
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Insulin amyloid fibril formation reduction by tripeptide stereoisomers†
Beatrice
Rosetti
 a,
Slavko
Kralj
bc,
Erica
Scarel
a,
Simone
Adorinni
a,
Barbara
Rossi
d,
Attilio V.
Vargiu
e,
Ana M.
Garcia
*f and
Silvia
Marchesan
*a
a,
Slavko
Kralj
bc,
Erica
Scarel
a,
Simone
Adorinni
a,
Barbara
Rossi
d,
Attilio V.
Vargiu
e,
Ana M.
Garcia
*f and
Silvia
Marchesan
*a
aChemical Pharmaceutical Science Department, University of Trieste, 34127 Trieste, Italy. E-mail: smarchesan@units.it
bMaterials Synthesis Department, Jožef Stefan, Institute, 1000 Ljubljana, Slovenia
cDepartment of Pharmaceutical Technology, Faculty of Pharmacy, University of Ljubljana, 1000 Ljubljana, Slovenia
dElettra Sincrotrone Trieste, Strada Statale 14 - km 163,5 Basovizza, 34149 Trieste, Italy
ePhysics Department, University of Cagliari, 09042 Monserrato, Cagliari, Italy
fFacultad de Ciencias y Tecnologías Químicas, Instituto Regional de Investigación Científica Aplicada (IRICA), Universidad de Castilla-La Mancha, 13071 Ciudad Real, Spain. E-mail: anam.garcia@uclm.es
First published on 6th May 2024
Abstract
Insulin fibrillation is a problem for diabetic patients that can occur during storage and transport, as well as at the subcutaneous injection site, with loss of bioactivity, inflammation, and various adverse effects. Tripeptides are ideal additives to stabilise insulin formulations, thanks to their low cost of production and inherent cytocompatibility. In this work, we analysed the ability of eight tripeptide stereoisomers to inhibit the fibrillation of human insulin in vitro. The sequences contain proline as β-breaker and Phe–Phe as binding motif for the amyloid-prone aromatic triplet found in insulin. Experimental data based on spectroscopy, fluorescence, microscopy, and calorimetric techniques reveal that one stereoisomer is a more effective inhibitor than the others, and cell live/dead assays confirmed its high cytocompatibility. Importantly, in silico data revealed the key regions of insulin engaged in the interaction with this tripeptide, rationalising the molecular mechanism behind insulin fibril formation reduction.
Introduction
The pathological fibrillation of amyloids is an unsolved challenge in medicine. At least 50 proteins or peptides have been found to aggregate into amyloid structures with various adverse effects on human health. The most widely known amyloidoses are linked to neurodegenerative processes, such as the Alzheimer's, Huntington's, and Parkinson's diseases.1 However, many more exist, and amyloids’ new roles are continuously being found, for instance affecting the heart,2 the kidney,3 or the gut.4The tendency of these proteins or peptides to fibrillate is not directly associated to their primary amino acidic sequence, as it is a general feature of a wide number of polypeptides under appropriate conditions.5–9 However, those polypeptides normally share common physico-chemical properties, such as high hydrophobicity with low net charge, and high propensity to convert from an α-helix towards a β-sheet. Amyloid aggregation typically involves protein unfolding or re-folding into a non-functional form, which then can stack to give rise to the cross-β hallmark found in fibrils.10 In fact, in neurodegenerative diseases, soluble oligomers demonstrated higher toxicity than mature fibrils, showing the importance to tackle the early stages of aggregation.11,12 As a result of fibrillation, physiological disfunctions occur that can lead to a cascade of adverse effects, especially when the protein or peptide is a hormone or it is involved in its transport.13–16
In particular, insulin fibrillation can cause cytotoxicity and inflammation,17 which can be exacerbated by its ability to promote fibrillation of other proteins, such as amylin.18 Besides, an increased risk of developing neurodegenerative syndromes has been linked to insulin dysfunction.19 Insulin fibrillation is a problem of particular relevance for diabetic patients. It can occur at the site of subcutaneous injection, or during transport and storage, being promoted by several factors, such as agitation, high ionic strength, high concentration, and low pH.20 Therefore, there is large scope for the identification of simple biomolecules that could serve as functional additives to stabilise insulin formulations. Several peptides have been studied for their ability to inhibit insulin fibrillation, with their design being typically based on insulin-binding sequences, for instance those found in the insulin receptor or in insulin itself.21
In this regard, peptides as short as two-to-three amino acids are particularly advantageous for their low-cost of production and generally high cytocompatibility, as demonstrated by their wide use in supplements, nutraceuticals, and medicinal products.22 The di- and tri-peptides used to inhibit insulin fibrillation were based on the Phe–Phe motif,23 which has a strong tendency to fibrillate and is found in insulin itself,24 coupled with a ferrocenyl unit (Fc).25,26 Best results were obtained with the tripeptides Fc–Phe–Phe–Phe and Fc–Phe–Phe–Tyr, which can be rationalised with the finding that tripeptides have the ideal number of non-hydrogen atoms for maximal efficacy of binding with receptors, hence for bioactivity.27 However, in those studies the effect of amino acid stereoconfiguration was not evaluated. Recently, we reported the detailed investigation of the supramolecular behaviour of the complete series of Pro–Phe–Phe stereoisomers, which contain proline as β-breaker and Phe–Phe as anchoring unit to inhibit amyloid fibrillation.28 In particular, L-Pro–D-Phe–L-Phe revealed to be the most effective inhibitor in vitro, with excellent cytocompatibility and resistance against enzyme-mediated hydrolysis.28 Therefore, in this study, we focussed on insulin as a target to assess whether their inhibitory activity can be extended to other amyloids, and whether appropriate choice of stereoconfiguration can enable target selectivity.
