
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficient plasmonic water splitting by heteroepitaxial junction-induced faceting of gold nanoparticles on an anatase titanium(IV) oxide nanoplate array electrode†
Shin-ichi
Naya
a,
Yoko
Morita
b,
Hisashi
Sugime
 bc,
Tetsuro
Soejima
bc,
Musashi
Fujishima
bc and
Hiroaki
Tada
*d
bc,
Tetsuro
Soejima
bc,
Musashi
Fujishima
bc and
Hiroaki
Tada
*d
aEnvironmental Research Laboratory, Kindai University, 3-4-1, Kowakae, Higashi-Osaka, Osaka 577-8502, Japan. E-mail: shinichi.naya@itp.kindai.ac.jp
bGraduate School of Science and Engineering, Kindai University, 3-4-1, Kowakae, Higashi-Osaka, Osaka 577-8502, Japan
cDepartment of Applied Chemistry, Faculty of Science and Engineering, Kindai University, 3-4-1, Kowakae, Higashi-Osaka, Osaka 577-8502, Japan
dInstitutes of Innovation for Future Society, Nagoya University, Furo-cho, Chikusa-ku, Nagoya, Aichi 464-8603, Japan. E-mail: hiroaki.tada@mirai.nagoya-u.ac.jp
First published on 12th June 2024
Abstract
Plasmonic photocatalysts represented by gold nanoparticle (NP)-loaded titanium(IV) oxide (Au/TiO2) can be promising solar-to-fuel converters by virtue of their response to visible-to-near infrared light. Hitherto, Au/rutile (R)-TiO2 has been recognized as exhibiting photocatalytic activity higher than that of Au/anatase (A)-TiO2. Herein, we demonstrate that the high potential of A-TiO2 as the Au NP support can be brought out through atomic level interface control. Faceting of Au NPs is induced by a heteroepitaxial junction on an A-TiO2(001) nanoplate array (Au/A-TiO2 NPLA). Photoexcitation towards the Au/A-TiO2 NPLA electrode generates current for the water oxidation reaction at λ < 900 nm with a maximum efficiency of 0.39% at λ = 600 nm, which is much larger than the values reported so far for the usual electrodes. The striking activity of the Au/A-TiO2 NPLA electrode was rationalized using a potential-dependent Fowler model. This study presented a novel approach for developing solar-driven electrodes for green and sustainable fuel production.
Introduction
An ultimate goal of science and technology is the construction of artificial photosynthesis systems with water splitting as a benchmark.1,2 Photoelectrochemical (PEC) water splitting assumes an advantage over the photocatalytic process using particles because of the inhibition of the back reaction and safety due to the spatial separation of the reaction fields where hydrogen (H2) and oxygen (O2) are generated. The key to achieving efficient PEC water splitting is the development of highly active photoanodes for the water oxidation reaction (WOR) with large activation energy.3 In a typical n-TiO2 photoanode, the performance is severely limited by the slow kinetics of the holes with a diffusion length of as small as ∼10 nm (ref. 4) and its response only to UV light accounting for ∼3% of the solar energy. In view of effective solar energy utilization, plasmonic photocatalysts represented by gold nanoparticle-loaded TiO2 (Au/TiO2) have recently attracted much interest on account of their wide spectral response from visible to near-infrared light.5–16 Also, the problem of sluggish hole transport in n-type semiconductor photoanodes can be eliminated in the WOR using the Au/TiO2 plasmonic electrode because water is oxidized by the holes generated in Au NPs via localized surface plasmon resonance (LSPR) excitation without requiring their diffusion. While intensive efforts have been devoted to increase the incident photon-to-current efficiency (IPCE) of the Au/TiO2 electrode through several approaches, such as Au–Ag alloying,17 lithium intercalation into TiO2,18 and loading promoters for the WOR,19,20 it is also a great challenge in materials sciences to enhance the performance of the prototype Au/TiO2 electrode itself for underpinning the development toward the practical applications in solar-to-fuel conversion. However, in the usual Au/TiO2 plasmonic electrode systems, the incident photon-to-current efficiency (IPCE) for the WOR remains below ∼0.03% (Table S1†).18,21–23 The IPCE is determined by the product of efficiencies in a series of physical processes including light harvesting, hot electron transfer (HET) from Au NPs to TiO2, charge separation, and the chemical process or WOR.24 All these processes can be affected by various factors including the size25 and shape26 of Au NPs, and the crystal form of TiO2.27 In particular, the IPCE for the WOR by the Au/TiO2 plasmonic electrode sharply increases when the Au particle size (dAu) is smaller than 10 nm.25 In addition, recent experimental and theoretical studies have indicated that strong electronic coupling between an electron acceptor (TiO2) and electron donors such as plasmonic metal NPs28,29 and CdS quantum dots30 through intimate contact can expedite the photoinduced interfacial electron transfer and extend the response light wavelength. Furthermore, the IPCEs of nanostructured TiO2 electrodes such as the nanotube array (NTA) and nanowire array (NWA), which are one to two orders magnitude greater than that of the conventional mesoporous (mp)-TiO2 electrode consisting of TiO2 nanocrystals,21–23 point to the importance of effective electron transport to the electron collecting electrode on the order of microns in addition to the charge separation via the space charge layer formed by the Schottky junction between Au NPs and TiO2.31 Since anatase (A)-TiO2 possesses electron mobility much larger than rutile (R)-TiO2,32 A-TiO2 films with large surface areas have been favourably used as the photoanodes of dye-sensitized solar cells and perovskite solar cells since their invention.33,34 Strangely, the IPCE for the WOR of Au/A-TiO2 photoanodes remains at a level comparable to that of Au/R-TiO2 photoanodes.21–23 On the other hand, in nanoparticulate Au/TiO2 plasmonic photocatalyst systems, Au/R-TiO2 exhibits much higher photocatalytic activity than Au/A-TiO2 for various reactions35–40 including the WOR.27 Recently, the superiority of Au/R-TiO2 has been found to originate from the (111)Au//(110)R-TiO2 heteroepitaxial (HEPI) junction-induced faceting of Au NPs facilitating the physical processes in the reaction41 and the increase in the peripheral distance of Au NPs at the interface with TiO2 or the catalytically active sites for the WOR.27 Thus, if faceted Au NPs with dAu < 10 nm can be formed on A-TiO2 by overcoming the general tendency to be hemispherical to minimize the surface area, the enhancement in the IPCE of the Au/A-TiO2 plasmonic electrode for the PEC WOR is highly expected.Here, we show that truncated octahedral (t-Oh) Au NPs with dAu < 10 nm can be formed on the A-TiO2 nanoplate array (NPLA) electrode with dominant (001) surfaces through a HEPI junction with the (111)Au//(001)A-TiO2 orientation, and this t-Oh-Au/A-TiO2 NPLA electrode exhibits activity for the PEC WOR that is much higher than that exhibited by hemispherical (HS)-Au/A-TiO2 NPLA and t-Oh Au/R-TiO2 NWA ones. Moreover, the reason is discussed on the basis of experimental and theoretical simulation results.
