
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceZwitterionic nanoparticles for thermally activated drug delivery in hyperthermia cancer treatment†
Camillo
Colli
 a,
Ilenia
Masi
b,
Emanuela
Jacchetti
a,
Silvia
Santoni
ac,
Mattia
Sponchioni
*a,
Bianca Maria
Colosimo
c,
Laura
Rosanò
b,
Manuela Teresa
Raimondi
a,
Emanuele
Mauri
*a and
Davide
Moscatelli
a
a,
Ilenia
Masi
b,
Emanuela
Jacchetti
a,
Silvia
Santoni
ac,
Mattia
Sponchioni
*a,
Bianca Maria
Colosimo
c,
Laura
Rosanò
b,
Manuela Teresa
Raimondi
a,
Emanuele
Mauri
*a and
Davide
Moscatelli
a
aDepartment of Chemistry, Materials and Chemical Engineering “G. Natta”, Politecnico di Milano, piazza L. da Vinci 32, 20133 Milan, Italy. E-mail: mattia.sponchioni@polimi.it; emanuele.mauri@polimi.it; Tel: +39 022399 3197 Tel: +39 022399 3397
bInstitute of Molecular Biology and Pathology, National Research Council (CNR), Rome 00185, Italy
cDepartment of Mechanical Engineering, Politecnico di Milano, Via La Masa, 1, 20156, Milan, Italy
First published on 7th June 2024
Abstract
Hyperthermia is considered a promising strategy to boost the curative outcome of traditional chemotherapeutic treatments. However, this thermally mediated drug delivery is still affected by important limitations. First, the poor accumulation of the conventional anticancer formulations in the target site limits the bioavailability of the active ingredient and induces off-site effects. In addition, some tumoral scenarios, such as ovarian carcinoma, are characterized by cell thermotolerance, which induces tumoral cells to activate self-protecting mechanisms against high temperatures. To overcome these constraints, we developed thermoresponsive nanoparticles (NPs) with an upper critical solution temperature (UCST) to intracellularly deliver a therapeutic payload and release it on demand through hyperthermia stimulation. These NPs were synthesized via reversible addition–fragmentation chain transfer (RAFT) emulsion polymerization and combine polyzwitterionic stabilizing segments and an oligoester-based biodegradable core. By leveraging the pseudo-living nature of RAFT polymerization, important physicochemical properties of the NPs were controlled and optimized, including their cloud point (Tcp) and size. We have tuned the Tcp of NPs to match the therapeutic needs of hyperthermia treatments at 43 °C and tested the nanocarriers in the controlled delivery of paclitaxel, a common anticancer drug. The NPs released almost entirely the encapsulated drug only following 1 h incubation at 43 °C, whereas they retained more than 95% of the payload in the physiological environment (37 °C), thus demonstrating their efficacy as on-demand drug delivery systems. The administration of drug-loaded NPs to ovarian cancer cells led to therapeutic effects outperforming the conventional administration of non-encapsulated paclitaxel, which highlights the potential of the zwitterionic UCST-type NPs as an innovative hyperthermia-responsive drug delivery system.
1. Introduction
Over the last few decades, the constant progress in polymer nanotechnology has revealed the pivotal role of nanoparticles (NPs) in addressing the main challenges of several tumoral applications, ranging from molecular imaging to controlled drug delivery and targeted therapy. The interest in polymer-based NPs has become two-fold.1 Firstly, like other nanocolloids, NPs are characterized by a high surface-to-volume ratio, which promotes their interfacial activity in an aqueous environment with either molecular or cellular processes, affecting cell response and growth. Secondly, the macromolecular structure can be tailored to specific therapeutic aims using different controlled polymerization methods.2 Recent works have demonstrated that the modern synthetic polymer chemistry ensures the covalent incorporation of monomers in desired sequences, thus providing an invaluable tool for tuning the physicochemical properties of the resulting NPs and their behavior in biological compartments (i.e., hydrophilic/hydrophobic, electrostatic, van der Waals, and π–π stacking interactions). A further degree of control of NP–cell interactions was introduced by materials capable of responding to environmental stimulation. Indeed, stimuli-responsive NPs have gained significant attention as efficient drug delivery vehicles in tumor immunotherapy, due to the opportunity of activating the drug release via specific endo- or exo-stimuli, including intracellular pH variations, a high concentration of glutathione, overexpression of enzymes and reactive oxygen species, and temperature changes.3,4 Thermoresponsive polymers are appealing for thermal therapies. In biomedicine, hyperthermia is regarded as a promising adjuvant strategy to counteract the progression of cancer in combination with radiotherapy or chemotherapy. Heat can directly alter the physical properties of cellular components, affecting multiple intracellular mechanisms and sensitizing cells to radiation and administration of chemotherapeutics.5,6 Promising clinical outcomes have been achieved in the treatment of breast cancer, cervical carcinoma, bladder cancer, head and neck tumors, soft tissue sarcoma and melanoma7–9 without significant side-effects in healthy cells.10,11 However, in some cases, the synergistic effects of heating and antineoplastic agents may be drastically reduced due to the activation of cell thermotolerance. In fact, tumor cells can develop the ability to protect themselves from high temperatures through upregulation of heat shock protein (HSP) and post-hyperthermia adaptation processes, such as cell cycle arrest in the G2-phase and changes in cell metabolism.12 This response decreases the therapeutic benefits, even though partial cell apoptosis still occurs.13 For these reasons, the curative treatment is still looking for novel approaches to overcome the current limitations.In this direction, NPs enabling the delivery of antineoplastic drugs in response to thermal activation could represent an important advance in tumor treatment. Several thermoresponsive nanoscaffolds are proposed in the literature, with efforts towards the development of lower critical solution temperature (LCST) nanocarriers.4,14,15 Indeed, the polymer chains of LCST-type nanocarriers shrink at temperatures above their LCST, inducing the release of the encapsulated drug. However, the drug is usually not entirely ejected from the NPs, due to a residual miscibility of the hydrophobic core with the payload beyond the LCST.16,17 This constrains the control of drug release performances.
On the other hand, the use of polymers characterized by an upper critical solution temperature (UCST) has recently emerged as a promising strategy to design NPs for thermic treatments. These polymers show a hydrophobic–hydrophilic transition and a single phase exists above their cloud point (Tcp). By exploiting this dynamic behavior, the UCST-type NPs can dissociate or swell, simultaneously releasing the loaded drug. Few UCST-type polymeric nanocarriers have been proposed so far for hyperthermia-mediated drug release to treat cancer, essentially focused on breast cancer18 and hepatocellular carcinoma.19 These NPs are composed of poly(acrylamide-co-acrylonitrile)-based copolymers,17,20,21 poly(N-acryloyl glycinamide) (PNAGA),22,23 ureido-derivatized polymers24 or polypeptides.15,25 However, in these cases, the poor control over the polymer microstructure is associated with a sub-optimal drug delivery system, which results in the current lack of appealing strategies for hyperthermia-mediated tumor treatments. For this reason, researchers have started developing zwitterionic NPs exhibiting the UCST transition. In fact, zwitterionic polymers are proved to enhance biocompatibility, reduce immune response, and can promote the cellular uptake of therapeutic agents.26–28 In addition, these polymers were reported to significantly reduce the protein adsorption, allowing the realization of stealth NPs with a prolonged circulation time. This is a crucial feature, as it enables the exploitation of these polymers as alternatives to polyethylene glycol (PEG), most often employed in this direction but recently proved to induce an accelerated blood clearance following repeated administrations due to the production of specific anti-PEG immunoglobulins.29–31
In this work, we propose the synthesis of UCST-type zwitterionic NPs and their validation as hyperthermia-activated drug delivery platforms for ovarian cancer treatment, a representative critical scenario characterized by cell thermotolerance up to 43 °C.32,33 The NPs were obtained through the self-assembly of poly(sulfobetaine-co-sulfabetaine) (p(SB-co-ZB)) copolymers chain-extended with a hydrophobic block of vinyl oligoesters. The block copolymers were produced via reversible addition–fragmentation chain transfer (RAFT) emulsion polymerization, which allowed us to modulate: (i) the Tcp of the NPs by changing the composition of the zwitterionic p(SB-co-ZB) segments and (ii) the size of the NPs by tuning the length of the hydrophobic portion. In this way, we have obtained a library of NPs exhibiting a UCST phase transition in the temperature range of 14–43 °C in physiological solution (i.e., 0.9% w/w NaCl), which can be optimized according to the specific therapeutic needs. As a case study to showcase the potential of these NPs, we have considered the in vitro hyperthermia treatment of ovarian carcinoma. SKOV3 cells exhibit thermal-resistant behavior, preserving ca. 98.8% viability, upon incubation at 43 °C, the conventional temperature of the hyperthermic intraperitoneal chemotherapy (HIPEC). We therefore selected the NP formulation with a Tcp close to 43 °C and validated these nanocarriers as on-demand release systems for paclitaxel (PTX), a known chemotherapeutic agent. Following the administration of PTX-loaded NPs to SKOV3, we demonstrated that the drug release could be induced by hyperthermia activation only, significantly affecting the metabolic activity of the target cells and outperforming the conventional administration of PTX in a non-encapsulated form. On the other hand, the drug was selectively retained at the typical physiological temperature of 37 °C.