Results and discussion
Tripeptide series preparation
The eight Pro–Phe–Phe stereoisomers (Scheme 1) were prepared by solid-phase peptide synthesis using Fmoc-protection strategy and standard protocols, as previously reported.28 Their purification by reverse-phase high-performance liquid chromatography (RP-HPLC) led to pure peptides, as confirmed by LC-MS analyses, 1H- and 13C-NMR data, which were in agreement with the literature.28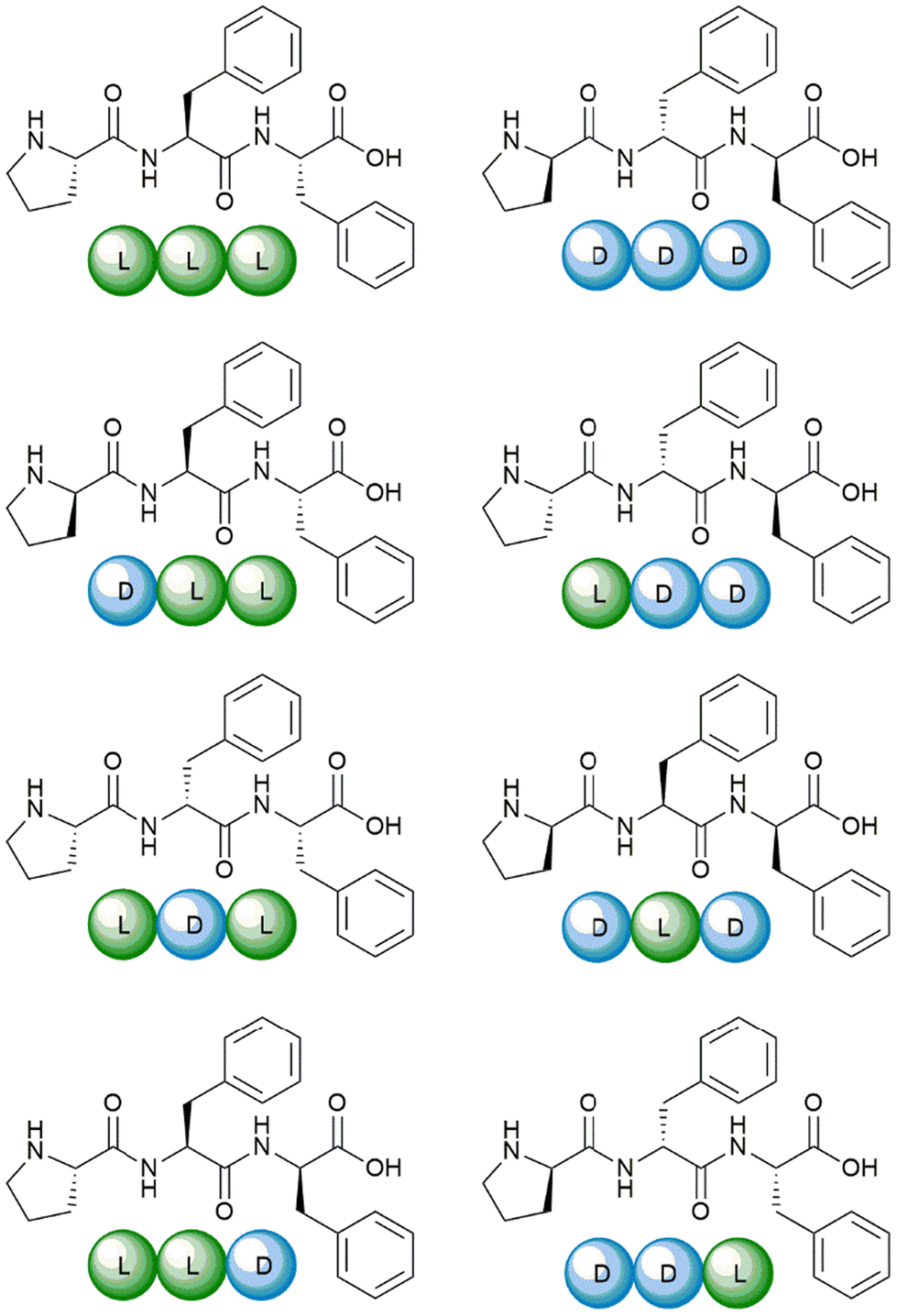 | ||
| Scheme 1 The eight Pro–Phe–Phe stereoisomers synthesized and studied as inhibitors of insulin fibrillation. | ||
Insulin fibrillation inhibition
Insulin is a heterodimeric α-helical protein, which is composed of two chains, A and B. At physiological pH, insulin is in a stable, zinc-bound hexameric form (Fig. 1a), while the bioactive monomer can undergo partial unfolding to expose aggregation-prone hydrophobic surfaces (Fig. 1b).20 Among these, the segment Leu–Val–Glu–Ala–Leu–Tyr–Leu (LVEALYL in Fig. 1b) of chain B plays a key role in insulin fibrillation. X-ray diffraction data revealed that it is central to the cross-β spine of insulin fibrils, as two adjacent LVEALYL segments interlock in a dry steric zipper, as a result of their conversion from the native α-helical conformation into extended β-strands.29 Another hydrophobic segment is the “aromatic triplet” Phe–Phe–Tyr (FFY in Fig. 1b) that is found near the C-terminus of chain B. This conserved motif creates an extended apolar surface, and it can establish intermolecular aromatic interactions with other triplets of insulin molecules.24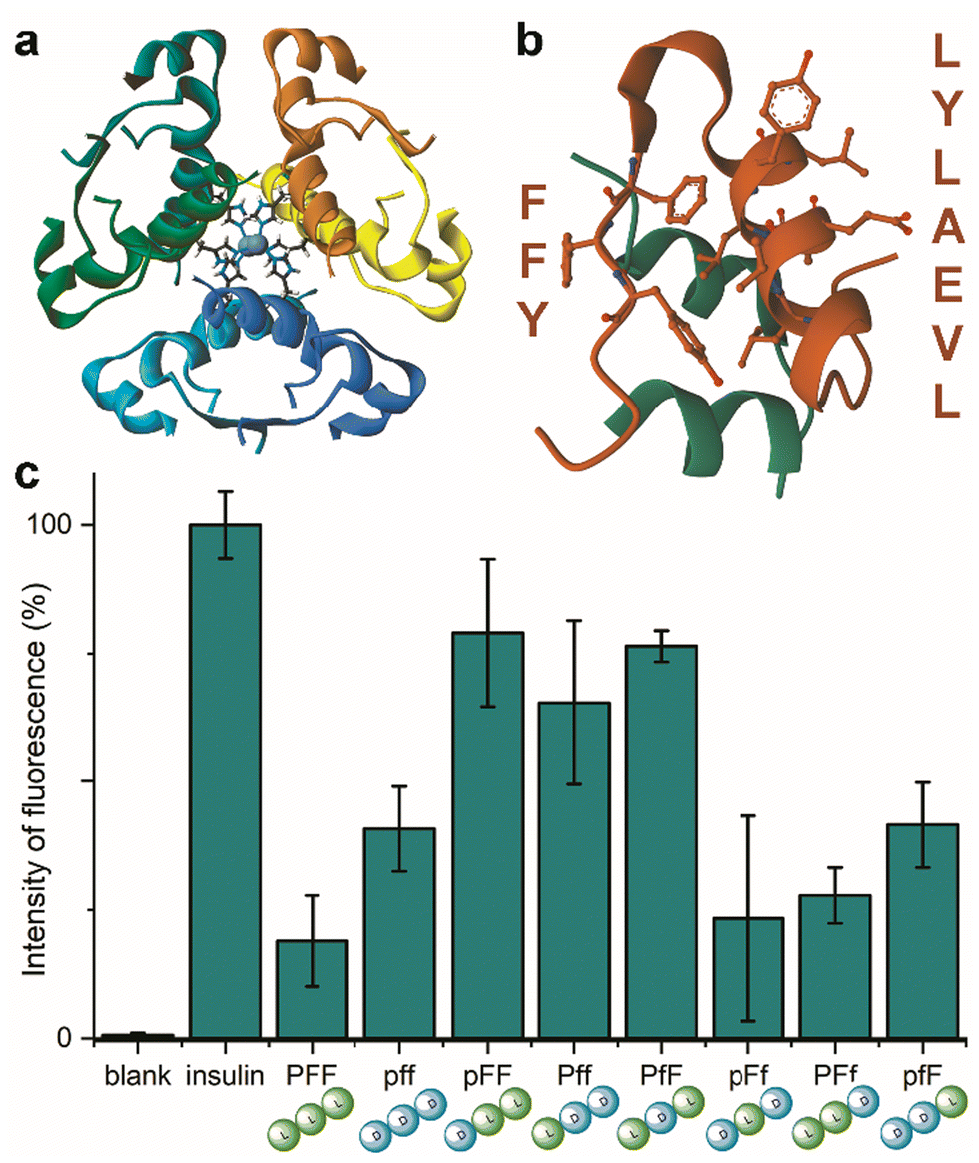 | ||
| Fig. 1 (a) Zinc-bound insulin hexamer with coordinating His residues shown in ball and stick (PDB entry 3AIY). (b) insulin monomer (PBD entry 3I40) showing the chains A (green) and B (orange) with the fibrillation-prone aromatic triplet PheB24–PheB25–TyrB26 (FFY) and LeuB11–ValB12–GluB13–AlaB14–LeuB15–TyrB16–LeuB17 (LVEALYL) shown in ball and stick. (c) ThT assay for human insulin fibrillation inhibition in the presence of the eight Pro–Phe–Phe stereoisomers. | ||
There are several protocols to reproduce in vitro insulin fibrillation, which is not trivial and it is influenced by a number of factors, such as temperature, agitation, ionic strength and insulin concentration.30 Under physiological conditions, insulin fibrillation in vitro can take several days, therefore acidic conditions are typically used to conveniently reduce the lag time.25,26,31–34 In particular, for this study we chose the use of NaCl, strong agitation, 37 °C and a pH 1.6 that are ideal experimental conditions to reduce variability amongst insulin concentration values and reduce the fibrillation lag time.35
Indeed, initial attempts to quantitatively monitor the kinetics of fibrillation led to reproducibility issues especially over the first hours, therefore we opted for 48 h end-points instead. In this manner, it was possible to note that the most effective inhibitor was the homochiral L-Pro–L-Phe–L-Phe, followed by D-Pro–L-Phe–D-Phe and L-Pro–L-Phe–D-Phe stereoisomers (Fig. 1c). Surprisingly, among the worst performers with negligible inhibitory activity, there was L-Pro–D-Phe–L-Phe that was previously found to be an effective inhibitor for amyloid β fibrillation in vitro.28 This finding is important as it is a promising indication of the possibility to strategically choose amino acid stereoconfiguration as a determinant for selectivity, thus giving scope for further studies on these simple biomolecules as therapeutics’ formulation additives.
