
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
PLGA micro/nanoparticle vaccination elicits non-tumor antigen specific resident memory CD8+ T cell protection from hepatocellular carcinoma†
Pan
Li‡
 *ab,
Zihe
Zhai‡
c,
Jiawen
Fang‡
a,
Ruo
Wang
b,
Weiqi
Li
a,
Beiduo
Wang
c,
Jinglei
Wang
a,
Jiaqi
Zhu
b,
Feng
Bing
b,
Qiaoling
Pan
b,
ChangYou
Gao
*c and
ShaoHong
Lu
*a
*ab,
Zihe
Zhai‡
c,
Jiawen
Fang‡
a,
Ruo
Wang
b,
Weiqi
Li
a,
Beiduo
Wang
c,
Jinglei
Wang
a,
Jiaqi
Zhu
b,
Feng
Bing
b,
Qiaoling
Pan
b,
ChangYou
Gao
*c and
ShaoHong
Lu
*a
aEngineering Research Center of Novel Vaccine of Zhejiang Province, Hangzhou Medical College, Hangzhou, 310051, China. E-mail: llsshh2003@163.com; panli0706@zju.edu.cn; 881012022054@hmc.edu.cn; 17662433651@163.com; 21818015@zju.edu.cn; 13305470636@163.com
bState Key Laboratory for the Diagnosis and Treatment of Infectious Diseases, Collaborative Innovation Center for Diagnosis and Treatment of Infectious Diseases, The First Affiliated Hospital, Zhejiang University School of Medicine, 79 Qingchun Rd., Hangzhou City, 310003, China. E-mail: wangruo1998@zju.edu.cn; 11818073@zju.edu.cn; qiaolingpan@zju.edu.cn
cMOE Key Laboratory of Macromolecular Synthesis and Functionalization, Department of Polymer Science and Engineering, Zhejiang University, Hangzhou, 310027, China. E-mail: cygao@zju.edu.cn; zhaizh@zju.edu.cn; bdwang10@zju.edu.cn
First published on 4th June 2024
Abstract
Together, tumor and virus-specific tissue-resident CD8+ memory T cells (TRMs) of hepatocellular carcinoma (HCC) patients with Hepatitis B virus (HBV) infection can provide rapid frontline immune surveillance. The quantity and activity of CD8+ TRMs were correlated with the relapse-free survival of patients with improved health. However, HBV-specific CD8+ TRMs have a more exhausted phenotype and respond more actively under anti-PDL1 or PD1 treatment of HBV+HCC patients. Vaccination strategies that induce a strong and sustained CD8+ TRMs response are quite promising. Herein, a biodegradable poly(D,L-lactide-co-glycolide) microsphere and nanosphere particle (PLGA N.M.P) delivery system co-assembled by anti-PD1 antibodies (aPD1) and loaded with ovalbumin (OVA-aPD1 N.M.P) was fabricated and characterized for size (200 nm and 1 μm diameter), charge (−15 mV), and loading efficiencies of OVA (238 μg mg−1 particles) and aPD1 (40 μg mg−1 particles). OVA-aPD1 N.M.P could stimulate the maturation of BMDCs and enhance the antigen uptake and presentation by 2-fold compared to free OVA. The nanoparticles also induced the activation of macrophages (RAW 264.7) to produce a high level of cytokines, including TNF-α, IL-6 and IL-10. In vivo stimulation of mice using OVA-aPD1 N.M.P robustly enhanced IFN-γ-producing-CD8+ T cell infiltration in tumor tissues and the secretion of IgG and IgG2a/IgG1 antibodies. OVA-aPD1 N.M.P delivered OVA to increase the activation and proliferation of OVA-specific CD8+ TRMs, and its combination with anti-PD1 antibodies promoted complete tumor rejection by the reversal of tumor-infiltrating CD8+ T cell exhaustion. Thus, PLGA N.M.P could induce a strong CD8+ TRMs response, further highlighting its therapeutic potential in enhancing an antitumor immune response.
1 Introduction
Chronic hepatitis B virus (cHBV) infection is one of the main causes of HCC (HBV+HCC).1,2 Authorities estimate that between 2015 and 2030, approximately 257–300 million cases of cHBV infection and 5 million deaths from HCC are projected globally.3,4 Despite great progress in chemoembolization (TACE) and targeted therapy, HCC is often diagnosed at advanced stages of disease, for which highly effective therapies are insufficient.5,6 Immune checkpoint blockade (ICB) immunotherapies based on anti-PD1/L1 antibodies have shown great antitumor activity in HCC; however, response rates of HBV+HCC were quite limited.7,8 Thus, novel strategies for increasing tumor-specific T-cell infiltration and improving the efficacy of immunotherapy in HBV+HCC are urgently needed. Previously, we found that the HBV infection had more effect on shaping immune compositions in tumor borders (TBs) than in tumors, with a significant enrichment of HBV-specific PD1+CD8+TRMs of HBV+HCC patients. Furthermore, the enrichment of PD1+CD8+TRMs in TB tissues was tightly associated with hepatic damage and fibrosis.9,31Liver CD8+TRMs are unique T cells residing in the liver that exert local immune surveillance including cytolytic activity and secretion of proinflammatory cytokines such as IFN-γ and TNF-α.10,11 They act as the first line of defense by directly lysing target cells and also confer protective functions through the recruitment of circulating T cells or other immune cells via chemokine production. CD8+TRMs generally express adhesion and retention molecules including CD103, CD69, CD49a, CD44, and chemokine receptors CXCR3 and CXCR6, but not all of these markers are expressed in all populations of TRMs, indicating the nuance among different TRMs subsets.12,13 CD8+TRMs were highly enriched in the setting of HBV infection compared with healthy livers, with a mean 3-fold increase in frequency, accounting for up to 68.5% of all intrahepatic memory CD8+ T cells.14
Increasing studies find the intra-tumoral HBV-specific CD103+CD69+CD8+ TRMs correlated with the improved prognosis of HBV+HCC patients. Furthermore, the degree of infiltration of CD8+ TRMs is associated with good prognosis of malignant tumors such as lung cancer15 and melanoma.16 Thus, a feasible immune intervention therapy approach is to reverse the immune microenvironment by targeting the activation of the non-tumor specific CD8+ TRMs, and then further improving the tumor-specific T cells to activate and proliferate through the crosstalk between cytokines or chemical factors among immune cells.
Poly(lactic-co-glycolic acid) (PLGA) is a widely used material with good biodegradable and biocompatibility.17 Particles in the nano (NP) and micron (MP) range are the most commonly used and extensively studied vaccine carrier polymers. Antigens can be adsorbed and encapsulated into the particle core or the polymer matrix, as well as conjugated to the building blocks or the particle surface.18,19 This biomaterial-based delivery system can enhance the uptake of either antigens or adjuvants by antigen-presenting cells (APCs), and is associated with better immune responses than those obtained with the soluble counterparts. Meanwhile, the prolonged release of antigens can provide more effective immune responses, and also avoid the risk of tolerance and substitute the need of several boosting administrations typically required to induce protective immunity.20,21
HBV is a strict hepatotropic virus, and mice are not permissive to HBV infection. Thus, the lack of an appropriate mouse model remains a major hurdle for studying the immunotolerance and immunopathogenesis induced by hepatitis B virus (HBV) infection-related HCC. Here, we assumed OVA (257–264) specific CD8+ TRMs as non-tumor antigen specific CD8+ TRMs in a HCC mouse model, and then prepared OVA-aPD1 N.M.P as a vaccine delivery system that was constructed by PLGA nano and microparticles. The inside of the particles was encapsulated with anti-mouse PD1 antibody (aPD1) and the surface was loaded with OVA(257–264) peptides, providing the OVA-aPD1 N.M.P with the ability to activate hepatic OVA-specific (non-tumor antigen specific) CD8+ TRMs. The function of the exhausted HCC specific CD8+ TRMs is then restored and the PD1 immunotherapy response is improved by cross-talking with other immune cells, which can then be available as a potential immune-therapeutic target in future HBV-related HCC.
2 Methods and materials
2.1 Preparation and characterization of particles
The nanoparticles (NPs) and microparticles (MPs) were prepared via a water-in-oil-in-water (W/O/W) double emulsion method with slight modification. Briefly, to prepare the empty particles, 25 mg PLGA (lactic acid![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :glycolic acid = 50:50, MW = 10000) (Jinan Daigang Biotech Co., Ltd, China) was dissolved in 2 mL CH2Cl2, into which 400 μL PBS was added. The mixture was then sonicated using a probe sonicator (45 W, JY92-IIDN, Scientz, China) for 1 min under an ice bath to obtain the first emulsions (W/O). This was followed by its addition to 5 mL aqueous solution (w/v, 2%) of polyvinyl alcohol (PVA) 1788 (alcoholysis degree = 87%–89% with MW 83000) (Aladdin Chemistry Co., Ltd, China), and sonication under an ice bath for 1.5 min using a probe sonicator and high-speed tissue homogenate machine (IKA T18 basic ULTRA-TURRAX, IKA, Germany) to obtain the second emulsions (W/O/W) of NPs and MPs, respectively. For the preparation of the antibody and antigen loaded particles, 400 μL aPD1 (0.9 mg) (Bio X Cell, USA) was added to 2 mL PLGA solution (25 mg mL−1, CH2Cl2) and sonicated using the probe sonicator for 1 min under an ice bath to obtain the W/O emulsions. This was followed by its addition to a mixture of OVA (0.5%, w/v) and sodium cholate (1%, w/v), and sonication under an ice bath for 1.5 min using a probe sonicator or high speed tissue homogenate machine to obtain the W/O/W emulsions of NPs or MPs, respectively.
