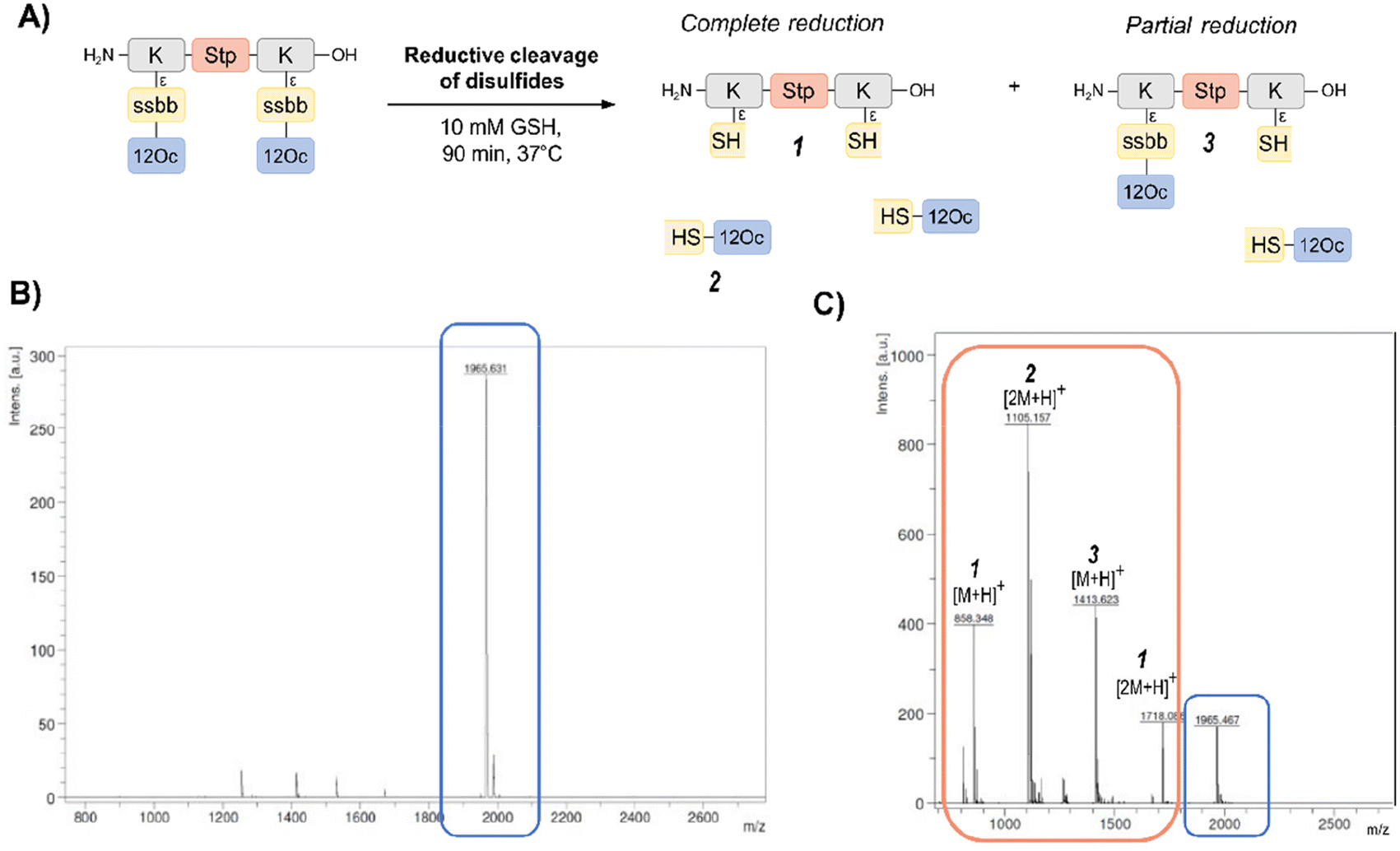 | ||
| Fig. 1 MALDI analysis before and after incubation with glutathione (GSH). (A) Scheme of disulfide-containing LAF-oligomer 1821 with 12Oc-U1-1:2 topology before and after GSH-induced reduction of disulfides with possible cleavage products upon complete or partial reduction. Representative MALDI mass spectra of disulfide-LAF-oligomer 1821 before (B) and after (C) treatment with 10 mM GSH for 90 min at 37 °C. The mass spectrum after GSH-treatment contains additional peaks (1, 2 and 3) assigned to cleavage products upon disulfide reduction. | ||
3.2 Physico-chemical evaluation of pDNA and mRNA polyplexes
pDNA. Polyplexes were formed with pCMVLuc DNA at N/P ratios 6, 12, 18 and in the case of bundle LAF carriers also 24, to identify the N/P ratio necessary for polyplex formation (Fig. S2A, ESI†). N/P ratio of 6 was not sufficient for most carriers to form stable and nano-sized polyplexes, independent of their topology, accompanied by mostly negative zeta potential which was not favorable for attachment to negatively charged cell membranes (Fig. S2B, ESI†). For the carriers following the 12Oc-U1-1
:2 topology (1611 and its analogs), N/P 12 was sufficient for polyplex formation, and a further decrease of particle size was observed for 1611 by increasing the N/P ratio to 18. In the U1-series, most comparable particles regarding their size and zeta potential were obtained at N/P 18. For 8Oc-B2-1:4 (1621 series), a minimum N/P 18 was required to obtain stable polyplexes for all analogs. In comparison to 1621, 1730 contains a second Stp unit, which is considered to support DNA compaction.38 This is underlined by slightly reduced particle sizes compared with 1621 and its analogs at N/P 18. In addition, the 1730 series formed stable nanoparticles at N/P 12. This finding is in line with previous work by Nie and co-workers, demonstrating the importance of a well-balanced lipophilic–hydrophilic ratio of bioreducible nucleic acid carrier constructs comprised of large cationic headgroups and lipophilic tails.46
mRNA. The whole set of xenopeptides was also evaluated regarding its ability to form mRNA polyplexes (Fig. S3, ESI†). In previous studies, 1611 and 1621 proved to form stable and reproducible particles at N/P 18.30 For the new disulfide and C6C4 spacer analogs, N/P 12 was also evaluated (Fig. S3A and B, ESI†). The 12Oc-U1-1
:2 analogs, 1821 (ssbb) and 1822 (C6C4), formed stable mRNA particles already at N/P 12, with positive zeta potential of 15–18 mV. Comparable to the original structure 1621, the 8Oc-B2-1:4-analogs formed nanosized polyplexes at N/P ≥ 18, except for the mono-ssbb analog 1791 which was unable to generate stable mRNA polyplexes at any N/P ratio. This observation was confirmed by an agarose gel shift assay, where free mRNA was detected for formulation with 1791, but not for other LAF-carriers (Fig. S3C, ESI†). The corresponding C6C4 spacer analog 1793 showed no aggregation at N/P ≥ 18. All 8Oc-B2-2:4 carriers of the 1730 series formed desirable mRNA nanoparticles at N/P 12, which were smaller in size than 1621.