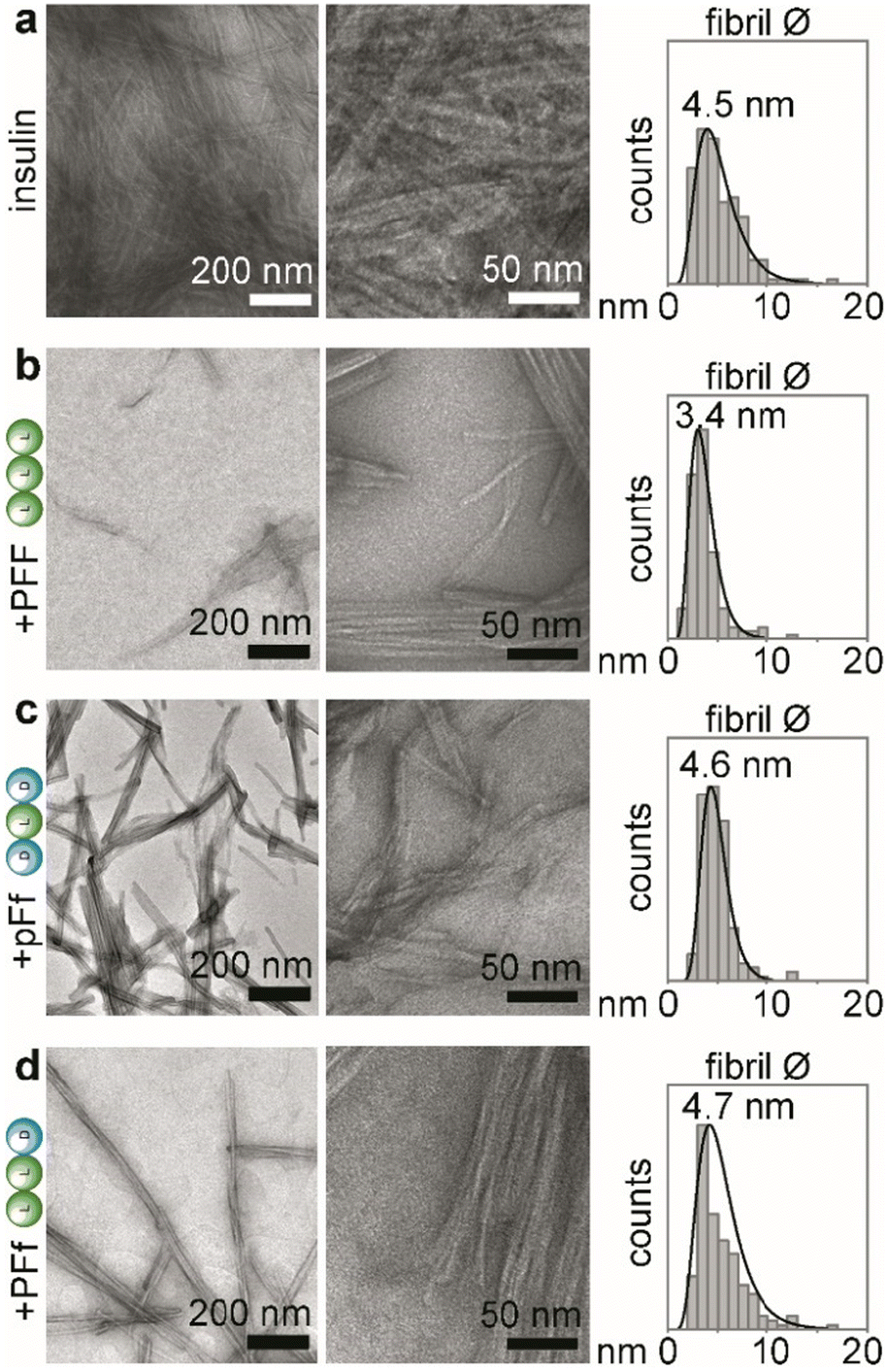 | ||
| Fig. 2 TEM analyses of insulin fibrillation inhibition, showing TEM micrographs at lower (left panels) and higher (middle panels) magnification, and statistical distribution of fibrils’ diameters with the reported median value (right panels, n = 150 counts). (a) insulin alone; (b) insulin + PFF; (c) insulin + pFf; (d) insulin + PFf. | ||
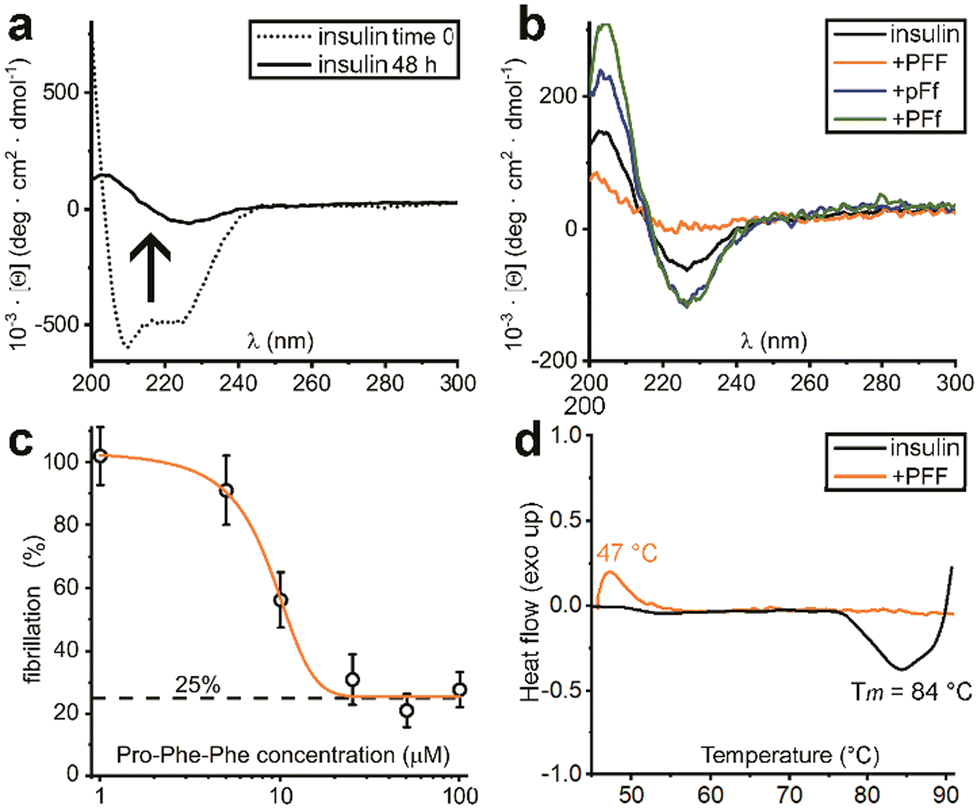 | ||
| Fig. 3 (a) CD spectra of insulin before (dotted line) and after 48 h of fibrillation (solid line). (b) CD spectra of insulin fibrils treated with each one of the three Pro–Phe–Phe inhibitors (L–L–L, orange; D–L–D blue; L–L–D green) or untreated (black) after 48 h. (c) Dose–response ThT assay with increasing concentrations of L-Pro–L-Phe–L-Phe. (d) DSC data of insulin fibrils (black) and treated with L-Pro–L-Phe–L-Phe (orange). | ||
UV resonance Raman spectroscopy (UVRR)
UVRR has been demonstrated to be a powerful tool for probing amyloid fibril structures in proteins at all stages of aggregation.43–46 With respect to conventional visible Raman technique, UV enhancement provides a significant increment of the detection limit, being particularly useful for investigating diluted samples. Additionally, the use of different excitation wavelengths allows for a selective resonance enhancement of specific portions in proteins. In particular, the use of an excitation wavelength at 226 nm is convenient to probe the aromatic signals of Phe and Tyr residues, thanks to the close resonance of this excitation energy with their electronic transitions.47,48 The consequent strong enhancement of the UV Raman cross-section of some vibrational modes attributed to Phe and Tyr makes these signals the predominant ones in the UVRR spectra, as confirmed by analysis of amino acid precursors devoid of the amide bond that is normally present in visible Raman spectra.49The UVRR spectra showed that fibrillation markedly reduced the Raman signals of Phe and Tyr at about 1610 cm−1 and 1180 cm−1 in insulin, which were partially restored in the presence of L-Pro–L-Phe–L-Phe (Fig. 4a). This is evident by looking at the difference spectra (DUVRR, Fig. 4b), which clearly display the effect of PFF to partially inhibit the fibrillation of insulin. The decrease in the intensity of aromatic bands in the DUVRR was observed during insulin aggregation,43,44,46 suggesting that the local environment of these amino acids is changed upon fibrillation. In particular, the strong reduction of the Raman signal at 1610 cm−1 could indicate that Tyr residues are in a more hydrophobic environment than in the native protein.44
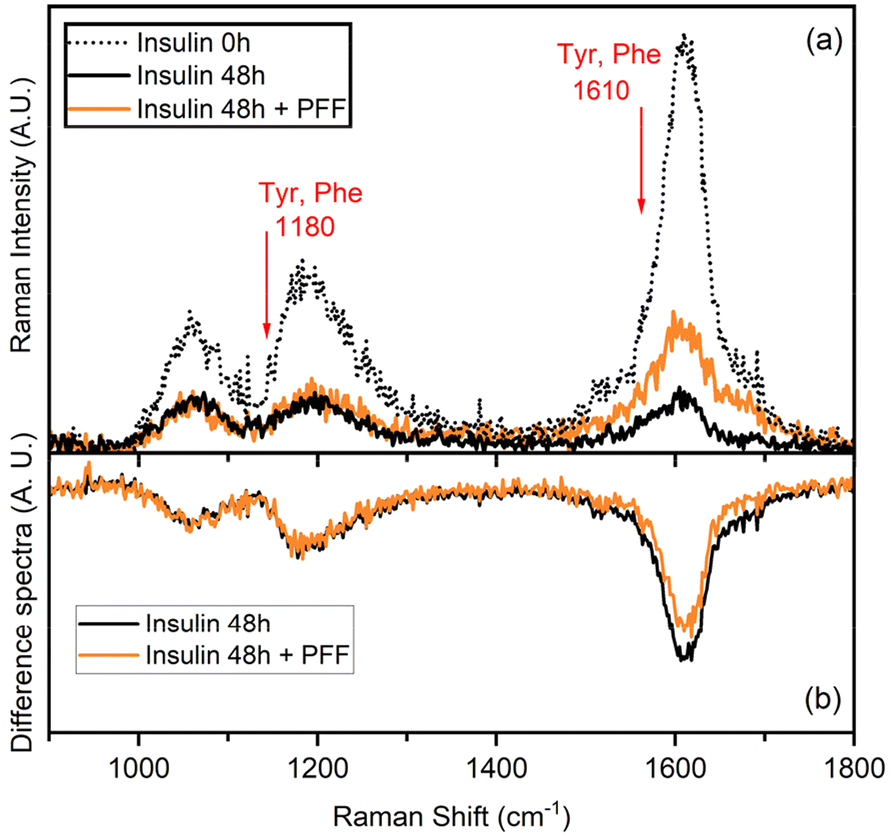 | ||
| Fig. 4 (a) Raman spectra of insulin in the native form (dotted black line) and after 48 h of fibrillation (black line) either alone or in the presence of L-Pro–L-Phe–L-Phe (PFF, orange line). The spectra have been subtracted by the spectral contribution of the solvent and solvent + PFF. (b) Difference spectra [insulin 48 h − insulin 0 h] and [(insulin 48 h + PFF) − insulin 0 h] highlighting that the local environment of Tyr residues changes in the presence or absence of PFF. | ||