To the best of our knowledge, a single application of UCST-type NPs is reported in the literature for hyperthermia ovarian treatment. These NPs were composed of poly(acrylamide-co-acrylonitrile)-b-poly[(oligo(ethylene glycol)methyl ether methacrylate)] and used as doxorubicin delivery systems to treat the OVCAR-3 cell line.20 However, these cells are more thermosensitive than SKOV3 and already exhibit a notable decrease in cell viability (ca. 21%) following the hyperthermia treatment only.34,35 Moreover, some limitations were debated by the authors regarding the therapeutic activity of the nanosystems, which required a higher dosage of the encapsulated drug compared to the free drug administration, under hyperthermia conditions, to achieve 50% inhibitory concentration (IC50).20
Here, we propose the innovative administration of zwitterionic thermoresponsive NPs to treat more hostile ovarian cancer cells combining hyperthermia and on-demand drug release, and provide preliminary inspiring outcomes towards the definition of a new strategy to counteract the progression of the disorder, still lacking an efficient therapeutic approach.
2. Experimental
2.1 Materials
2-Hydroxyethyl methacrylate (HEMA, 97%, MW = 130.14), ε-caprolactone (CL, 97%, MW = 114.14), tin(II) octoate (Sn(Oct)2, 92.5–100%, MW = 405.12), sodium sulfate (Na2SO4, ≥99%, MW = 142.04), 4,4′-azobis(4-cyanovaleric acid) (ACVA, ≥98%, MW = 280.28), 4-cyano-4-(phenylcarbonothioylthio)pentanoic acid (CPA, ≥97%, MW = 279.38), [2-(methacryloyloxy)ethyl]dimethyl-(3-sulfopropyl)ammonium hydroxide (SB, 97%, MW = 279.35), 2-(dimethylamino)ethyl methacrylate (DMAEMA, 98%, MW = 157.21), 1,3-propanediol cyclic sulfate (PCS, 98%, MW = 138.14), and sodium chloride (NaCl, ≥99.5%, MW = 58.44) were purchased from Merck KGaA (Darmstadt, Germany) and used as received without any further purification. Cyanine 7 NHS ester (Cy7, MW = 733.64) was purchased from Lumiprobe GmbH (Hannover, Germany), whereas the anticancer drug paclitaxel (PTX, MW = 853.91) was purchased form D.B.A. Italia s.r.l. (Segrate, Italy). Solvents were of analytical grade purity and used as received. NPs were stored at 4 °C.2.2 Characterization techniques
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 v/v solution. Polyethylene glycol standards were used to calibrate the system. The sample was prepared by dissolving 4 mg of polymer in the mobile phase, followed by filtration with a 0.45 μm nylon membrane.
:20 v/v solution. Polyethylene glycol standards were used to calibrate the system. The sample was prepared by dissolving 4 mg of polymer in the mobile phase, followed by filtration with a 0.45 μm nylon membrane.
2.3 Synthesis of zwitterionic copolymers with a UCST phase transition
The polyzwitterion was synthesized through RAFT copolymerization between sulfabetaine methacrylate (ZB) and sulfobetaine methacrylate (SB). According to our previous work,36 the procedure consists of two steps. The first stage concerned the synthesis of the ZB monomer via an addition reaction. Briefly, DMAEMA (2.5 g, 15.9 mmol) and PCS (2 g, 14.5 mmol) were separately dissolved in 2.5 and 17.5 mL of dry acetonitrile at RT, respectively. The methacrylate solution was added to the PCS one and the resulting mixture was kept under stirring for 72 h at 50 °C under inert conditions (N2 atmosphere). After cooling to RT, cold acetone (30 mL) was added to precipitate the monomer and the solid was collected by vacuum filtration and further washing with acetone. The product was dried in a vacuum oven at 30 °C to remove the residual solvent and characterized via1H-NMR using deuterium oxide (D2O) as a solvent to evaluate the purity of the synthesized ZB monomer (yield 94%).In the second stage, the p(SB-co-ZB) copolymer was synthesized by dissolving ZB and SB in a 1:4 v/v mixture of ethanol:acetic buffer (pH = 4.5) containing NaCl (1 M, with respect to the buffer volume) to promote the “salting in” effect. ACVA was used as the initiator and CPA was used as the chain transfer agent (CTA). The total monomer concentration (i.e., sum of ZB and SB) was set to 20% w/w and the initiator to CTA molar ratio was equal to 1:3. Different SB/ZB molar ratios were used in the synthesis of the zwitterionic copolymer, maintaining its degree of polymerization (DP) fixed to 200. In particular, the amount of SB was calculated to obtain a homopolymer DP (i.e., DPSB) equal to 105, 110, 115, 120 and 125, providing five different formulations of the final p(SB-co-ZB) (ESI†). This choice relies on preliminary evaluations of the impact of DPSB and DPZB on the UCST and the size of the NPs. As mentioned in a previous work,36 higher DPSB results in polymer phase separation at higher temperatures, which would shift the UCST of the NPs out of the range of interest for this application and lead to larger NPs.
As an example, the copolymer characterized by DPSB = 105 and DPZB = 95 (labelled as p(105SB-co-95ZB)) was synthesized by dissolving 254 mg (0.91 mmol) of SB and 243 mg (0.82 mmol) of ZB in 2.5 mL of an ethanol:acetic buffer mixture and adding 810 μg (2.88 μmol) of ACVA and 2.42 mg (8.66 μmol) of CPA. The resulting mixture was purged for 10 min by bubbling nitrogen and left under magnetic stirring at 65 °C (pre-heated oil bath) for 24 h. Successively, an aliquot was withdrawn and dried in a vacuum oven to evaluate the monomer conversion via1H NMR (D2O 0.9% w/w NaCl, 30 °C) and the molecular weight distribution via GPC analysis (ESI†). The polymer was purified by precipitation in acetone, followed by centrifugation to enhance the recovery. Finally, the product was dried in a vacuum oven at 35 °C to remove all residual solvent and stored at −20 °C.
2.4 Synthesis of caprolactone-based macromonomers
Ring opening polymerization (ROP) of ε-caprolactone (CL) was conducted to obtain the lipophilic macromonomer used in the NP synthesis. HEMA was chosen as the initiator and Sn(Oct)2 as the catalyst. Two oligomers, with different lengths of the aliphatic chain, were synthesized by using a molar ratio monomer:initiator equal to 3:1 and 5:1 to obtain HEMACL3 and HEMACL5, respectively, where the subscript refers to the average number of CL units in the macromonomer. According to the procedure discussed in our previous work,37 HEMACL5 was obtained as follows: CL (20 g, 175.2 mmol) and Na2SO4, used to keep the environment anhydrous, were mixed and heated at 125 °C. Then, a mixture of the initiator (4.56 g, 35 mmol) and the catalyst (70.9 mg, 0.175 mmol), pre-heated at the same temperature, was added dropwise to the monomer by means of a syringe under inert conditions (N2 flow). The reaction system was left under stirring for 3 h at 125 °C. The polymerization process was assessed by withdrawing an aliquot from the batch system and performing 1H-NMR (CDCl3, 25 °C) and GPC analyses to confirm the formation of the macromonomer. Finally, the system was cooled down to RT and stored at 4 °C. The same procedure was performed to obtain HEMACL3, varying the amount of the monomer (12 g, 105 mmol).
2.5 HEMA-Cy7 functionalization
To ensure the traceability of the polymeric nanoscaffolds in in vitro studies, HEMA was conjugated to Cy7 NHS ester. HEMA (9 mg, 69.15 μmol) and the cyanine compound (10 mg, 12.07 μmol) were dissolved in anhydrous DCM (1 mL) and left under stirring for 30 min in the dark at RT. The resulting solution was stored at 4 °C.2.6 Synthesis of UCST-type zwitterionic NPs
The thermoresponsive NPs were synthesized by chain-extending p(SB-co-ZB)200 with the caprolactone-derived macromonomer via RAFT emulsion polymerization. This strategy allowed the simultaneous formation of amphiphilic block copolymers and their self-assembly. The DP of the lipophilic block was set equal to 20 and different combinations were tested by changing the zwitterionic copolymers (i.e., different DPSB and DPZB) and selecting either HEMACL3 or HEMACL5 as the monomer. In a typical synthesis, p(SB-co-ZB)200 (160.2 mg, 2.79 μmol) and NaCl (190 mg) were dissolved in acetic buffer (pH = 4.5, 5.17 mL), whereas HEMACL3 (27.8 mg, 56 μmol) or HEMACL5 (39.5 mg, 56 μmol) was dissolved in DMSO (1 mL). The latter was added dropwise to the zwitterionic mixture and the resulting emulsion was purged with N2 for 10 min and heated to 65 °C. ACVA (260 μg, 0.93 μmol) was finally added to the system and left under stirring for 24 h. The obtained product was purified through dialysis (SpectraPor, regenerated cellulose membrane, Mw cut-off = 6–8 kDa) against 0.9% w/w NaCl solution for 1 day at RT and stored as suspension at 4 °C. The NP concentration in a physiological solution was estimated by drying aliquots (3 × 100 μL) at 50 °C and weighing the solid residue.The synthesis of Cy7-labelled NPs was performed following the same procedure by just adding 3 drops of HEMA-Cy7 to the polymer mixture prior to the addition of ACVA.