Experimental
Materials
Fluorine-doped tin(IV) oxide film-coated glass (FTO, TEC7) and titanium(IV) fluoride (TiF4) were purchased from Aldrich. Tetrabutyl orthotitanate (Ti(OC4H9)4 > 97.0%), hydrochloric acid (HCl > 98.0%), sodium hydroxide (NaOH > 97.0%), hydrogen tetrachloroaurate(III) tetrahydrate (HAuCl4·4H2O > 99%), urea (CH4N2O > 99.0%), and sodium borohydride (NaBH4 > 92.0%) were purchased from Kanto Chemical Co. All chemicals were used as-received without further purification.Synthesis of the anatase TiO2 nanoplate array
The anatase TiO2 nanoplate array (A-TiO2 NPLA) was synthesized by following reported procedures with slight modification.42 TiF4 (124 mg) was dissolved in a solution of Ti(OBu)4 (0.17 mL) in 6 M HCl (10 mL) and stirred at room temperature for 15 min. The solution was poured into a Teflon container (volume 25 mL), and FTO (3 cm × 2 cm, 3 pieces) was immersed in the solution. The Teflon container was sealed in a stainless-steel cylinder and heated at 423 K for 18 h. The resulting sample was washed with 0.1 M aqueous NaOH, distilled water, and acetone, and dried in vacuo at room temperature to obtain the A-TiO2 NPLA.Synthesis of the rutile TiO2 nanowire array
According the literature,43 the rutile TiO2 nanowire array (A-TiO2 NWA) was hydrothermally synthesized. A solution of Ti(OBu)4 (0.40 mL) in 6 M HCl (10 mL) was poured into a Teflon container (volume 25 mL), and FTO (3 cm × 2 cm, 3 pieces) was immersed in the solution. The Teflon container was sealed in a stainless-steel cylinder and heated at 423 K for 8 h. The resulting sample was washed with distilled water and acetone, and dried in vacuo at room temperature. The sample was calcined at 773 K for 8 h in air to obtain the R-TiO2 NWA.Au nanoparticle loading
Au nanoparticles were loaded on a TiO2 electrode by the modified deposition–precipitation method using urea as the neutralizer.44 Urea (146 mg) was added to an aqueous solution of HAuCl4 (2.43 mM, 10 mL). The TiO2 electrode was immersed in the solution and heated at 353 K for 18 h. The resulting sample was washed with distilled water and acetone, and dried in vacuo at room temperature to obtain an Au3+-adsorbed TiO2 electrode. The sample was calcined at 673 K for 1 h in air to obtain the Au/TiO2 electrode. Also, Au NPs were loaded on the TiO2 electrode by chemical reduction. The Au3+-adsorbed TiO2 electrode was immersed in an ethanol solution of NaBH4 (1 mM), and left at room temperature for 5 min. The resulting sample was washed with distilled water and acetone. The chemical reduction procedures were repeated 3 times to completely reduce the adsorbed Au3+ ions.Characterization
Scanning electron microscopy (SEM) images and energy dispersive spectroscopic (EDS) elemental mapping were conducted using a Hitachi SU8230 instrument at an applied voltage of 20 kV. Raman spectra were recorded using a Raman spectrometer (JASCO FP-1000) with a green laser (532 nm) as an excitation source. Transmission electron microscopy (TEM) images were obtained using a JEOL JEM-2100F instrument at an applied voltage of 200 kV. X-ray diffraction (XRD) patterns were recorded at 40 kV and 100 mA using a Rigaku SmartLab X-ray diffractometer. The specific surface area was quantified by means of a Micromeritics automatic surface area and porosimetry analyzer (TriStar 3000, Shimadzu) via nitrogen adsorption–desorption isotherms at 77 K. The samples were measured after degassing at 423 K for 1 h under vacuum. To quantify the Au loading amount, Au NPs were dissolved in aqua regia, and the concentration was determined by inductively coupled plasma spectroscopy (iCAP7600, Shimadzu). UV-Vis-NIR absorption spectra were obtained by the diffuse reflectance method using a UV-2600 spectrometer (Shimadzu) with an integrating sphere unit (Shimadzu, ISR-2600Plus). As the reference, BaSO4 was used for the reflectance (R∞). The Kubelka–Munk function [F(R∞) = (1 − R∞)2/2R∞] was used for expressing the relative absorption coefficient. X-ray photoelectron (XP) spectra were measured using a Kratos Axis Nova X-ray photoelectron spectrometer with applied voltage of 15 kV and current of 10 mA using Al Kα as the X-ray source. For the energy reference, the peak of C1s (284.6 eV) was used.Photoelectrochemical measurements
PEC measurements were carried out using the standard three-electrode electrochemical cell with the structure of Au/A-TiO2 NPLA/FTO (working electrode) | 0.1 M NaClO4 aqueous solution | Ag/AgCl (reference electrode) | glassy carbon (counter electrode). The active area of the working electrode was 4 cm2 (2 × 2 cm). The electrolyte solution was deaerated by Ar bubbling for 30 min. The working electrode was illuminated using a solar simulator (PEC-L12, Peccell Tech. Inc.) through an optical filter Y-45 (AM 1.5, 1 sun, λex > 430 nm). The linear sweep voltammetry measurements were performed using a galvanostat/potentiostat (HZ-7000, Hokuto Denko). To evaluate the incident photon-to-current efficiencies (IPCEs), the working electrode on the same cells was irradiated by the monochromatic light of a xenon lamp with a monochromator (full width at half-maximum, 10 nm) (HM-5, JASCO). The IPCEs were calculated using eqn (1):| IPCE[%] = (Jph(E)NAhc/IFλ) × 100 | (1) |
Also, the solar-to-current efficiency (SCE) was calculated using eqn (2):
| SCE[%] = ∫IPCE(λ) × Is(λ)dλ/∫Is(λ)dλ | (2) |
PEC impedance spectroscopy (PECIS)
PECIS measurements were carried out using the same cells under the illumination of simulated sunlight (λex > 430 nm, 1 sun). Nyquist plots were obtained from EIS using a frequency response analyzer (HZA-FRA1, Hokuto Denko) built into the galvanostat/potentiostat. The measurements were carried out by applying a 10 mV AC sinusoidal signal over the frequency range between 100 mHz and 100 kHz. Charge transfer resistance (Rct) values were estimated by a curve fitting for Nyquist plots assuming an RC-equivalent circuit.Finite difference time domain (FDTD) simulations