Instead, in the presence of the tripeptide, the signal intensity is mostly, but not completely, restored, in agreement with the ThT data. Therefore, UVRR data further supported the bioactivity of the tripeptide in markedly reducing fibrillation of insulin.
Insulin-tripeptide interactions
The interaction between insulin and L-homochiral Pro–Phe–Phe (PFF) or L-Pro–D-Phe–L-Phe (hereafter PfF) was studied by means of all-atom MD simulations. Studies were performed at pH 1.6, closer to the conditions of the experiments, and at physiological pH (7.4). Each amino acid was assigned the most likely protonation state at the corresponding pH values. In both cases, one protein and ten peptides (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 ratio) were simulated in 0.1 M NaCl water solution at room temperature and pressure. At pH 1.6, MD simulations identified long-lasting interactions formed by one or more homochiral peptides with both the “aromatic triplet” FFY sequence located at the insulin C-terminus and the hydrophobic LVEALYL segment, which is notoriously prone to fibrillation. Examples of molecular arrangements establishing these interactions are reported in Fig. 5 (see also ESI, section S9†). In contrast, not more than one single heterochiral PfF peptide was able to establish stable interactions with the LVEALYL segment in four independent MD simulations (Fig. 5 and ESI section S9†). In addition, the interactions between insulin and the homochiral PFF peptides have a longer lifetime than those established with the heterochiral PfF (see the captions of Fig. 5 and ESI section S9†). A similar situation is seen at pH 7.4, although in this case also PfF establishes interactions with the FFY motif, while the tripeptide-insulin interactions remain stronger for homochiral PFF.
:10 ratio) were simulated in 0.1 M NaCl water solution at room temperature and pressure. At pH 1.6, MD simulations identified long-lasting interactions formed by one or more homochiral peptides with both the “aromatic triplet” FFY sequence located at the insulin C-terminus and the hydrophobic LVEALYL segment, which is notoriously prone to fibrillation. Examples of molecular arrangements establishing these interactions are reported in Fig. 5 (see also ESI, section S9†). In contrast, not more than one single heterochiral PfF peptide was able to establish stable interactions with the LVEALYL segment in four independent MD simulations (Fig. 5 and ESI section S9†). In addition, the interactions between insulin and the homochiral PFF peptides have a longer lifetime than those established with the heterochiral PfF (see the captions of Fig. 5 and ESI section S9†). A similar situation is seen at pH 7.4, although in this case also PfF establishes interactions with the FFY motif, while the tripeptide-insulin interactions remain stronger for homochiral PFF.