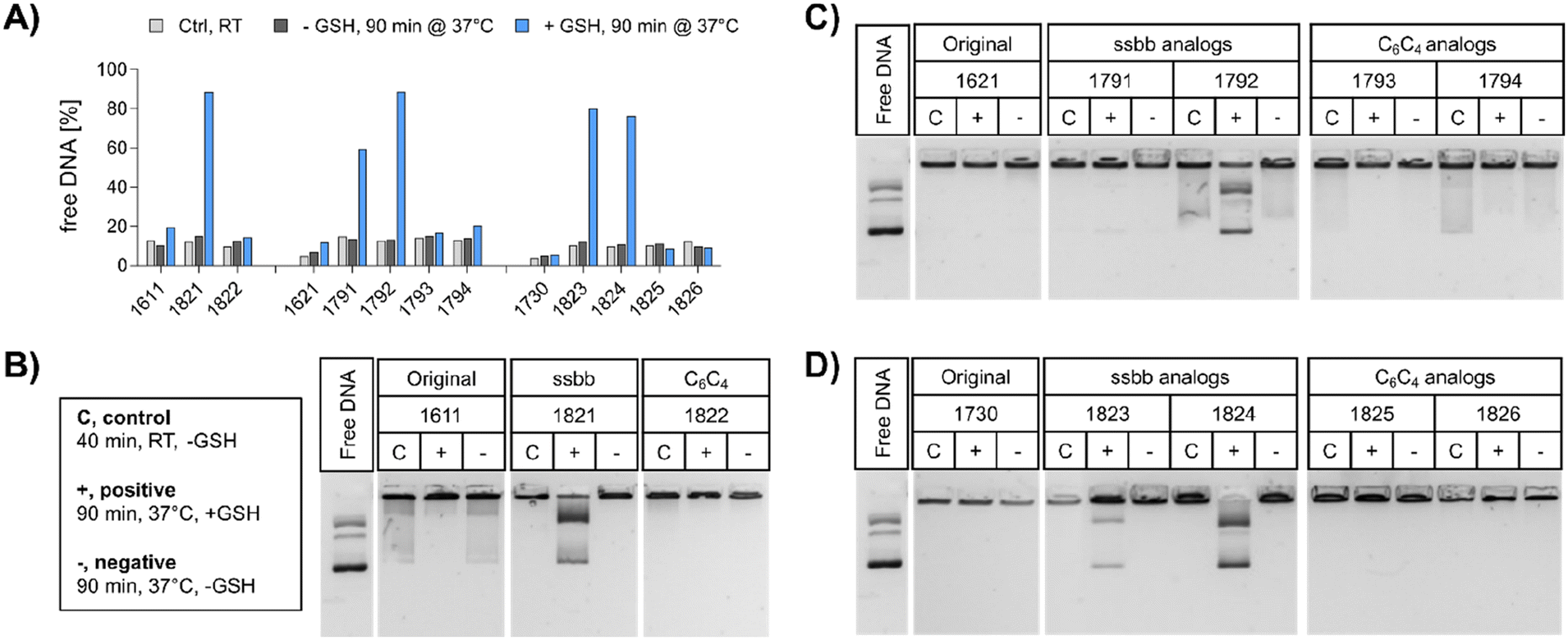 | ||
| Fig. 2 DNA compaction ability and cargo release of LAF–Stp carriers before and after GSH treatment. (A) Ethidium bromide assay of pDNA polyplexes formed with the indicated carriers at N/P 18. The carriers were incubated with GSH (blue) or with HBG (dark grey) for 90 min at 37 °C prior to EtBr assay or incubated in absence of GSH for 40 min at RT (light grey). The fluorescence intensity of EtBr correlates with the amount of free and non-compacted pDNA, as pDNA only in this state is accessible for EtBr intercalation. 100% EtBr fluorescence refers to the fluorescence of free pDNA without carrier in the same concentration as used for polyplex formation. (B–D) DNA compaction and release ability of polyplexes formed at N/P 18 was determined in an agarose gel shift assay (1% w/v agarose) after 90 min incubation at 37 °C under reducing (+) and non-reducing (−) conditions. Lanes ‘C’ refer to standard control polyplexes without preincubation at 37 °C. | ||
pDNA. The compaction ability was determined by an ethidium bromide (EtBr) exclusion assay and agarose gel shift assay (Fig. 2). A pre-experiment confirmed absence of disturbing effects like potential DNA degradation as a result of the treatment conditions (37 °C for 90 min, ±10 mM GSH) on free DNA (Fig. S4, ESI†). Under standard conditions, all polyplex formulations showed favorable DNA compaction ability under non-reductive conditions, with less than 20% of free DNA detected (Fig. 2A). In case of 1730 (two Stp units), compaction was even stronger than for 1621 (one Stp unit). After exposure to a reducing environment (10 mM GSH, 90 min, 37 °C), decompaction of DNA was detected exclusively for disulfide-containing analogs, while no effect on DNA compaction was observed for non-reducible xenopeptides. This was found for all reducible analogs of the 1621 B2 bundle series (mono-disulfide 1791, bis-disulfide 1792), 1730 B2 bundle series (1823 and 1824), and the U-shape disulfide analog 1821.
Agarose gel shift assays (Fig. 2B–D) were performed to confirm the findings of the ethidium bromide exclusion assay. For 12Oc-U1-1:2 polyplexes, original 1611 polyplexes showed a minor release of DNA, but like polyplexes of the C6C4 spacer analog 1822, were not affected by reductive GSH. In contrast, the disulfide analog 1821 showed GSH-dependent destabilization of polyplexes (Fig. 2B). DNA release was also observed for all disulfide containing B2 bundle xenopeptides after exposure to reductive environment. This effect was stronger for bis-disulfide analogs 1792 and 1824 than mono-disulfide analogs (Fig. 2C and D). It has to be noted that the gel shift assay indicates free pDNA that is not enclosed into nanoparticles. In contrast the EtBr assay detects also barely compacted pDNA by intercalation of the dye also in loose particles.
mRNA. Encapsulation efficiency (ee) of mRNA polyplexes was determined via RiboGreen assay for a selected set of 1621 and the bis-modified analogs 1792 (ssbb2) and 1794 ((C6C4)2) at N/P 24 (Fig. S5, ESI†). Under standard conditions, the ee for all tested formulations was >85%, in case of 1621 and 1792 even >90%. Subsequent to incubation with the reductive GSH for 90 min at 37 °C, ee of the disulfide-containing analog 1792 was about 30% reduced, verifying polyplex destabilization under bioreducible conditions, while ee of non-reducible analogs, 1621 and 1794, remained unchanged.
3.3 Biological and transfection activity
:4 1730 series showed about 30% increase in lytic activity over 1621 series, despite the less lipophilic Stp/LAF ratio of 2:4 instead of 1:4. However, the negative charge of erythrocyte membranes must be noted, where cationic amphiphiles display advantageous leakage effects.62–64 Similar tendencies as the increase of lytic activity at pH 6.5 among the modified, both reducible and non-reducible LAF carriers, were observed also for the B2-2:4 series. These findings are consistent with our previous work investigating the lytic activity of different U-shapes, where not only an increase of the lipidic domain, but also the amount of Stp caused an increase of lytic activity (U1-1:2 < U1-2:2 < U1-2:4).30 In case of disulfide-containing structures, the lytic activity was abolished after reductive cleavage by incubation with GSH for 90 minutes. This effect might have positive consequences on biocompatibility at later time points beyond entry.
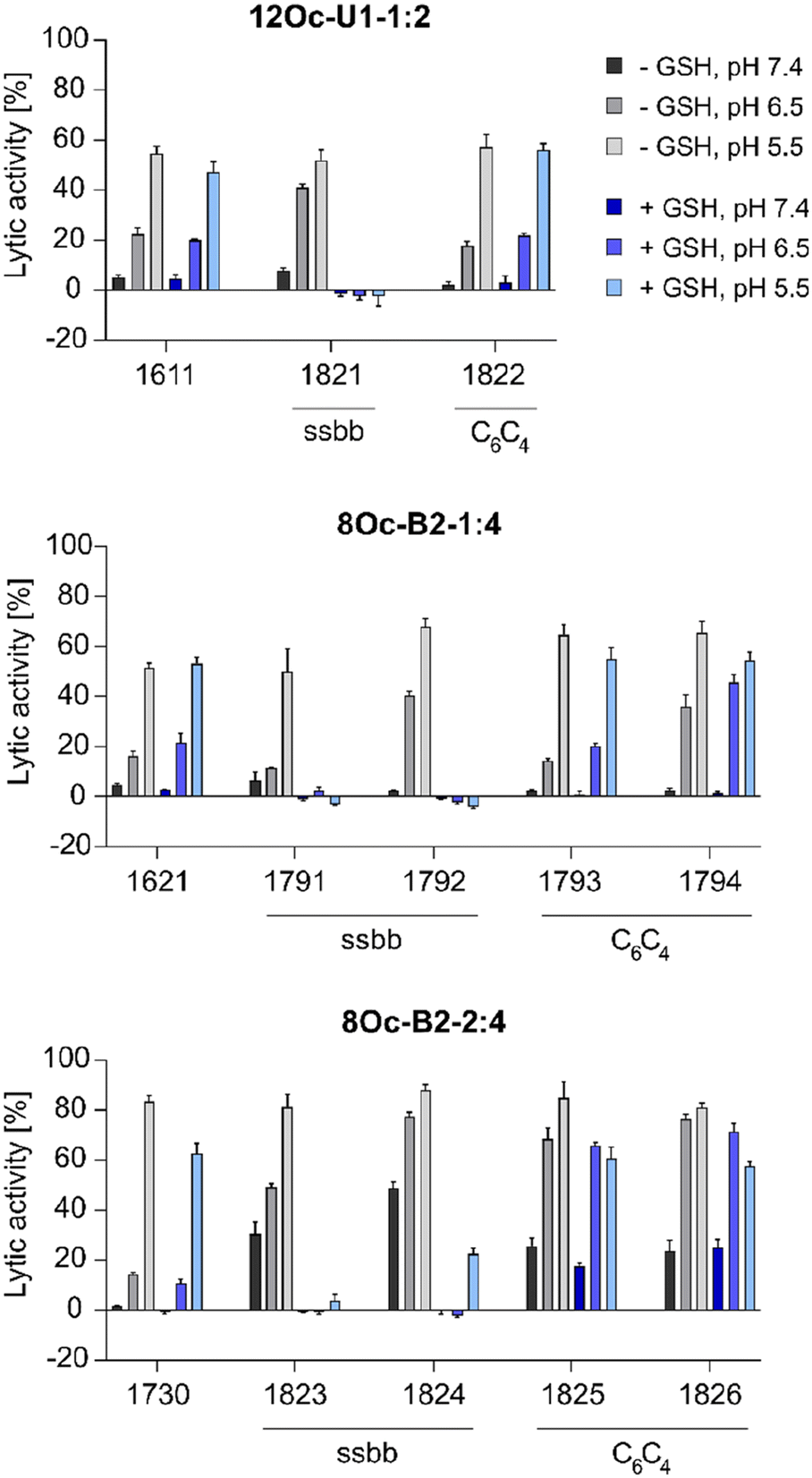 | ||
| Fig. 3 Lytic potential of free LAF xenopeptides evaluated in an erythrocyte leakage assay (n = 4; mean ± SD) at a concentration of 1.25 μM LAF carrier at three different physiological relevant pH values (pH 7.4, 6.5, 5.5). GSH treated xenopeptides were incubated with 10 mM GSH in PBS adjusted to the indicated pH at 37 °C for 90 min. PBS-treated erythrocytes were set to 0% lytic activity. 1% Triton X-100 served as positive control and was set to 100% lytic potential. | ||
:4 bundle structure 1730 a superior pDNA carrier was identified, whereas 1730 cannot meet the known high mRNA transfection efficiency of 8Oc-B2-1:4 bundle structure 1621.
