
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-assembled viologens on HOPG: solid-state NMR and AFM unravel the location of the anions†
Jean
Joseph
a,
Jésus
Raya
 a,
Frank
Palmino
b,
Judicaël
Jeannoutot
b,
Mathilde
Berville
a,
Jean
Weiss
a,
Frédéric
Chérioux
*b and
Jennifer A.
Wytko
*a
a,
Frank
Palmino
b,
Judicaël
Jeannoutot
b,
Mathilde
Berville
a,
Jean
Weiss
a,
Frédéric
Chérioux
*b and
Jennifer A.
Wytko
*a
aInstitut de Chimie, UMR 7177 CNRS-Université de Strasbourg, 4 rue Blaise Pascal, 67008 Strasbourg, France. E-mail: jwytko@unistra.fr
bUniversité Franche-Comté, CNRS, FEMTO-ST, 25000 Besançon, France. E-mail: frederic.cherioux@femto-st.fr
First published on 14th June 2024
Abstract
The controlled growth of self-assembled networks on surfaces based on viologen salts is a major scientific challenge due to their unique electronic properties. The combination of solid-state NMR spectroscopy and atomic force microscopy at ambient conditions can unravel the fine organization of the supramolecular network on a graphitic surface by positioning the counter-ions relative to the viologen cation.
Adsorption of supramolecular networks is a useful technique to tune the electronic properties of the underlying surfaces.1–3 Organic molecules with the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) at a suitable position with respect to the Fermi level of underlying surfaces can be used to dope surfaces.4 Among all π-conjugated molecules, redox-active viologens, with their low-lying LUMOs, have emerged as promising candidates to address this major challenge.5,6 Viologens are aromatic species built from a 4,4′-bipyridinyl core in which the two nitrogen atoms are quaternized to generate an organic dication.
Atomic force microscopy (AFM) is a commonly used method to investigate the self-organization of molecules on surfaces under ambient conditions at the sub-molecular level.7,8 However, for organic salts like viologens, the position of the organic cations is well determined whereas the position of corresponding anions has been investigated less, mainly because these anions are often smaller than the organic cations.9,10 The position of the anions has been studied using scanning tunneling microscopy.6,11–14 However, apart from the case of chloride anions on a Cu(100) surface,11 the position of the anions has not been clearly identified.
The role of electrostatic interactions between anions and cations is essential to understanding on-surface organization. These electrostatic interactions strongly depend on the relative positions of these charged species and require the determination of the positions of both cations and anions. In rare seminal works, the combination of solid-state NMR (ssNMR) with optical microscopy was successfully reported for liquid crystalline viologen derivatives.15 In addition, ssNMR and AFM have been employed separately as analytical methods to characterize self-assembling materials with low solubility.3 However, the complementarity of ssNMR and AFM was neither established nor reported. This work describes an original strategy to unravel the position of anions and cations in organic salts deposited on a highly-oriented pyrolytic graphite (HOPG) surface. This simple strategy shows that ssNMR can quickly indicate the tendency of building blocks to organize efficiently prior to AFM studies. As shown hereafter, ssNMR can be combined with AFM to understand fully the self-organization of viologen salts on HOPG.