:glycolic acid = 50:50, MW = 10000) (Jinan Daigang Biotech Co., Ltd, China) was dissolved in 2 mL CH2Cl2, into which 400 μL PBS was added. The mixture was then sonicated using a probe sonicator (45 W, JY92-IIDN, Scientz, China) for 1 min under an ice bath to obtain the first emulsions (W/O). This was followed by its addition to 5 mL aqueous solution (w/v, 2%) of polyvinyl alcohol (PVA) 1788 (alcoholysis degree = 87%–89% with MW 83000) (Aladdin Chemistry Co., Ltd, China), and sonication under an ice bath for 1.5 min using a probe sonicator and high-speed tissue homogenate machine (IKA T18 basic ULTRA-TURRAX, IKA, Germany) to obtain the second emulsions (W/O/W) of NPs and MPs, respectively. For the preparation of the antibody and antigen loaded particles, 400 μL aPD1 (0.9 mg) (Bio X Cell, USA) was added to 2 mL PLGA solution (25 mg mL−1, CH2Cl2) and sonicated using the probe sonicator for 1 min under an ice bath to obtain the W/O emulsions. This was followed by its addition to a mixture of OVA (0.5%, w/v) and sodium cholate (1%, w/v), and sonication under an ice bath for 1.5 min using a probe sonicator or high speed tissue homogenate machine to obtain the W/O/W emulsions of NPs or MPs, respectively.
The obtained emulsions were magnetically stirred under room temperature for 2 h to completely evaporate the CH2Cl2. Finally, the particles were collected by centrifugation and washed with ddH2O for 3 times. The NPs and MPs were re-dispersed in PBS, mixed at a ratio of 1:1 (w/w) to obtain the empty NPs and MPs (Emp. N.M.P), and antibody and antigen loaded NPs and MPs (OVA-aPD1 N.M.P), respectively. The particles were kept at −20 °C before use. To prepare the fluorescent-labelled particles, OVA-FITC (Beijing Solarbio Science & Technology Co., Ltd, China) was added to the PLGA solution, and the following procedure was the same as that for the empty PLGA particles.
The morphology of the particles was observed by scanning electron microscopy (SEM). The particle suspension (10 μL) was dripped onto a glass slide and dried at room temperature overnight, and then observed by SEM (S-4800, Hitachi, Japan) at an acceleration voltage of 3 kV. The size and zeta potential of the particles were measured by dynamic light scattering (DLS) (Nano-ZS, Malvern, UK).
The content of OVA coated on the particle surface was measured by the Bradford Assay Kit (Thermo Scientific, USA) according to the manufacturer's instructions. Briefly, the loaded particles (10 mg) were suspended in 1 ml 1 M sodium hydroxide (NaOH), and agitated at 250 rpm using an electronic shaker for 3 h at room temperature (RT) to hydrolyse the polymer. A 3 ml volume of PBS (pH 7.4) and 1 ml 1 M hydrochloride acid (HCl) were added to the PLGA–NaOH mixture. The resultant sample (0.1 ml) was mixed with 2 ml BCA reagents A and B (50:1). The absorbance of the above solution was measured using UV-vis spectroscopy at 557 nm. Each measurement was repeated in triplicate. A calibration curve was prepared at BSA concentrations of 20, 40, 80, 120, 160 and 200 mg ml−1.
The aPD1 loading contents were calculated according to the unloaded antibody in the centrifugal supernatant measured by ELISA. The loading capacity (L.C.) and encapsulation efficiency (E.E.) of the aPD1-encapsulated nanoparticles were determined by measuring the amount of nonencapsulated IgG through a mouse monoclonal antibody ELISA. The IgG Rat ELISA Kit was purchased from Thermo Fisher Scientific Inc. LC and EE were calculated as LC = (A − B)/C, EE = (A − B)/A, where A was the expected encapsulated amount of antibody, B was the free amount of antibody in the collection solution, and C was the total weight of the particles.
2.2 Cytotoxicity and immunogenicity evaluation of PLGA N.M.P in vitro
The cytotoxicity of the particles was determined by CCK-8 assay (Topscience Co., Ltd, China). AML12 cells were incubated with Emp. N.M.P or OVA-aPD1 N.M.P, respectively. After 24 h, the particles were removed and further cocultured with a new medium containing 10% (v/v) CCK-8 for another 3 h. The OD450 nm was measured by a microplate reader (Multiscan Sky, Thermoscientific, USA).Mouse BMDCs were prepared according to the previous method.22 Briefly, bone marrow cells were isolated from the femur and tibia of female C57BL/6 mice (6–8 weeks), and then cultured in RPMI-1640 medium with 10% fetal bovine serum (FBS) and 1% penicillin–streptomycin (Hyclone) supplemented with GM-CSF (20 ng mL−1) and IL-4 (10 ng mL−1). The culture media were replaced every 2 days. On day 6, non-adherent immature DCs (iDCs) were harvested for further evaluation.
To quantitatively determine the effect of PLGA N.M.P on the cellular uptake of the antigen, iDCs were cultured with free FITC-OVA, or that formulated in nano/micro-particles, and the OVA loading for the particle formulations was set at 238 μg OVA per 1 mg particles. The antigen endocytosis and presentation were analyzed by measuring OVA-FITC positive DCs (CD11c+CD11b+) using flow cytometry (BD LSRFortessa). For intracellular antigen localization, iDCs were cultured with free OVA-FITC or N.M.P-formulated OVA-FITC. After 24 h, cell membranes were labeled with Alexa FluorVR 594 phalloidin, and intracellular localization of OVA was examined by a confocal laser scanning microscope (CLSM, Leica TCS SP8).
To evaluate the iDCs activation and maturation, iDCs were stimulated with OVA-aPD1 N.M.P, medium, equal amounts of free OVA and aPD1 (OVA-aPD1), Emp. N.M.P, and PMA/Ionomycin for 24 h and 48 h, respectively. Then, the iDCs were collected and stained with fluorophore-labeled antibodies, including Brilliant Violet 510™ anti-mouse CD11b (Biolegend, cat. #101263), FITC anti-mouse CD80 (Biolegend, cat. #104706), APC anti-mouse CD86 (Biolegend, cat. #v105012), PE/Cyanine7 anti-mouse CD11c (Biolegend, cat. #117318), BD Horizon™ V500 Rat Anti-Mouse I-A/I-E (BD Bioscience, cat. #562366) and life/dead Zombie Aqua™ Fixable Viability Kit (Biolegend, cat. #423101). Finally, the redundant antibodies were removed through centrifugation, and the stained cells were analyzed using flow cytometry (CytoFlex LX).
For the inflammatory activation level of the vaccine towards DCs and RAW264.7 macrophages, the cytokine concentrations of IL-12p70, IL-10, TNF-α of DCs, and TNF-α, IL-6, IL-10 of RAW264.7 cells were determined by ELISA. Briefly, RAW264.7 cells and BMDCs were seeded into 96-well plates separately, and then incubated with naked or particles formulated OVA-aPD1, 24 h and 48 h later. The concentrations of different cytokines in the supernatant of each sample were determined according to the manufacturer's protocol.
2.3 Immunization, blood collection, and lymphocytes isolation
C57BL/6 mice (6 to 8 weeks old) were purchased from Nanjing Biomedical Research Institute of Nanjing University. All mice were maintained under specific pathogen-free conditions. All animal procedures were performed in accordance with the Guidelines for the Care and Use of Laboratory Animals of the First Affiliated Hospital of Zhe Jiang University, and approved by the Animal Ethics Committee of the First Affiliated Hospital of Zhejiang University.For the establishment of the liver cancer mouse, mice were hydrodynamically injected with a 5:1:1 molar ratio of transposon to transposase-encoding plasmid (26 μg total DNA) via the tail vein. Two weeks later, the high dose OVA antigen immunization was continued for 14 days. Both the activity and quantity of the tumor specific and non-tumor (OVA) specific CD8+ TRMs of these mice were confirmed by flow cytometry (CytoFlex LX). Then, these mice were randomly divided into four groups with 10 mice in each group, and immunized with various formulations, including PBS, Emp. N.M.P, OVA-aPD1, and OVA-aPD1 N.M.P by tail vein injection with a pre-determined immunization dose of 3 times at 1 week intervals. To test the effect of particles on the CD8+ TRMs, half of the mice in each group were administered i.v. with 50 μg FTY720 (SML0700, SIGMA), which dissolved in 0.9% NaCl, and at a time period starting 1 week before the fourth vaccine immunization.