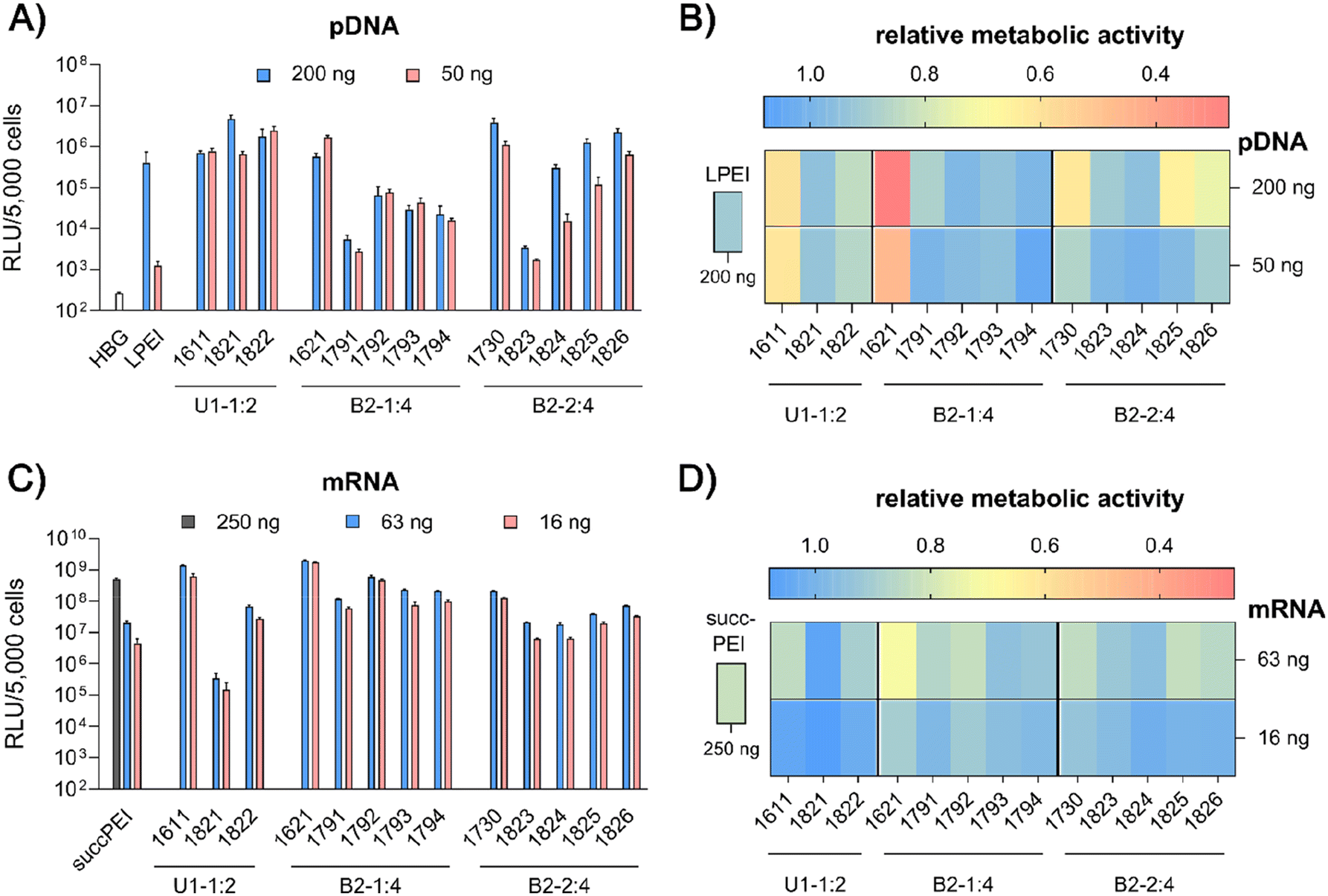 | ||
| Fig. 4
In vitro screening of the library of LAF–Stp xenopeptides applied in pDNA polyplexes (A and B) or mRNA polyplexes (C and D) on HeLa cells. pDNA polyplexes were formed at N/P 18 and mRNA polyplexes were formed at N/P 18 (12Oc-U1-1:2 carriers), N/P 24 (8Oc-B2-1:4 carriers) and N/P 12 (8Oc-B2-2:4 carriers). LPEI polyplexes were formed at N/P 6 and succPEI polyplexes were formed at w/w ratio 4. Polyplexes were transfected at the indicated nucleic acid doses per well. (A and C) Transfection efficiency determined by luciferase expression assay after 24 hours of incubation (n = 3 ± SD). (B and D) Cell viability measured by CellTiter Glo® assay in relation to control wells treated with HBG (n = 3 ± SD). | ||
pDNA. pCMVLuc polyplexes were formed at N/P 18, since at this ratio all carriers form stable, nano-sized polyplexes (Fig. S2A, ESI†), and tested in 200 ng high and 50 ng low pDNA doses on HeLa cells (Fig. 4A and B). The ‘gold standard’ for pDNA transfections, LPEI,14,34 was included for comparison and mediated the expected high luciferase gene expression, but at the high dose only. At this dose, no toxicity was observed. The standard U1-shape 1611 shows similar high transfection levels at both high and low dose. Metabolic activity of 1611 polyplexes, as determined by CellTiter Glo® assay, however, was significantly reduced (Fig. 4B), but could be recovered to >80% relative metabolic activity with the novel reducible and non-reducible spacer xenopeptide analogs (1821 and 1822, respectively) while maintaining high transfection efficacy at both dose levels. In fact, the bioreducible carrier 1821 mediated a 11.6-fold higher gene transfer than LPEI at the same dose. The 1611 series was also screened for transfecting N2a cells as well as Huh7 cells at 200 ng pDNA dose (Fig. S6, ESI†). Here, neither standard 1611 nor the novel analogs did considerably affect metabolic activity of cells, and the 1611 series mediated a transfection activity outperforming LPEI by 5- to ∼100-fold. The novel 8Oc-B2-2
:4 carrier 1730 exceeded the transfection efficiency of both LPEI and 1621 on all investigated cell lines (HeLa, Fig. 4; N2a and Huh7 cells, Fig. S6, ESI†). Furthermore, 1730 reaches or even exceeds transfection efficiencies of 1611 polyplexes. The disulfide-spacer analogs (1823 and 1824) loose transfection activity on HeLa cells, whereas the non-reducible spacer analogs (1825 and 1826) display activity similar as 1730 (Fig. 4). Spacer effects are less pronounced in the other cell lines (Fig. S6, ESI†). The 1621 series was least suitable for pDNA apart from standard 1621, which however displayed cytotoxicity (Fig. 4 and Fig. S6, ESI†). Analogs with disulfide or non-reducible spacer improved metabolic activity at the expense of reduced pDNA transfection efficiency (Fig. 4B and Fig. S6D, ESI†).