2.7 Encapsulation and release of paclitaxel
Paclitaxel was encapsulated into the NPs via flash nanoprecipitation.38 Based on the UCST behavior of the selected NPs, PTX loading was performed at 45 °C (above the UCST transition) and the instruments used in this stage (i.e., a 5 mL syringe, a needle and glass vials) were heated and equilibrated at the same temperature before usage. A mass ratio of 0.05 mgPTX mgNP−1 was set for the procedure. 200 μg μL−1 drug solution (10.5 mg PTX in 52.5 μL of DMSO) and NP suspension (30 mg mL−1) were heated to 45 °C and 7 mL of the latter were withdrawn and added to the former solution. The final mixture was aspired and ejected 5 times from a needle-equipped syringe to provide adequate turbulent conditions to lead to mixing and drug encapsulation. The amount of entrapped drug, i.e. the encapsulation efficiency (EE%), was determined as follows: aliquots (3 × 100 μL) of the resulting system were collected and dried under air flow, re-dispersed in 0.5 mL of acetonitrile, vortex-stirred for 60 s, and the supernatant was analyzed by HPLC. EE% was calculated as (eqn (1)): | (1) |
 | (2) |
The drug release profile was estimated at two different temperatures, 37 °C and 43 °C, to assess the NP UCST-activated drug delivery performances. In both cases, 1 mL of PTX-loaded NP suspension was added to a dialysis membrane (Float-A-Lyzer dialysis cassette, 1 mL, Mw cut-off: 8–10 kDa) and submerged in 0.9% w/w NaCl solution (250 mL) kept at 37° or 43 °C over time according to the chosen experimental conditions. At defined time points, aliquots (100 μL) were withdrawn from the dialysis cassette, dried under air flow at RT, and the residue was dissolved in acetonitrile (500 μL) for HPLC analysis. Based on the PTX calibration curve, the amount of released drug was estimated as reported in the following equation (eqn (3)):
 | (3) |
2.8 Cell culture
The human ovarian serous adenocarcinoma cell line SKOV3 was obtained from the American Type Culture Collection (ATCC) (LGC Standards, Teddington, UK) and cultured in McCoy's 5A medium (Cat# 26600-023; Thermo Fisher Scientific) supplemented with 10% fetal bovine serum, 10000 U mL−1 penicillin, and 10 mg mL−1 streptomycin. The cells were incubated at 37 °C under a humidified atmosphere with 5% CO2.
2.9 Hyperthermia cell treatment
The hyperthermia sensitivity of SKOV3 was assessed to identify the thermal tolerance range ensuring a cell viability at least equal to 95% and the lethal temperature producing 50% cell death (i.e., LT50). The cells were seeded at a density of 6.5 × 103 cells per cm2 into a 96-well plate for 24 h and then exposed to different temperatures in the range of 39–53 °C with a 2 °C step increase (i.e., 39 °C, 41 °C, 43 °C, 45 °C, 47 °C, 49 °C, 51 °C and 53 °C) for 1 h. Afterwards, the media were replaced with fresh aliquots and the cells were incubated under normal culture conditions (37 °C, 5% CO2) for 24 h. Effects of the hyperthermia treatment on cell metabolic activity were evaluated through the MTT assay according to the manufacturer's instructions. Briefly, the activity of mitochondrial dehydrogenases in living cells was measured in terms of absorbance at 570 nm (TECAN Spark) after 3 h of exposure to MTT solution (0.5 mg mL−1) in PBS at 37 °C and 5% CO2. The absorbance values obtained in the absence of the cells were used for background subtraction. The cells not exposed to hyperthermia were used as a control for normalization. LT50 was calculated via curve fitting. Experiments were performed in triplicate.2.10 NP biocompatibility
A preliminary evaluation of the potential UCST NP side-effect on cell biological activity was conducted through the MTT assay. 6.5 × 103 cells per cm2 were seeded into a 96-well plate and incubated for 24 h. The NP suspension was heated to 43 °C and filtered twice: firstly, through a 0.45 μm nylon filter and then through a 0.2 μm sterile nylon filter. The filtration process did not lead to a significant mass loss of NPs and a stock suspension at a concentration of 25 mg mL−1 was prepared. Serial dilutions of the latter, ranging from 2.5 mg mL−1 to 25 μg mL−1, were administered to SKOV3 and incubated for 24 h at 37 °C and 5% CO2. Then, the metabolic activity assay was performed as discussed in the previous section.2.11 Fluorescence staining and confocal microscopy for NP internalization
Fluorescence microscopy was used to visualize the cellular internalization of the NPs. SKOV3 cells were seeded at a concentration of 6.5 × 103 cells per cm2 into an 8-well chamber slide (Nunc Lab-Tek-8-chamber wells, ThermoFisher Scientific, Italy) and incubated for 24 h. Then, NPs were filtered as described in the previous section and administered at a concentration of 1.25 mg mL−1. After 24 h, the cells were stained with calcein-AM to confirm the cell viability following NP internalization or fixed and stained with phalloidin-FITC and Hoechst 2232 to observe the cell morphology and NP localization. In the first case, the cells were incubated with 1 μM calcein-AM (ThermoFisher Scientific, Italy) for 5 min and then rinsed with fresh medium. In the second case, the cells were fixed in paraformaldehyde (4% in PBS) for 15 min at RT and then incubated 45 min with 0.5 μg ml−1 phalloidin-FITC dye (Sigma Aldrich – Merck, Italy). After washing three times with Tween (0.1% in PBS), cell nuclei were labeled with 1 μM Hoechst 2232 (10 min incubation). Micrographs were collected using a Nikon AR1+ equipped with an incubator and diode lasers (λexcitation 405/488/561/640 nm). The stained cells were imaged with a 40× water immersion objective (A.N.1.15, WD 0.60). The pinhole was set to 0.9 Airy unit. 1024 × 1024 pixel micrographs were acquired as z-stack images with a stem equal to 0.3 μm.2.12 Metabolic activity assay
Cell metabolic activity was evaluated following the administration of the PTX-loaded NPs under hyperthermic conditions to validate the potential therapeutic effect of drug release mediated by the NP UCST behavior. SKOV3 cells were seeded at the density of 6.5 × 103 cells per cm2 in growth medium and incubated for 24 h. PTX-loaded NPs (4.4 ngPTX mgNP−1) were prepared as follows: the NP suspension was filtered through a 0.45 μm nylon filter and a 0.2 μm sterile nylon filter at 43 °C. The NP loading was performed as described in section 2.7, starting from a polymer concentration of 25 mg mL−1.The resulting suspension was added to the cells at a concentration of 1.25 mg mL−1 and incubated at 37 °C for 24 h. Successively, the cells were exposed to 43 °C hyperthermia treatment for 1 h, followed by medium exchange and incubation at 37 °C for 12 h or 24 h. The cells incubated with pristine (i.e., non-encapsulated) PTX solution represented the reference group. All conditions were set to have the same final concentration of 1.3 nM drug in the culture medium corresponding to the IC50 value (i.e., the concentration at which the drug produces a 50% cell viability reduction, ESI†) under hyperthermia conditions. Additionally, the cells treated with the same volume of DMSO (1% v/v) required for the preparation of the drug solution are set as the internal control to demonstrate the absence of evident cytotoxic effects related to the administration of the organic solvent. The MTT assay was performed as previously mentioned, 12 h and 24 h post-treatment Additionally, SKOV3 cells were incubated with PTX-loaded NPs at 37 °C without the hyperthermia stage following the same procedure. Experimental data were collected from three independent experiments for each condition.