Blood was collected before every injection. Whole blood was centrifuged at 1000g to isolate the serum, which was then collected and stored at −80 °C until use. Fourteen days after the last immunization, the mice were sacrificed. The liver lymphocytes were isolated, and then the liver was washed with PBS and minced after perfusion. This was followed by enzymatic digestion with collagenase IV (Sigma, V900893), deoxyribonuclease type I (Sigma, D5025), and hyaluronidase type V (Sigma, H3506) at 37 °C for 30 min. Finally, the sample was filtered through the 70-mesh filter screen. After being washed with cell staining buffer (CSB, PBS containing 0.5% OVA and 0.02% NaN3), the cell pellets were resuspended into ACK lysing buffer to remove the red blood cells. The rest of the single cells were processed using the Percoll density gradient media (GE Healthcare) to get rid of fats and debris, followed by washing twice with wash buffer.
2.4 Evaluation of specific antibody responses and T cell responses in the liver
To study the in vivo inhibition of tumor growth of HCC, mice were sacrificed on the 14th day after the last treatment, and the tumor-bearing livers were photographed and weighed. The liver-to-body-weight ratio was also calculated. Blood was collected two weeks after the last immunization, and the total IgG (Genie, cat. #MOFI00923), IgG1 (Alpha Diagnostic International, cat. #600-110-OG1) and IgG2a (Alpha Diagnostic International, cat. #600-120-O2A) in serum were determined using ELISA kits. The concentrations of the IFN-γ, TNF-α, IL-2 and IL-10 cytokines in serum were also measured according to the manufacturer's instructions (Dakewe Biotech Company, Shen Zhen, China). For the cellular immunity assay, all of the antibodies for flow cytometry were purchased from BioLegend, unless otherwise stated. Single lymphocytes were pre-incubated with blocking buffer for 20 min on ice. For CD8+ TRMs staining, cells were stained with PE-594-conjugated anti-mouse CD3e (clone KT3.1.1), PerCP-cy5.5-conjugated anti-mouse CD4 (clone GK1.5), PE-conjugated anti-mouse CD45 (clone 30-F11), PE-Cy7-conjugated anti-mouse CD19 (clone 6D5), PE-Cy7-conjugated anti-mouse CD11b (clone M1/70), PB-conjugated anti-mouse CD8 (DAKO; clone M7103), FITC-conjugated anti-mouse PD1 (clone 29F.1A12), APC-conjugated anti-mouse CD103 (clone 2E7) and life/dead Zombie Aqua™ Fixable Viability Kit (Biolegend). Cells were stained 30 min on ice and analyzed by flow cytometry. Aqua+CD45−CD19+CD11b+CD4+ cells were excluded for analysis. For tetramer staining, PE-conjugated H-2Kb-restricted OVA257–264-peptide tetramer (MBL Life Science, TS-5001-1C) and APC-Cy7-conjugated anti-human CD45 were used. For cell sorting, Aqua−CD45+CD19−CD11b−CD3+CD4−CD8+CD103+ and PD-1+ cells were sorted into CSB for RNA isolation.Intracellular cytokine staining TILs were stimulated in vitro with Phorbol 12-Myristate 13-Acetate (PMA) (50 ng mL−1) and Ionomycin (1 mg mL−1; Sigma-Aldrich) or soluble OVA257–264-peptide (50 mg mL−1) separately in the presence of GolgiPlug (BD Biosciences; 1:1000) and GolgiStop (BD Biosciences; 1:1500) for 3–4 hours. Cells were washed twice and resuspended in FACS wash buffer for staining with cell surface monoclonal antibodies (mAbs), and subsequently fixed, permeabilized and stained with anti-mouse IFN-γ and TNF-α monoclonal antibody (mAb) cocktail for flow cytometry analysis.
2.5 Histopathological analysis
Tumor tissue histology and immunofluorescence analysis: for tissue section staining, tumors were fixed in 4% paraformaldehyde solution for 24 h, and then dehydrated, embedded, sectioned, and stained with H&E for immunofluorescence (IF) according to the manufacturer's instruction. The slice thickness was 5 μm. For IF staining of tumor cell proliferation, the sections were immersed in boiling citrate solution and then covered by primary antibody solutions of Ki67. The fluorescein isothiocyanate-conjugated secondary antibody was used to incubate the sections. The nuclei were counterstained by DAPI. Stained sections were observed by CLSM (Axio Scope.A1, Zeiss), and the semi-quantitative analyses were performed using ImageJ software. Hematoxylin–eosin (H&E) staining was applied to visualize the tumor histopathology following a standard protocol procedure.2.6 The immune mechanism analysis of vaccination
Mouse liver lymphocytes from OVA-aPD1 N.M.P and control were used for scRNA-seq. The pulmonary immune cells were purified by CD45 microbeads (Miltenyi Biotec, Germany), and loaded on the Chromium Controller. After single cell cDNA libraries were prepared, sequencing was performed on Illumina platforms. After quality control and filtering, the sequencing data were generated for subsequent analysis. For clustering of cells by gene expression, the graph-based clustering algorithm was implemented in Seurat after principal component analysis. For visualization, t-SNE was used to reduce the dimensionality. GO and KEGG enrichment analysis between the two groups was performed using the C5 GO gene dataset and C2 KEGG gene dataset (v7.2) of the MSigDB data (https://www.gsea-msigdb.org/gsea/msigdb) by GSEA. Monocle (an R package) was applied to perform pseudotime analysis. Differentially expressed genes were characterized as those with a fold change of >1.5 and P value <0.05. CellChat, an open source R package (https://github.com/sqjin/CellChat), was used to infer, visualize and analyze intercellular communications from the scRNA-seq data. We used a modified version of the SCENIC (Single-Cell rEgulatory Network Inference) approach for constructing GRNs from the single-cell RNaseq data.2.7 Statistical analysis
The dotplot and heatmap were generated in the R package. All data were presented as mean ± standard deviations (SDs). Differences between two groups were evaluated by two-tailed Student's t-test in Graphpad Prism. Significance levels were defined as *, P < 0.05; **, P < 0.01; ***, P < 0.001.3 Results and discussion
3.1 Characterization analysis of PLGA particles
Particles were prepared using a W/O/W double emulsion-solvent evaporation method23 (ESI Fig. 1A†). Particles exhibited two size ranges (Fig. 1A, ESI Fig. 1B†), in which the size of the nanoparticles was around 200 nm (peak 1) and that of the microparticles was around 1 μm (peak 2). Compared to Emp.N.M.P, OVA-aPD1 N.M.P possessed a higher ζ potential of −15 mV as a result of the OVA coating (Fig. 1B, Table 1). Both Emp. N.M.P and OVA-aPD1 N.M.P had no toxicity to AML12 cells within the investigation range of 5 to 100 μg mL−1 (Fig. 1C). SEM studies showed that both Emp.N.M.P and OVA-aPD1 N.M.P had a spherical morphology (Fig. 1D) with some collapse. This was due to the volatilization of the internal water phase during natural drying at room temperature. The OVA-aPD1 N.M.P had a rougher surface, revealing the successful coating of OVA. The loading content of OVA and aPD-1 in OVA-aPD1 N.M.P was 238 μg mg−1 and 40 μg mg−1 particles as determined by Bradford Assay Kit and ELISA Kit, respectively. The L.C. and E.E. of the OVA-encapsulated particles were determined to be 1.58% (w/w) and 90.00 ± 0.02%, respectively, by ELISA, and the aPD1-encapsulated particles were correspondingly (Table 1).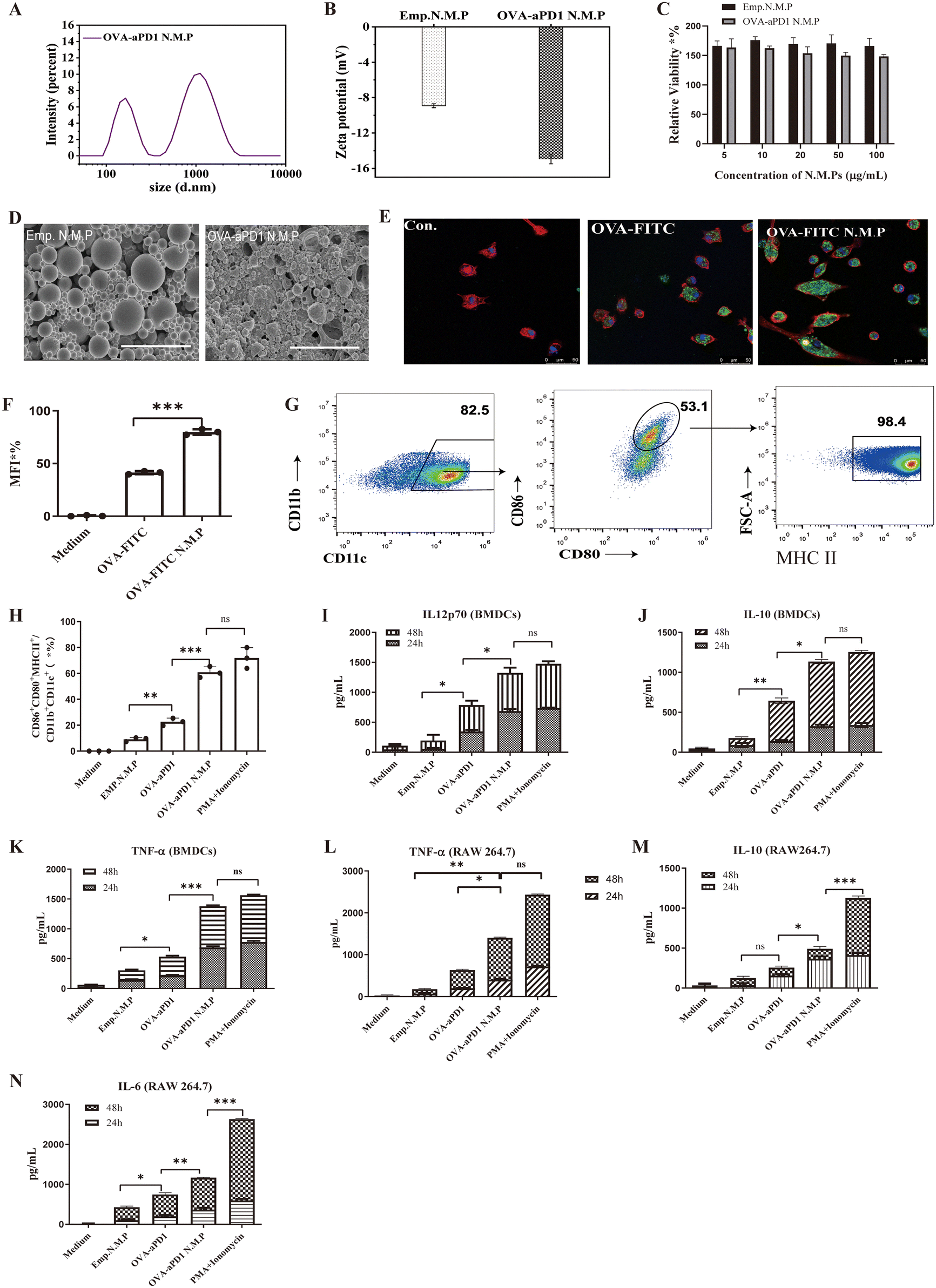 | ||