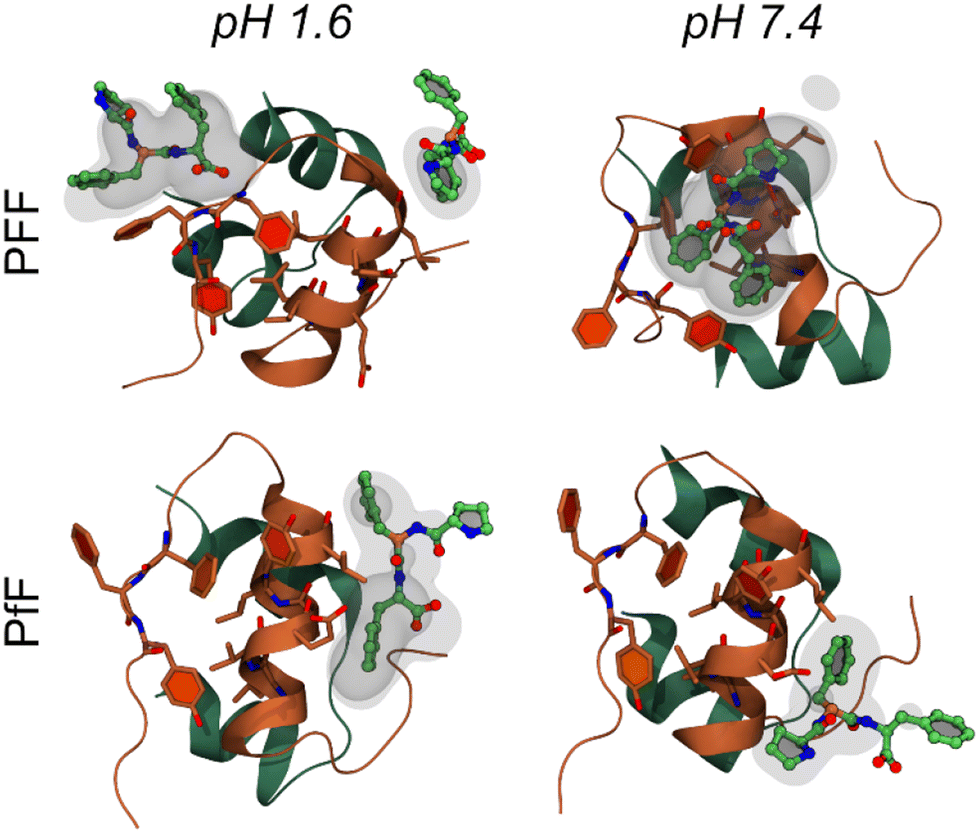 | ||
| Fig. 5 Molecular interactions between the insulin monomer and PFF (first row) or PfF (second row) stereoisomers. Reported are four examples of highly sampled conformations at pH 1.6 (left column) and pH 7.4 (right), taken from the MD simulations associated to the largest density values calculated among all replicas for each system (form upper-left to bottom-right: 16.8, 11.5, 6.4, and 9.1 respectively, see Methods and ESI section S9†). Insulin is coloured as in Fig. 1b, while peptides are coloured by atomic element (C, O, N in green, red, and blue respectively). Transparent and semi-transparent surfaces represent isosurfaces corresponding respectively to density values 2 and 3 times higher than the value of water at standard conditions. | ||
From the MD simulations, it is evident that both tripeptide stereoisomers exploit multiple interaction patterns to bind insulin (see ESI section S9†). In most cases, tripeptide Phe residues interact directly with the sidechains of the hydrophobic FFY triplet and LVEALYL stretch. However, in several dynamics, Pro is also involved in direct contacts with insulin, and one case (i.e., PFF at pH 1.6, replica 4) there is a strong interaction between the terminal carboxyl group and the insulin backbone.
Overall, these results clearly indicate that regions of the insulin polypeptide involved in the early stages of fibrillation have a strong affinity to homochiral PFF, pointing to a clear putative mechanism of inhibition. The lower inhibitory activity of heterochiral PfF can be partly explained by the different affinity of this tripeptide towards the protein, although further investigations are required to clearly identify all the molecular determinants behind the peculiar behaviour of the homochiral stereoisomer.
Cytocompatibility of Pro–Phe–Phe
Cytocompatibility of L-Pro–L-Phe–L-Phe was tested in vitro on fibroblast cell culture with live/dead and metabolic assays (Fig. 6). No significant difference between the untreated and treated samples was found either in the number of cells, or in their viability or morphology, for all the tested tripeptide concentrations up to 1 mM (Fig. 6a). Similarly, the metabolic MTT assay showed insignificant differences in metabolic activity between untreated and treated fibroblast cells, upon exposure for 24 hours to L-Pro–L-Phe–L-Phe up to 0.5 mM. Metabolic activity was reduced to 70% at the highest concentration of 1 mM, which is remarkably high (Fig. 6b). Overall, these data indicate a promising profile for the tripeptide in terms of cytocompatibility.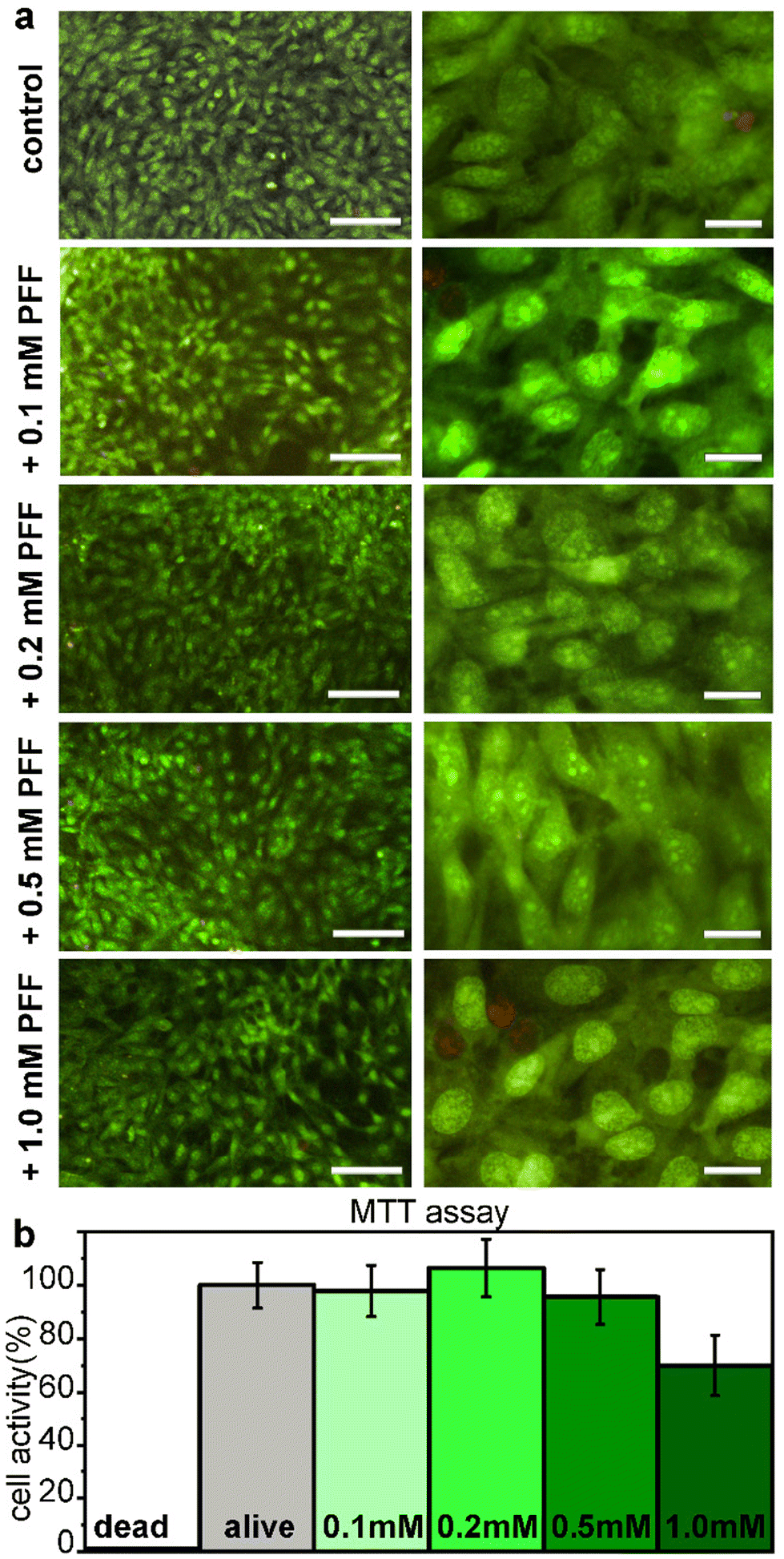 | ||
| Fig. 6 (a) Microscopy images at low (10×, left panels) or high (40×, right panels) magnification for the live (green)/dead (red) assay for L-Pro–L-Phe–L-Phe on fibroblast cell culture. Scalebars = 100 μm (left panels) and 20 μm (right panels). (b) MTT assay after 24 h exposure to L-Pro–L-Phe–L-Phe at various concentrations. | ||
Experimental
Materials and methods
Recombinant human insulin produced in yeast (Roche) was purchased from Merck. Fmoc-amino acids, 2-chlorotrityl resin, and coupling agents were obtained from GL Biochem (Shanghai) Ltd. All other reagents and solvents were acquired at analytical grade from Merck. An in-line Millipore RiOs/Origin system provided high-purity Milli-Q-water with resistivity >18 MΩ cm. 1H-NMR and 13C-NMR spectra were recorded on a Varian Innova at 400 MHz with chemical shift reported as ppm (with tetramethylsilane as internal standard).An Agilent 6120 LC-MS system was used for ESI-MS spectra. Peptide synthesis, purification, and characterisation, as well as DSC measurements, MTT and live/dead cell assays were carried out as previously described.28
ThT assay for insulin fibrillation inhibition
Eppendorf tubes were prepared containing a stock solution of insulin at 172 μM (NaCl 0.1 M and HCl 25 mM, pH 1.6). For the fibrillation inhibition test, insulin stock solutions were transferred to another Eppendorf tube and diluted to 14.3 μM (with NaCl 0.1 M and HCl 25 mM, pH 1.6, 351 μL), to which 8.6 μL of dimethylsulfoxide (DMSO) without or with the tripeptides were added (final concentration 286.6 μM) with a total volume of 300 μL per sample. The tripeptide/insulin molar ratio 20:1 was chosen for the initial screenings. Samples were incubated in a thermoblock for the fibrillation process for 48 hours at 980 rpm and 37 °C. Fluorescence emission was measured in triplicate on two independent experiments using a TECAN Infinite M1000 PRO plate reader instrument, selecting an excitation wavelength of 446 nm and an emission wavelength of 490 nm, with a bandwidth of 20 nm. All the samples (50 μL) were added to the wells of Corning 96 flat- and transparent-bottom black polystyrene plates. Thioflavin-T was dissolved separately in a 50 mM glycine-NaOH buffer at pH 6.5 and filtered (0.2 μm). The concentration of Thioflavin-T was calculated with the absorbance measurement (ε = 36000 cm−1, Abs ≈ 0.7, ∼21 μM). 150 μl of ThT solution were added on top of samples in the 96-well plate. After 15 minutes, fluorescence was recorded using the parameters indicated above. Average values and standard deviations (n = 6) are plotted. For the dose–response assay, the same procedure was applied using different concentrations of Pro–Phe–Phe.