2.13 Live/dead assay
The cell viability was evaluated through live/dead assay following the cell uptake of the drug-loaded NPs and the hyperthermia treatment. 6.5 × 103 cells per cm2 were seeded into a 12-well plate and allowed to adhere for 24 h (37 °C, 5% CO2). The investigated conditions were the same as described in section 2.12 (i.e., administration of PTX solution, PTX-loaded NPs, and DMSO 1% v/v as internal control). After 24 h treatment incubation, 1 h hyperthermia was performed and the culture medium was replaced with fresh aliquots. Then, the cells were incubated for 24 h at 37 °C. Subsequently, the Cyto3DTM live–dead assay kit (TheWell Bioscience, Inc., North Brunswick, New Jersey, USA) was used to determine the live/dead nucleated cells through a dual-fluorescence system of acridine orange (AO) and propidium iodide (PI), both nuclear staining (nucleic acid binding) dyes. Live nucleated cells fluoresced green and dead nucleated cells fluoresced red. The samples were analyzed using a Nikon Eclipse Ti2 microscope with a 10× (1.4 NA) objective equipped with a CrestOptics X-Light V3 confocal spinning disk and a back illuminated Kinetix sCMOS camera. Images were acquired with NIS Elements 5.4 software and analyzed with the FIJI image processing package using the mean gray value of the green and red channels separately, and then the green/red ratio was calculated as a representative of the cell viability. Zoomed insets were obtained using FIJI “zoom in images and stacks action tool”.2.14 Statistical analysis
Data were analyzed using Prism ver. 10.1.1 (GraphPad Software, San Diego, CA) and reported as mean ± SD if not otherwise specified. The statistical significance, set at the 0.05 level, was evaluated through one-way analysis of variance (one-way ANOVA) followed by Tukey's multiple comparison test.3. Results and discussion
3.1 Characterization of p(SB-co-ZB) copolymers and thermoresponsive behavior
First, the ZB monomer was synthesized in house by reacting PCS and DMAEMA. The monomer purity and composition were confirmed by NMR analysis (Fig. 1a). Then, the synthesis of p(SB-co-ZB) copolymers with different monomer ratios was performed via RAFT polymerization to ensure high control on the polymer interchain composition and living end-group fidelity. The synthesis of the zwitterionic copolymers was confirmed by NMR and ATR-IR analyses. Fig. 1c shows the 1H-NMR spectrum of p(110SB-co-90ZB), as the representative copolymer, where the characteristic peaks of SB and ZB are highlighted. In particular, the signals related to the vinyl hydrogens of the monomers (at 6.09 and 5.71 ppm for ZB and at 6.13 and 5.75 ppm for SB, Fig. 1a and b) are not detectable, indicating the effective polymerization. Additionally, a slight shift of the methylene ester peak of each monomer is visible. The monomer conversion (χ) was estimated considering the integral of the vinyl protons and the methyl protons of the dimethylammonium groups (–N+(CH3)2) according to the following equation (eqn (4)): | (4) |
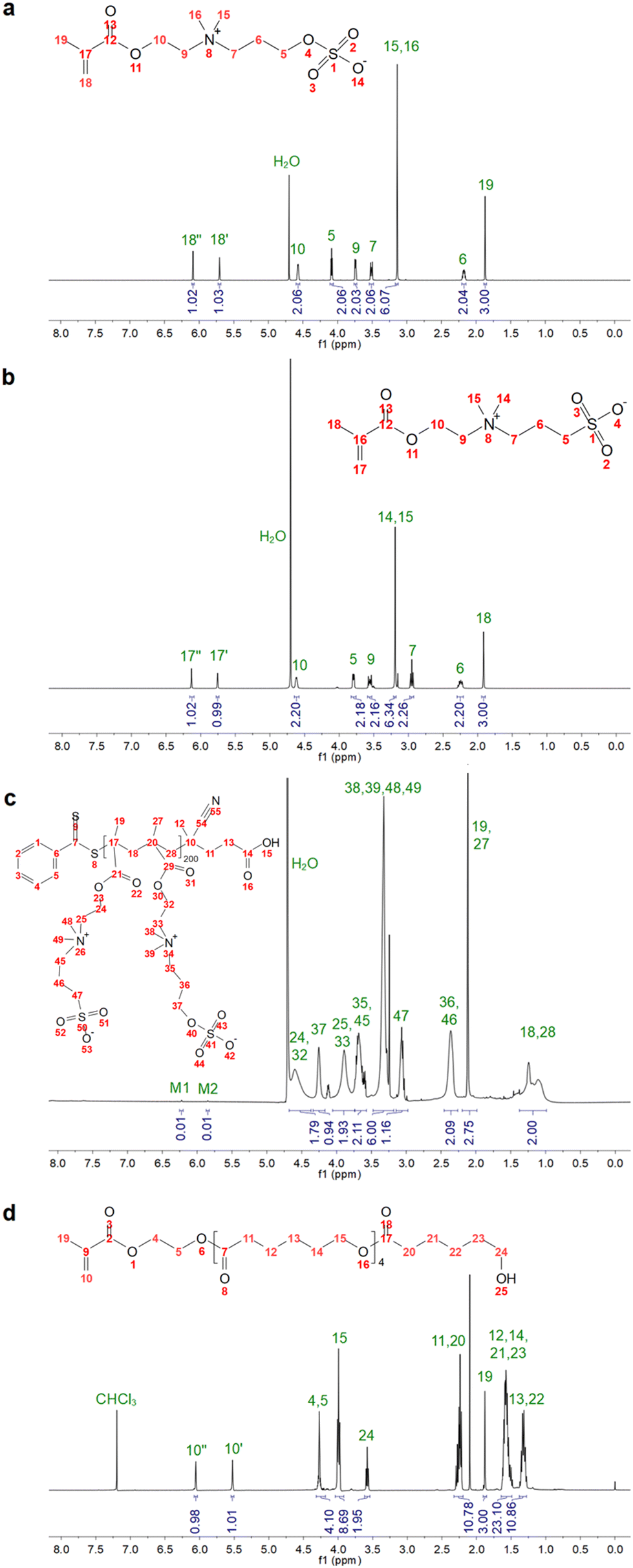 | ||
| Fig. 1 1H-NMR spectra of the synthesized methacrylate ZB (a), SB (b), zwitterionic copolymer p(110SB-co-90ZB) (c), and HEMACL5 (d). Peaks M1 and M2 are the residual vinyl groups of ZB and SB. | ||
The SB and ZB molar fraction (r) and the corresponding DP in the synthesized copolymers were estimated by considering the ratio between the integral (i.e., AUC) of the methylene peak linked to the sulfate or sulfonate moiety (i.e., 47 and 37 for –CH2CH2SO3− and –CH2CH2OSO3−, respectively) and their sum according to eqn (5)–(8):
 | (5) |
 | (6) |
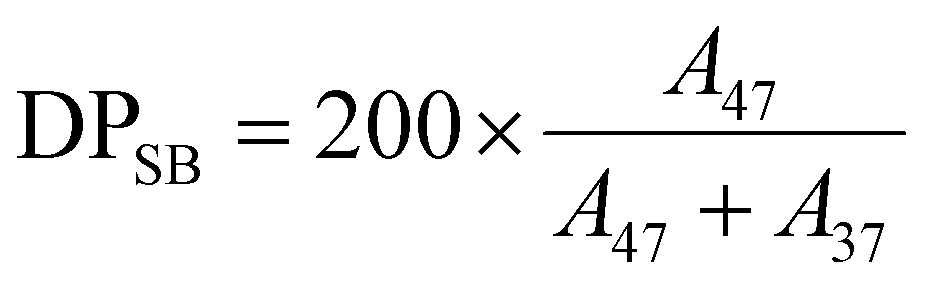 | (7) |
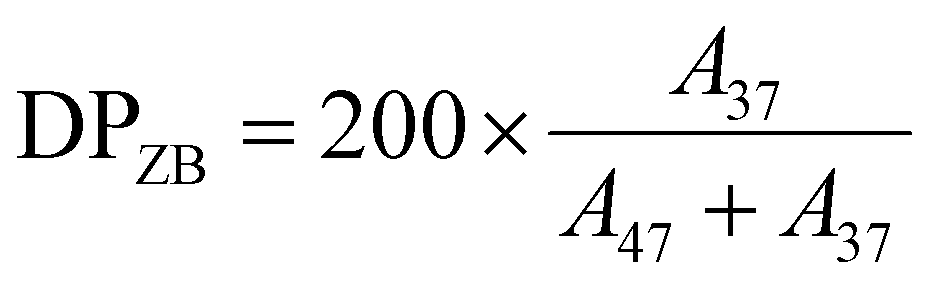 | (8) |
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O at 1170 cm−1; νs SO at 1036 cm−1 in the SB spectrum), the SO4− stretching (νas SO at 1222 cm−1; νs SO at 1021 cm−1 in the ZB spectrum), and the CC stretching (1640 cm−1). In the spectrum of the zwitterionic copolymer, the absence of the CC absorption band and the presence of the –OH stretching at 3432 cm−1, attributable to the RAFT agent, confirming the conjugation between the two monomers (the secondary peak at 1659 cm−1 could be ascribed to the CO stretching of the CPA carboxylic acid group).
O at 1170 cm−1; νs SO at 1036 cm−1 in the SB spectrum), the SO4− stretching (νas SO at 1222 cm−1; νs SO at 1021 cm−1 in the ZB spectrum), and the CC stretching (1640 cm−1). In the spectrum of the zwitterionic copolymer, the absence of the CC absorption band and the presence of the –OH stretching at 3432 cm−1, attributable to the RAFT agent, confirming the conjugation between the two monomers (the secondary peak at 1659 cm−1 could be ascribed to the CO stretching of the CPA carboxylic acid group).
 | ||
| Fig. 2 ATR-FTIR spectra of ZB (a), SB (b), the zwitterionic copolymer p(110SB-co-90ZB) (c), HEMACL5 (d), and UCST-type NPs (e). The main characteristic peaks of the specimens are highlighted: CO stretching (light blue), CC stretching (light red), the quaternary ammonium group (orange) and the distinctive bands of the ZB sulfate group, the SB sulfonate group and HEMACL5 C–C(O)–O (violet). | ||
The synthesized copolymers were characterized in terms of UCST behavior. Their phase transition in a physiological solution occurred in the temperature range of 13–21 °C and resulted in the formation of liquid droplets (Fig. 3a) which, through coalescence, gave rise to a homogeneous polymer-rich phase. Upon UCST transition, the intra- and intermolecular interactions of the polymer chains (i.e., hydrogen bonding and electrostatic interactions) promoted the formation of polymer-rich droplets, which exhibited a superficial hydration layer, due to the solvation of the charged terminal groups and the interfacial hydrogen bonding.41 The hydration shell minimizes the adsorption of macromolecules from the external environment and enhances the stability of the separated phase. This surface resistance, linked to the material hydrophilicity, is a pivotal feature enabling the use of p(SB-co-ZB) for the design of nanocarriers for drug delivery aims, thanks to the advantage of preventing protein adsorption from the biological media and reducing blood clearance and adverse immunological responses.42–44 This behavior is not found in other UCST-type polymers such as poly(acrylamide-co-acrylonitrile) copolymers, ureido derivatives and poly(N-acryloyl glycinamide), highlighting the potential specific benefits of betaine-based polymers.22,45,46
 | ||
| Fig. 3 (a) Brightfield images of p(110SB-co-90ZB), as a representative zwitterionic copolymer, above (45 °C) and below (25 °C) the Tcp. Scale bar = 50 μm. (b) Correlation of DPZBvs. Tcp for the synthesized zwitterionic copolymers. | ||
To characterize the thermoresponsive behavior for our samples, we performed turbidimetry studies by cooling down the polymer solutions (5 mg mL−1) from 35 °C, and we defined as the cloud point (Tcp) the temperature leading to a reduction to 50% of the initial transmittance, indicative of the UCST behavior. We observed that by reducing the units of ZB in the copolymer (and hence DPZB), the temperature for this polymer phase separation decreased with a linear trend (Fig. 3b and ESI†). This provided us with a powerful tool for tuning the phase separation of these zwitterionic copolymers by simply acting on the stoichiometry of the RAFT copolymerization of SB and ZB.
This tuning can be therefore advantageously exploited to adapt the material response to the specific drug delivery application of interest.