| Fig. 1 Characterization of particles and the effect of the PLGA N.M.P formulation on phagocytosis by macrophages and BMDCs. (A) Size distribution of the OVA-aPD1 N.M.P. (B) The ζ potential of particles measured by DLS, n = 3. (C) Relative cytotoxicity of particles, n = 5. (D) TEM image of OVA-aPD1 N.M.P and Emp. N.M.P, scale bars = 200 nm. (E) The effect of PLGA particles on the cellular uptake by CLSM. Scale bar, 50 μm. (F) The flow cytometry profile of OVA-FITC uptake by BMDCs; data are presented as the MFI. (G) Gating strategy for the mature DCs. (H) The effect of particles on BMDCs maturation by flow cytometry; the percentage of CD80+CD86+MHC-II+CD11c+ was calculated, n = 3. (I, J and K) The concentration of IL-12p70 (I), IL-10 (J) and TNF-α (K) secreted by BMDCs after incubation with PBS, Emp. N.M.P, OVA-aPD1 N.M.P or PMA + Ionomycin, respectively. (L, M and N) The levels of cytokines: TNF-α (L), IL-10 (M) and IL-6 (N) secreted by RAW264.7 after incubation with PBS, Emp. N.M.P, OVA-aPD1 N.M.P and PMA + Ionomycin, respectively. Data are shown as the mean ± SD. Statistical significance was analyzed using the two-tailed Student's t-test between two groups, *p < 0.05, **p < 0.01, ***p < 0.001. | ||
| Particles | Particle size (nm, mean ± SD) | Polydispersive index (PDI, mean ± SD) | Loading content (μg mg−1 particles) | Zeta potential (mean ± SD) | |
|---|---|---|---|---|---|
| Peak 1 size (nm) & intensity (%) | Peak 2 size (nm) & intensity (%) | ||||
| OVA + aPD1 N.M.P. | 203 ± 68 nm, 7.07 | 1.3 ± 0.6 μm, 10.1 | 0.194 ± 0.026 | OVA (238); aPD1 (40) | −8.92 ± 0.22 |
| Emp. N.M.P. | 295 ± 25 nm, 8.23 | 1.3 ± 0.4 μm, 12.9 | 0.128 ± 0.115 | — | −14.93 ± 0.55 |
3.2 OVA-aPD1 N.M.P exhibited a high adjuvant effect on DCs and macrophages in vitro
T-cell activation plays a significant role in the initiation and regulation of the immune response. It involves interaction with antigen-presenting cells (APCs) and related signals.24 Antigen uptake and processing by DCs is a crucial signal. PLGA particles can mimic the particulate nature of pathogens, which may facilitate the antigen uptake by DCs.25 To examine the adjuvant effect of PLGA N.M.P on antigen internalization, iDCs were incubated with free OVA-FITC, or OVA-FITC formulated in PLGA N.M.P, and cell membranes were labeled with Alexa Fluor 594 phalloidin. OVA delivered by PLGA N.M.P was mainly localized in the cytosol, as shown by the bright green FITC fluorescence signals. The uptake of fluorescent antigen was observed using confocal laser-scanning microscopy (Fig. 1E) and quantified by flow cytometry (Fig. 1F). It is evident that iDCs contained low levels of fluorescence, indicating that soluble OVA-FITC was poorly phagocytosed. In contrast, punctate regions of green fluorescence were displayed in cells treated with the nano/micro particle formulation, suggesting enhanced FITC-OVA uptake by DCs. These observations were confirmed by flow cytometry. The cells that were exposed to OVA-FITC N.M.P exhibited enhanced phagocytosis with an increase in both the number of fluorescent cells and fluorescence intensity as high as 82.5%, which were mostly 2-fold higher than that of the free OVA-treated group.Another important signal is mediated by the engagement of co-stimulatory molecules, such as B7.1 (CD80) and B7.2 (CD86), on APC and CD28 on the T cells. These two signals start a cross-talk between the T cells and APCs, which both release cytokines that collectively will define the inflammatory milieu. DCs are the main type of phagocytic cells, and play an important role in initiating adaptive immune responses. On sensing dangerous signals through their pattern recognition receptors (PRRs), iDCs undergo maturation while migrating to the secondary lymphoid organs to prime T cells.25,26 So, the effect of PLGA N.M.P on the activation and maturation of BMDCs was also assessed. The activation of iDCs is commonly accompanied with the expression of several co-stimulatory factors. Upon stimulation with different antigen formulations and controls, the expression of surface markers of iDCs (including CD86, CD80 and MHCII) was examined by FACS. The loop gate rule of mature DCs is shown in Fig. 1G. The OVA-aPD1 N.M.P significantly enhanced the proportion of CD86+CD80+MHCII+ DCs (Fig. 1H). Persistent production of IL-12 by iDCs during active infection was indispensable for the polarization and maintenance of the Th1 response.27 Here, the concentration of IL-12p70 in the supernatant of DCs incubated with different stimulators was determined. Although free OVA-aPD1 significantly enhanced the secretion of IL-12p70, the OVA-aPD1 N.M.P-treated group even displayed a similar IL-12p70 level in comparison with the positive control of PMA/Ionomycin. Based on these results, we can conclude that the PLGA micro/nanoparticles facilitated the endocytosis of the antigen, and the production of inflammatory cytokines significantly promoted the activation and maturation of iDCs (Fig. 1I). Cytokine secretion of IL-10 (Fig. 1J) and TNF-α (Fig. 1K) in the culture supernatant are up-regulated compared to the control cells, indicating the activation and maturation of iDCs.
Apart from DCs, macrophages also play a pivotal role in the initiation of adaptive immune responses by recruiting and activating NK cells and T and B lymphocytes.28 The cytokine production by RAW 264.7 macrophages was determined in vitro after stimulation with different stimulators. The concentration of TNF-α (Fig. 1L), IL-10 (Fig. 1M) and IL-6 (Fig. 1N) was detected, and OVA-aPD1 N.M.P greatly increased the cytokine secretion. These results indicate that the nano and micro particles were able to assist the M1 polarization.
3.3 OVA-aPD1 N.M.P enhanced the antibody and Th1 cytokine secretion
Nano and micro-particles have attracted much attention as carriers for vaccine delivery due to their interesting advantages, such as a large surface-to-volume ratio, multifunctional surface, and targeting characteristics by enhanced permeability and retention (EPR) effect.29 Before immunization of OVA-aPD1 N.M.P, the quantity changes of CD8+ TRMs and OVA-specific CD8+ TRMs in the spleen and liver were tested, respectively, which followed the plasmid injection or chronic antigen stimulation. Using the FACS gating rule (as shown in ESI Fig. 1C†), two weeks after hydrodynamically injecting the plasmids, the percentage of CD8+ TRMs in the liver was increased but not in the spleen (ESI Fig. 1D†). Furthermore, with the OVA(257–264) peptide long-term stimulus, the ratio of OVA-specific CD8+ TRMs also dramatically increased in the liver (ESI Fig. 1E†), suggesting that the cell plays an important role in the anti-tumor immunity of the liver. Taken together, these results provide a solid basis for the immune efficacy of vaccine evaluation.The mouse modeling and therapy scheme are shown in Fig. 2A. Mice were randomly divided into four groups with 10 mice in each group, and immunized with various formulations, including PBS, Emp. N.M.P, OVA-aPD1, and OVA-aPD1 N.M.P, by tail vein injection with a pre-determined immunization dose 3 times at 1 week interval. To test the effect of particles on CD8+ TRMs, half of the mice in each group was administered i.v. with 50 μg FTY720 (SML0700, SIGMA), which dissolved in 0.9% NaCl, and starting 1 week before the fourth vaccine immunization.