Simulation models consist of an Au hemisphere (HS-Au) and an Au truncated octahedron (t-Oh-Au) loaded on a TiO2 slab (650 × 650 × 50 nm3), respectively. Au particle sizes were assessed on the basis of the experimental values obtained from TEM observations. Complex refractive indices taken from the literature were used to reproduce the optical responses of these models. The light scattering behaviour of the models was simulated using a total-field scattered-field (TFSF) source. An x-polarized plane wave with wavelength from 300 to 1200 nm (f = 250–1000 THz) was generated to illuminate the top of the Au NPs (the zenith of the HS NP or the top plane of the t-Oh NP) from the z-axis direction perpendicular to the TiO2 slab. E-field monitors were placed on the xy plane (z = 0) and the xz plane (y = 0) to collect local electric field images. Considering the balance between computational cost and accuracy, mesh refinement and perfectly matched layer (PML) with symmetric and/or anti-symmetric boundary conditions were imposed on the models. All of the FDTD simulations were carried out with an Ansys Lumerical FDTD simulation program package.Results
TiO2 films were hydrothermally grown on fluorine-doped tin oxide (FTO) substrates from a hydrochloric acid solution of Ti(OBu)4 in the absence and the presence of TiF4 at 423 K. X-ray diffraction (XRD) patterns of the samples synthesized without and with TiF4 exhibited peaks at 2θ = 36.1° and 25.3° due to diffraction from the (101) planes of R-TiO2 and A-TiO2, respectively (Fig. 1a). The abnormally strong (101) diffraction peaks of R-TiO2 and A-TiO2 indicate that TiO2 grows with high orientation with respect to the FTO surface in each sample. FTO has weak Raman signals at 634.3 cm−1 and 770.6 cm−1 assignable to the A1g and B2g modes, respectively (Fig. 1b).45 The three signals for the sample synthesized without TiF4 at 234.6 cm−1, 445.6 cm−1, and 609.6 cm−1 are assigned to the B1u + Eu, Eg, and A1g modes of R-TiO2, respectively.46 In the spectrum of the sample synthesized with TiF4, four signals are observed at 145.8 cm−1, 406.0 cm−1, 513.7 cm−1, and 630.8 cm−1 due to the Eg, B1g, A1g, and Eg modes of A-TiO2, respectively.46 Clearly, the addition of TiF4 to the reaction solution completely changes the crystal form of TiO2 from rutile to anatase.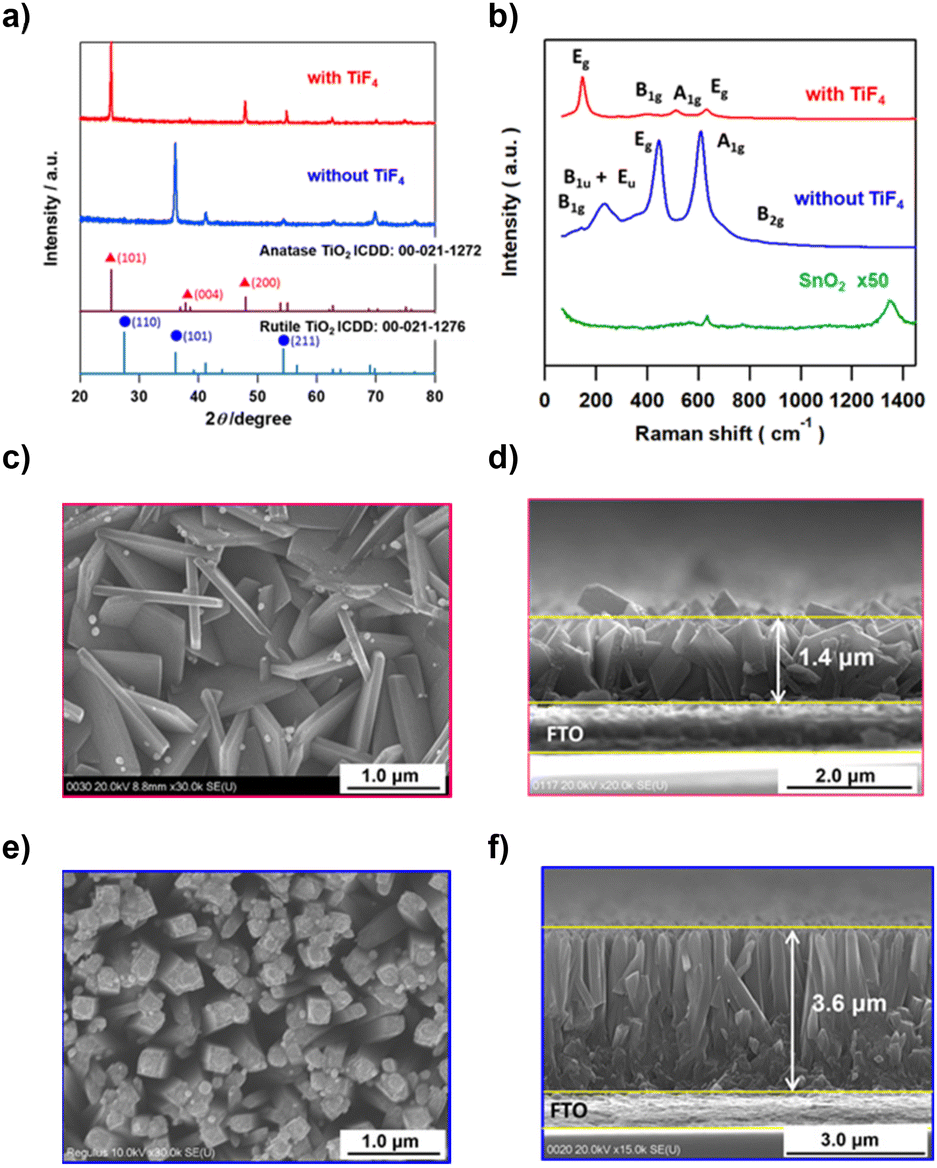 | ||
| Fig. 1 XRD patterns (a) and Raman spectra (b) of the samples hydrothermally synthesized without and with TiF4. Top view (c) and side view (d) SEM images of the DP-Au/A-TiO2 NPLA. Top view (e) and side view (f) SEM images of the DP-Au/R-TiO2 NWA. | ||
Au NPs were loaded on A-TiO2 by the deposition–precipitation (DP) and chemical reduction (CR) methods. The top view (left) and side view (right) scanning electron microscopy (SEM) images of the samples show that the A-TiO2 NPLA (Fig. 1c and d) and the R-TiO2 nanowire array (R-TiO2 NWA, Fig. 1e and f) are grown vertically or obliquely on the FTO films by the hydrothermal processes with and without TiF4, respectively. The A-TiO2 NPLA is composed of NPLs with length ∼1.4 μm and thickness ∼100 nm, while the dimensions of the R-TiO2 NWs are ∼3.6 μm in length and ∼200 nm in diameter. In the F− ion-free system, the Cl− ions are adsorbed on the R-TiO2(110) surface, and HEPI crystal growth from the FTO substrate occurs in the [001] direction to yield the R-TiO2 NWA.47 On the other hand, in the F− ion-added system, TiF4 with a sheet structure can be transformed into an A-TiO2 NPL through hydrolysis.42 During the growth of A-TiO2 NPLs, the 2D structure is maintained due to the lowering of the surface energy by the preferential adsorption of F− ions on the (001) facets of the A-TiO2 NPL.48,49
In order to study the effects of the Au NP shape, the TiO2 support, and the state of their interface on the activity of the Au/TiO2 plasmonic electrodes for the WOR, the size of Au NPs should be controlled to be approximately equal between samples because the activity is sensitive to NP size. As shown by the transmission electron microscopy (TEM) images, Au NPs are highly dispersed on the A-TiO2 NPLA with dAu of 7.6 ± 1.8 nm by the DP method (Fig. 2a) and 7.9 ± 1.6 nm by the CR method (Fig. S1†). In the high resolution (HR)-TEM image of the DP-Au/A-TiO2 NPL (Fig. 2b), the d-spacings of 1.84 Å and 2.37 Å observed in TiO2 are in agreement with the values of the A-TiO2(200) and (004) crystal planes, respectively. Also, the d-spacings of 2.07 Å and 2.33 Å in the Au NP are close to the values of the Au(100) and (111) crystal planes, respectively. Interestingly, faceted Au NPs with a truncated octahedral (t-Oh) shape are formed on A-TiO2 with an atomically commensurate interface between Au(111) and A-TiO2(001). As recently reported for the Au/R-TiO2 NWA,22 t-Oh Au NPs with dAu = 6.2 ± 1.2 nm are also formed on the (110) side walls of the R-TiO2 NW by the DP method (Fig. S2†). On the other hand, most Au NPs of a CR-Au/A-TiO2 NPL are near-hemispherical (HS) polyhedra (Fig. S1†), and the Au NP-TiO2 interface of the CR-Au/A-TiO2 NPL appears to be of lower quality at an atomic level than the interfaces of the DP-Au/A-TiO2 NPL and DP-Au/R-TiO2 NWA.
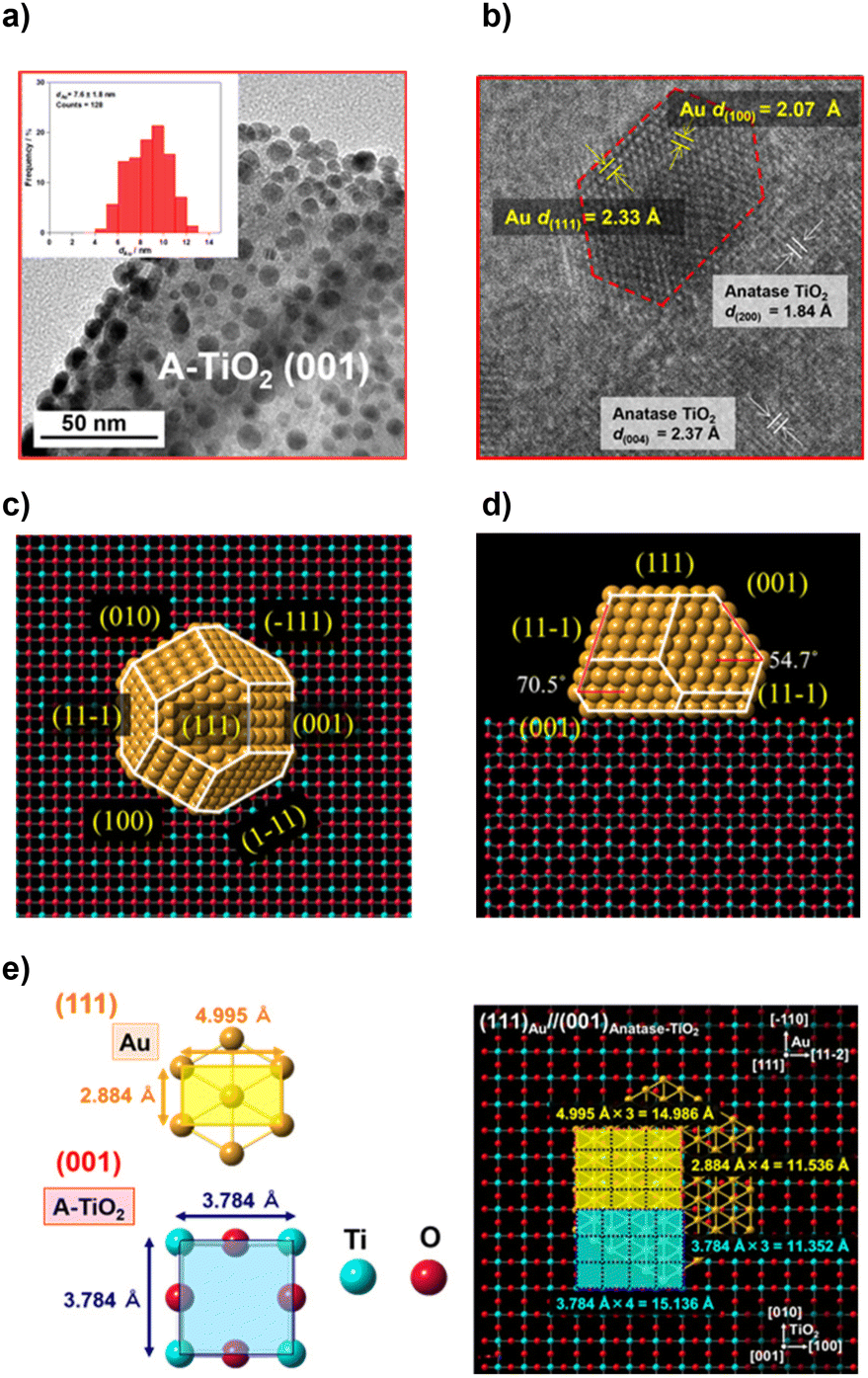 | ||
| Fig. 2 TEM (a) and HR-TEM (b) images of the DP-Au/A-TiO2 NPLA. Top view (c) and side view (d) of faceted Au NPs on A-TiO2(001) constructed by the Wulff theorem. (e) An interface junction model with the (111)Au//(001)A-TiO2 orientation. | ||
The equilibrium shape of Au NPs on the A-TiO2(001) plane was constructed to be a t-Oh by the Wulff theorem using the surface energies of Au(111) (0.72 J m−2), Au(100) (0.87 J m−2), A-TiO2(001) (0.90 J m−2), and the interface energy.50,51 The interface energy was estimated to be 1.23 J m−2 from the contact angle (θ ≈ 116°) using the Young equation (Fig. S3†). The top view (Fig. 2c) and side view (Fig. 2d) are very similar to those observed in the TEM image (Fig. 2a) and HR-TEM image (Fig. 2b), respectively. The 2D-rectangular lattices of Au(111) and A-TiO2(001) have the unit dimensions of 4.995 Å × 2.884 Å and 3.784 Å × 3.784 Å, respectively (Fig. 2e). Fourfold of the d-spacing of Au(−110) crystal planes (dAu(−110)) overlaps with threefold of dA-TiO2(010), and threefold of dAu(11−2) overlaps with fourfold of dA-TiO2(100) with small mismatches of +1.6% [{(2.884 Å × 4 − 3.784 Å × 3)/3.784 Å × 3} × 100] and −1.0% [{(4.995 Å × 3 − 3.784 Å × 4)/3.784 Å × 4} × 100], respectively, which strongly suggests the formation of the domain-matching epitaxial junction. In the Au/R-TiO2 NW system, a HEPI junction with the orientation of (111)Au//(110)R-TiO2 has been reported to provoke the faceting of Au NPs.41,52 In a similar manner, the faceting Au NPs can be induced by a domain-matching HEPI junction with the (111)Au//(001)A-TiO2 orientation. The HEPI junction with the same orientation has recently been reported in the system consisting of Au NPs and A-TiO2 nanosheets with dominant (001) surfaces and thickness of ∼5 nm.53