To achieve the formation of large-scale networks of organic salts on an HOPG surface, we designed salts intended to favor intermolecular interaction and molecule-surface interaction. The targeted salts ArV-C8, ArV-C12 and MV-C18 (Fig. 1) consist of a dicationic viologen core bearing peripheral length-adjustable alkoxy chains (Fig. 1). Two hexafluorophosphate anions act as counter-ions for these organic dications. Alkoxy chains are well known to promote molecule–surface interactions on an HOPG surface and molecule–molecule interactions due to interdigitation of adjacent chains.8,16,17
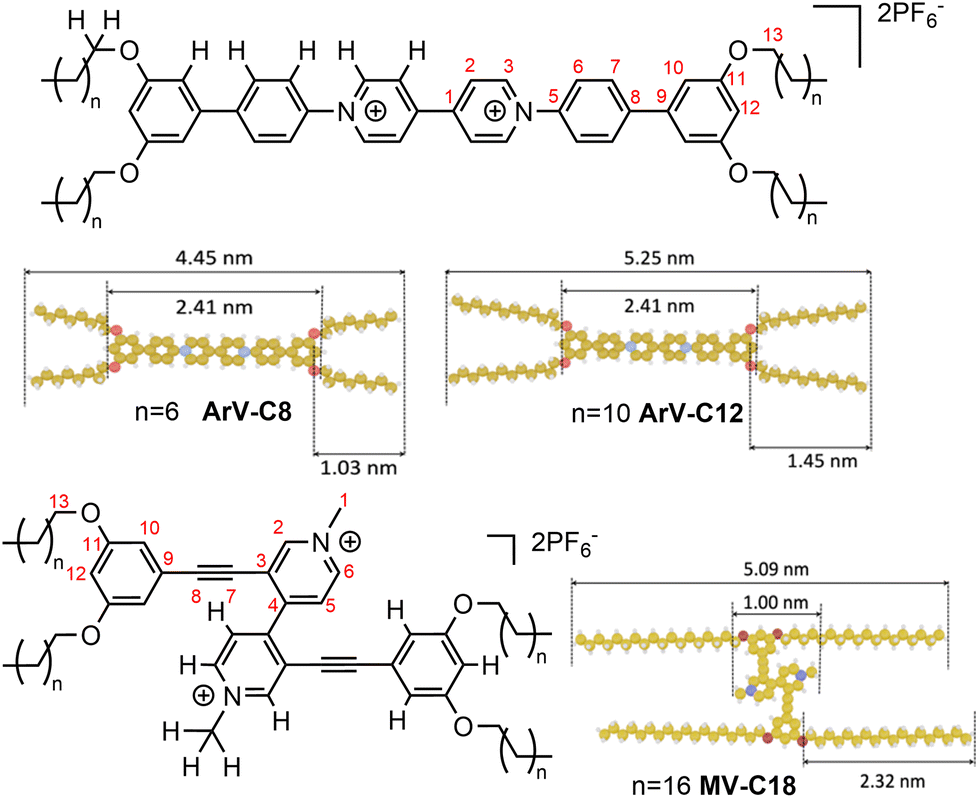 | ||
| Fig. 1 Structures and CPK models of the three viologens studied in this work. | ||
Compounds ArV-C8 and ArV-C12 are N-arylviologens that differ by the length of their alkoxy chains. MV-C18 is an N-methylviologen functionalized at the 3,3′-positions with C18 alkoxy chains that were chosen to compensate for the dihedral angle of the 3,3′-substituted pyridinium rings. These structural variations modulate the degree of molecule–surface interactions on HOPG. The length of their aromatic cores varies from 1.00 to 2.41 nm and their estimated total length, when considering linear gauche alkyl chain conformations, varies from 4.45 to 5.25 nm (Fig. 1).
ArV-C8 and ArV-C12 were prepared using strategies based on Zincke reactions, whereas MV-C18 was synthesized by quaternization of its bipyridine precursor18 (see ESI† for experimental details). All new compounds were fully characterized and 1H and 13C resonances in solution were assigned (see ESI†) with the help of 2D correlations. Both ArV-C8 and ArV-C12 showed a tendency to precipitate in chloroform at NMR concentrations (10−3 M). This observation prompted us to characterize these compounds by ssNMR, taking advantage of the latter to gain information on the nature of the aggregates.
In the past years, ssNMR has demonstrated its ability to provide structural information at the atomic level owing to the various interactions taking place within the spin systems under study, notably the so-called dipolar interaction. The latter occurs through space with a 1/r3 dependency where r is the inter-nuclear distance. This property can be exploited to measure inter-nuclear distances by rotational-echo double-resonance (REDOR)-type experiments19 or to determine which nuclei are in close contact (intra- or intermolecularly) using the cross polarization/magic angle spinning experiment (CP/MAS).20 In our case, if the 31P of PF6− anions can be polarized by protons, the mere observation of a 1H–31P CP/MAS spectrum would constitute irrefutable proof that these hexafluorophosphate anions are part of the supramolecular structure. The only protons available for transfer of polarization to the 31P of the PF6− anions are located on either the alkyl side chains or the viologen moieties. To elucidate further the structure of each compound, two-dimensional ssNMR was used. First, the 1H resonances of the viologens were assigned using 1H–13C through-space correlations (dipolar interaction also).21 Then the same type of experiments were performed with 31P instead (1H–31P through-space correlations) to locate precisely the anions within the supramolecular assembly.