mRNA. Carrier demands were significantly different for mRNA delivery. The novel carriers were tested at the low doses of 63 ng or 16 ng FLuc mRNA. SuccPEI was used as established positive control20,52 also including a higher, non-toxic 250 ng mRNA dose. The carriers 1611 (12Oc-U1-1
:2) and 1621 (8Oc-B2-1:4) had outstanding potential for the delivery of mRNA at low and ultra-low doses, with RLU values greater than 109 on N2a and Huh7 cells (Fig. S7A and B, ESI†), consistent with previous findings,30 whereas 1730 (B2-2:4) showed slightly reduced mRNA transfection efficacies along all cell lines. Luciferase expression was diminished after modification of the 12Oc-U1-1:2 1611 carrier with both, reducible and non-reducible building blocks on HeLa and N2a cells (Fig. 4C and Fig. S7A, B, ESI†), probably as a consequence of the enhanced spacing between the LAF residues and Stp domain. For transfections to Huh7 cells, the non-reducible spacer was beneficial (Fig. S6B and S7D, ESI†). Even though the modifications had beneficial effects on cell viability of HeLa cells, the disulfide xenopeptide 1821 and the non-reducible analog 1822 was not or less suitable for mRNA delivery (Fig. 4C, D and Fig. S7A, ESI†). In sharp contrast, regarding the B2 bundles, the 8Oc-B2-1:4 1621 series showed overall favorable mRNA transfection efficiency compared to 8Oc-B2-2:4 1730 series, making 1621 and its analogs the most promising candidates for mRNA delivery in this study (Fig. 4C and Fig. S7A, C, ESI†). As observed also for pDNA polyplexes, luciferase activity of 1791 (B2-1:4-ssbb) was substantially reduced, but for 1792 (B2-1:4-ssbb2) transfection efficacy was high both on HeLa as well as N2a cells (Fig. 4C and Fig. S7A, C, ESI†). The C6C4 spacer analogs of the bundles were comparable to the disulfide analogs; dose reduction to 16 ng mRNA per well had no large impact on transfection efficacy on HeLa cells. Relative metabolic activity (Fig. 4D) was improved for disulfide- and also non-reducible spacer analogs over 1621 on HeLa and Huh7 cells (Fig. 4D and S7D, ESI†). The mRNA transfection efficiency of 1730 was lower than 1621 across all cell lines, and additional spacing, either by disulfide or non-reducible spacer, leads to slight decrease of transfection efficacy. The disulfide-oligomers 1823 and 1824 showed reduced cytotoxicity against HeLa over both, original 1730 and non-reducible spacer-analogs 1825 and 1826. Overall, less effects on metabolic activity were observed than for pDNA polyplexes, which might be due to overall lower doses of mRNA.
In summary, the particular characteristics of each cell line influenced both transfection efficiency and metabolic activity of cells. The reducible and non-reducible analogs of 1611 (12Oc-U1-1:2) and 1730 (8Oc-B2-2:4) were identified as promising candidates for efficient pDNA delivery with improved metabolic cell activity. In case of mRNA, 1792 and 1794, the bis-modified analogs to 1621, were defined as encouraging oligomers with improved cell viability. A strong benefit of the disulfide spacer was most clearly found on HeLa cells, which are known for their high intracellular GSH levels. In the case of Huh7 and N2a cells other intracellular mechanisms causing or preventing cytotoxicity may apply which are independent of bioreduction. This hypothesis is further stressed by the positive impact of the non-reducible spacers on both transfection efficacy and cell viability.