X-ray photoelectron (XP) spectra of the DP-Au/A-TiO2 NPLA, DP-Au/R-TiO2 NWA, and CR-Au/A-TiO2 NPLA have two signals at binding energies of 83.7 ± 0.1 eV and 87.4 ± 0.1 eV due to emission from the Au4f7/2 and 4f5/2 orbitals, respectively (Fig. S4†), which are somewhat smaller than the values for the bulk Au (83.9 eV and 87.6 eV).54 These results indicate that the Au NPs are in a metallic state in every Au/TiO2 system, and the interfacial electron transfer from TiO2 to Au NPs takes place to form the Schottky junction in the dark.31
In the Kubelka–Munk-transformed absorption spectra, the A-TiO2 NPLA and R-TiO2 NWA have the absorption edges of 390 nm and 410 nm, which are in agreement with the values for A-TiO2 and R-TiO2, respectively (Fig. 3a).55 When Au NPs were loaded on TiO2, the absorption due to the LSPR appears in the visible region. The absorption of the DP-Au/A-TiO2 NPLA as well as the DP-Au/R-TiO2 NWA is much stronger and broader than that of the CR-Au/A-TiO2 NPLA. Also, the peak wavelengths (λp) of the DP-Au/A-TiO2 NPLA (549 nm) and DP-Au/R-TiO2 NWA (538 nm) are shorter than that of the CR-Au/A-TiO2 NPLA (564 nm). The absorption spectra of CR-Au/A-TiO2 and DP-Au/A-TiO2 NPLAs were calculated for HS and t-Oh Au NPs placed on A-TiO2 employed as their models, respectively, by the finite difference time domain (FDTD) method (Fig. S5†). The spectra indicate that the faceting of Au NPs engenders the enhancement of the LSPR absorption, and the λp blueshifts with respect to the value for HS Au NPs. The strong absorption of the DP-Au/R-TiO2 NW with the (110) side walls has recently been attributed to the (111)Au//(110)R-TiO2 HEPI junction-induced faceting of Au NPs.41 The broadening of the absorption spectrum for the DP-Au/A-TiO2 NPLA is not well reproduced by the simulations. However, the simulated spectrum of t-Oh Au/A-TiO2 possesses several absorption peaks corresponding to different resonance modes (Fig. S6†), while only one resonance peak is observed in the spectrum of the HS-Au/A-TiO2. Thus, the extended LSPR absorption of the DP-Au/A-TiO2 NPLA and DP-Au/R-TiO2 NWA can result from the overlapping of each resonance peak broadened by the polydispersibility of the size and shape of Au NPs, and the significant electronic interaction between Au NPs and TiO2 through the HEPI junction.40,56
 | ||
| Fig. 3 (a) Kubelka–Munk-transformed absorption spectra of the DP-Au/A-TiO2 NPLA, CR-Au/A-TiO2 NPLA, and DP-Au/R-TiO2 NWA. (b) Current (J)–potential (E) curves in the dark (dotted lines) and under visible light irradiation (λex > 430 nm, solid lines). The current density was calculated using the apparent electrode surface area. (c) IPCE action spectra for the WOR with the solar spectrum for comparison (E = 0.8 V vs. RHE). (d) Nyquist plots for the plasmonic electrodes under visible light irradiation (λex > 430 nm). | ||
The current (J)–potential (E) curves of three-electrode PEC cells with the structure of the working electrode | 0.1 M NaClO4 aqueous solution | Ag/AgCl (reference electrode) | glassy carbon (GC, counter electrode) using the DP-Au/A-TiO2 NPLA, CR-Au/A-TiO2 NPLA, and DP-Au/R-TiO2-NWA as the working electrodes were measured (Fig. 3b). The electrode potential (E) is shown with respect to the reversible hydrogen electrode (RHE) below. In each system, current hardly flows in the dark at +0.4 V ≤ E ≤ +1.2 V. The illumination of visible light (AM-1.5, light intensity = 100 mW cm−2, λ > 430 nm) generates anodic current due to the WOR at E > +0.54 ± 0.02 V.25 Surprisingly, the DP-Au/A-TiO2 NPLA supplies photocurrent far exceeding that of the CR-Au/A-TiO2 NPLA and DP-Au/R-TiO2 NWA, whereas Au/A-TiO2 particles usually show plasmonic photocatalytic activity for the WOR that is much lower than the Au/R-TiO2 ones.25,41 The specific surface areas of the A-TiO2 NPLA and R-TiO2 NWA were determined to be 5.1 m2 g−1 and 13.6 m2 g−1, respectively, by the Brunauer–Emmett–Teller (BET) method. The photocurrent normalized by the actual surface area of the DP-Au/A-TiO2 NPLA reaches as much as ∼6 times that of the DP-Au/R-TiO2 NWA (Fig. S7†). Clearly, the HEPI junction-induced faceting of Au NPs engenders a drastic increase in the plasmonic WOR activity of Au/A-TiO2 to far surpass even that of the Au/R-TiO2 electrode.
The photocurrents of the DP-Au/A-TiO2 NPLA and DP-Au/R-TiO2 NWA plasmonic electrodes for the WOR were measured under the irradiation of monochromatic light with varying wavelengths (Fig. 3c). The photocurrent flows at λ < 900 nm in each electrode system, and the DP-Au/A-TiO2 NPLA electrode affords IPCEs much greater than those of the DP-Au/R-TiO2 NWA electrode in the whole wavelength region. The maximum IPCE of the former reaches 0.39% at λ = 600 nm, while the value of the latter is 0.18% in accordance with the reported value.22 Recently, a maximum IPCE of ∼0.06% at λ = 550 nm has been reported for the brookite (B)-TiO2 nanorod array electrode by selectively depositing Au NPs on the lateral {210} surfaces although the interface structure and shape of Au NPs are unclear.57 Also, each IPCE action spectrum well traces the absorption spectrum, indicating that the photocurrent mainly caused by the 5d–6sp interband excitation of Au NPs26,58 has to be further enhanced by the LSPR excitation. The redshift in the photocurrent peak of ∼50 nm with respect to the LSPR peak (λp) is ascribable to the large dielectric constant of water (80.2 at 293 K)59 since λp is proportional to (1 + 2εm)1/2, where εm is the dielectric constant of the surroundings.60 Furthermore, the broad IPCE action spectra of the DP-Au/A-TiO2 NPLA and DP-Au/R-TiO2 NWA electrodes well matches the solar spectrum to afford the solar-to-current efficiency at λ > 430 nm of 0.15% and 0.08%, respectively.
To gain insight into the origin for the high plasmonic photocatalytic activity of the DP-Au/A-TiO2 NPLA for the WOR, PEC impedance analysis was performed for the plasmonic electrodes with an equivalent circuit used for the analysis in which charge transfer resistance (Rct) coupled in parallel with the constant phase element (CFE) is connected in series with ohmic resistance (Rs) (Fig. 3D and Table S2†). In the Nyquist plots under the illumination of visible light (AM-1.5, light intensity = 100 mW cm−2, λ > 430 nm), the Rct corresponding to the diameter of the semicircle decreases in the order of CR-Au/A-TiO2 NPLA (2.4 × 105 Ω) ≫ DP-Au/R-TiO2 NWA (9.0 × 103 Ω) > DP-Au/A-TiO2 NPLA (1.3 × 103 Ω). Evidently, the rate of interfacial charge transfer between the plasmonic electrode and water increases in the order of CR-Au/A-TiO2 NPLA ≪ DP-Au/R-TiO2 NWA < DP-Au/A-TiO2 NPLA.