Before exploring the possible connections between protons and phosphorus atoms, a series of classical and quantitative experiments known as direct polarization/magic angle spinning (DP/MAS) were carried out to characterize the environment of the anions. The relatively high chemical shift anisotropy (CSA) of 31P means that it is highly sensitive to very small variations in chemical environment. In these direct polarization experiments, ArV-C12 displays only one multiplet at ∂iso = −145.4 ppm, whereas ArV-C8 exhibits two well-resolved multiplets at ∂iso = −146.9 ppm and ∂iso = −147.9 ppm. Scalar couplings with 19F account for the multiplicity of these signals (J19F–31P = 713 Hz). For MV-C18, a superposition of a dozen multiplets of varying intensity centred at ∂iso = −145 ppm and separated by small chemical shift gaps (<0.5 ppm) is observed. These quick initial results indicate that ArV-C8 and ArV-C12 are rather well-structured with only one position of the PF6− anions for ArV-C12 and two different positions for the anions of ArV-C8 in proportions of 27% (∂iso = −146.9 ppm) and 73% (∂iso = −147.9 ppm). These results also indicate that MV-C18 exhibits a finite series of small structural variations that can be assigned, in a first approximation, to variations in the dihedral angle between the two pyridinium units.
Cross-polarization experiments in which only the proton is directly polarized and its magnetization is transferred to the heteronucleus by through-space dipolar interaction yielded resonance patterns almost identical to those obtained by direct polarization. For ArV-C8, the 31P multiplets are reproduced, with variations in intensity depending on the different isotropic peaks present (Fig. 2).‡ In addition, the amount of signal obtained is as great, if not greater, than with simple direct polarization. Based on both observations, we can immediately conclude that the polarization transfer is not limited to a fraction of the anions and that all of the PF6− anions are polarized by viologen protons. Consequently, the PF6− anions of ArV-C8 are an integral part of the supramolecular structure.
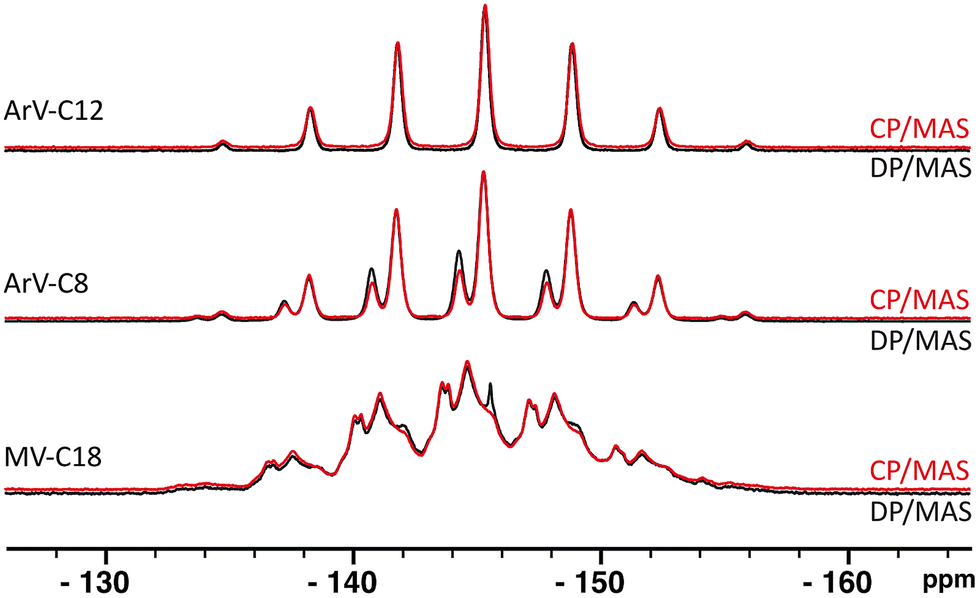 | ||
| Fig. 2 Direct polarization (black) and cross polarization (red)/magic angle spinning (DP/MAS and CP/MAS) {1H} 31P spectra of ArV-C12 (top traces), ArV-C8 (middle) and MV-C18 (bottom). Each individual spin-system appears with the multiplicity given by the 19F–31P scalar coupling (seven lines centered at δiso). Only one PF6− site detected for ArV-C12, two for ArV-C8 (27% and 73%) and at least 12 slightly different sites for MV-C18. The latter variability is ascribed to small differences in the dihedral angle of the two pyridinium subunits. Comparisons of CP/MAS to DP/MAS directly demonstrate spatial proximity between viologen protons and PF6− anions. | ||
To go a step further and locate the anions within the molecular framework, the protons near the phosphorus must be identified. This task requires proton spectra with satisfactory resolution that is hardly ever possible in the solid state. The same is not true for 13C. When the systems under study are not too structurally heterogeneous, 1H–13C CP/MAS spectra can be acquired readily with a resolution high enough to identify the various carbons that constitute the backbone of organic molecules. As the resolution of the phosphorus spectra indicated very good structural homogeneity of the viologen-PF6− assemblies, it is immediately apparent from the CP/MAS spectra obtained for each of the three viologens that the use of 1H–X FSLG HETCOR experiments,21 where X can be either 13C or 31P, is indeed justified (Fig. 3). In these experiments, where the protons correlate with heteronuclei through space, the proton is not directly observed; thus, it can easily undergo homonuclear decoupling. Indeed, the proton resolution, which is rather poor in a direct spectrum, becomes sufficient in the indirect dimension. Fig. 4 shows the example of ArV-C12, where the various correlation peaks are resolved well enough to be assigned (see ESI Fig. S25†). These experiments, repeated for the other two viologens (see ESI Fig. S26 and S27†) allowed us first to conclude that the chemical shifts in the solid state are very close to those observed in solution, and second and above all, to identify the protons by their chemical shifts (see ESI Tables S1–S3† for assignments). One notable difference is that in solution, the protons of ArV-C8 at positions 10 and 10′, and also at 13 and 13′, are dynamically equivalent but in the bulk, these carbon atoms and protons are differentiated because interactions between the alkyl chains perturb the free rotation of the terminal aryl nuclei.