Transmission electron microscopy (TEM)
Solutions of insulin at 172 μM (NaCl 0.1 M and HCl 25 mM, pH 1.6) with or without the tripeptides (1.7 mM) were prepared in Eppendorf tubes and incubated in a thermoblock at 980 rpm and 37 °C for 48 hours. Copper-lacey carbon film grids were exposed under UV in a UV-Ozone cleaner (UV-Ozone Procleaner Plus) for 5 minutes to increase their surface hydrophilicity. Samples were then immediately deposited on the grids, dried at room temperature and stained with uranyl acetate. TEM micrographs were acquired on a Jeol JEM 2100 instrument at 100 kV. Fibril diameters were measured using ImageJ.Circular dichroism
Solutions of insulin at 172 μM (NaCl 0.1 M and HCl 25 mM, pH 1.6) without or with the tripeptides (344 μM), were prepared in Eppendorf tubes. Samples were incubated in a thermoblock at 980 rpm and 37 °C for 48 hours. Samples were then transferred into 0.1 mm quartz cuvettes and measured in a Jasco J815 spectropolarimeter, with one accumulation, D.I.T. 2 s, bandwidth of 1 nm, data pitch 0.5 nm, scanrate 50 nm min−1 and a range of wavelength from 200 to 300 nm. Spectra were averaged (n = 10) and plotted.UV resonance Raman spectroscopy (UVRR)
UVRR spectra were collected on the BL10.2-IUVS beamline of Elettra Synchrotron Trieste (Italy)49 using an excitation wavelength at 226 nm provided by the synchrotron radiation (SR). The excitation energy was set by regulating the undulator gap aperture and monochromatizing the incoming SR light with a Czerny-Turner monochromator (Acton SP2750, Princeton Instruments, Acton, MA, USA) equipped with a holographic grating with 3600 groves per mm. All UVRR spectra were recorded in back-scattered geometry by using a single pass of a Czerny-Turner spectrometer (Trivista 557, Princeton Instruments, 750 mm of focal length) equipped with a holographic grating at 1800 grooves per mm. The spectral resolution was set at 1.6 cm−1 per pixel. The calibration of the spectrometer was standardized using cyclohexane (spectroscopic grade, Sigma Aldrich). Any possible photodamage effect due to prolonged exposure of the sample to UV radiation (the maximum beam power measured on the sample was tens of μW) was avoided by continuously spinning the sample cell during measurements. All samples were freshly dissolved in buffer for assembly and placed in quartz cuvettes for UVRR measurements. The temperature was set at 298 K. Samples were prepared as described above, with insulin at 172 μM (NaCl 0.1 mM, HCl 25 mM, pH 1.6) at time 0 (native form) and after fibrillation at 37 °C and 980 rpm for 48 hours without or with L-Pro–L-Phe–L-Phe (1.7 mM). Peptide controls did not contain insulin.Molecular modelling
All-atom MD simulations and analyses were performed with the AMBER20 and AmberTools21 software packages.50 The structure of insulin was taken from the PDB databank (PDB entry 3I40,51 while the conformation of PFF was generated using the sequence command of the tleap module and further optimising the structure. The conformation of PfF was generated from that of PFF by inverting the chirality if the central amino acid using VMD1.9.352 and further optimising the structure with tleap. Next, 10 peptides were randomly placed around the protein and the system was solvated with 0.1 M NaCl water solution (OPC model for water). Two conditions, corresponding to the most likely protonation state of each amino acid at pH 1.6 and 7.4, were used to mimic experimental and physiological conditions. In the first case, both Asp and Glu sidechain carboxyl group were protonated at one oxygen, and the His were also protonated on both nitrogens. Finally, all C-termini (protein and tripeptides) were protonated.The ff19SB force field was used for the peptides and the proteins.53 All the simulations were performed as follows. First, three consecutive restrained structural optimizations (up to 25000 steps) were performed in the presence of harmonic restraints (k = 1 kcal mol−1 Å−1) applied to: (a) all non-hydrogenous atoms of the system; (b) backbone atoms; (c) Cα atoms. Reference structures at steps (b) and (c) were the final ones from the previous step. Next, up to 50000 cycles of unrestrained optimization were performed. Each system was then heated to 298.15 K in 1 ns via constant-pressure-temperature (NTP) MD simulations (using the isotropic Berendsen barostat and the Langevin thermostat) followed by an equilibration phase of 10 ns. Starting from the equilibrated structure, four MD simulation replicas, each of 0.5 μs in length, were performed for each system using a time step of 2 fs. Periodic boundary conditions were employed, and electrostatic interactions were estimated using the Particle Mesh Ewald scheme with a cutoff of 9.0 Å for the short-range evaluation in direct space and for Lennard–Jones interactions (with a continuum model correction for energy and pressure).