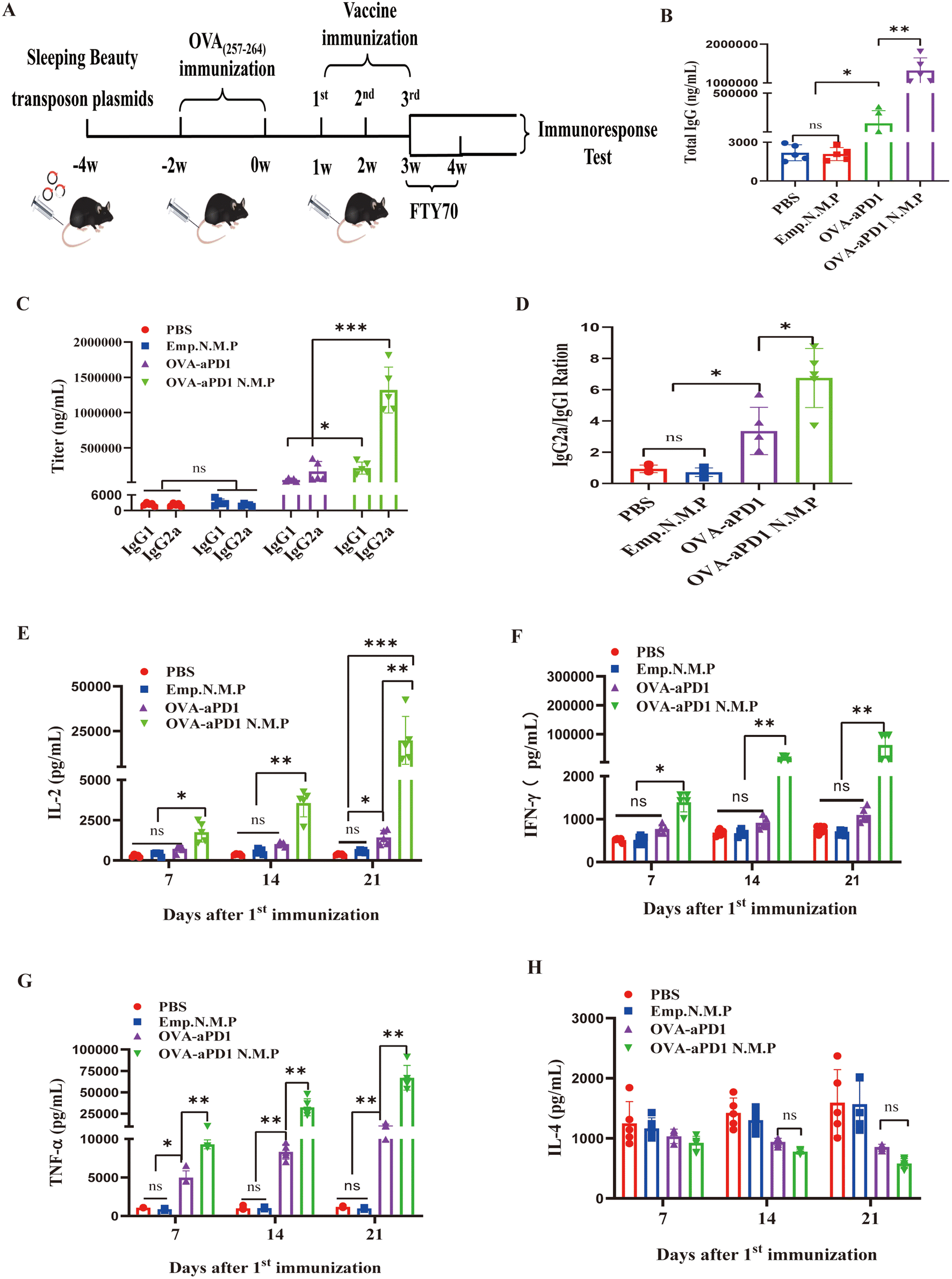 | ||
| Fig. 2 OVA-aPD1 N.M.P enhanced the antibody and Th1 cytokine secretion. (A) Experimental scheme of the OVA-aPD1 N.M.P treatment of HCC. (B) The serum total IgG antibody response, n = 5. (C) IgG1 and IgG2a subclass antibody response in C57BL/6 mice immunized with different vaccine formulations, n = 5. (D) The IgG2a and IgG1 ratio of different groups, n = 5. (E, F, G and H) The cytokine level of IL-2 (E), IFN-γ (F), IL-4 (G) and TNF-α (H) in the mouse serum, n = 5. | ||
The level of cellular and humoral immune response was detected based on IgG titer and cytokine concentration by ELISA. OVA-aPD1 N.M.P induced a significantly higher IgG response at 7 days after the last immunization. Free OVA-aPD1 also elicited an IgG response, but it was much lower than that of the immunotherapy group (Fig. 2B). The OVA-specific IgG1 and IgG2a levels were also measured to determine the types of T helper (Th) cell immune responses. In comparison with IgG1, more IgG2a is produced from the OVA-aPD1 N.M.P and free OVA-aPD1 groups (Fig. 2C). Furthermore, the IgG2a/IgG1 ratio is statistically significantly higher in the OVA-PD1 N.M.P group, indicating a mixed Th1/Th2 response when the Th1 bias response was aroused (Fig. 2D).
The concentrations of the cytokines in serum, which was collected at 7, 14, and 21 days after the first immunization, were evaluated. IL-2 emerged to be a key cytokine in regulating the survival, proliferation, and differentiation of activated T cells. IFN-γ and TNF-α play key roles in cellular immune processes and could promote Th1 responses. The secretion of IL-2 (Fig. 2E), IFN-γ (Fig. 2F), and TNF-α (Fig. 2G) was enhanced with the boost and third immunization. The OVA-aPD1 N.M.P induced a significantly higher level of Th1-type cytokines (IL-2, IFN-γ and TNF-α) compared to the other groups (P < 0.05). IL-4 acted directly on tumor cells as a tumor promoting cytokine. Furthermore, IL-4 contributed to the establishment and maintenance of Th2-polarized immune responses, reduced the tumoricidal activity of CD8+ T cells, and indirectly impaired the antitumor immunity in tumor-bearing animals or cancer patients. The two formulations of OVA-aPD1 induced a lower production of IL-4 (Fig. 2H). These results suggest that OVA-aPD1 N.M.P robustly augmented both humoral and cellular immune responses, especially a potent Th1 immune response in vivo.