 | ||
| Fig. 3 {1H} 1H–13C CP/MAS spectrum of ArV-C12 (non-quantitative, room temperature). Rather narrow linewidths, ranging from 0.3 to 0.5 ppm allow assignments by means of 1H to 13C 2D NMR (Fig. 5). Carbons 10 and 10′ differ due to their variable distance from PF6− in the 3D scaffold. | ||
 | ||
| Fig. 4 1H–13C FSLG HETCOR spectrum of ArV-C12. Blue arrows indicate polarization pathways from 1H to 13C. With short contact time (50 μs), only CH and CH2 signals are detected. Longer contact times (≥500 μs) allow detection and assignment of CH3 and quaternary carbons. | ||
Finally, by repeating the heteronuclear correlation experiments between proton and phosphorus, using the same experimental conditions for the proton dimension, the 2D maps obtained are directly superimposable with the 2D 1H–13C spectra because both experiments share exactly one of their dimensions, namely the proton dimension. Fig. 5 clearly shows that for ArV-C12, only the protons labelled 2 and 3 are closest to 31P. For viologen MV-C18, all phosphorus populations correlate with protons 1 and 2, but not with 10 (Fig. 6). Thus, the anions can be precisely positioned for ArV-C12 and MV-C18 based on ssNMR results. For both compounds, stacked arrangements comprised of alternating layers of cations and anions, are consistent with these observations.
 | ||
| Fig. 5 Superposition of 1H–13C (blue) and 1H–31P (red) correlation experiments of ArV-C12 at short contact times (55 μs and 100 μs respectively). The F2 scale is for 13C (31P omitted) while 1H scale is common. The polarization pathways are shown for nuclei of interest (1H to 13C in blue, 1H to 31P in red). Strong through-space interaction detected between phosphorus and protons 2 and 3. With protons 6 and 7, a weaker correlation is detected and the PF6− anions may be placed as depicted. | ||
 | ||
| Fig. 6 Superposition of 1H–13C (blue) and 1H–31P (red) correlation experiments of MV-C18 at short contact times (55 μs and 100 μs, respectively). F2 scale is for 13C (31P omitted) and 1H scale is common. Polarization pathways shown for any correlation displayed (blue arrows for 1H to 13C, red arrows for 1H to 31P). Methyl 1 and quaternary C3 exhibit low intensity correlations due to short contact time (thinner arrows). Carbons 2, 3 and 10 and protons 2 and 10 exhibit small chemical shift dispersions due to the dozen similar structures present. Close contacts with phosphorus are only detected with protons 1 and 2 and eventually with 6, but not with proton 10. If proton 6 was in close contact with PF6− a correlation peak should be detected at this position. As nothing is observed, we can exclude proton 6. Thus, 31P atoms may be unambiguously positioned as shown. | ||
The case of ArV-C8 differs slightly from the others (Fig. 7). The isotropic 31P peak of the predominant form at −147.9 ppm correlates with protons 3, 6, 7 and 10, whereas for the less abundant form (−146.9 ppm), the detected 31P proximities concern protons 2, 6, 7 and 10. These data suggest two geometries, one major and one minor, in the stacking of 2D planes in the bulk (vide infra).