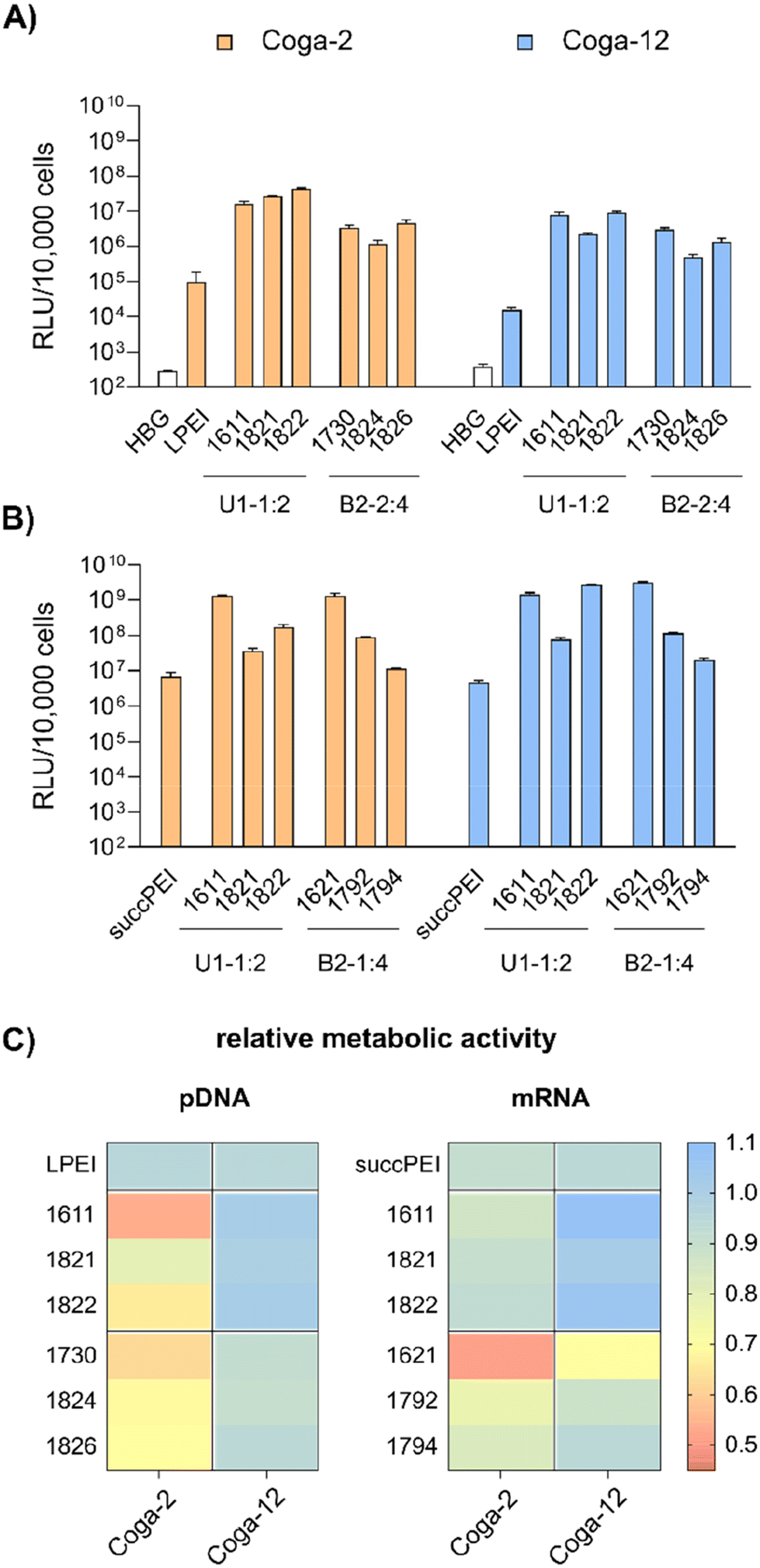 | ||
| Fig. 5 Transfections of LAF carriers on Coga-2 and Coga-12 colon carcinoma cells. (A) Luciferase gene expression of colon carcinoma cells at 24 hours after transfection of pDNA polyplexes (n = 3, mean + SD). The polyplexes were formed at N/P 18 and contained 50 ng pDNA per well with a concentration of 10 μg mL−1 pDNA. As control, LPEI polyplexes (N/P 6) were used. (B) Luciferase gene expression of colon carcinoma cells at 24 hours after transfection of mRNA polyplexes (n = 3, mean + SD). The cells were treated with polyplexes formed at N/P 18 (12Oc-U1-1:2) or N/P 24 (8Oc-B2-1:4) containing 31 ng mRNA per well with a concentration of 12.5 μg mL−1. As control, succPEI (w/w ratio 4) was used. (C) Relative metabolic activity of Coga-2 and Coga-12 cells treated with pDNA- (left) or mRNA-polyplexes (right) at 24 hours after transfection determined by CellTiter Glo® assay (n = 3). | ||
3.4 Cellular mechanisms of transfection
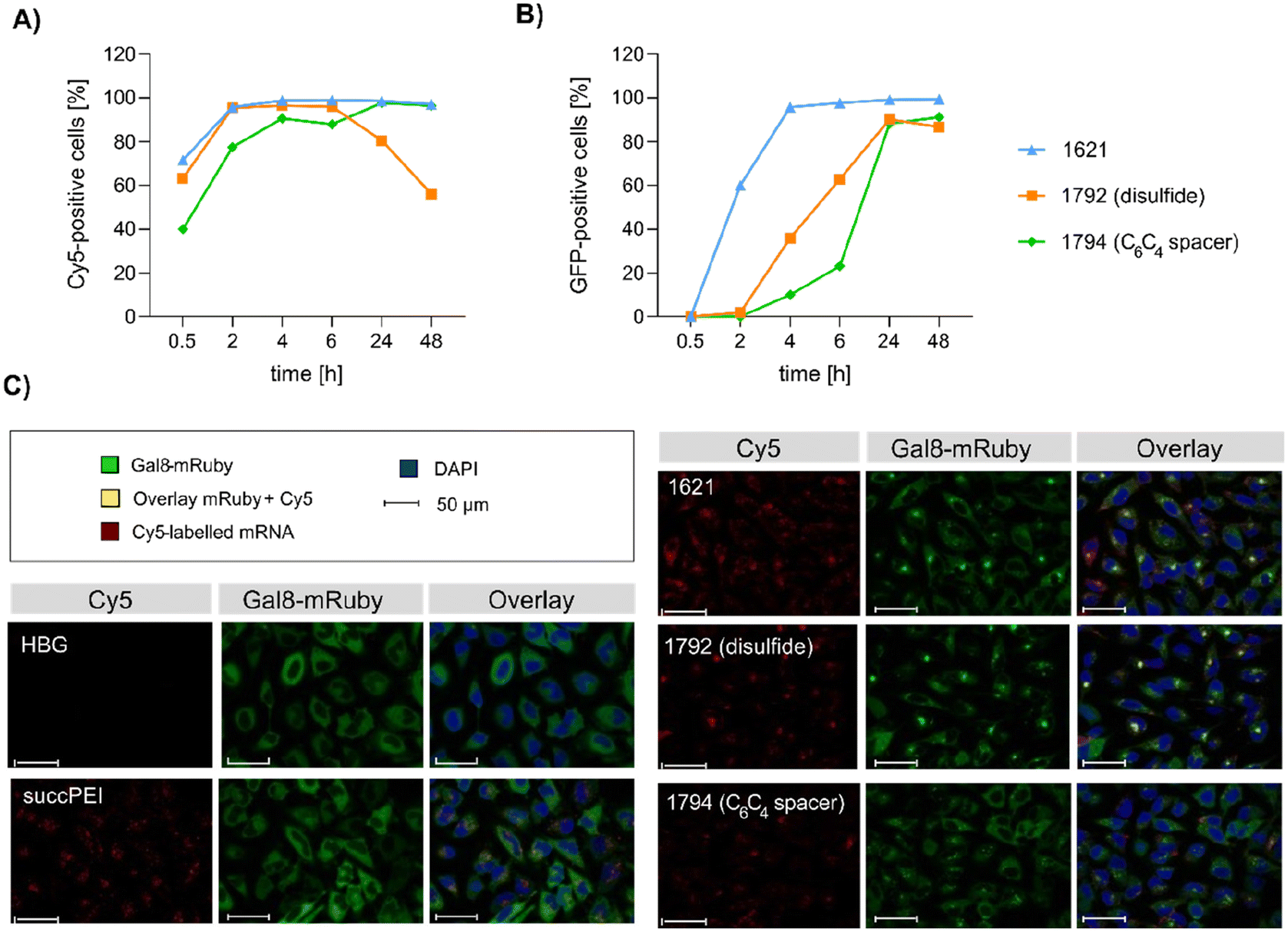 | ||
| Fig. 6 Mechanistic studies. (A and B) Flow cytometry analysis of HeLa cells treated with mRNA polyplexes. The polyplexes were formed with the indicated xenopeptides and 100 ng per well of cargo containing Cy5-Luc mRNA (20% (w/w)) and GFP-mRNA (80% (w/w)) with an N/P ratio of 24 (12.5 μg mL−1 total mRNA) and transfected to 18000 HeLa cells per well. Cells were analyzed by flow cytometry at the indicated time points. (A) Cell association and cellular uptake of mRNA polyplexes by HeLa cells presented by Cy5-positive cells. (B) GFP expression as measure of transfection efficacy. (C) Endosomal disruption evaluated in HeLa-Gal8-mRuby cells treated with mRNA polyplexes formed with the indicated xenopeptide in comparison to succPEI (w/w 4) at a mRNA dose of 47 ng at 4 hours after transfection by confocal laser scanning microscopy (CLSM). Redistribution of cytosolic Gal8-mRuby fluorescence (green) from cytosol (see HBG control) to an intracellular punctuate vesicle pattern represents endosomal membrane disruption. | ||
:4 bundle 1730 showed high sensitivity to BafA1, with a 324-fold decreased gene transfer activity in presence of BafA1, whereas 1621 (B2-1:4) was independent of the pH value. This observation is in accordance with the results of the erythrocyte leakage assay, where the lytic activity of B2-2:4 carriers was also strongly pH dependent (compare Fig. 3). 1730 has a stronger interaction and consequently denser compaction of DNA than 1621, due to the additional protonatable Stp unit (as supported by the EtBr exclusion assay, Fig. 2). This, in turn, leads to a greater dependence on lower pH values for xenopeptide solubilization and efficient release of the cargo. In accordance with the lytic potential towards erythrocytes, the analogs of 1730 were less sensitive to BafA1. 1611 polyplexes were moderately dependent of endosomal acidification (3-fold inhibition by bafilomycin), as previously observed also in other cell lines.301611 and 1621 spacer analogs, however, were more dependent on endosomal acidification, especially the disulfide-containing xenopeptides (39- and 502-fold inhibition by bafilomycin).