3.4 OVA-aPD1 N.M.P inhibited the tumor by activating CD8+ TRMs
CD8+ T cell exhaustion is often associated with the inefficient control of persisting infections and cancers. However, re-invigoration of exhausted T cells with an inhibitory receptor blockade can promote improved immunity and disease outcomes.30 Two weeks after the last immunization, lymphocytes of the liver were harvested and analyzed by FACS. Compared to the immunotherapy group, the percentage of exhausted CD8+ T cells of both PBS and PLGA N.M.P was more than 60% of all lymphocytes based on the expression of Tim3 and PD1 (Fig. 3A, ESI Fig. 2A†). Although the total IFN-γ or TNF-α positive CD8+ T cells have no significant differences, the quantity of IFN-γ and TNF-α double positive CD8+ T cells from OVA-aPD1 or OVA-aPD1 N.M.P was higher than others, with the averages at 33% and 39%, respectively (Fig. 3B, ESI Fig. 2A†).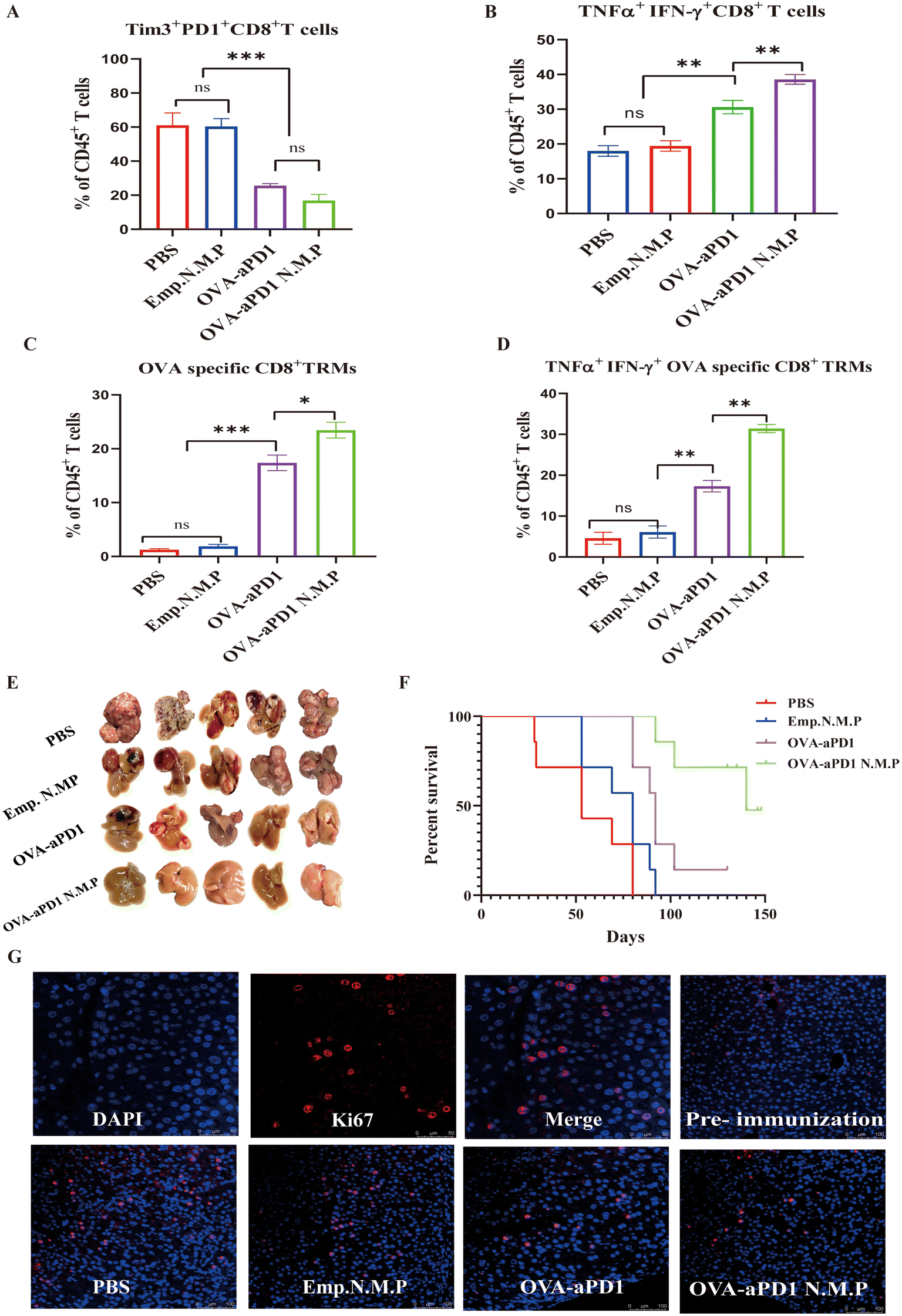 | ||
| Fig. 3 OVA-aPD1 N.M.P inhibited the tumor by activating CD8+ TRMs. (A) and (B) Frequency comparison of Tim3+PD-1+CD8+ T cells (A) and TNF-α+IFN-γ+CD8+ T cells (B) in liver tissues of different groups. (C) and (D) Frequency comparisons of OVA specific CD8+ TRMs (C) and TNF-α+IFN-γ+OVA specific CD8+ TRM (D) in liver tissues with different treatments. (E) Photographs of resected tumor-bearing livers after treatments. (F) Kaplan–Meier survival curves. Significance was determined by log-rank (Mantel-Cox) test; n = 10 per group except PBS, n = 4. (G) Immunofluorescence staining of Ki67 in tumor sections, scale bar = 200 μm. | ||
CD8+ TRMs acted as a facilitator that can secrete various cytokines, such as IFN-γ, TNF-α, and IL-2, with triggering adaptive and innate immune responses rapidly including DC maturation, NK cell activation, and B cell recruitment.10 Here, mice were treated with FTY720 to block the memory lymphocyte migration from the lymph nodes to the infected tissue. After the fourth immunization, we find that both OVA-aPD1 and OVA-aPD1 N.M.P groups elicited the increase of OVA-specific CD8+ TRMs, although there were also significant differences between the two groups (Fig. 3C, ESI Fig. 2B†). Furthermore, the OVA-specific CD8+ TRMs of the OVA-aPD1 N.M.P with a high level of TNF-α and INF-γ were compared to other groups (Fig. 3D, ESI Fig. 2B†). Consistent with the stronger CD8+ T cells and CD8+ TRMs immune response of the OVA-aPD1 N.M.P, a robust tumor regression was also observed (Fig. 3E). In line with these results, the liver-to-body-weight ratio was significantly higher in the PBS and Emp N.M.P groups compared with the other two groups (ESI Fig. 2C†). As the survival curves show, in the majority of cases, the curves were consistent with the inhibition of tumor growth, whereby the groups with smaller tumor sizes survived longer. Such survival was particularly notorious in two of the groups in which the tumor was not initially detectable. Only the OVA-aPD1 N.M.P group maintained 100% survival of the animals for more than five months after vaccine immunization (Fig. 3F). The H&E (ESI Fig. 2D†) and Ki67 (cell proliferation) (Fig. 3G) images showed more serious cell apoptosis with extensively damaged areas and cell proliferation areas in the control groups than the OVA-aPD1 N.M.P groups under the same conditions. These findings clearly demonstrated that OVA-aPD1 N.M.P could effectively boost strong systemic antitumor immune responses for synergistically suppressing the tumor growth.
Tumor-specific CD8+ TRMs that infiltrate tumor sites often fail to control tumor growth due to exhaustion or dysfunction sculpted by the immunosuppressive TME. Thus, the circuitous immunotherapy route that first activates the non-tumor antigen-specific TRMs and then activates the tumor antigen-specific T cells by intercellular cross-talk may be a viable route. Overall, OVA-aPD1 N.M.P immunization could highly activate the local CD8+ TRMs, and then indirectly activate T cell immunity through cytokine crosstalk. With the PD1 antibody and the adjuvant effect of the PLGA nano/microparticles, OVA-aPD1 N.M.P immunization can further improve the immune effects for the synergistic inhibition of both the primary and distal HCC tumor growths, which might provide a promising therapeutic vaccination strategy for potentiating HCC immunotherapy.
3.5 The anti-tumor immune mechanism of OVA-aPD1 N.M.P
To better understand the immunotherapy effect of OVA-aPD1 N.M.P, we applied single-cell RNA sequencing (scRNA-seq) to characterize the transcriptomic status of intrahepatic immune cells. After quality control and filtering, we obtained 14386 transcriptomes of single cells (ESI Fig. 3†). Based on the top differentially expressed genes (DEGs), we identified unique markers for each of the 16 clusters (ESI Fig. 4A and B†). Seven major cell clusters and corresponding proportion were identified, including T cells, NK cells, B cells, plasma cells, macrophage, DCs and neutrophils (Fig. 4A). To investigate the distinctive immune profiles of the immunotherapy groups, we analyzed the difference of the immune cell composition between OVA-aPD1 N.M.P and the control. Both B and T cells were significantly increased with immunized OVA-aPD1 N.M.P. However, the proportion of macrophages and neutrophils was reduced in contrast to the control (Fig. 4B). For T cells, we further identified 1 subcluster for the CD4+ T cells, 4 for the CD8+ T cells, 1 of the γδ T cells, and 1 of the NK T cells (Fig. 4C).