 | ||
| Fig. 7 Superposition of 1H–13C (blue) and 1H–31P (red) correlation experiments of ArV-C8 at short contact times (55 and 100 μs respectively). F2 scale is for 13C (31P omitted) and 1H scale is common. The polarization pathways are shown only for nuclei of interest (blue arrows for 1H to 13C, red arrows for 1H to 31P). For the sake of clarity only the major form is depicted (δiso31P = −147.9 ppm). In this case the 31P is in close contact with protons 3, 6, 7 and 10 whereas in the minor form (δiso31P = −146.9 ppm) proximities are detected with the same protons except proton 3, which is replaced by proton 2. | ||
The organized character of each viologen in the bulk documented by the ssNMR data can be taken into account when analyzing the AFM images. Together, these observations show interesting correlations between the 2D and the 3D organizations of these charged species. Large-scale AFM images (>170 × 170 nm2) obtained after the deposition of ArV-C8, ArV-C12 and MV-C18 on the HOPG surface (see Fig. S28 in ESI†) are shown in Fig. 8 (see ESI† for detailed procedure). For each molecule, we chose solutions with concentrations of 1 × 10−6 M in CHCl3 to avoid aggregate formation before deposition. This concentration was selected because the three compounds only form H-aggregates at concentrations exceeding 1 × 10−5 M, as determined by UV-Visible spectroscopy (see Fig. S29–S31 in ESI†). In all cases, compact domains with a thickness of 0.9 ± 0.3 nm were observed in the topography images (see Fig. S32 in ESI†). In addition, adhesion images show different signals for the uncovered areas and the areas covered by compact domains (see Fig. S33 in ESI†). Therefore, the observed compact domains correspond to monolayers of self-assembled organic salts. In addition, adhesion images support that the compact domains for both ArV-C8 and ArV-C12 are composed of parallel bright nanolines separated by darker strips (Fig. 8a and b). The bright lines correspond to the aromatic viologen cores whereas the darker strips represent areas of the surface on which the alkyl chains are adsorbed through C–H/π interactions. The periodicity between adjacent bright lines in Fig. 8a and b is 4.51 and 4.14 nm, respectively (see Fig. S32a and b in ESI†). For MV-C18, linear bright nanolines, separated by darker stripes are observed but some folded nanolines are also clearly visible (Fig. 8c). The periodicity between adjacent bright nanolines is 4.00 nm (see Fig. S32c in ESI†). The curved sections of the supramolecular lines, observed in the case of the deposition of MV-C18, appear to be rotated by 120° (Fig. 8c), suggesting that they align with the main axes of the HOPG surface. Thus, the curved features stem from strong molecule–surface interactions that compel the supramolecular lines initially aligned with one axis to align with another of the three mains axes of the HOPG surface. Based on these data, we assumed that the monolayers observed in AFM images are constituted of self-assembled organic salts. There is no anchoring group in the skeleton of these organic salts and thus no covalent bond to the surface. Therefore, the three organic salts are adsorbed with the aromatic viologen core parallel to the HOPG surface.
 | ||
| Fig. 8 AFM images on a HOPG surface after the deposition of (a) ArV-C8 (250 × 250 nm2), (b) ArV-C12 (200 × 200 nm2) and (c) MV-C18 (170 × 170 nm2). In all cases, supramolecular networks appear as bright nanolines. | ||
The adsorption models depicted in Fig. 9d, 10d and 11b for the three monolayered interfaces are the only ones that match the periodicities determined in the AFM images and the atom correlations revealed by ss-NMR experiments, as we have suggested. Other possible models, such as edge-on or more compact ones, do not fit with all of the experimental data. The large-scale expansion and the compactness of the monolayers are mainly governed by molecule–molecule and molecule–surface interactions.22 Concerning the molecule–surface interactions, the adsorption of molecules bearing n-alkyloxy chains on an HOPG surface leads to the formation of close-packed 2D parallel-aligned nanolines. This adsorption is driven by the correspondence between the zigzag alternation of the methylene groups of the n-alkyloxy chains and the 〈100〉 direction of HOPG.23 The molecule–molecule interactions originate from the interdigitation of the n-alkyloxy chains of molecules within two parallel nanolines. As observed in the AFM images, the periodicity between adjacent bright nanolines is shorter for ArV-C12 and MV-C18 (4.14 and 4.00 nm, respectively) than for ArV-C8 (4.54 nm). The longer n-alkoxy chains of ArV-C12 and MV-C18 (12 and 18 carbon atoms, respectively) than of ArV-C8 (only eight carbon atoms) promote molecule–molecule interactions17,24 and account for this difference in periodicity.