Spatial density of the tripeptides around insulin were calculated over the production trajectory with the grid command of the cpptraj tool of AMBER20, using a spacing of 0.5 Å in each direction. Figures were prepared with VMD1.9.3.52
Conclusions
In conclusion, this work analysed the complete stereoisomer series of eight Pro–Phe–Phe tripeptides as inhibitors for human insulin fibrillation. The L-homochiral compound was the best inhibitor leading to a reduction of fibrillation in vitro by 75% when an excess peptide was used, and inhibiting 50% of the fibrillation when 1 molar equivalent relative to insulin was present. The inhibitory activity was confirmed by a series of spectroscopic, microscopic, and calorimetric techniques, and lack of cytotoxicity was evident with live/dead assays on fibroblast cells in vitro, confirming the potential for this compound as additive to stabilise insulin formulations. In silico data confirmed that the design, which was based on the combination of Pro as a β-breaker, and Phe-Phe as a binding motif for the “aromatic triplet” at the insulin C-terminus was successful. Importantly, another key interaction was found with the hydrophobic LVEALYL segment, which is notoriously prone to fibrillation. Considering the high potential for such low-cost and biocompatible short peptides as additives for therapeutic formulations,54 this work paves the way to further developments in this exciting research area.Author contributions
B. R., S. K., E. S., S. A., A. V. V., A. M. G. – investigation; S. A., B. R., A. M. G., S. M. – supervision; B. R., A. V. V., A. M. G., S. M. – visualization; A. M. G., S. M. – conceptualization; A. M. G. – initial draft writing; all authors – writing and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. M. acknowledges funding from the University of Trieste (FRA2023) and from Fondazione Cassa di Risparmio Pistoia e Pescia (Italy). A. M. G. acknowledges the María Zambrano program under the grant agreement UNI/551/2021, and also Junta de Comunidades de Castilla-La Mancha (JCCM-FEDER, project SBPLY/21/180501/000206). A. V. V. acknowledges funding from Fondazione di Sardegna (annualità 2020, FdS grant no. F75F21001250007). S. K. acknowledges funding from the Slovenian Research and Innovation Agency – ARIS (P2-0089, J2-3043, J3-3079, J7-4420 and bilateral ARIS projects BI-FR/23-24-PROTEUS-005), and the CENN Nanocenter for TEM access. We acknowledge Elettra Sincrotrone Trieste for providing access to its synchrotron radiation facilities and we thank Fatima Matroodi for assistance in using beamline IUVS.References
- M. G. Iadanza, M. P. Jackson, E. W. Hewitt, N. A. Ranson and S. E. Radford, Nat. Rev. Mol. Cell Biol., 2018, 19, 755–773 CrossRef CAS PubMed.
- K. Sud, N. Narula, E. Aikawa, E. Arbustini, P. Pibarot, G. Merlini, R. S. Rosenson, S. V. Seshan, E. Argulian, A. Ahmadi, F. Zhou, A. L. Moreira, N. Côté, S. Tsimikas, V. Fuster, S. Gandy, R. O. Bonow, O. Gursky and J. Narula, Nat. Rev. Cardiol., 2023, 20, 418–428 CrossRef CAS PubMed.
- S. Karam, M. Haidous, V. Royal and N. Leung, Kidney Int., 2023, 103, 473–484 CrossRef CAS PubMed.
- M. G. Herrera and V. I. Dodero, Biophys. Rev., 2021, 13, 1147–1154 CrossRef CAS PubMed.
- F. Chiti, P. Webster, N. Taddei, A. Clark, M. Stefani, G. Ramponi and C. M. Dobson, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 3590–3594 CrossRef CAS PubMed.
- E. Gazit, Angew. Chem., Int. Ed., 2002, 41, 257–259 CrossRef CAS.
- F. Hane, FEBS Lett., 2013, 587, 3617–3619 CrossRef CAS PubMed.
- P. C. Ke, R. Zhou, L. C. Serpell, R. Riek, T. P. J. Knowles, H. A. Lashuel, E. Gazit, I. W. Hamley, T. P. Davis, M. Fändrich, D. E. Otzen, M. R. Chapman, C. M. Dobson, D. S. Eisenberg and R. Mezzenga, Chem. Soc. Rev., 2020, 49, 5473–5509 RSC.
- A. M. Garcia, C. Giorgiutti, Y. El Khoury, V. Bauer, C. Spiegelhalter, E. Leize-Wagner, P. Hellwig, N. Potier and V. Torbeev, Chem. – Eur. J., 2020, 26, 9889–9899 CrossRef CAS PubMed.
- F. Chiti, M. Stefani, N. Taddei, G. Ramponi and C. M. Dobson, Nature, 2003, 424, 805–808 CrossRef CAS PubMed.
- S. Ghosh, R. Ali and S. Verma, Int. J. Biol. Macromol., 2023, 239, 124231 CrossRef CAS PubMed.
- K. Kulenkampff, A. M. Wolf Perez, P. Sormanni, J. Habchi and M. Vendruscolo, Nat. Rev. Chem., 2021, 5, 277–294 CrossRef CAS PubMed.
- M. Spiess, N. Beuret, C. Prescianotto Baschong and J. Rutishauser, in Vitamins and Hormones, ed. G. Litwack, Academic Press, 2020, vol. 113, pp. 55–77 Search PubMed.
- S. Sen, R. Ali, A. Onkar, S. Ganesh and S. Verma, ChemBioChem, 2022, 23, e202100678 CrossRef CAS PubMed.
- F. Manganelli, G. M. Fabrizi, M. Luigetti, P. Mandich, A. Mazzeo and D. Pareyson, Neurol. Sci., 2022, 43, 595–604 CrossRef PubMed.
- A. K. Bishoyi, P. H. Roham, K. Rachineni, S. Save, M. A. Hazari, S. Sharma and A. Kumar, Biol. Chem., 2021, 402, 133–153 CAS.
- B. E. Lewis, A. Mulka, L. Mao, R. Sharafieh, Y. Qiao, S. Kesserwan, R. Wu, D. Kreutzer and U. Klueh, J. Diabetes Sci. Technol., 2023, 17, 163–171 CrossRef CAS PubMed.
- W. Cui, J.-W. Ma, P. Lei, W.-H. Wu, Y.-P. Yu, Y. Xiang, A.-J. Tong, Y.-F. Zhao and Y.-M. Li, FEBS J., 2009, 276, 3365–3371 CrossRef CAS PubMed.
- A.-V. Lupaescu, M. Iavorschi and M. Covasa, Pharmaceutics, 2022, 14, 235 CrossRef CAS PubMed.
- M. Akbarian, R. Yousefi, F. Farjadian and V. N. Uversky, Chem. Commun., 2020, 56, 11354–11373 RSC.