| Cargo | Carrier [ID] | Dosis [ng] | Fold decrease −BafA1/+BafA1 |
|---|---|---|---|
| HeLa cells were pre-incubated with BafA1 30 min prior to polyplex treatment and transfected with the indicated LAF–Stp polyplexes (mRNA: LAF polyplexes at N/P 24, succPEI w/w 4; pDNA: LAF polyplexes at N/P 18, LPEI N/P 6). Luciferase activity of cells treated with mRNA polyplexes was determined at 4 hours after transfection. pDNA polyplexes were allowed to incubate for further 20 h in BafA1-free medium (after 4 hours incubation with BafA1) before read-out by luciferase expression assay. The fold decrease of relative light units detected in cells treated with BafA1 compared to cells without BafA1 treatment was calculated. | |||
| mRNA | succPEI | 250 | 156 |
| 1621 | 63 | 1 | |
| 1792 | 39 | ||
| 1794 | 11 | ||
| pDNA | LPEI | 200 | 2384 |
| 1611 | 100 | 3 | |
| 1821 | 502 | ||
| 1822 | 12 | ||
| 1730 | 100 | 324 | |
| 1824 | 122 | ||
| 1826 | 39 | ||
3.5 Investigations on cell viability and proliferation
Preceding screening experiments revealed that the outstanding potential of LAF–Stp carriers is often associated with reduced metabolic activity of transfected cells. While cells treated with HBG and LPEI exhibited unaltered cell proliferation, those transfected with polyplexes of the original xenopeptides 1611 and 1621 demonstrated a reduced confluency after a 24-hour incubation period (Fig. S10†). In contrast, cells treated with disulfide spacer-containing xenopeptide polyplexes showed similar confluency as the non-toxic controls HBG and LPEI, supporting favorable effects of the biodegradable carriers, which became only moderately evident by the metabolic activity assay thus far. In addition to the initial evaluation of cell viability with the results gleaned from CellTiter-Glo® assays, supplementary assays were performed to develop a deeper understanding about the influence of the LAF–Stp xenopeptides on cell viability. | ||
| Fig. 7 LDH release after transfection of HeLa cells with pDNA polyplexes (12Oc-U1-1:2 and 8Oc-B2-2:4 series) formed at N/P 18 at a dose of 100 ng pDNA per well (A and B) and with mRNA polyplexes (8Oc-B2-1:4 analogs) formed at N/P 24 at a mRNA dose of 63 ng per well (C). LDH release was detected at 8, 12, 16 and 24 hours after transfection by an enzymatic reaction with iodonitrotetrazoliumchlorid leading to formation a purple formazan product. Each polyplex was transfected as triplicate. Absorbance of the formazan product was determined at 490 nm. LDH release was calculated in relation to cells treated with lysis buffer (representing maximum LDH release). | ||
Intriguingly, no LDH release was detected at 8 hours, suggesting that the carriers do not induce immediate membrane damage upon cellular exposure, or shortly after endosomal escape which started after about 4 hours, as indicated by CLSM analysis (Fig. 6C). For 12Oc-U1-1:2-pDNA polyplexes lacking bio-degradable domains, i.e., 1611 and the spacer analog 1822, LDH release became detectable at 12 hours after transfection. While LDH release continued to rise for 1611 up to the 24 hours-mark, cells treated with the 1822 (12Oc-U1-1:2-spacer) exhibited a decline after reaching a peak at 16 hours. In contrast, for the bio-reducible polyplexes formed with 1821, no release of LDH was prompted, emphasizing the beneficial effect of the disulfide spacer (Fig. 7A). For the 8Oc-B2-2:4 analogs, LDH release was absent in case of the disulfide analog 1824, but to some extent measurable for polyplexes with the non-reducible 1730 and 1826 xenopeptides after 24 hours post-transfection (Fig. 7B). Notably, cells treated with mRNA polyplexes of the 1621 series demonstrated an LDH release profile comparable to those transfected with the structurally similar 1730 series, even though they delivered different cargo molecules. Within the 1621 series, the original xenopeptide exhibited the highest LDH release, and modification with disulfide spacers led to a moderate reduction in cytotoxicity, while the non-reducible spacer displayed no membrane damage at all (Fig. 7C). These findings support prior observations derived from the CellTiter-Glo® assay. By the LDH release assay, a disulfide dependent benefit on cell viability became evident at least for cells treated with pDNA polyplexes. Additionally, cell viability was improved for spacer-modified carriers in mRNA polyplexes, irrespective of reducible or non-reducible chemistry.
:4) or mRNA (8Oc-B2-1:4), cell growth was inhibited as observed by an about 8 to 20 hours shifted increase in confluency after transfection. This finding supports the hypothesis that toxicity occurs not directly upon polyplex exposure but after internalization and probably endosomal escape, starting after 4 hours incubation for 1621 and 1792. The xenopeptide spacer analogs of 1621 showed also a decelerated, almost stagnated cell proliferation. Overall, the proliferation of cells incubated with pDNA polyplexes formed with disulfide-analogs of either 8Oc-B2-2:4 or 12Oc-U1-1:2 occurred in a similar manner as HBG-treated control cells with a constant increase. An exception is the 1792 mRNA polyplex (8Oc-B2-1:4-ssbb2), which, in contrast to other disulfide analogs, showed a decrease in confluence at 8 hours post transfection, but recovered after 16 hours, highlighting the results of the LDH assay. Second, at 24 hours after transfection, viable cells were counted by flow cytometry (Fig. 8B). The analysis revealed an about three to four-fold increase of healthy cells for disulfide-spacer xenopeptide pDNA polyplexes formed with 12Oc-U1-1:2 or 8Oc-B2-2:4 carriers. For mRNA polyplexes, an about 10-fold increase in number of healthy cells was found upon transfection with 1792 (8Oc-B2-1:4-ssbb2). In all cases, a slight increase (approx. 2-fold) of viable cell numbers was detected for non-reducible spacer xenopeptide analogs of each topology, independent of the nucleic acid cargo.
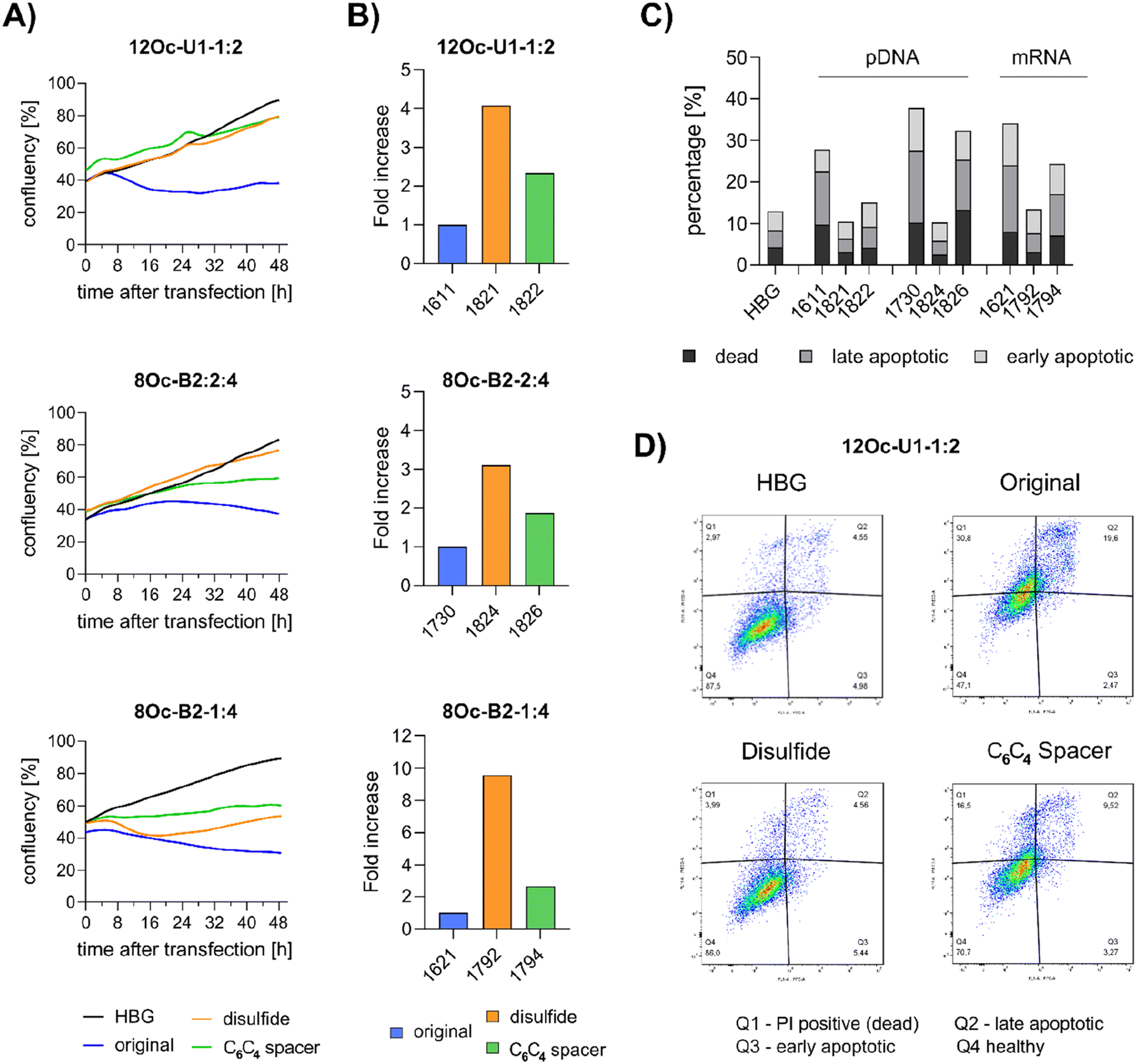 | ||