The local electric field was calculated for HS-Au(dAu = 7.9 nm)/A-TiO2 and t-Oh-Au(dAu = 7.6 nm)/A-TiO2 as the models for the CR-Au/A-TiO2 NPLA and DP-Au/A-TiO2 NPLA, respectively, by the 3D-FDTD method (Fig. 4a and b). In each case, Au NPs were placed on the xy plane of an A-TiO2 slab, and light was irradiated from the z direction perpendicular to the xy plane with the excited localized electromagnetic fields monitored at the xz and xy planes. In the HS-Au/A-TiO2 system, the electric field is uniformly distributed around the periphery of the HS-Au NPs at the interface with A-TiO2 (Fig. 4c). On the other hand, in the t-Oh-Au/A-TiO2 system, an intense electric field is generated around the corners and edges of t-Oh-Au NPs at the interface with A-TiO2 (Fig. 4d and Fig. S6†). The enhancement factor (EF) defined by the square of the ratio of the local electric field intensity to the electric field intensity of incident light was calculated (Table S3†). For the HS-Au/A-TiO2 system, the maximum EFxz of 2.5 × 103 and EFxy of 3.6 × 103 are obtained at λp = 702 nm. In the t-Oh-Au/A-TiO2 system, the maximum EFxz and EFxy value reaches 1.1 × 105 at λp = 650 nm, which is greater than the values for the HS-Au/A-TiO2 system by factors of 44 and 31, respectively. The calculations were further performed for t-Oh-Au(dAu = 6.2 nm)/R-TiO2 as the model for the DP-Au/R-TiO2 NWA (Fig. S8a†). An intense electric field localized near the corners and edges at the interface with R-TiO2 is induced by LSPR excitation of the faceted Au NPs (Fig. S8b†). The maximum EFxz and EFxy value of 1.2 × 105 at λp = 672 nm is comparable to the values of the t-Oh-Au/A-TiO2 system (Table S3†).
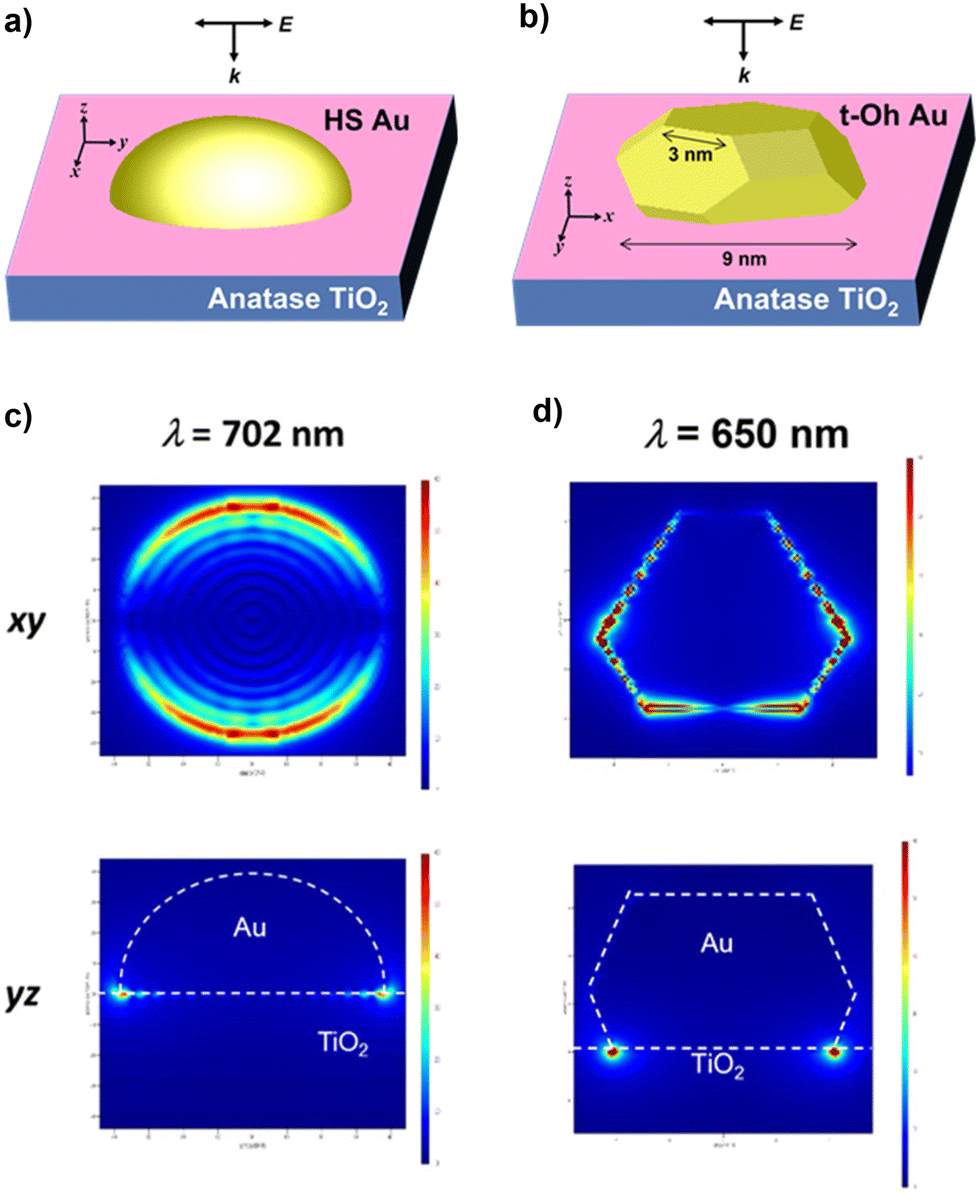 | ||
| Fig. 4 Local electric fields for HS-Au/A-TiO2 (a) and t-Oh-Au/A-TiO2 (b) calculated by the 3D-FDTD method. Local electric field distributions in HS-Au/A-TiO2 (c) and t-Oh-Au/A-TiO2 (d). | ||
Discussion
Truncated octahedral (t-Oh) Au NPs are formed on the large-area (110) side walls of R-TiO2 NWs with high probability by the DP method through the HEPI junction with the (111)Au//(110)R-TiO2 orientation.41 In a similar manner, faceting of Au NPs is induced on A-TiO2 NPLs with dominant (001) surfaces by the (111)Au//(001)A-TiO2 HEPI junction during the DP process with a heating temperature of 673 K. A recent in situ TEM study has also shown that t-Oh and round-shaped Au NPs are formed on A-TiO2(001) and (101) planes at 773 K, respectively.61 On the other hand, the room-temperature CR process produces HS-like Au NPs on A-TiO2 NPLs. Thus, preferential surface exposure of the TiO2 facets that can match the crystal lattice of Au(111) and heating at temperatures above 673 K are necessary for the formation of faceted Au NPs on TiO2 with a high-quality and large-area interface, and long peripheral distance of the Au NPs at the interface with TiO2. The Fermi energy of TiO2 (εF,TiO2) can be calculated using the equation of εF,TiO2 = εCBM + kT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(ND/NC), where εCBM is the conduction band (CB) minimum energy of TiO2, k is the Boltzmann constant, ND is the donor density of TiO2, and NC is the effective density of states near the CBM of TiO2. The εF,TiO2 value is estimated to be −5.1 ± 0.1 eV by substituting εCBM = −5.0 ± 0.1 eV (ref. 62) and NC = 2.5 × 1019 cm−3(ref. 63) for the usual ND value of 1017 cm−3. When the Au NP having a large work function (ΦW, Au = 5.31–5.47 eV)59 comes into direct contact with TiO2 (ΦW,TiO2 = 5.1 ± 0.1 eV), the interfacial electron transfer proceeds from TiO2 to the Au NP until their Fermi energies are equal. Consequently, a Schottky junction is formed between them at the equilibrium state in the dark.31
ln(ND/NC), where εCBM is the conduction band (CB) minimum energy of TiO2, k is the Boltzmann constant, ND is the donor density of TiO2, and NC is the effective density of states near the CBM of TiO2. The εF,TiO2 value is estimated to be −5.1 ± 0.1 eV by substituting εCBM = −5.0 ± 0.1 eV (ref. 62) and NC = 2.5 × 1019 cm−3(ref. 63) for the usual ND value of 1017 cm−3. When the Au NP having a large work function (ΦW, Au = 5.31–5.47 eV)59 comes into direct contact with TiO2 (ΦW,TiO2 = 5.1 ± 0.1 eV), the interfacial electron transfer proceeds from TiO2 to the Au NP until their Fermi energies are equal. Consequently, a Schottky junction is formed between them at the equilibrium state in the dark.31
The IPCE action spectra indicate that the WOR on the Au/TiO2 plasmonic electrode in the present systems is mainly driven via the LSPR-induced hot electron transfer (HET) mechanism (Fig. 5).22,26 When the Au NPs of Au/TiO2 are excited by photons with energy (ħω) larger than the Schottky barrier (Δε), a number of the hot electrons are injected into the CB of TiO2.25 In the present cases, this energetic requirement is indeed fulfilled because the photocurrent flows at ħω > 1.38 eV (Fig. 3c), which is sufficiently larger than the Δε of ∼0.5 eV reported for the Au/TiO2 system.31 The electrons injected into the CB of TiO2 can be pulled away from the interface to be transported to the FTO electrode, and the holes are accumulated at the interface according to the potential gradient in the space charge layer. As a result of charge separation, the holes effectively oxidize water at the Au NP-TiO2-liquid triphase interface.26
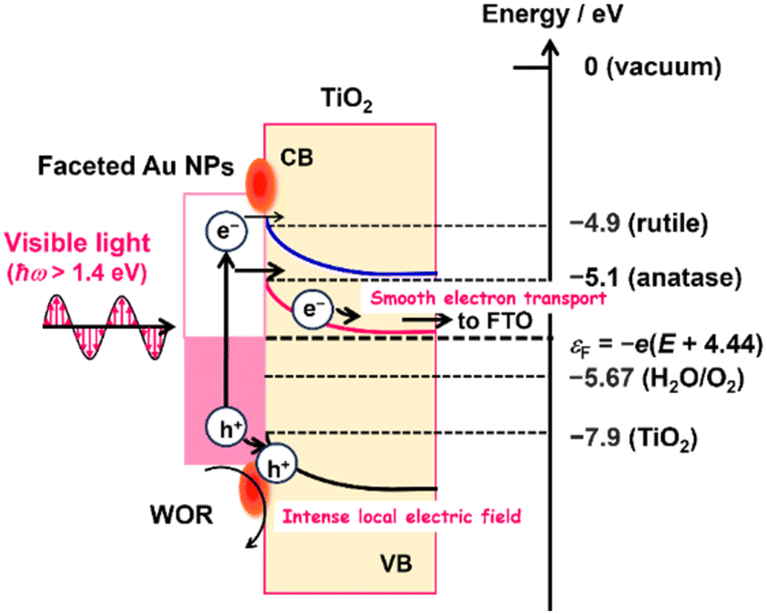 | ||
| Fig. 5 Action mechanism of the DP-Au/A-TiO2 NPLA and DP-Au/R-TiO2 NWA plasmonic electrodes for the WOR. | ||