3.2 Characterization of UCST-type NPs
After having assessed the UCST behavior for the betaine-based copolymers, we synthesized a library of thermoresponsive zwitterionic NPs through the combination of the p(SB-co-ZB) copolymers and the HEMACLn macromonomers (where n is the length of the oligocaprolactone chain, equal to 3 or 5). The latter were produced via ROP and used as chain extenders of the zwitterionic counterpart (which works as a macromolecular chain transfer agent) via RAFT emulsion polymerization, grafting hydrophobic motifs to the polymeric chain (Fig. 4a). The 1H-NMR spectrum of the caprolactone-based macromonomer HEMACL5 is shown in Fig. 1d, where the signals related to the vinyl protons are clearly visible at 6.07 ppm and 5.53 ppm; additionally, IR analysis (Fig. 2d) shows the characteristic peaks of the C–C(O)–O band in the range of 1100–1250 cm−1, CO stretching at 1720 cm−1, C–H stretching at 2850–3000 cm−1 (νas CH2 at 2930 cm−1; νs CH2 at 2875 cm−1) and –OH stretching at 3400 cm−1. Similar considerations can be inferred from the HEMACL3 spectrum (ESI†).
 | ||
| Fig. 4 (a) RAFT emulsion polymerization using the generic zwitterionic copolymer (DP = 200) as a macromolecular chain transfer agent and HEMACLn as a monomer and the consequent NP formation. (b) TEM image of the 110-90-5 NPs recorded by staining the warm polymer suspension above its Tcp. (c) Representative curve of the hydrodynamic diameter of the NPs as a function of the temperature. The Tcp is highlighted in red (39.6 °C) and evaluated as an inflection point of the curve. According to the UCST behavior, the NPs resulted in micrometric clusters below Tcp, due to the phase separation, whereas they were well-dispersed at temperatures higher than Tcp. The graph is related to the NPs composed of p(110SB-co-90ZB) and HEMACL5, as an example. (d) Correlation between the Tcp of NPs and the units of ZB in the zwitterionic portion of the polymer: upon increasing ZB, the UCST threshold increases. Data related to the NPs containing HEMACL3 are reported in blue, whereas the data of HEMACL5-based NPs are reported in black. | ||
The chemical characterization of the synthesized NPs was performed via IR analysis. As reported in Fig. 2e, the distinctive peaks of the starting materials can be identified (i.e., CO stretching, the quaternary ammonium group, sulfate and sulfonate bands), confirming the comb-like structure of the final block copolymer.
Following RAFT emulsion polymerization, we obtained NPs characterized by a spherical morphology (Fig. 4b) with a narrow particle size distribution (Table 1). In particular, the average hydrodynamic diameter (DH) was affected by the number of caprolactone units in the copolymer. Indeed, DLS analysis showed the NP size in the range of 16–26 nm when HEMACL3 was used as the macromonomer, whereas the hydrodynamic diameter was in the range of 170–300 nm for HEMACL5-based NPs. Considering that the DP of HEMACLn was set to 20 for all specimens, the effect on the NP size could be ascribed to the length and the steric hindrance of the lipophilic chain, which promoted a different self-assembling.36
| NPs name | Composition | D H (nm) | PDI (−) | ζ-Pot. (mV) | T cp (°C) | T hyp (°C) | |
|---|---|---|---|---|---|---|---|
| Copolymer | Macromonomer | ||||||
| 105-95-3 | p(105SB-co-95ZB) | HEMACL3 | 16.6 | 0.24 | −9.1 | 28.1 | 30 |
| 110-90-3 | p(110SB-co-90ZB) | 20.9 | 0.25 | −9.7 | 27.3 | 29 | |
| 115-85-3 | p(115SB-co-85ZB) | 23.3 | 0.25 | −9.2 | 21.4 | 24 | |
| 120-80-3 | p(120SB-co-80ZB) | 24.4 | 0.35 | −7.9 | 15.6 | 20 | |
| 125-75-3 | p(125SB-co-75ZB) | 26.3 | 0.31 | −8.1 | 13.5 | 15 | |
| 105-95-5 | p(105SB-co-95ZB) | HEMACL5 | 174 | 0.22 | −9.3 | 42.8 | 48 |
| 110-90-5 | p(110SB-co-90ZB) | 185 | 0.24 | −9.2 | 39.6 | 43 | |
| 115-85-5 | p(115SB-co-85ZB) | 256 | 0.28 | −9.3 | 37.3 | 39 | |
| 120-80-5 | p(120SB-co-80ZB) | 289 | 0.22 | −9.3 | 36.4 | 40 | |
| 125-75-5 | p(125SB-co-75ZB) | 302 | 0.31 | −9.4 | 29.7 | 34 | |
The UCST behavior of the NPs was evaluated via DLS in physiological solution by recording the NP size as a function of temperature, setting a heat-up ramp of 1 °C/5 min. The Tcp was defined as the temperature corresponding to the inflection point of the obtained curves size vs. temperature. Below this critical temperature, the formation of a polymer-rich phase led to a cloudiness of the sample and a refractive index change detected by DLS measurement. As reported in the example of Fig. 4c (see also the ESI†), the NPs present a constant hydrodynamic diameter above the UCST threshold, indicating a stable nanosystem. This is ascribed to the exposure of the hydrophilic p(SB-co-ZB) to the NP surface, providing colloidal stability. On the other hand, a significant size increase occurs below the Tcp, with a decrease of the scattering intensity recorded by DLS, which indicates the formation of large NP clusters/agglomerates generating a separated phase over time (movie in the ESI†). This behavior can be justified considering the phase separation of the polyzwitterionic chains, which in this hydrophobic state are no longer able to provide colloidal stability and hence to prevent the aggregation of multiple NPs. This characteristic response for polymers exhibiting a UCST and grafted to hydrophobic chains has been confirmed by previous studies.20,36,47 All the investigated NPs exhibited this behavior, confirming the thermoresponsive feature inherited by the betaine-based copolymers. In fact, a linear correlation between the SB/ZB ratio and the Tcp already observed for p(SB-co-ZB) can be reconfirmed for these block copolymers (Fig. 4d). The Tcp decreased with reducing the ZB units, in analogy to the behavior of the pristine zwitterionic copolymer. However, the conjugation of HEMACLn led to an increase of the Tcp in all NPs compared to the values corresponding to the pristine zwitterionic copolymers, and this variation was higher with the use of the longer chain (i.e., HEMACL5). This might be related to the reinforced inter-/intra-molecular hydrogen bonding and van der Waals interactions between the hydrophobic moieties, due to the increased chain length.47 It can be speculated that higher heating temperatures are required to decrease the enthalpic attraction in combination with hydrophobic effects in chain–chain interactions,48 leading to an increase in Tcp value.
Table 1 summarizes the size, ζ-potential, and Tcp values for all synthesized NPs. The specimens are labelled referring to their zwitterionic-caprolactone composition: as an example, NPs 110-90-5 refers to the nanocarriers obtained by polymerization between p(110SB-co-90ZB) and HEMACL5.
Additionally, Thyp indicates the first temperature value at which NPs are well dispersed (lower plateau), with the zwitterionic portion extended in the water phase. This temperature represents the reference for the hyperthermia conditions. Indeed, at Thyp, the NPs are dispersed and the hydration of the zwitterionic layer as well as the drastic increase in the surface exposed to the buffer solution can promote the enhanced release of an encapsulated drug.
Referring to these considerations, the design of zwitterionic NPs with tunable UCST behavior and size can be performed through the rational combination of zwitterionic copolymers and lipophilic counterpart. Indeed, at a constant SB/ZB composition, changing the length of the caprolactone-based macromonomer results in nanocarriers with different Tcp values to address specific application needs. In this work, we have selected NPs exhibiting a Tcp closest to the thermotolerance value of the investigated cell line (i.e., 43 °C, as discussed in section 3.4). 110-90-5 NPs were characterized by a Tcp = 39.6 °C and a Thyp = 43 °C, equal to the boundary value of SKOV thermotolerance and chosen as a candidate for the evaluation of the therapeutic benefits.
It is worth highlighting that slightly negative surface potentials were invariably measured for all the samples. Although the zwitterionic monomers SB and ZB are expected to be electroneutral because of the balance between the negative sulfate/sulfonic groups and the positive ammonium groups, the negative ζ-potential was ascribed to the RAFT agent exploited in polymer synthesis. As shown in Fig. 4a, it displays a carboxyl group, which is expected to be dissociated at physiological pH. However, the low absolute value of the ζ-potential is expected not to play a major role in the NP uptake by the cells.