 | ||
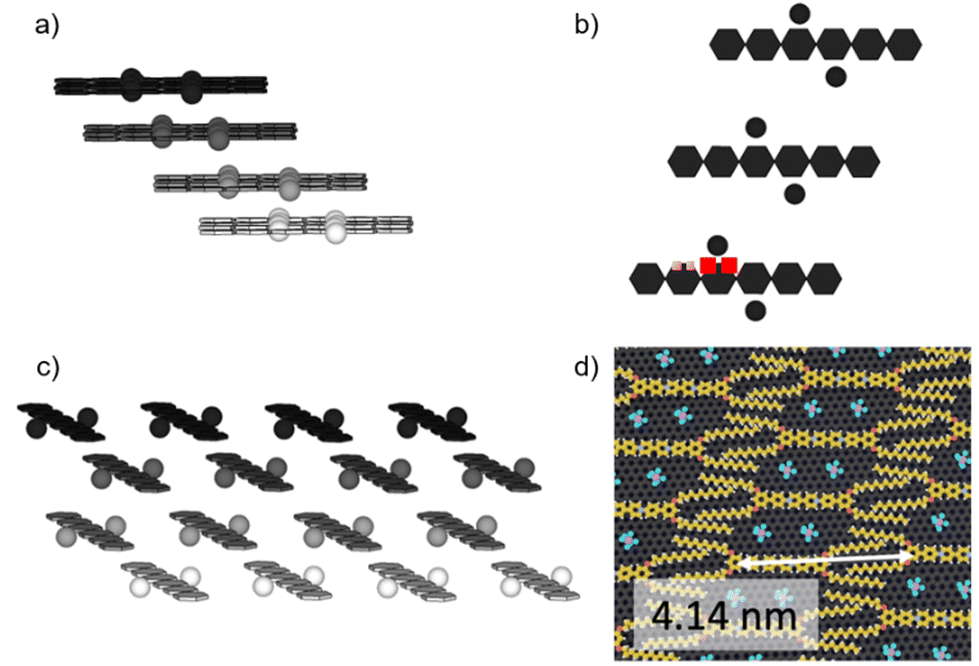
| Fig. 9 (a) Sketch of the 3D stacking arrangement of ArV-C8 in the solid state from ssNMR measurements. Top layer = black, second layer = dark grey, third layer = grey, bottom layer = light grey. Blue spheres represent the possible position of PF6− anions in the minor form; (b) red squares mark the positions involved in the polarization transfer in the major form and green squares mark the positions involved in the polarization transfer in the minor form; (c) side view of (a); (d) 2D layer model built from AFM measurements. | ||
 | ||
| Fig. 10 (a) Sketch of the 3D stacking of ArV-C12 based on ssNMR: top layer = black, second layer = dark grey, third layer = grey, bottom layer = light grey; (b) schematic model of a monolayer of ArV-C12 based on ssNMR: red squares mark the positions involved in the strong polarization transfer and yellow squares mark a weaker transfer; (c) side view of (a); (d) adsorption model of a monolayer of ArV-C12 on HOPG based on AFM measurements. | ||
 | ||
| Fig. 11 (a) Three possible stacking modes of non-planar viologens and PF6− anions of MV-C18 with large degrees of freedom in the bulk; top layer = black, second layer = dark grey, third layer = grey, bottom layer = light grey. Combinations of slipped and flipped are not shown. (b) Adsorption model of a monolayer of MV-C18 on HOPG based on AFM image. | ||
Despite the high resolution of the AFM images, we could not specify the position of the hexafluorophosphate anions in the monolayers. Interestingly, the positioning of the PF6− anions suggested by the ssNMR and the close contacts observed between 1H with 13C and 31P atoms in the 3D solid-state can be useful to determine their positioning in the 2D self-assembled monolayers. The fast response in ssNMR suggests a high degree of organization in the bulk that cannot be very different from a stack of organized planar assemblies. Thus, both the arrangement of organic matter on the surface and the information provided by the ssNMR strongly support the positioning of the anions in the pores defined by the viologens on the surface.
In the bulk, the supramolecular organization of ArV-C8 minimizes repulsion between anions and maximizes attraction between anions and cations. This interplay results in the sliding of each layer. ssNMR data indicate the existence of two possible arrangements in the solid state. The first one is a slipped quasi-planar arrangement (Fig. 9a and c) that minimizes most electrostatic repulsions. Therefore, the anions are located close to two neighboring protons on opposite sides of the viologen core (red squares in Fig. 9b). This positioning is the same as that observed in the major form in ss-NMR (73%). The cohesion in the bulk does not depend on the presence of the alkyl chains; hence, some degree of control is missing and defects are observed in ssNMR in 27% of a minor form. In this second form, the anions are probably squeezed out of a pore and trapped between two slipped layers (blue circles in Fig. 9a, b and c), which explains the simultaneous polarization transfer with the 2-position of the pyridinium (green squares in Fig. 9b).