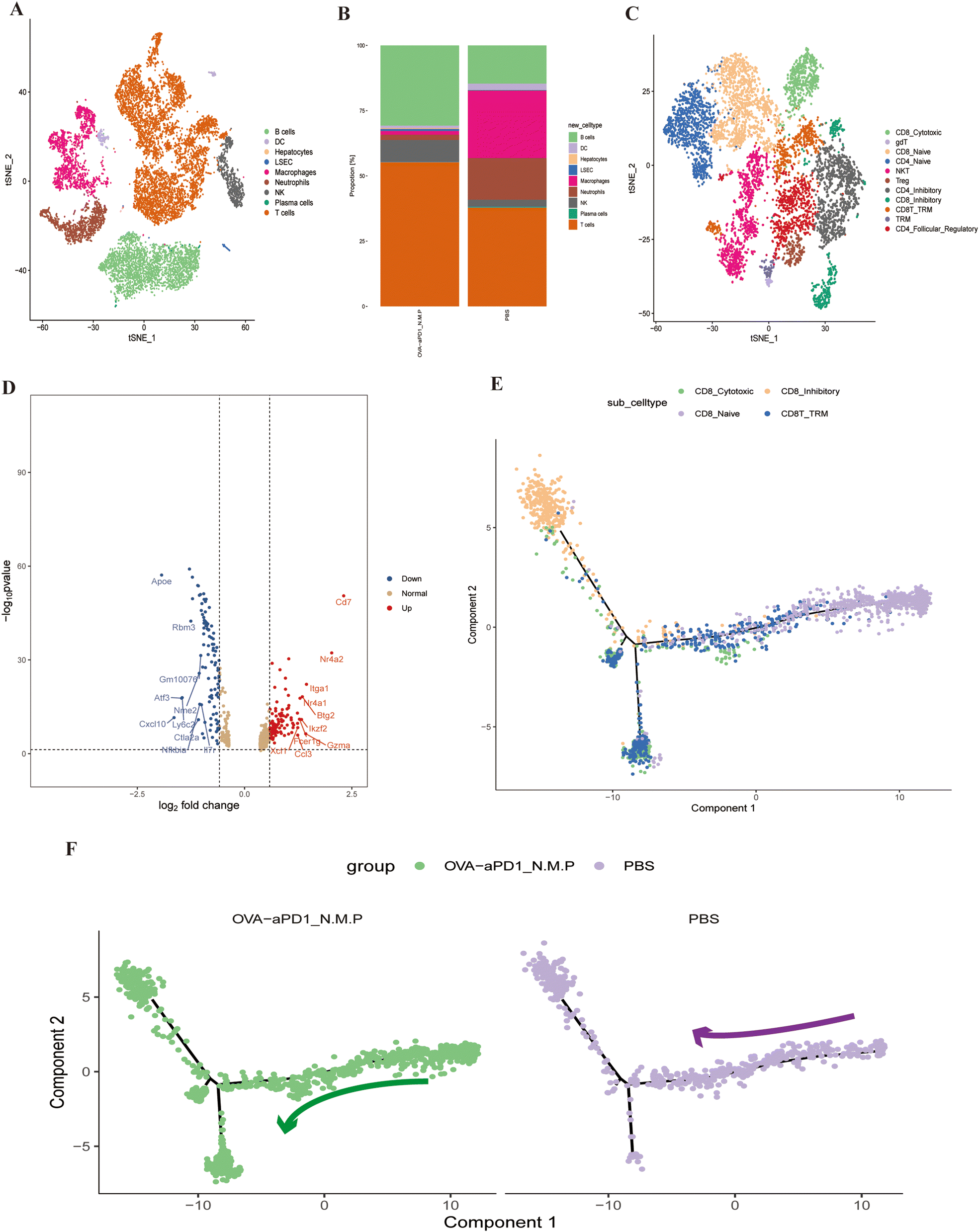 | ||
| Fig. 4 Liver local immune cell changes after OVA-aPD1 N.M.P immunization and related immune mechanism. (A) A t-SNE plot map showing the liver major immune cell subsets. (B) The fraction of the different cell types of the OVA-aPD1 N.M.P group and control. (C) A t-SNE plot map of the liver T cell subsets. (D) Volcano map of differentially expressed genes of CD8+ TRM; the red points represent the upregulated genes selected based on the corrected p-value <0.05 and logFC > 1, the blue points represent the downregulated genes selected based on the corrected p-value <0.05 and logFC < −1. The brown points represent genes with no significant difference. (E) Pseudo-time ordering of the T cell subtypes. (F) The differentiation trajectory of the T cell subtypes after immunotherapy. | ||
The scRNA-seq dataset of T cells was further investigated. T cells of the liver from OVA-aPD1 N.M.P had significantly higher expression genes of SATB1, CCL3, CCR7, LEF1, S1PR1, CD8B1, TCF7, FOXP1, NR4A2, CCL5, CTLA2 and KLF3, but lower expression genes of CXCL2, ATF3 and APOE (ESI Fig. 4D†). CD8+ T cells with a TRM phenotype can be identified as co-expressing transcripts for ITGAE, CXCR6, RUNX3 and CD69 (ESI Fig. 4B†), and there is evidence for these cells expressing high amounts of CD7, GZMA, NR4A2 and ITGA1 in the OVA-aPD1 N.M.P group (Fig. 4D). Heatmap analysis demonstrated that the immunotherapy group has a distinct set of up-regulated genes of CD8+ TRM, including that of different treatments (ESI Fig. 4D†).
Additionally, the Monocle 2 algorithm was used to perform pseudotime analysis. Two evolution fates of CD8+ T cells were found, one leading to inhibitory T cells and one leading to CD8+ TRMs (Fig. 4E, ESI Fig. 5A†). The trajectory began with CD8 naive T cells, followed by CD8 cytotoxic T cells. Then, some CD8 cytotoxic T cells ended with exhausted T cells, while some transformed into CD8+ TRMs (Fig. 4F).
CellChat is a tool based on gene expression and curated knowledge of communication information, such as receptors, ligands and their interactions, from known databases.32 Here, we analyzed the cell–cell interaction between T cells. Strikingly significant interactions were found between CD8+ TRMs, cytotoxic CD8+ T cells and NKT cells within immunotherapy. The number and strength of T cell subtype interactions of the OVA-aPD1 N.M.P group (Fig. 5A, ESI Fig. 5B†) were significant higher compared to the control (ESI Fig. 5C and D†). Moreover, the CCL, ICAM, ITGAL, MHC1, TNF-α signaling pathways were enriched in CD8+ TEMs and TRMs after infection (Fig. 5B). Transcription factors (TFs) and their downstream-regulated genes constitute a complex and intermingled network of gene regulation, which determines and maintains cell identity (Fig. 5C).
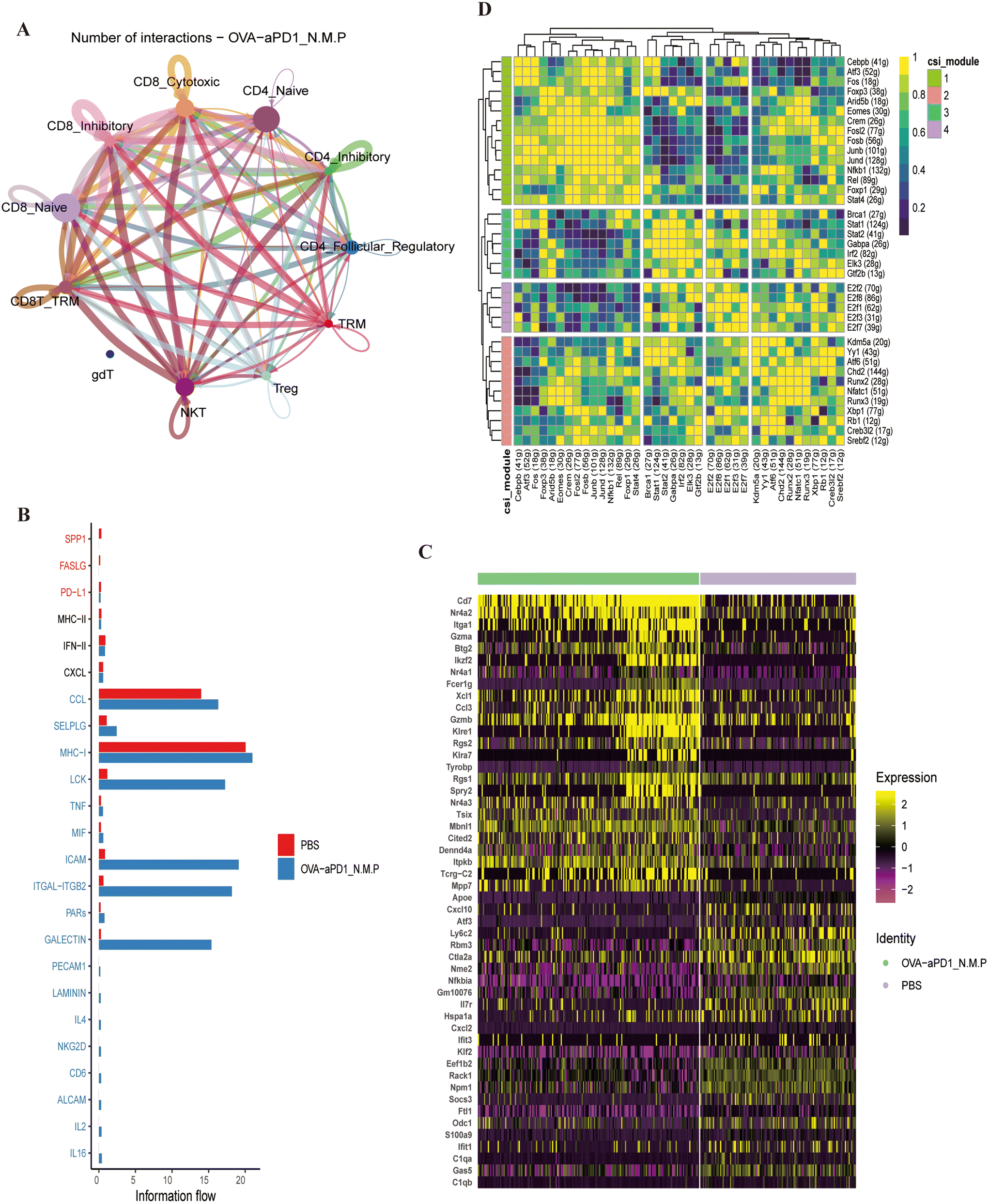 | ||
| Fig. 5 Liver local immune cell interactions after OVA-aPD1 N.M.P immunization and related immune mechanism. (A) The bubble plots show the number of interactions among T cell subtypes in liver tumor tissues of OVA-aPD1 N.M.P predicted by Cellphone DB; the node size represents the number of interactions. The width of the edge represents the number of significant ligand–receptor pairs between the two cell types. (B) Information flow of each signaling pathway from the OVA-aPD1 N.M.P group versus the HCC control. (C) Heatmap of the top 10 marker genes of CD8+ TRM with immunotherapy. (D) Heatmap of the regulon activity analyzed by SCENIC with default thresholds for binarization. The “regulon” refers to the regulatory network of TFs and their target genes. | ||
Single-cell regulatory network inference and clustering (SCENIC) analysis was performed to infer the activity of regulons (a TF together with its target genes comprise a regulon) for the CD8+ TRMs from the two groups. The regulon modules based on the regulon crosstalk (regulon-to-regulon correlation) were determined by the Connection Specificity Index (CSI) that ranks the regulon significance and mitigates the effects of nonspecific interactions. The analysis of the control and therapy group led to 16 regulons across four regulon modules (Fig. 5D). Comparison of the activity of the four modules revealed that module 1, including the TFs of CREB3L2, ARID5B, ELK3, ATF6, E2F1, KDM5A, SREBF2 and CREM, displayed the highest regulation activity in CD8+ TRMs from the OVA-aPD1 N.M.P group (ESI Fig. 5E†). Taken together, these findings indicated that OVA-aPD1 N.M.P may play a critical role in forming an activated liver local immunoenvironment by enhancing the immunoprotect effect of CD8+ TRMs and related genes.