3.3 On-demand drug release
The performance of our UCST-type NPs as thermoresponsive drug delivery systems was investigated using PTX as a candidate drug. PTX is a diterpenoid taxane derivative, commonly used as an antineoplastic agent in ovarian cancer treatment, to inhibit cell proliferation and metabolic activity.49 The drug loading was performed at 45 °C, a temperature that ensures the proper dispersion of the NPs and prevents the thermal degradation of the chemotherapeutic, preserving its effectiveness.32,50 The resulting encapsulation efficiency was 98.9% as detected by HPLC and the drug loading content equal to 4.95%. We investigated the PTX release at two different temperatures: 37 °C and 43 °C, below and above the Tcp of the NPs. These values are representative of the physiological and hyperthermia conditions, respectively. As reported in Fig. 5, at 43 °C, PTX release was almost completed in 2 h, with ca. 85% of the encapsulated amount released after 1 h, whereas at 37 °C, the drug was efficiently retained in the NPs for a longer time, with ca. 4.5% of PTX released up to 24 h. These results confirmed the opportunity to control the drug release by changing the thermal conditions according to the UCST behavior of the NPs and their potential as stimuli-responsive drug release systems. In fact, a fast release of PTX, largely driven by diffusion mechanisms,36 occurred above the Tcp (i.e. at 43 °C), whereas under physiological conditions, below Tcp (i.e., 37 °C), the NPs were capable of retaining the encapsulated drug over time. These considerations point out the proposed NPs as a switchable temperature-triggered drug delivery system. The NP suspension could be stored in the coagulated state below Tcp until use, then pre-heated above the UCST threshold to encapsulate the drug, and finally administered to the cells under physiological conditions. Indeed, only after hyperthermia stimulation, the NPs would rapidly release the drug, establishing an on-demand drug delivery strategy.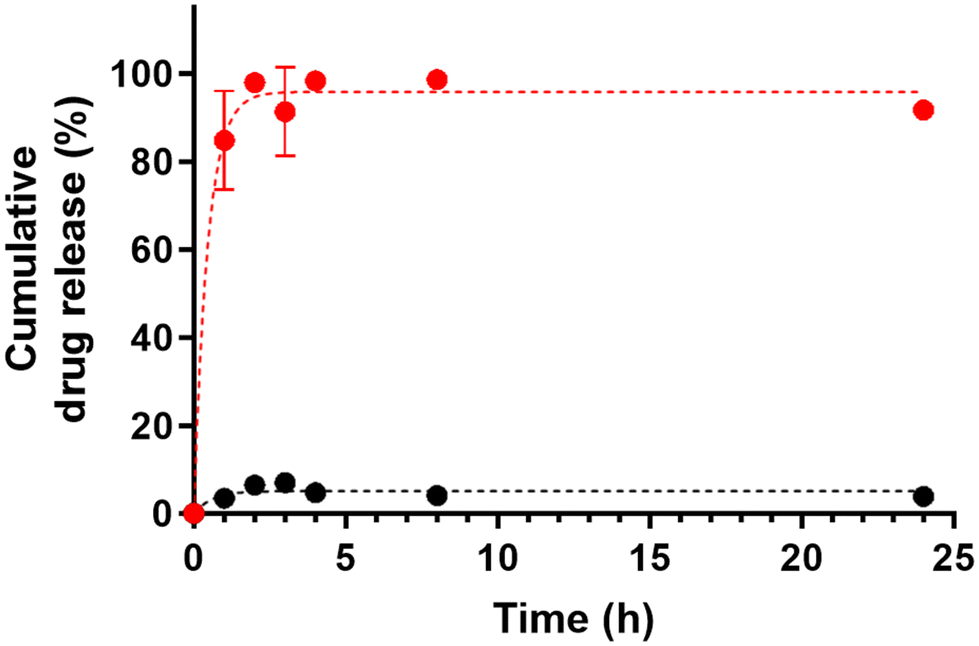 | ||
| Fig. 5 Release profiles of PTX delivered by p(110SB-co-90ZB)-b-HEMACL5 NPs at 43 °C (red) and 37 °C (black) measured by HPLC. | ||
3.4 Cell viability and NP internalization
SKOV3 cells were chosen as a representative ovarian cancer cell line exhibiting a significant tolerance to hyperthermia treatments.33 We exposed the cells to 1 h hyperthermia, equivalent to the duration of hyperthermic intraperitoneal chemotherapy (HIPEC), at different temperatures, and we evaluated the cell thermal sensitivity. SKOV3 cells preserved their viability and metabolic activity up to 43 °C (cell viability post-treatment 98.8%) and were characterized by a LT50 equal to 47.7 °C (Fig. 6a). This suggested that at 43 °C, a common temperature for hyperthermia treatments, the cells were resistant and no hyperthermia-related cytotoxic effects or necrosis could be induced. Evidently, the cell thermotolerance restricts the efficacy of the traditional HIPEC-based antitumor treatments and introduces the necessity of alternative strategies. In particular, the chosen 110-90-5 NPs represent a promising candidate for on-demand drug delivery stimulated by hyperthermia at 43 °C. Indeed, due to their UCST behavior, they would enable the intracellular release of PTX at the target temperature.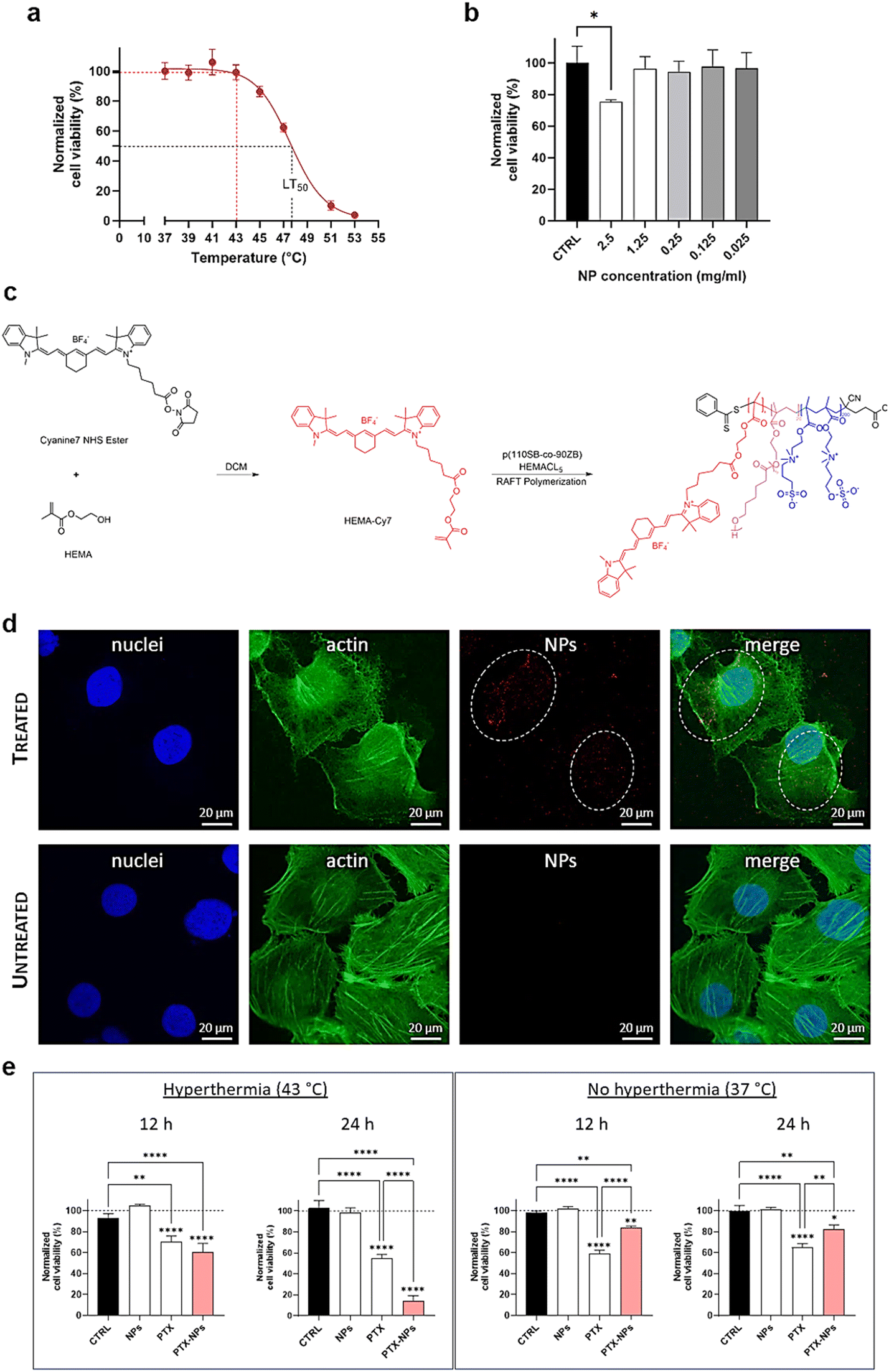 | ||
| Fig. 6 (a) Temperature–response curve of SKOV3 viability using the MTT assay. The solid line represents the fitted curve according to the four-parameter logistic curve, defining an LT50 of 47.7 °C. The hyperthermia conditions (43 °C) and the corresponding cell viability are marked in red dashed lines. (b) Cytocompatibility of the NPs by the MTT assay. Viability levels have been normalized to the CTRL group (i.e., untreated cells) and expressed as the mean ± SD. (c) Scheme for the synthesis of the fluorescent HEMA-Cy7 monomer and the structure of the resulting dye-labelled copolymer. (d) Representative confocal micrographs of the internalization of Cy7-labelled NPs (in red) in SKOV3 (treated) after 24 h from hyperthermia treatment. Micrographs of the untreated cells are also reported (untreated). Actin cytoskeleton (in green) was stained with FITC-labeled phalloidin and nuclei (in blue) were counterstained with Hoechst 2232. Scale bar = 20 μm. (e) PTX-induced cytotoxicity in SKOV3 at 12 h and 24 h after the administration of the free drug (PTX) and drug-loaded NPs (PTX-NPs) following exposure to hyperthermia treatment and without it, respectively. In all groups, the therapeutic effects are expressed in terms of cell viability levels normalized against their internal controls (i.e., untreated cells, dashed line) measured through the MTT assay. The cells treated with the same volume of DMSO (1% v/v) required for the preparation of the drug solution are set as an additional control (CTRL), highlighting the absence of relevant cytotoxic effects related to the organic solvent. The results are the mean ± SD of three independent experiments. Statistical analysis was performed using one-way ANOVA. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Non-significant outcomes (i.e., ns) are intended for all comparisons not highlighted in the graph; they are not reported for the sake of clarity. | ||
To evaluate the cytocompatibility of pristine NPs (without drug payload), a preliminary analysis was conducted via the MTT assay. NP suspensions at different concentrations were pre-heated to 43 °C and directly added to the cell medium. Their effect on cell viability was then assessed after 24 h. Fig. 6b reports that the NP-treated cells showed no significant differences compared to the control (i.e., cells without the NPs) up to a concentration of 1.25 mg mL−1, indicating that the NPs were biocompatible. A slight cytotoxic effect (cell viability 80%) was found at 2.5 mg mL−1 (p < 0.05) only. Dose-dependent cell responses are usually established to define the optimal concentration range which avoids lethal accumulation of NPs in tissues and organs.51,52
According to these data, we selected the concentration 1.25 mg mL−1 for the investigation of the NP-mediated therapeutic effects in hyperthermia. The NPs were labeled with Cy7 (Fig. 6c), as described in section 2.6, to ensure their in vitro tracking. The functionalization did not significantly affect the physicochemical properties of the nanosystems. The internalization process of NPs was assessed by fluorescence microscopy (Fig. 6d), with confocal images showing the NP distribution in the cytosol after 24 h administration (3D rendering of z-stack micrographs is provided in the ESI†). This confirmed the uptake of the nanocarriers and their potential as drug delivery systems in the hyperthermia treatment of ovarian carcinoma.