The photocurrent observed for the WOR (Jph) is proportional to the efficiencies of a series of physical processes involving light harvesting (LHE), hot-electron injection (ϕHEI), and charge separation/transport (ϕCS–CT) (eqn (3)).41 Furthermore, the ϕHEI can be approximated using the Fowler theory (eqn (4)):31
| Jph ∝ LHE × ϕHEI × ϕCS–CT. | (3) |
| ϕHEI(E) ≈C(ħω − Δε)2/ħω | (4) |
As indicated by the absorption spectra (Fig. 3a), the DP-Au/A-TiO2 NPLA and DP-Au/R-TiO2 NWA have LHEs significantly larger than that of the CR-Au/A-TiO2 NPLA, which are induced by the HEPI junctions in the former systems.24 Also, the ϕHEI in the LSPR-induced HET mechanism is proportional to the square of the absolute value of the electric field, that is, the EF.24,64 The EF values of the DP-Au/A-TiO2 NPLA and DP-Au/R-TiO2 NWA are several tens of times larger than that of the CR-Au/A-TiO2 NPLA. Furthermore, the high quality of the Au NP-TiO2 interface in the DP-Au/A-TiO2 NPLA and DP-Au/R-TiO2 NWA contributes to the increase in the ϕHEI.65 Therefore, the photocurrents of the DP-Au/A-TiO2 NPLA and DP-Au/R-TiO2 NWA electrodes far surpass that of the CR-Au/A-TiO2 NPLA electrode and are ascribable to the high LHE and extraordinarily large EF values in addition to the higher quality of the interface. However, the large difference in the photocurrent between the DP-Au/A-TiO2 NPLA and DP-Au/R-TiO2 NWA electrodes cannot be explained only in terms of these factors.
Previously, the electron affinity was determined to be 4.9 eV for R-TiO2 and 5.1 eV for A-TiO2 by means of photoemission electron microscopy.62 Thus, the energy barrier for the HET from Au NPs to TiO2 in the DP-Au/A-TiO2 NPLA electrode is ∼0.2 eV lower than that in the DP-Au/R-TiO2 NWA electrode regardless of the electrode potential (Fig. 5). In addition, A-TiO2 possesses an electron mobility of 4–20 cm2 V−1 s−1 at 300 K, which is much larger than that for R-TiO2 (0.1 cm2 V−1 s−1 at 300 K).32 In the DP-Au/A-TiO2 NPLA electrode, the electrons injected into the CB of TiO2 can be smoothly separated from the Au NPs and transported to the electron-collecting FTO electrode due to the large electron mobility of A-TiO2.32 As a result, the hot holes accumulated in the Au NPs are effectively concentrated around the Au NP–TiO2–solution triphase interface to be used for the WOR with the assistance of the catalytic activity,25,66 which leads to a drastic enhancement of the photocurrent through lowering the Rct. Consequently, the plasmonic activity of the DP-Au/A-TiO2 NPLA, which is much superior to that of the DP-Au/R-TiO2 NWA electrode, can stem from its higher HET efficiency and electric conductivity.
Furthermore, it is worth noting in the electrode system that Δε in eqn (2) is a function of the electrode potential (E vs. standard hydrogen electrode, SHE) or the Fermi energy of the electrode (εF = −e(E + 4.44) vs. under vacuum). As a result of the increase in E, the Δε increases, while the charge separation is enhanced due to the increase in potential gradient and width of the space charge layer. These considerations predict the presence of the optimum potential in the Au/TiO2 plasmonic electrode. Actually, the maximum photocurrents are observed in the J–E curves at 0.83 V in the DP-Au/R-TiO2 NWA electrode and 0.96 V in the DP-Au/A-TiO2 NPLA electrode (Fig. 3b).
In a particulate photocatalytic system, Chen and Tan have recently reported that a nanohybrid plasmonic photocatalyst consisting of Pt cuboctahedra and radial WO3 mesocrystals provides an apparent quantum efficiency greater than 7% at λex = 680 nm for the overall water splitting, which is much larger than that of spherical Pt NP-loaded WO3.67 This remarkable activity can also arise from the LSPR-induced strong local electric field around the interface between the faceted Pt NPs and WO3. Also, this study points to the importance of the high catalytic activities of Pt NPs for the hydrogen evolution reaction and the oxygen evolution reaction. In this case, it should be noted that both of these catalytic activities can only work simultaneously through the separation of the functions of the Pt NPs as oxidation and reduction sites due to the large absorption of the Pt cuboctahedra and the hierarchical structure of WO3.68
In summary, this study aims at enhancing the activity of the prototype plasmonic electrode of Au/TiO2 for the PEC WOR without changing the composition of each component or surface modification by promoters. The t-Oh Au NPs with dAu < 10 nm are formed on the A-TiO2 NPLA with dominant (001) surfaces through the (111)Au//(001)A-TiO2 HEPI junction by the DP process. The resulting DP-Au/A-TiO2 NPLA possesses features including the faceting of the Au NPs and a high-quality interface. The DP-Au/A-TiO2 NPLA electrode has been found to provide an IPCE of 0.39% at λ = 600 nm for the PEC WOR, which is greater than the values reported so far for the usual Au/TiO2 electrodes by more than one order of magnitude. The remarkable activity of the DP-Au/A-TiO2 NPLA plasmonic electrode is ascribable to the intense local electric field generated around the edges and corners of the faceted Au NPs, the high-quality interface between Au NPs and TiO2, and the large electron mobility in A-TiO2, which enhance the physical and chemical processes in the plasmonic WOR. Furthermore, we shed light on the origin of the large difference in the plasmonic activity for the WOR between the DP-Au/A-TiO2 NPLA and DP-Au/R-TiO2 NWA, and the unique features in the photocurrent–potential curves in terms of a potential-dependent Fowler model. This marked progress in the performance of the prototype Au/TiO2 plasmonic electrode could contribute to its practical use for solar-to-fuel energy conversion.