For a monolayer of ArV-C8 on HOPG, the small size of the pore is not governed by the inter-digitation of the alkyl chains but is mostly controlled by electrostatic interactions and repulsion between the viologen units because ArV-C8 bears short alkyl chains. Therefore, in both 3D and 2D assemblies, the positioning of the anions results from two parameters, namely the size of the pore and the electrostatic forces. Because the species are small and the interactions between the alkyl chains are weak, electrostatic attraction and repulsion also dictate the 2D supramolecular organisation on HOPG. Therefore, in the 2D monolayer, the hexafluorophosphate anions are located in the nanopores and interact with the same four protons as in the major form in the bulk.
When longer alkyl chains are used, as in ArV-C12, hydrophobic interactions become dominant both on the surface and in the bulk. The ssNMR experiments on ArV-C12 clearly show a single position for the anions in the 3D bulk as only one set of resonances is observed. Thus, in the solid state, a perfect balance between electrostatic interactions and alkyl chain interdigitation controls not only the size of the pores but also the bulk arrangement of alternating stacked viologen planes and therefore the positioning of the anions. On the surface, the model of ArV-C12 shows that the alkyl chains interdigitate (Fig. 10d). The size of the pore, which is mostly governed by the inter-digitation of the alkyl chains, allows distancing of like-charges in the monolayer while promoting coulombic attractions between anions and cations. Based on the contacts detected by ssNMR in the bulk, the anions can be precisely placed in close proximity to the viologen core to form a rather dense network on the surface with pores between the viologen cores.
Finally, the case of MV-C18 deserves attention and completes the previous observations and hypotheses. The main characteristic of this viologen is that, due to the 3,3′ substitution of the bipyridine core, the viologen deviates more from planarity than ArV-C8 and ArV-C12.18 In the bulk, strong hydrophobic interactions between the long alkyl chains align the viologen units. The strong non-planarity of MV-C18 now defines 3D pores instead of the 2D pores observed for the flatter ArV-C8 or ArV-C12 species. The multiple yet very similar chemical shifts in ssNMR show that all PF6− anions are located in a very similar environment, which suggests the presence of large pores and more freedom for the positioning of the anions near the organic dications. Three examples of possible cation–anion arrangements in the bulk are proposed in Fig. 11a. In the 2D assembly, the anions of MV-C18 are positioned on both sides of the viologen species in the pore defined by the alkyl chains and the short bipyridinium unit. Each pore contains only one PF6− anion and thus the position of the latter is only controlled by coulombic attractions (Fig. 11b).
By combining ssNMR and AFM data, it has been possible to complete fully the adsorption model of three organic salts. As the hexafluorophosphate anions are close to the C–H group of the aromatic moieties of the three organic salts and do not interact with the n-alkoxy chains, the data suggest that the anions are located in the nanopores of the assemblies, close to the central viologen core of each of the three salts (Fig. 9–11 and S34 in ESI†). In this manner, the hexafluorophosphate anions are included within the bright nanolines observed in AFM images and the 2D assembly represents a layer of the corresponding 3D solid-state arrangement.
In conclusion, the combination of ssNMR spectroscopy and AFM topography images provides a complete picture of the supramolecular self-organization of organic salts on an HOPG surface. This first example of combined 3D and 2D analysis using solid state NMR and AFM provides insight into the programming of self-assembly and organization of charged species on surfaces. The location of counter-ions is controlled by the degree of hydrophobic interactions, the size of the pores and the mobility of the counter-ions on the surface. These complementary approaches also show that a quick way to evaluate the odds of observing 2D assemblies is to perform ssNMR experiments with any available nucleus. The extension of the method to other cations and anions as well as neutral species is in progress.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the French National Research Agency HITS grant (ANR-20-CE09-0016) and PRIMO grant (ANR-12-BS07-0014-01), and from the Pays de Montbéliard Agglomération. JJ thanks the French MNERT for a PhD fellowship.References
- B. Lüssem, C.-M. Keum, D. Kasemann, B. Naab, Z. Bao and K. Leo, Chem. Rev., 2016, 116, 13714 CrossRef PubMed.
- K. S. Mali, J. Greenwood, J. Adisoejoso, R. Phillipson and S. De Feyter, Nanoscale, 2015, 7, 1566 RSC.
- A. Narita, I. A. Verzhbitskiy, W. Frederickx, K. S. Mali, S. A. Jensen, M. R. Hansen, M. Bonn, S. De Feyter, C. Casiraghi, X. Feng and K. Muellen, ACS Nano, 2014, 8, 11622 CrossRef CAS PubMed.