3.5 Effect of hyperthermia-induced release of PTX in SKOV3
The validation of our drug-loaded thermoresponsive system was performed in vitro, comparing the therapeutic effect of NP-mediated PTX release with the conventional administration route of the drug under hyperthermia conditions. We incubated both drug-loaded NPs and free PTX (i.e., non-encapsulated) for the same time and then treated cells with 1 h hyperthermia at 43 °C. As a further comparison, an in vitro evaluation was conducted also at 37 °C, without hyperthermia treatment, to demonstrate the potential of the proposed NPs to act as drug reservoir under physiological conditions and limit the cytotoxic effects in the absence of thermal activation. The drug concentration was set equal to 1.3 nM, corresponding to the sublethal value IC50, as estimated through the dose–response curve at 24 h post-hyperthermia on SKOV3 cells cultured in adhesion using the MTT assay (ESI†). Fig. 6e shows the evaluation of the cell metabolic activity through the MTT assay up to 24 h. Following hyperthermia, the NP UCST behavior promoted an intracellular release of PTX, resulting in an enhanced therapeutic effect of the drug.In detail, after 24 h, the NP-mediated drug delivery (PTX-NPs) reduced the cell viability to 14% (p < 0.0001 vs. CTRL), indicating an effective impact on cell metabolism which outperformed the conventional drug administration route (p < 0.0001 vs. PTX) characterized by a 3.5-fold higher cell viability. This confirmed that the administration of PTX-loaded NPs, thanks to their thermoresponsive behavior, boosted the therapeutic effect of the drug. Furthermore, the cells ordinarily incubated with our nanosystems (i.e., 37 °C, without hyperthermia) did not show significant PTX-mediated effects (81% cell viability, at 24 h) in accordance with the NP capability to retain the drug below the UCST. Under these conditions, the administration of PTX in the free form resulted in a more pronounced effect on cell metabolism (reduction of viability to 60%, p < 0.01 vs. PTX-NPs), which was, however, less effective than the outcome related to our drug-loaded NPs in hyperthermia treatment. This verified that our NPs could retain the payload until exposure to hyperthermia (i.e., temperature above UCST), resulting in a smart on-demand drug delivery system capable of switching its behavior according to the external thermal stimuli.
These results were further validated by a live/dead assay performed on the cells following the hyperthermia treatment. As reported in Fig. 7, the confocal micrograph related to PTX-NP administration shows a lower cell survival, as clearly visible by the high number of red-stained cells (dead nucleated cells) compared to SKOV3 cells treated with a non-encapsulated drug (p < 0.05 vs. PTX).
 | ||
| Fig. 7 Representative images from the live (green)/dead (red) assay in SKOV3 cells after 24 h administration of the NPs, free drug (PTX), and drug-loaded NPs (PTX-NPs) following exposure to hyperthermia treatment using a dual-fluorescence system. The cells treated with the same volume of DMSO (1% v/v) required for the preparation of the drug solution are set as internal the control (CTRL) and the untreated cells (dashed line) are set as the reference. Scale bar, 100 μm. The results are the mean ± SD of the live/dead cell ratio of three independent experiments. Statistical analysis was performed using one-way ANOVA. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Non-significant results (i.e., ns) are intended for the comparisons not highlighted in the graph; they are not reported for the sake of clarity. | ||
Overall, our results confirmed the efficacy of NP-encapsulated PTX, demonstrating a clear advantage of the thermoresponsive zwitterionic NPs as carriers of the chemotherapeutic agent, which overcome the limitations in hyperthermic treatments of thermoresistant cells. In particular, these outcomes can be considered a preliminary in vitro validation of the proposed nanosystems and represent a reliable basis for their test in in vivo tumor models looking for anticancer hyperthermia-based therapies. Indeed, the assessment of the formulation in a three-dimensional tumor environment mimicking the effective tumor heterogeneity and architecture, including the presence of pathophysiological barriers, ensures a more in-depth evaluation of the therapeutic benefits.53 This could lead to the definition of novel treatments in diseases still lacking a therapy, such as ovarian carcinoma.
4. Conclusion
In this work, we have proposed the synthesis of zwitterionic UCST-type NPs and their innovative use under conditions mimicking the tumoral hyperthermia treatments. NPs with tunable composition, size and UCST behavior can be produced through the combination of RAFT and ROP polymerization to meet specific thermoresponsivity. We have demonstrated that by changing the stoichiometry of the polymerization reactions, i.e., the SB/ZB ratio and the length of the lipophilic oligoester, it is possible to modulate the macromolecular architecture of the NPs and their phase separation/dispersion as a function of temperature. Thanks to the synthetic route, the NPs can be synthesized ad hoc to be responsive at the desired temperature, such as the one involved in hyperthermia therapeutic treatments. We have validated our NPs as drug delivery systems in the hyperthermic treatment of SKOV3, an ovarian cancer cell line exhibiting thermoresistance up to 43 °C. The NPs could be internalized by the cells and trigger drug release at the target temperature (43 °C) according to their UCST. The NP-mediated intracellular release of PTX, a common chemotherapy drug, led to an enhanced therapeutic effect outperforming the conventional non-encapsulated drug administration route. Overall, these results suggest that zwitterionic UCST NPs may represent a promising and innovative hyperthermia-based approach to counteract the progression of ovarian carcinoma, in particular overcoming the constraint of the thermotolerance of specific cell lines, which significantly reduce the potential of hyperthermic intraperitoneal chemotherapy.Author contributions
C. C.: investigation, data curation, and formal analysis; I. M.: investigation and formal analysis; E. J.: investigation and data curation; S. S.: investigation; M. S.: conceptualization, methodology, and writing – review and editing; B. M. C.: funding acquisition, L. R.: resources, funding acquisition, and visualization; M. T. R.: resources and funding acquisition; E. M.: conceptualization, methodology, supervision, and writing – original draft; D. M.: supervision and funding acquisition.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
M. T. R. was supported by the European Research Council (call ERC-AdG-2021, project BEACONSANDEGG, G. A. 101053122). L. R. was supported by Associazione Italiana Ricerca sul Cancro (AIRC) [grant number AIRC 21372].References
- P. Yadav, J. Jain and A. P. Sherje, React. Funct. Polym., 2021, 165, 104970 CrossRef CAS.
- J. P. Coats, R. Cochereau, I. A. Dinu, D. Messmer, F. Sciortino and C. G. Palivan, Macromol. Biosci., 2023, 2200474 CrossRef CAS PubMed.
- J. Zhang, Y. Lin, Z. Lin, Q. Wei, J. Qian, R. Ruan, X. Jiang, L. Hou, J. Song, J. Ding and H. Yang, Adv. Sci., 2022, 9, 2103444 CrossRef CAS PubMed.
- M. Sponchioni, U. Capasso Palmiero and D. Moscatelli, Mater. Sci. Eng., C, 2019, 102, 589–605 CrossRef CAS PubMed.
- A. L. Oei, H. P. Kok, S. B. Oei, M. R. Horsman, L. J. A. Stalpers, N. A. P. Franken and J. Crezee, Adv. Drug Delivery Rev., 2020, 163–164, 84–97 CrossRef CAS PubMed.
- G. Y. Yi, M. J. Kim, H. I. Kim, J. Park and S. H. Baek, Antioxidants, 2022, 11, 625 CrossRef CAS PubMed.
- N. R. Datta, S. G. Ordóñez, U. S. Gaipl, M. M. Paulides, H. Crezee, J. Gellermann, D. Marder, E. Puric and S. Bodis, Cancer Treat. Rev., 2015, 41, 742–753 CrossRef CAS PubMed.
- J. Qi, W. Li, K. Lu, F. Jin, D. Liu, X. Xu, X. Wang, X. Kang, W. Wang, G. Shu, F. Han, X. Ying, J. You, J. Ji and Y. Du, Nano Lett., 2019, 19, 4949–4959 CrossRef CAS PubMed.
- J. C. Peeken, P. Vaupel and S. E. Combs, Front. Oncol., 2017, 7, 132 CrossRef PubMed.
- M. Mallory, E. Gogineni, G. C. Jones, L. Greer and C. B. Simone, Crit. Rev. Oncol. Hematol., 2016, 97, 56–64 CrossRef PubMed.
- S. Di Agostino, V. Canu, S. Donzelli, C. Pulito, A. Sacconi, F. Ganci, F. Valenti, F. Goeman, S. Scalera, F. Rollo, A. Bagnato, M. G. Diodoro, E. Vizza, M. Carosi, B. Rufini, O. Federici, M. Giofrè, F. Carboni, P. Muti, G. Ciliberto, S. Strano, M. Valle and G. Blandino, Cell Death Dis., 2023, 14, 535 CrossRef CAS PubMed.