| Fig. 8 Cell proliferation after polyplex treatment. (A) Time-dependent monitoring of cell growth using Cell Watcher M (PHIO Scientific, Munich, Germany) over 48 hours after transfection of polyplexes. 12Oc-U1-1:2 and 8Oc-B2-2:4: pDNA polyplexes at N/P 18 with 100 ng pDNA per well for LAF polyplexes and 200 ng pDNA per well for LPEI polyplexes (N/P 6), 8Oc-B2-1:4: mRNA polyplexes at N/P 24 with a nucleic acid dose of 63 ng, compared to succPEI (w/w 4, 250 ng per well). Confluency of cells was determined automatically every 30 minutes. Black: HBG control; blue: original xenopeptide; orange: disulfide-spacer; green: non-reducible spacer xenopeptide. (B) Flow cytometer analysis of HeLa cells at 24 hours after transfection. Cells were treated with pDNA-polyplexes formed with analogs of 12Oc-U1-1:2 topology or 8Oc-B2-2:4 carriers at N/P 18 or mRNA polyplexes formed with 8Oc-B2-1:4 carriers at N/P 24. HeLa cells were analyzed for a constant time of 60 seconds and a flow rate of 170 μL min−1. Only DAPI-negative cells were counted. (C and D) Annexin/PI assay on HeLa cells (40000 cells per well in a 24-well plate) at 24 hours after treatment with pDNA polyplexes formed with either 12Oc-U1-1:2-analogs or 8Oc-B2-2:4-analogs at N/P 18 in comparison to HBG-buffer treated cells (pDNA dose = 125 ng per well) or mRNA polyplexes formed with 8Oc-B2-1:4-analogs at N/P 24 in comparison to HBG-buffer treated cells (mRNA dose = 78 ng per well). (C) Overview of the complete data set. (D) Exemplarily dot plot of selected data set of 12Oc-U1-1:2 polyplexes at a pDNA dose of 125 ng per well. | ||
4 Conclusions
Three different classes of LAF–Stp xenopeptides with proven high efficacy for either pDNA or mRNA delivery were further optimized by inserting bioreducible disulfide spacers and isosteric non-reducible spacers. Interestingly, both types of linkages were found to be effective in modulating transfection efficacy and cellular metabolic responses. The initial physico-chemical and biological screening highlighted the differing demands of either pDNA or mRNA cargo on the xenopeptide Stp/LAF ratio, sequence and topology for polyplex performance. The potency of xenopeptides and their effects on cellular metabolic activity were also found to strongly depend on the selected cell lines. Favorable individual xenopeptide/nucleic acid cargo/target cell combinations were identified for five different cancer cell lines, including low passage number human colorectal carcinoma cells. In some cases, high transfection efficacy was strictly coupled with cytotoxicity at higher dose; modifications relieving toxicity also abolished potency of transfection, suggesting an underlying toxic delivery mechanism. In other cases, inserted spacers improved metabolic cell activity but also maintained or even improved transfection activity. Closer analysis revealed topology-dependent differences between unmodified and spacer-modified carriers regarding cellular association, uptake, dependence on endosomal acidification, and expression kinetics. Spacer xenopeptide based polyplexes showed reduced post-transfection cell membrane damage as detected by LDH release assays. Time-resolved cell growth monitoring detected post-transfection inhibition of cellular proliferation with original LAF xenopeptides and apoptotic events that could be attenuated by spacer integration. Thus, this study gained insights in mechanisms of potent LAF–Stp xenopeptide carriers and options for improving their biocompatibility, thereby paving the way for the development of safe and effective nucleic acid delivery systems.Author contributions
Conceptualization, RCS and EWa; methodology, RCS, ST, EWe, MH; investigation, RCS, ST, EWe, JS, PF, MH, EWa; data curation, RCS, ST, EWe, EWa; formal analysis, RCS, ST, EWe; validation, RCS, ST, EWe, MH, EWa; visualization, RCS, ST, EWe, MH, EWa; writing – original draft, RCS, EWa; writing – review and editing, RCS, ST, EWe, EWa; funding acquisition, project administration, resources, supervision, EWa. All authors have read and agreed to the submitted version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge support by the German Research Foundation (DFG) SFB1032 (project-ID 201269156) sub-project B4 (to E. Wa.), and BMBF Cluster for Future ‘CNATM – Cluster for Nucleic Acid Therapeutics Munich’ project-ID 03ZU1201AA (to E. Wa.). We thank Melina Grau, Lun Peng and Janin Germer for providing previously established transfection reagents, Melina Grau and Tobias Burghardt for MALDI measurements, Simone Berger, Janin Germer and Mina Yazdi for scientific discussions, Wolfgang Rödl and Olga Brück for technical and organizational support.Notes and references
- P. L. Felgner, Y. Barenholz, J. P. Behr, S. H. Cheng, P. Cullis, L. Huang, J. A. Jessee, L. Seymour, F. Szoka, A. R. Thierry, E. Wagner and G. Wu, Hum. Gene Ther., 1997, 8, 511–512 CrossRef CAS.
- P. R. Cullis and M. J. Hope, Mol. Ther., 2017, 25, 1467–1475 CrossRef CAS.
- J. Witten, Y. Hu, R. Langer and D. G. Anderson, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2307798120 CrossRef CAS.
- C. A. Tsuchida, K. M. Wasko, J. R. Hamilton and J. A. Doudna, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2307796121 CrossRef CAS.
- S. Berger, U. Lächelt and E. Wagner, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2307799120 CrossRef CAS PubMed.
- D. W. Pack, A. S. Hoffman, S. Pun and P. S. Stayton, Nat. Rev. Drug Discovery, 2005, 4, 581–593 CrossRef CAS.
- K. Miyata, M. Oba, M. Nakanishi, S. Fukushima, Y. Yamasaki, H. Koyama, N. Nishiyama and K. Kataoka, J. Am. Chem. Soc., 2008, 130, 16287–16294 CrossRef CAS.
- C. Scholz and E. Wagner, J. Controlled Release, 2012, 161, 554–565 CrossRef CAS.
- C. Goncalves, S. Akhter, C. Pichon and P. Midoux, Mol. Pharm., 2016, 13, 3153–3163 CrossRef CAS PubMed.
- U. Lächelt and E. Wagner, Chem. Rev., 2015, 115, 11043–11078 CrossRef.
- R. Kumar, C. F. Santa Chalarca, M. R. Bockman, C. V. Bruggen, C. J. Grimme, R. J. Dalal, M. G. Hanson, J. K. Hexum and T. M. Reineke, Chem. Rev., 2021, 121, 11527–11652 CrossRef CAS.
- O. Boussif, F. Lezoualc'h, M. A. Zanta, M. D. Mergny, D. Scherman, B. Demeneix and J. P. Behr, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 7297–7301 CrossRef CAS PubMed.
- P. Neuberg and A. Kichler, Adv. Genet., 2014, 88, 263–288 CAS.
- L. Wightman, R. Kircheis, V. Rossler, S. Carotta, R. Ruzicka, M. Kursa and E. Wagner, J. Gene Med., 2001, 3, 362–372 CrossRef CAS PubMed.
- M. A. Gosselin, W. Guo and R. J. Lee, Bioconjugate Chem., 2001, 12, 989–994 CrossRef CAS PubMed.
- D. Fischer, Y. Li, B. Ahlemeyer, J. Krieglstein and T. Kissel, Biomaterials, 2003, 24, 1121–1131 CrossRef CAS PubMed.
- M. Thomas, Q. Ge, J. J. Lu, J. Chen and A. M. Klibanov, Pharm. Res., 2005, 22, 373–380 CrossRef CAS.
- J. Kloeckner, S. Bruzzano, M. Ogris and E. Wagner, Bioconjugate Chem., 2006, 17, 1339–1345 CrossRef CAS.
- M. Breunig, U. Lungwitz, R. Liebl and A. Goepferich, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 14454–14459 CrossRef CAS.
- A. Zintchenko, A. Philipp, A. Dehshahri and E. Wagner, Bioconjugate Chem., 2008, 19, 1448–1455 CrossRef CAS.
- V. Russ, H. Elfberg, C. Thoma, J. Kloeckner, M. Ogris and E. Wagner, Gene Ther., 2008, 15, 18–29 CrossRef CAS PubMed.
- V. Knorr, M. Ogris and E. Wagner, Pharm. Res., 2008, 25, 2937–2945 CrossRef CAS.
- H. Yu, V. Russ and E. Wagner, AAPS J., 2009, 11, 445–455 CrossRef CAS.
- M. Chiper, N. Tounsi, R. Kole, A. Kichler and G. Zuber, J. Controlled Release, 2017, 246, 60–70 CrossRef CAS.