Author contributions
S. N., Y. M., H. S. and T. S. prepared and characterized the electrodes, and carried out PEC experiments and data analysis; M. F. performed MD and FDTD calculations; and H. T. supervised the experimental work and data analysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge K. Miyazaki, R. Kuma, T. Sento, and M. Shima (Nippon Shokubai Co., Ltd) for helpful discussions. This work was financially supported by JSPS KAKENHI Grant-in-Aid for Scientific Research (C) no. 21K05236, the Futaba Foundation, Nippon Sheet Glass Foundation for Materials Science and Engineering, Sumitomo Foundation, Takahashi Industrial and Economic Research Foundation, and the Kato Foundation for the Promotion of Science.References
- A. Fujishima and K. Honda, Electrochemical photolysis of water at a semiconductor electrode, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- T. Hisatomi and K. Domen, Reaction systems for solar hydrogen production via water splitting with particulate semiconductor photocatalysts, Nat. Catal., 2019, 2, 387–399 CrossRef CAS.
- M. W. Kanan and D. G. Nocera, In situ formation of an oxygen-evolving catalyst in natural water containing phosphate and Co2+, Science, 2008, 321, 1072–1075 CrossRef CAS PubMed.
- L. Zhang, L. Sun, Z. Guan, S. Lee, Y. Li, H. D. Deng, Y. Li, N. L. Ahlborg, M. Boloor, N. A. Melosh and W. C. Chueh, Quantifying and elucidating thermally enhanced minority carrier diffusion length using radius-controlled rutile nanowires, Nano Lett., 2017, 17, 5264–5272 CrossRef CAS PubMed.
- M. Xiao, R. Jiang, F. Wang, C. Fang, J. Wang and J. C. Yu, Plasmon-enhanced chemical reactions, J. Mater. Chem. A, 2013, 1, 5790–5805 RSC.
- K. Ueno and H. Misawa, Surface plasmon-enhanced photochemical reactions, J. Photochem. Photobiol., C, 2013, 15, 31–52 CrossRef CAS.
- X. Lang, X. Chen and J. Zhao, Heterogeneous visible light photocatalysis for selective organic transformations, Chem. Soc. Rev., 2014, 43, 473–486 RSC.
- C. Wang and D. Astruc, Nanogold plasmonic photocatalysis for organic synthesis and clean energy conversion, Chem. Soc. Rev., 2014, 43, 7188–7216 RSC.
- R. Jiang, B. Li, C. Fang and J. Wang, Metal/semiconductor hybrid nanostructures for plasmon-enhanced applications, Adv. Mater., 2014, 26, 5274–5309 CrossRef CAS PubMed.
- H. Cheng, K. Fuku, Y. Kuwahara, K. Mori and H. Yamashita, Harnessing single-active plasmonic nanostructures for enhanced photocatalysis under visible light, J. Mater. Chem. A, 2015, 3, 5244–5258 RSC.
- X. Li, J. Zhu and B. Wei, Hybrid nanostructures of metal/two-dimensional nanomaterials for plasmon-enhanced applications, Chem. Soc. Rev., 2016, 45, 3145–3187 RSC.
- Y. Zhang, S. He, W. Guo, Y. He, J. Huang, J. R. Mulcahy and W. D. Wei, Surface-plasmon-driven hot electron photochemistry, Chem. Rev., 2018, 118, 2927–2954 CrossRef CAS PubMed.
- N. Wu, Plasmonic metal–semiconductor photocatalysts and photoelectrochemical cells: a review, Nanoscale, 2018, 10, 2679–2696 RSC.
- A. Gelle, T. Jin, L. de la Garza, G. D. Price and L. V. Besteiro, Applications of plasmon-enhanced nanocatalysis to organic transformations, Chem. Rev., 2020, 120, 986–1041 CrossRef CAS PubMed.
- S. Luo, X. Ren, H. Lin, H. Song and J. Ye, Plasmonic photothermal catalysis for solar-to-fuelconversion: current status and prospects, Chem. Sci., 2021, 12, 5701–5719 RSC.
- M. Sayed, J. Yu, G. Liu and M. Jaroniec, Non-noble plasmonic metal-based photocatalysts, Chem. Rev., 2022, 122, 10484–10537 CrossRef CAS PubMed.
- R. S. Haider, S. Wang, Y. Gao, A. S. Malik, N. Ta, H. Li, B. Zeng, M. Dupuis, F. Fan and C. Li, Boosting photocatalytic water oxidation by surface plasmon resonance of AgxAu1-x alloy nanoparticles, Nano Energy, 2021, 87, 106189 CrossRef CAS.
- H. Li, S. Wang, M. Wang, Y. Gao, J. Tang, S. Zhao, H. Chi, P. Zhang, J. Qu, F. Fan and C. Li, Enhancement of Plasmon-Induced Photoelectrocatalytic Water Oxidation over Au/TiO2 with Lithium Intercalation, Angew. Chem., Int. Ed., 2022, 61, e202204272 CrossRef CAS PubMed.
- R. Negishi, S. Naya, H. Kobayashi and H. Tada, Gold(core)-lead(shell) nanoparticle-loaded titanium(IV) oxide prepared by underpotential photodeposition: Plasmonic water oxidation, Angew. Chem., Int. Ed., 2017, 56, 10347–10351 CrossRef CAS PubMed.
- M. Okazaki, Y. Suganami, N. Hirayama, H. Nakata, T. Oshikiri, T. Yokoi, H. Misawa and K. Maeda, Site-selective deposition of a cobalt cocatalyst onto a plasmonic Au/TiO2 photoanode for improved water oxidation, ACS Appl. Energy Mater., 2020, 3, 5142–5146 CrossRef CAS.
- P. A. DeSario, J. J. Pietron, D. E. DeVantier, T. H. Brintlinger, R. M. Stroud and D. R. Rolison, Plasmonic enhancement of visible-light water splitting with Au–TiO2 composite aerogels, Nanoscale, 2013, 5, 8079–8083 RSC.
- T. Onishi, M. Teranishi, S. Naya, M. Fujishima and H. Tada, Electrocatalytic effect on the photon−to−current conversion efficiency of gold−nanoparticle−loaded titanium(IV) oxide plasmonic electrode for water oxidation, J. Phys. Chem. C, 2020, 124, 6103–6109 CrossRef CAS.
- S.-F. Hung, F.-X. Xiao, Y.-Y. Hsu, N.-T. Suen, H.-B. Yang, H. M. Chen and B. Liu, Iridium oxide-assisted plasmon-induced hot carriers: Improvement on kinetics and thermodynamics of hot carriers, Adv. Energy Mater., 2016, 6, 1501339 CrossRef.
- H. Tada, Rational design for gold nanoparticle−based plasmonic catalysts and electrodes for water oxidation towards the artificial photosynthesis, Dalton Trans., 2022, 51, 3383–3393 RSC.
- M. Teranishi, M. Wada, S. Naya and H. Tada, Size-dependence of the activity of gold nanoparticle-loaded titanium(IV) oxide plasmonic photocatalyst for water oxidation, ChemPhysChem, 2016, 17, 2813–2817 CrossRef CAS PubMed.
- Y. Nishijima, K. Ueno, Y. Kotake, K. Murakoshi, H. Inoue and H. Misawa, Near-infrared plasmon-assisted water oxidation, J. Phys. Chem. Lett., 2012, 3, 148–1252 Search PubMed.
- S. Wang, Y. Gao, S. Miao, T. Liu, L. Mu, R. Li, F. Fan and C. Li, Positioning the water oxidation reaction sites in plasmonic photocatalysts, J. Am. Chem. Soc., 2017, 139, 11771–11778 CrossRef CAS PubMed.
- K. Wu, J. Chen, J. R. McBride and T. Lian, Efficient hot-electron transfer by a plasmon-induced interfacial charge-transfer transition, Science, 2015, 349, 632–635 CrossRef CAS PubMed.
- J. Ma and S. Gao, Plasmon-induced electron-hole separation at the Ag/TiO2(110) interface, ACS Nano, 2019, 13, 13658–13667 CrossRef CAS PubMed.
- M. Yoshii, H. Kobayashi and H. Tada, Sub-bandgap excitation-induced electron injection from CdSe quantum dots to TiO2 in a directly coupled system, ChemPhysChem, 2015, 16, 1846–1851 CrossRef CAS PubMed.
- M. W. Knight, H. Sobhami, P. Nordlander and M. J. Halas, Photodetection with active optical antennas, Science, 2011, 332, 702–704 CrossRef CAS PubMed.
- M. Voïnov and J. Augustynski, Heterogeneous Photocatalysis, ed. M. Schiavello, John Wiley & Sons, Baffins Lane, 1997 Search PubMed.
- B. O'Regan and M. Grätzel, A low-cost, high-efficiency solar cell based on dye-sensitized colloidal TiO2 films, Nature, 1991, 353, 737–740 CrossRef.
- M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Efficient hybrid solar cells based on meso-superstructured organometal halide perovskites, Science, 2012, 338, 643–647 CrossRef CAS PubMed.
- E. Kowalska, R. Abe and B. Ohtani, Visible light-induced photocatalytic reaction of gold-modified titanium(IV) oxide particles: action spectrum analysis, Chem. Commun., 2009, 45, 241–243 RSC.
- E. Kowalska, O. O. P. Mahaney and B. Ohtani, Visible-light-induced photocatalysis through surface plasmon excitation of gold on titania surfaces, Phys. Chem. Chem. Phys., 2010, 12, 2344–2355 RSC.