- K. Pei, Surf. Interfaces, 2022, 30, 101887 CrossRef CAS.
- D. Kiriya, M. Tosun, P. Zhao, J. S. Kang and A. Javey, J. Am. Chem. Soc., 2014, 136, 7853 CrossRef CAS PubMed.
- T. M. T. Huynh, T. H. Phan and S. De Feyter, J. Phys. Chem. C, 2022, 126, 6413 CrossRef CAS.
- Y. Gan, Surf. Sci. Rep., 2009, 64, 99 CrossRef CAS.
- W. Hourani, S. Lamare, Y. Makoudi, F. Palmino and F. Chérioux, Nanotechnology, 2016, 27, 425601 CrossRef CAS PubMed.
- J. M. Black, M. Zhu, P. Zhang, R. R. Unocic, D. Guo, M. B. Okatan, S. Dai, P. T. Cummings, S. V. Kalinin, G. Feng and N. Balke, Sci. Rep., 2016, 6, 32389 CrossRef CAS PubMed.
- H. Li, R. J. Wood, F. Endres and R. Atkin, J. Phys.: Condens. Matter, 2014, 26, 284115 CrossRef PubMed.
- M. Jiang, E. Sak, K. Gentz, A. Krupski and K. Wandelt, ChemPhysChem, 2010, 11, 1542 CrossRef CAS PubMed.
- Z. Li, B. Han, G. Meszaros, I. Pobelov, T. Wandlowski, A. Blaszcyk and M. Mayor, Faraday Discuss., 2006, 131, 121 RSC.
- R. C. Quardokus, Y. Lu, N. A. Wasio, C. S. Lent, F. Justaud, C. Lapinte and S. A. Kandel, J. Am. Chem. Soc., 2011, 134, 1710 CrossRef PubMed.
- T. Kudernac, N. Shabelina, W. Mamdouh, S. Höger and S. De Feyter, Beilstein J. Nanotechnol., 2011, 2, 674 CrossRef CAS PubMed.
- (a) G. Casella, V. Causin, F. Rastrelli and G. Saielli, Phys. Chem. Chem. Phys., 2014, 16, 5048 RSC; (b) M. Bonchio, M. Carraro, G. Casella, V. Causin, F. Rastrelli and G. Saielli, Phys. Chem. Chem. Phys., 2012, 14, 2710 RSC; (c) V. Causin and G. Saielli, J. Mater. Chem., 2009, 19, 9153 RSC.
- J. A. A. W. Elemans, S. Lei and S. De Feyter, Angew. Chem., Int. Ed., 2009, 48, 7298 CrossRef CAS PubMed.
- D. Bléger, D. Kreher, F. Mathevet, A.-J. Attias, G. Schull, A. Huard, L. Douillard, C. Fiorini-Debuischert and F. Charra, Angew. Chem., Int. Ed., 2007, 46, 7404 CrossRef PubMed.
- J. Richard, J. Joseph, C. Wang, A. Ciesielski, J. Weiss, P. Samorì, V. Mamane and J. A. Wytko, J. Org. Chem., 2021, 4, 3356 CrossRef PubMed.
- T. Gullion and J. Schaefer, J. Magn. Reson., 1989, 81, 196 CAS.
- A. Pines, M. G. Gibby and J. S. Waugh, J. Chem. Phys., 1972, 56, 1776 CrossRef CAS.
- B.-J. van Rossum, H. Förster and H. J. M. de Groot, J. Magn. Reson., 1997, 124, 516 CrossRef CAS.
- G. Copie, F. Cleri, Y. Makoudi, C. Krzeminski, M. Berthe, F. Chérioux, F. Palmino and B. Grandidier, Phys. Rev. Lett., 2015, 114, 066101 CrossRef CAS PubMed.
- A. J. Groszek, Proc. R. Soc. London, Ser. A, 1970, 314, 473 Search PubMed.
- A. T. Rösch, R. Reynaerts, B. A. G. Lamers, K. S. Mali, S. De Feyter, A. R. A. Palmans and E. W. Meijer, Chem. Mater., 2021, 33, 8800 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental and synthetic methods, solution NMR spectra, ssNMR 1H–31P FSLG HETCOR spectra, concentration dependent UV-visible spectra, Z-profile from AFM topography images, adsorption models. See DOI: https://doi.org/10.1039/d4nr00894d |
| ‡ The amount of signal obtained during CP depends on several parameters, including the intensity of dipolar coupling, T1rho relaxation and the contact time used in the experiment. Thus, CP spectra are usually not quantitative. |
| This journal is © The Royal Society of Chemistry 2024 |