- F. Freitag and E. Wagner, Adv. Drug Delivery Rev., 2021, 168, 30–54 CrossRef CAS PubMed.
- D. Schaffert, N. Badgujar and E. Wagner, Org. Lett., 2011, 13, 1586–1589 CrossRef CAS PubMed.
- D. Schaffert, C. Troiber, E. E. Salcher, T. Fröhlich, I. Martin, N. Badgujar, C. Dohmen, D. Edinger, R. Klager, G. Maiwald, K. Farkasova, S. Seeber, K. Jahn-Hofmann, P. Hadwiger and E. Wagner, Angew. Chem., Int. Ed., 2011, 50, 8986–8989 CrossRef CAS.
- C. Scholz, P. Kos, L. Leclercq, X. Jin, H. Cottet and E. Wagner, ChemMedChem, 2014, 9, 2104–2110 CrossRef CAS.
- U. Lächelt, P. Kos, F. M. Mickler, A. Herrmann, E. E. Salcher, W. Rodl, N. Badgujar, C. Brauchle and E. Wagner, Nanomedicine, 2014, 10, 35–44 CrossRef PubMed.
- S. Thalmayr, M. Grau, L. Peng, J. Pöhmerer, U. Wilk, P. Folda, M. Yazdi, E. Weidinger, T. Burghardt, M. Hohn, E. Wagner and S. Berger, Adv. Mater., 2023, 35, e2211105 CrossRef.
- F. Haase, J. Pöhmerer, M. Yazdi, M. Grau, Y. Zeyn, U. Wilk, T. Burghardt, M. Hohn, C. Hieber, M. Bros, E. Wagner and S. Berger, Eur. J. Pharm. Biopharm., 2024, 194, 95–109 CrossRef CAS PubMed.
- S. M. Moghimi, P. Symonds, J. C. Murray, A. C. Hunter, G. Debska and A. Szewczyk, Mol. Ther., 2005, 11, 990–995 CrossRef CAS.
- A. Hall, U. Lächelt, J. Bartek, E. Wagner and S. M. Moghimi, Mol. Ther., 2017, 25, 1476–1490 CrossRef CAS PubMed.
- J. L. Coll, P. Chollet, E. Brambilla, D. Desplanques, J. P. Behr and M. Favrot, Hum. Gene Ther., 1999, 10, 1659–1666 CrossRef CAS.
- G. Grandinetti, N. P. Ingle and T. M. Reineke, Mol. Pharm., 2011, 8, 1709–1719 CrossRef CAS.
- G. Grandinetti and T. M. Reineke, Mol. Pharm., 2012, 9, 2256–2267 CrossRef CAS.
- G. Grandinetti, A. E. Smith and T. M. Reineke, Mol. Pharm., 2012, 9, 523–538 CrossRef CAS.
- C. Scholz, P. Kos, L. Leclercq, X. Jin, H. Cottet and E. Wagner, ChemMedChem, 2014, 9, 2104–2110 CrossRef CAS PubMed.
- A. Hall, J. Bartek, E. Wagner, U. Lächelt and S. M. Moghimi, J. Controlled Release, 2023, 361, 115–129 CrossRef CAS.
- H. Uchida, K. Itaka, T. Nomoto, T. Ishii, T. Suma, M. Ikegami, K. Miyata, M. Oba, N. Nishiyama and K. Kataoka, J. Am. Chem. Soc., 2014, 136, 12396–12405 CrossRef CAS.
- M. L. Forrest, J. T. Koerber and D. W. Pack, Bioconjugate Chem., 2003, 14, 934–940 CrossRef CAS.
- V. Knorr, V. Russ, L. Allmendinger, M. Ogris and E. Wagner, Bioconjugate Chem., 2008, 19, 1625–1634 CrossRef CAS.
- J. Liu, J. Chang, Y. Jiang, X. Meng, T. Sun, L. Mao, Q. Xu and M. Wang, Adv. Mater., 2019, 31, e1902575 CrossRef PubMed.
- Z. Shen, C. Liu, Z. Wang, F. Xie, X. Liu, L. Dong, X. Pan, C. Zeng and P. G. Wang, Pharmaceutics, 2023, 15, 477 CrossRef CAS.
- X. Chen, J. Yang, H. Liang, Q. Jiang, B. Ke and Y. Nie, J. Mater. Chem. B, 2017, 5, 1482–1497 RSC.
- H. Liang, A. Hu, X. Chen, R. Jin, K. Wang, B. Ke and Y. Nie, J. Mater. Chem. B, 2019, 7, 915–926 RSC.
- J. Guo, T. Wan, B. Li, Q. Pan, H. Xin, Y. Qiu and Y. Ping, ACS Cent. Sci., 2021, 7, 990–1000 CrossRef CAS.
- Y. Wang, B. Ma, A. A. Abdeen, G. Chen, R. Xie, K. Saha and S. Gong, ACS Appl. Mater. Interfaces, 2018, 10, 31915–31927 CrossRef CAS PubMed.
- G. Chen, B. Ma, Y. Wang and S. Gong, ACS Appl. Mater. Interfaces, 2018, 10, 18515–18523 CrossRef CAS.
- Z. Chen, Y. Tian, J. Yang, F. Wu, S. Liu, W. Cao, W. Xu, T. Hu, D. J. Siegwart and H. Xiong, J. Am. Chem. Soc., 2023, 145, 24302–24314 CrossRef CAS.
- P. M. Klein, S. Reinhard, D. J. Lee, K. Müller, D. Ponader, L. Hartmann and E. Wagner, Nanoscale, 2016, 8, 18098–18104 RSC.
- A. Krhac Levacic, S. Berger, J. Müller, A. Wegner, U. Lächelt, C. Dohmen, C. Rudolph and E. Wagner, J. Controlled Release, 2021, 339, 27–40 CrossRef CAS.
- P. M. Klein and E. Wagner, Antioxid. Redox Signal., 2014, 21, 804–817 CrossRef CAS.
- S. Berger, A. Krhac Levacic, E. Hörterer, U. Wilk, T. Benli-Hoppe, Y. Wang, O. Özturk, J. Luo and E. Wagner, Biomacromolecules, 2021, 22, 1282–1296 CrossRef CAS.
- Y. Lin, U. Wilk, J. Pöhmerer, E. Hörterer, M. Höhn, X. Luo, H. Mai, E. Wagner and U. Lächelt, Small, 2023, 19, e2205318 CrossRef.
- Y. Rui, D. R. Wilson, S. Y. Tzeng, H. M. Yamagata, D. Sudhakar, M. Conge, C. A. Berlinicke, D. J. Zack, A. Tuesca and J. J. Green, Sci. Adv., 2022, 8, eabk2855 CrossRef CAS PubMed.
- M. Lyu, M. Yazdi, Y. Lin, M. Höhn, U. Lächelt and E. Wagner, ACS Biomater. Sci. Eng., 2024, 10, 99–114 CrossRef CAS PubMed.
- B. Vecsey-Semjen, K. F. Becker, A. Sinski, E. Blennow, I. Vietor, K. Zatloukal, H. Beug, E. Wagner and L. A. Huber, Oncogene, 2002, 21, 4646–4662 CrossRef CAS.
- L. Hartmann, S. Häfele, R. Peschka-Süss, M. Antonietti and H. G. Börner, Macromolecules, 2007, 40, 7771–7776 CrossRef CAS.
- A. Meister, J. Biol. Chem., 1988, 263, 17205–17208 CrossRef CAS PubMed.
- F. Q. Schafer and G. R. Buettner, Free Radicals Biol. Med., 2001, 30, 1191–1212 CrossRef CAS.
- S. Boeckle, E. Wagner and M. Ogris, J. Gene Med., 2005, 7, 1335–1347 CrossRef CAS.
- C. Plank, B. Oberhauser, K. Mechtler, C. Koch and E. Wagner, J. Biol. Chem., 1994, 269, 12918–12924 CrossRef CAS.
- M. Ogris, R. C. Carlisle, T. Bettinger and L. W. Seymour, J. Biol. Chem., 2001, 276, 47550–47555 CrossRef CAS PubMed.
- J. Pelisek, L. Gaedtke, J. DeRouchey, G. F. Walker, S. Nikol and E. Wagner, J. Gene Med., 2006, 8, 186–197 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr01357c |
| This journal is © The Royal Society of Chemistry 2024 |