- K. Kimura, S. Naya, Y. Jin-nouchi and H. Tada, TiO2 crystal form-dependence of the Au/TiO2 plasmon photocatalyst's activity, J. Phys. Chem. C, 2012, 116, 7111–7117 CrossRef CAS.
- S. Naya, K. Kimura and H. Tada, One-step selective aerobic oxidation of amines to imines by gold nanoparticle-loaded rutile titanium(IV) oxide plasmon photocatalyst, ACS Catal., 2013, 3, 10–13 CrossRef CAS.
- J. B. Priebe, J. Radnik, A. J. J. Lennox, M.-M. Pohl, M. Karnahl, D. Hollmann, K. Grabow, U. Bentrup, H. Junge, M. Beller and A. Bruckner, Solar hydrogen production by plasmonic Au-TiO2 catalsyt: impact of synthesis protocol and TiO2 phase on charge transfer efficiency and H2 evolution rates, ACS Catal., 2015, 5, 2137–2148 CrossRef CAS.
- S. Naya, T. Niwa, T. Kume and H. Tada, Visible-light-induced electron transport from small to large nanoparticles in bimodal gold nanoparticle-loaded titanium(IV) oxide, Angew. Chem., Int. Ed., 2014, 53, 7305–7309 CrossRef CAS PubMed.
- S. Naya, A. Akita, Y. Morita, M. Fujishima and H. Tada, Crystallographic interface control of the plasmonic photocatalyst consisting of gold nanoparticles and titanium(IV) oxide, Chem. Sci., 2022, 13, 12340–12347 RSC.
- S. Feng, J. Yang, H. Zhu, M. Liu, J. Zhang, J. Wu and J. Wan, Synthesis of single crystalline anatase TiO2 (001) tetragonal nanosheet-array films fluorine-doped tin oxide substrate, J. Am. Ceram. Soc., 2011, 94, 310–315 CrossRef CAS.
- B. Liu and E. S. Aydil, Growth of oriented single-crystalline rutile TiO2 nanorods on transparent conducting substrates for dye-sensitized solar cells, J. Am. Chem. Soc., 2009, 131, 3985–3990 CrossRef CAS PubMed.
- R. Zenella, S. Giorgio, C. R. Henry and C. Louis, Alternative methods for the preparation of gold nanoparticles supported on TiO2, J. Phys. Chem. B, 2002, 106, 7634–7642 CrossRef.
- A. Dieguez, A. Romano-Rodriguez, A. Vila and J. R. Morante, The complete Raman spectrum of nanometric SnO2 particles, J. Appl. Phys., 2001, 90, 1550–1557 CrossRef CAS.
- O. Frank, M. Zukalova, B. Laskova, J. Kürti, J. Koltai and L. Kavan, Raman spectra of titanium dioxide (anatase, rutile) with identified oxygen isotopes (16, 17, 18), Phys. Chem. Chem. Phys., 2012, 14, 14567–14572 RSC.
- A. Akita, H. Kobayashi and H. Tada, Action of chloride ions as a habit modifier in the hydrothermal crystal growth of rutile TiO2 nanorod from SnO2 seed crystal, Chem. Phys. Lett., 2020, 761, 138003 CrossRef CAS.
- H. G. Yang, C. H. Sun, S. Z. Qiao, J. Zou, G. Liu, S. C. Smith, H. M. Chen and G. Q. Liu, Anatase TiO2 single crystals with a large percentage of reactive facets, Nature, 2008, 453, 638–641 CrossRef CAS PubMed.
- S. Liu, J. Yu and M. Jaroniec, Tunable photocatalytic selectivity of hollow TiO2 microspheres composed of anatase polyhedra with exposed {001} facets, J. Am. Chem. Soc., 2010, 132, 11914–11916 CrossRef CAS PubMed.
- D. Holec, P. Dumitraschkewitz, D. Vollath and F. D. Fischer, Surface energy of Au nanoparticles depending on their size and shape, Nanomaterials, 2020, 10, 484 CrossRef CAS PubMed.
- G. Liu, H. G. Yang, J. Pan, Y. Q. Yang, G. Q. Lu and H.-M. Cheng, Titanium dioxide crystals with tailored facets, Chem. Rev., 2014, 114, 9559–9612 CrossRef CAS PubMed.
- F. Cosandey, L. Zhang and T. E. Madey, Effect of substrate temperature on the epitaxial growth of Au on TiO2(110), Surf. Sci., 2001, 474, 1–13 CrossRef CAS.
- W. Yuan, B. Zhu, K. Fang, X.-Y. Li, T. W. Hansen, Y. Ou, H. Yang, J. B. Wagner, Y. Gao, Y. Wang and Z. Zhang, In situ manipulation of the active Au-TiO2 interface with atomic precision during CO oxidation, Science, 2021, 371, 517–521 CrossRef CAS PubMed.
- K. Tanaka, S. Tanuma, K. Dohmae, Y. Nagoshi and A. Nisawa, X-ray photoelectron spectroscopy, ed. T. Sawada, S. Tanuma and K. Tanaka, Maruzen, Tokyo, 1998 Search PubMed.
- H. Kisch, Semiconductor Photocatalysis-Principles and ApplicationsWiley-VCH, Weinheim, 2015 Search PubMed.
- H. Hovel, S. Fritz, A. Hilger and V. Kreibig, Width of cluster plasmon resonances: Bulk dielectric functions and chemical interface damping, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 18178–18188 CrossRef PubMed.
- A. Naldoni, T. Montini, F. Malara, M. M. Mroz, A. Beltram, T. Virgili, C. L. Boldrini, M. Marelli, I. Romero-Ocana, J. J. Delgado, V. Dal Santo and P. Fornasiero, Hot electron collection on brookite nanorods lateral facets for plasmon-enhanced water oxidation, ACS Catal., 2017, 7, 1270–1278 CrossRef CAS.
- L. Liu, P. Li, B. Adisak, S. Ouyang, N. Umezawa, J. Ye, R. Kodiyath, T. Tanabe, G. V. Ramesh, S. Ueda and H. Abe, Gold photosensitized SrTiO3 for visible-light water oxidation induced by Au interband transitions, J. Mater. Chem. A, 2014, 2, 9875–9882 RSC.
- Handbook of Chemistry and Physics, ed. D. R. Lide, CRC Press, Boca Raton, 83rd edn, 2002 Search PubMed.
- U. Kreibig and M. Vollmer, Optical properties of metal clusters, Springer, Berlin, 1996 Search PubMed.
- W. Yuan, D. Zhang, Y. Ou, K. Fang, B. Zhu, H. Yang, T. W. Hansen, J. B. Wagner, Z. Zhang, Y. Gao and Y. Wang, Direct in situ TEM visualization and insight into the facet-dependent sintering behaviours of gold on TiO2, Angew. Chem., Int. Ed., 2018, 57, 16827–16831 CrossRef CAS PubMed.
- G. Xiong, R. Shao, T. C. Droubay, A. G. Joly, K. M. Beck, S. A. Chambers and W. P. Hess, Photoemission electron microscopy of TiO2 anatase films embedded with rutile nanocrystals, Adv. Funct. Mater., 2007, 17, 2133–2138 CrossRef CAS.
- H. Tang, K. Prasad, R. Sanjinès, P. E. Schmid and F. Lévy, Electrical and optical properties of TiO2 anatase thin films, J. Appl. Phys., 1994, 75, 2042–2047 CrossRef CAS.
- A. O. Govorov, H. Zhang, H. V. Demir and Y. K. Gun'ko, Photogeneration of hot plasmonic electrons with metal nanocrystals: Quantum description and potential applications, Nano Today, 2014, 9, 85–101 CrossRef CAS.
- S. Naya, T. Kume, R. Akashi, M. Fujishima and H. Tada, Red−Light−Driven Water Splitting by Au(Core)−CdS(Shell) Half−Cut Nanoegg with Heteroepitaxial Junction, J. Am. Chem. Soc., 2018, 140, 1251–1254 CrossRef CAS PubMed.
- E. Thimsen, F. Le Formal, M. Grätzel and S. C. Warren, Influence of plasmonic Au nanoparticles on the photoactivity of Fe2O3 electrodes for water splitting, Nano Lett., 2011, 11, 35–43 CrossRef CAS PubMed.
- Q. Chen and Y. Tan, Enhanced plasmonic absorption of Pt cuboctahedra-WO3 nanohybrids used as visible light photocatalysts for overall water splitting, Nano Res., 2023, 16, 5919–5928 CrossRef CAS.
- R. Kojima, S. Naya and H. Tada, Three-dimensional plasmonic photocatalyst consisting of faceted gold nanoparticles and radial titanium(IV) oxide heteromesocrystal, J. Phys. Chem. C, 2023, 127, 3478–3485 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr01013b |
| This journal is © The Royal Society of Chemistry 2024 |