- B. Hildebrandt, P. Wust, O. Ahlers, A. Dieing, G. Sreenivasa, T. Kerner, R. Felix and H. Riess, Crit. Rev. Oncol. Hematol., 2002, 43, 33–56 CrossRef PubMed.
- A. L. B. Seynhaeve, M. Amin, D. Haemmerich, G. C. van Rhoon and T. L. M. ten Hagen, Adv. Drug Delivery Rev., 2020, 163–164, 125–144 CrossRef CAS PubMed.
- N. Manfredini, G. Gardoni, M. Sponchioni and D. Moscatelli, Eur. Polym. J., 2023, 198, 112421 CrossRef CAS.
- A. Bordat, T. Boissenot, J. Nicolas and N. Tsapis, Adv. Drug Delivery Rev., 2019, 138, 167–192 CrossRef CAS PubMed.
- N. Vanparijs, L. Nuhn and B. G. De Geest, Chem. Soc. Rev., 2017, 46, 1193–1239 RSC.
- M. Le, W. Huang, K.-F. Chen, C. Lin, L. Cai, H. Zhang and Y.-G. Jia, Chem. Eng. J., 2022, 432, 134354 CrossRef CAS.
- Z. Yang, R. Cheng, C. Zhao, N. Sun, H. Luo, Y. Chen, Z. Liu, X. Li, J. Liu and Z. Tian, Theranostics, 2018, 8, 4097–4115 CrossRef CAS PubMed.
- W. Li, L. Huang, X. Ying, Y. Jian, Y. Hong, F. Hu and Y. Du, Angew. Chem., Int. Ed., 2015, 54, 3126–3131 CrossRef CAS PubMed.
- A. Bordat, N. Soliman, I. Ben Chraït, K. Manerlax, N. Yagoubi, T. Boissenot, J. Nicolas and N. Tsapis, Eur. J. Pharm. Biopharm., 2019, 142, 281–290 CrossRef CAS PubMed.
- D. Huang, H. Dai, K. Tang, B. Chen, H. Zhu, D. Chen, N. Li, Y. Wang, C. Liu, Y. Huang, J. Yang, C. Zhang, R. Lin and W. He, Nanoscale, 2020, 12, 20002–20015 RSC.
- F. Käfer, A. Lerch and S. Agarwal, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 274–279 CrossRef.
- Y. Deng, F. Käfer, T. Chen, Q. Jin, J. Ji and S. Agarwal, Small, 2018, 14, 1802420 CrossRef PubMed.
- Y. Morita, K. Kobayashi, Y. Toku, Y. Kimura, Q. Luo, G. Song and Y. Ju, Biomater. Adv., 2022, 139, 213026 CrossRef CAS PubMed.
- J. R. McDaniel, D. C. Radford and A. Chilkoti, Biomacromolecules, 2013, 14, 2866–2872 CrossRef CAS PubMed.
- Q. Jin, Y. Chen, Y. Wang and J. Ji, Colloids Surf., B, 2014, 124, 80–86 CrossRef CAS PubMed.
- L.-Y. Zhou, Y.-H. Zhu, X.-Y. Wang, C. Shen, X.-W. Wei, T. Xu and Z.-Y. He, Comput. Struct. Biotechnol. J., 2020, 18, 1980–1999 CrossRef CAS PubMed.
- S. Peng, H. Wang, W. Zhao, Y. Xin, Y. Liu, X. Yu, M. Zhan, S. Shen and L. Lu, Adv. Funct. Mater., 2020, 30, 2001832 CrossRef CAS.
- T. Ishida and H. Kiwada, Int. J. Pharm., 2008, 354, 56–62 CrossRef CAS PubMed.
- H. Koide, T. Asai, K. Hatanaka, S. Akai, T. Ishii, E. Kenjo, T. Ishida, H. Kiwada, H. Tsukada and N. Oku, Int. J. Pharm., 2010, 392, 218–223 CrossRef CAS PubMed.
- H.-J. Im, C. G. England, L. Feng, S. A. Graves, R. Hernandez, R. J. Nickles, Z. Liu, D. S. Lee, S. Y. Cho and W. Cai, ACS Appl. Mater. Interfaces, 2016, 8, 17955–17963 CrossRef CAS PubMed.
- X.-X. Kong, S. Jiang, T. Liu, G.-F. Liu and M. Dong, Biomed. Pharmacother., 2020, 132, 110907 CrossRef CAS PubMed.
- H. Hatakeyama, S. Y. Wu, Y. A. Lyons, S. Pradeep, W. Wang, Q. Huang, K. A. Court, T. Liu, S. Nie, C. Rodriguez-Aguayo, F. Shen, Y. Huang, T. Hisamatsu, T. Mitamura, N. Jennings, J. Shim, P. L. Dorniak, L. S. Mangala, M. Petrillo, V. A. Petyuk, A. A. Schepmoes, A. K. Shukla, M. Torres-Lugo, J.-S. Lee, K. D. Rodland, A. Fagotti, G. Lopez-Berestein, C. Li and A. K. Sood, Cell Rep., 2016, 17, 1621–1631 CrossRef CAS PubMed.
- A. Sukovas, V. Cesna, A. Jasukaitiene, G. Barauskas, R. J. Nadisauskiene, Z. Dambrauskas, S. Paskauskas and A. Gulbinas, Anticancer Res., 2017, 37, 5011–5018 CAS.
- A. Sukovas, G. Silkuniene, S. Trumbeckaite, A. Jasukaitiene, L. Degutyte-Fomins, V. Mildaziene, A. Gulbinas, R. Baniene, Z. Dambrauskas and S. Paskauskas, J. Bioenerg. Biomembr., 2019, 51, 301–310 CrossRef CAS PubMed.
- M. Sponchioni, P. Rodrigues Bassam, D. Moscatelli, P. Arosio and U. Capasso Palmiero, Nanoscale, 2019, 11, 16582–16591 RSC.
- R. Ferrari, L. Talamini, M. B. Violatto, P. Giangregorio, M. Sponchioni, M. Morbidelli, M. Salmona, P. Bigini and D. Moscatelli, Mol. Pharm., 2017, 14, 124–134 CrossRef CAS PubMed.
- R. Auriemma, M. Sponchioni, S. Lotti, L. Morosi, M. Zucchetti, M. Lupi, D. Moscatelli and U. Capasso Palmiero, Ind. Eng. Chem. Res., 2021, 60, 10699–10709 CrossRef CAS.
- J. Wu, Z. Xiao, A. Chen, H. He, C. He, X. Shuai, X. Li, S. Chen, Y. Zhang, B. Ren, J. Zheng and J. Xiao, Acta Biomater., 2018, 71, 293–305 CrossRef CAS PubMed.
- R. Lalani and L. Liu, Polymer, 2011, 52, 5344–5354 CrossRef CAS.
- J. Ladd, Z. Zhang, S. Chen, J. C. Hower and S. Jiang, Biomacromolecules, 2008, 9, 1357–1361 CrossRef CAS PubMed.
- K. P. García, K. Zarschler, L. Barbaro, J. A. Barreto, W. O'Malley, L. Spiccia, H. Stephan and B. Graham, Small, 2014, 10, 2516–2529 CrossRef PubMed.
- J. Zhao, Z. Qin, J. Wu, L. Li, Q. Jin and J. Ji, Biomater. Sci., 2018, 6, 200–206 RSC.
- H. Sun, L. Yan, R. Zhang, J. F. Lovell, Y. Wu and C. Cheng, Biomater. Sci., 2021, 9, 5000–5010 RSC.
- J. Seuring and S. Agarwal, Macromolecules, 2012, 45, 3910–3918 CrossRef CAS.
- N. Shimada, H. Ino, K. Maie, M. Nakayama, A. Kano and A. Maruyama, Biomacromolecules, 2011, 12, 3418–3422 CrossRef CAS PubMed.
- S. Zhu, L. Wen, Y. Xiao and M. Lang, Polym. Chem., 2020, 11, 5173–5180 RSC.
- Z. Zhang, H. Li, S. Kasmi, S. Van Herck, K. Deswarte, B. N. Lambrecht, R. Hoogenboom, L. Nuhn and B. G. De Geest, Angew. Chem., Int. Ed., 2019, 58, 7866–7872 CrossRef CAS PubMed.
- A. Madariaga, S. Garg, J. P. Bruce, S. Thiryayi, V. Mandilaras, P. Rath, A. M. Oza, N. C. Dhani, D. W. Cescon, Y. C. Lee, E. Chen, L. Wang, B. Clarke and S. Lheureux, Gynecol. Oncol., 2020, 159, 539–545 CrossRef CAS PubMed.
- J. Zhang, L. Li, J. Yin, X. Zhang, Y. Zheng and R. Feng, BMC Pharmacol. Toxicol., 2023, 24, 13 CrossRef CAS PubMed.
- A. Sukhanova, S. Bozrova, P. Sokolov, M. Berestovoy, A. Karaulov and I. Nabiev, Nanoscale Res. Lett., 2018, 13, 44 CrossRef PubMed.
- L. Yildirimer, N. T. K. Thanh, M. Loizidou and A. M. Seifalian, Nano Today, 2011, 6, 585–607 CrossRef CAS PubMed.
- T. R. Abreu, M. Biscaia, N. Gonçalves, N. A. Fonseca and J. N. Moreira, in Bio-Nanomedicine for Cancer Therapy, ed. F. Fontana and H. A. Santos, Springer International Publishing, Cham, 2021, pp. 271–299, DOI:10.1007/978-3-030-58174-9_12.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr00723a |
| This journal is © The Royal Society of Chemistry 2024 |