- B. Rosetti and S. Marchesan, Int. J. Mol. Sci., 2023, 24, 1306 CrossRef CAS PubMed.
- S. Santos, I. Torcato and M. A. R. B. Castanho, Biopolymers, 2012, 98, 288–293 CrossRef CAS PubMed.
- M. Reches and E. Gazit, Science, 2003, 300, 625–627 CrossRef CAS PubMed.
- N. K. Mishra, R. N. V. Krishna Deepak, R. Sankararamakrishnan and S. Verma, J. Phys. Chem. B, 2015, 119, 15395–15406 CrossRef CAS PubMed.
- P. Yao, J. Zhang, S. You, W. Qi, R. Su and Z. He, J. Mater. Chem. B, 2020, 8, 3076–3086 RSC.
- J. Zhang, P. Yao, S. You, W. Qi, R. Su and Z. He, J. Mater. Chem. B, 2022, 10, 7780–7788 RSC.
- P. Ung and D. A. Winkler, J. Med. Chem., 2011, 54, 1111–1125 CrossRef CAS PubMed.
- A. M. Garcia, M. Melchionna, O. Bellotto, S. Kralj, S. Semeraro, E. Parisi, D. Iglesias, P. D'Andrea, R. De Zorzi, A. V. Vargiu and S. Marchesan, ACS Nano, 2021, 15, 3015–3025 CrossRef CAS PubMed.
- M. I. Ivanova, S. A. Sievers, M. R. Sawaya, J. S. Wall and D. Eisenberg, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 18990–18995 CrossRef CAS PubMed.
- C. Thorlaksen, M. B. Neergaard, M. Groenning and V. Foderà, in Protein Aggregation: Methods and Protocols, ed. A. S. Cieplak, Springer US, New York, NY, 2023, pp. 297–309 Search PubMed.
- M. K. Siddiqi, N. Majid, P. Alam, S. Malik, A. Alam, S. Rajan, M. R. Ajmal and R. H. Khan, Int. J. Biol. Macromol., 2020, 143, 102–111 CrossRef CAS PubMed.
- B. N. Ratha, A. Ghosh, J. R. Brender, N. Gayen, H. Ilyas, C. Neeraja, K. P. Das, A. K. Mandal and A. Bhunia, J. Biol. Chem., 2016, 291, 23545–23556 CrossRef CAS PubMed.
- B. N. Ratha, R. K. Kar, S. Kalita, S. Kalita, S. Raha, A. Singha, K. Garai, B. Mandal and A. Bhunia, Biochim. Biophys. Acta, Proteins Proteomics, 2019, 1867, 405–415 CrossRef CAS PubMed.
- V. Banerjee, R. K. Kar, A. Datta, K. Parthasarathi, S. Chatterjee, K. P. Das and A. Bhunia, PLoS One, 2013, 8, e72318 CrossRef CAS PubMed.
- L. Nielsen, R. Khurana, A. Coats, S. Frokjaer, J. Brange, S. Vyas, V. N. Uversky and A. L. Fink, Biochemistry, 2001, 40, 6036–6046 CrossRef CAS PubMed.
- N. Amdursky, Y. Erez and D. Huppert, Acc. Chem. Res., 2012, 45, 1548–1557 CrossRef CAS PubMed.
- M. Ziaunys, A. Sakalauskas and V. Smirnovas, Biomacromolecules, 2020, 21, 4989–4997 CrossRef CAS PubMed.
- B. Sarmento, A. Ribeiro, F. Veiga and D. Ferreira, Colloids Surf., B, 2006, 53, 193–202 CrossRef CAS PubMed.
- B. Morel, L. Varela and F. Conejero-Lara, J. Phys. Chem. B, 2010, 114, 4010–4019 CrossRef CAS PubMed.
- E. Judy and N. Kishore, Biochimie, 2023, 207, 20–32 CrossRef CAS PubMed.
- D. Sharma, S. Arora and J. Singh, Int. J. Polym. Mater., 2020, 69, 1054–1068 CrossRef CAS PubMed.
- W. Dzwolak, R. Ravindra, J. Lendermann and R. Winter, Biochemistry, 2003, 42, 11347–11355 CrossRef CAS PubMed.
- M. Pachetti, F. D'Amico, L. Pascolo, S. Pucciarelli, A. Gessini, P. Parisse, L. Vaccari and C. Masciovecchio, Biophys. J., 2021, 120, 4575–4589 CrossRef CAS PubMed.
- D. Kurouski, J. Washington, M. Ozbil, R. Prabhakar, A. Shekhtman and I. K. Lednev, PLoS One, 2012, 7, e36989 CrossRef CAS PubMed.
- S. Venturi, B. Rossi, M. Tortora, R. Torre, A. Lapini, P. Foggi, M. Paolantoni and S. Catalini, Int. J. Biol. Macromol., 2023, 242, 124621 CrossRef CAS PubMed.
- D. Kurouski, R. A. Lombardi, R. K. Dukor, I. K. Lednev and L. A. Nafie, Chem. Commun., 2010, 46, 7154–7156 RSC.
- Z. Chi and S. A. Asher, Biochemistry, 1998, 37, 2865–2872 CrossRef CAS PubMed.
- S. A. Asher, M. Ludwig and C. R. Johnson, J. Am. Chem. Soc., 1986, 108, 3186–3197 CrossRef CAS.
- E. Scarel, O. Bellotto, P. Rozhin, S. Kralj, M. Tortora, A. V. Vargiu, R. De Zorzi, B. Rossi and S. Marchesan, Soft Matter, 2022, 18, 2129–2136 RSC.
- D. A. Case, K. Belfon, I. Y. Ben-Shalom, S. R. Brozell, D. S. Cerutti, T. E. Cheatham III, V. W. D. Cruzeiro, T. A. Darden, R. E. Duke, G. Giambasu, M. K. Gilson, H. Gohlke, A. W. Goetz, R. Harris, S. Izadi, S. A. Iz-mailov, K. Kasavajhala, A. Kovalenko, R. Krasny, T. Kurtzman, T. S. Lee, S. LeGrand, P. Li, C. Lin, J. Liu, T. Luchko, R. Luo, V. Man, K. M. Merz, Y. Miao, O. Mikhailovskii, G. Monard, H. Nguyen, A. Onufriev, F. Pan, S. Pantano, R. Qi, D. R. Roe, A. Roitberg, C. Sagui, S. Schott-Verdugo, J. Shen, C. L. Simmerling, N. R. Skrynnikov, J. Smith, J. Swails, R. C. Walker, J. Wang, L. Wilson, R. M. Wolf, X. Wu, Y. Xiong, Y. Xue, D. M. York and P. A. Kollman, AMBER 2020, University of California, San Francisco, 2020 Search PubMed.
- V. I. Timofeev, R. N. Chuprov-Netochin, V. R. Samigina, V. V. Bezuglov, K. A. Miroshnikov and I. P. Kuranova, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2010, 66, 259–263 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- C. Tian, K. Kasavajhala, K. A. A. Belfon, L. Raguette, H. Huang, A. N. Migues, J. Bickel, Y. Wang, J. Pincay, Q. Wu and C. Simmerling, J. Chem. Theory Comput., 2020, 16, 528–552 CrossRef PubMed.
- O. Bellotto and S. Marchesan, in Peptide Bionanomaterials: From Design to Application, ed. M. A. Elsawy, Springer International Publishing, Cham, 2023, pp. 401–429, DOI:10.1007/978-3-031-29360-3_12.
Footnote |
| † Electronic supplementary information (ESI) available: TEM micrographs and UVRR spectra. See DOI: https://doi.org/10.1039/d4nr00693c |
| This journal is © The Royal Society of Chemistry 2024 |