4 Conclusion
In summary, we demonstrated a novel synergistic combination strategy for augmented anti-tumor angiogenesis therapy in HCC. Co-delivery of OVA (257–264) and anti-programmed cell death protein 1 (PD1) antibodies in PLGA nano and microparticles effectively facilitate the antigen uptake and maturation of DCs, promote the activation of iDCs and Raw264.7 macrophages, and significantly activate the local humoral and cellular immune response. Importantly, the OVA-aPD1 N.M.P elicited the activation of CD8+ TRMs, and then indirectly activated T cell immunity through cytokine crosstalk. Together with the anti-PD1 antibody and the adjuvant effect of the PLGA nano/microparticles, OVA-aPD1 N.M.P further improved the immune effects for synergistic inhibition of both primary and distal HCC tumor growths. Moreover, OVA-aPD1 N.M.P plays a critical role in forming an activated liver local immune-environment by enhancing the immunoprotect effect of CD8+ TRMs and related genes. The present study offers an efficient and promising strategy toward HBV-reduced HCC therapy by combining non-tumor specific CD8+ TRM activation and anti-PD1-mediated CD8+ TIL activation and dominant intratumor antitumor immunity in TME.Author contributions
All authors have contributed to the manuscript and have given approval to the final version of the manuscript.Ethics approval and consent to participate
All animal experimental procedures were conducted according to a protocol approved by the Ethics Committee of The First Affiliated Hospital of Zhejiang University.Data availability
All data generated or analyzed during this study are included in this article.Conflicts of interest
All authors declare no competing interests.Acknowledgements
We would like to thank the staff of OE Biotech Co., Ltd (Shanghai, China) for their help in single-cell RNA sequencing analysis. This work was supported by the National Natural Science Foundation of China (No. 82003015), Natural Science Foundation of Zhejiang Province of China (No. 2024KY931), The Fundamental Research Funds for the Central Universities (No. 2022ZFJH003), and the Key Discipline of Zhejiang Province in Public Health and Preventive Medicine (First Class, Category A), Hangzhou Medical College.References
- A. G. Singal, P. Lampertico and P. Nahon, Epidemiology and surveillance for hepatocellular carcinoma: New trends, J. Hepatol., 2020, 72(2), 250–261 CrossRef CAS PubMed.
- H. Sung, et al., Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries, CA Cancer J. Clin., 2021, 71(3), 209–249 CrossRef PubMed.
- Y. Chen and Z. Tian, HBV-Induced Immune Imbalance in the Development of HCC, Front. Immunol., 2019, 10, 2048 CrossRef CAS PubMed.
- G. E. M. Rizzo, G. Cabibbo and A. Craxì, Hepatitis B Virus-Associated Hepatocellular Carcinoma, Viruses, 2022, 14(5), 986 CrossRef CAS PubMed.
- Y. Jiang, et al., The Mechanisms of HBV-Induced Hepatocellular Carcinoma, J. Hepatocell. Carcinoma, 2021, 8, 435–450 CrossRef PubMed.
- S. H. Yeh, et al., Hepatitis B Virus DNA Integration Drives Carcinogenesis and Provides a New Biomarker for HBV-related HCC, Cell. Mol. Gastroenterol. Hepatol., 2023, 15(4), 921–929 CrossRef CAS PubMed.
- X. Hu, et al., Interaction between baseline HBV loads and the prognosis of patients with HCC receiving anti-PD-1 in combination with antiangiogenic therapy undergoing concurrent TAF prophylaxis, BMC Infect. Dis., 2022, 22(1), 614 CrossRef CAS PubMed.
- M. K. He, et al., Comparison of HBV reactivation between patients with high HBV-DNA and low HBV-DNA loads undergoing PD-1 inhibitor and concurrent antiviral prophylaxis, Cancer Immunol. Immunother., 2021, 70(11), 3207–3216 CrossRef CAS PubMed.
- Y. H. Lee, et al., Combinational Immunotherapy for Hepatocellular Carcinoma: Radiotherapy, Immune Checkpoint Blockade and Beyond, Front. Immunol., 2020, 11, 568759 CrossRef CAS PubMed.
- E. Menares, et al., Tissue-resident memory CD8+ T cells amplify anti-tumor immunity by triggering antigen spreading through dendritic cells, Nat. Commun., 2019, 10(1), 4401 CrossRef PubMed.
- R. Casalegno Garduño and J. Däbritz, New Insights on CD8(+) T Cells in Inflammatory Bowel Disease and Therapeutic Approaches, Front. Immunol., 2021, 12, 738762 CrossRef PubMed.
- K. Okła, D. L. Farber and W. Zou, Tissue-resident memory T cells in tumor immunity and immunotherapy, J. Exp. Med., 2021, 218(4), e20201605 CrossRef PubMed.
- J. V. Stein, N. Ruef and S. Wissmann, Organ-Specific Surveillance and Long-Term Residency Strategies Adapted by Tissue-Resident Memory CD8(+) T Cells, Front. Immunol., 2021, 12, 626019 CrossRef CAS PubMed.
- M. Enamorado, et al., Enhanced anti-tumour immunity requires the interplay between resident and circulating memory CD8+ T cells, Nat. Commun., 2017, 8(1), 16073 CrossRef CAS PubMed.
- N. Suryadevara, et al., A molecular signature of lung-resident CD8+ T cells elicited by subunit vaccination, Sci. Rep., 2022, 12(1), 19101 CrossRef CAS PubMed.
- I. Visan, Specific help for TRM cells, Nat. Immunol., 2015, 16(1), 64–64 Search PubMed.
- C. Puricelli, et al., Use of Poly Lactic-co-glycolic Acid Nano and Micro Particles in the Delivery of Drugs Modulating Different Phases of Inflammation, Pharmaceutics, 2023, 15(6), 1772 CrossRef CAS PubMed.
- S. Adepu and S. Ramakrishna, Controlled Drug Delivery Systems: Current Status and Future Directions, Molecules, 2021, 26(19), 5905 CrossRef CAS PubMed.
- C. Backlund, et al., Biomaterials-Mediated Engineering of the Immune System, Annu. Rev. Immunol., 2023, 41, 153–179 CrossRef CAS PubMed.
- Q. N. Ye, et al., Biomaterials-Based Delivery of Therapeutic Antibodies for Cancer Therapy, Adv. Healthc. Mater., 2021, 10(11), e2002139 CrossRef PubMed.
- A. Malek-Khatabi, et al., Recent progress in PLGA-based microneedle-mediated transdermal drug and vaccine delivery, Biomater. Sci., 2023, 5390–5409 RSC.
- P. Li, et al., Self-assembled PEG-b-PDPA-b-PGEM copolymer nanoparticles as protein antigen delivery vehicles to dendritic cells: preparation, characterization and cellular uptake, Regener. Biomater., 2017, 4(1), 11–20 CrossRef CAS PubMed.
- J. P. Rao and K. E. Geckeler, Polymer nanoparticles: Preparation techniques and size-control parameters, Prog. Polym. Sci., 2011, 36(7), 887–913 CrossRef CAS.
- M. Corrado and E. L. Pearce, Targeting memory T cell metabolism to improve immunity, J. Clin. Invest., 2022, 132(1), e148546 CrossRef PubMed.
- N. K. Al-Nemrawi, et al., Polymeric Nanoparticles for Inhaled Vaccines, Polymers, 2022, 14(20), 4450 CrossRef CAS PubMed.
- J. Goc, et al., Dendritic cells in tumor-associated tertiary lymphoid structures signal a Th1 cytotoxic immune contexture and license the positive prognostic value of infiltrating CD8+ T cells, Cancer Res., 2014, 74(3), 705–715 CrossRef CAS PubMed.
- G. Trinchieri, Interleukin-12 and the regulation of innate resistance and adaptive immunity, Nat. Rev. Immunol., 2003, 3(2), 133–146 CrossRef CAS PubMed.
- P. M. Kou and J. E. Babensee, Macrophage and dendritic cell phenotypic diversity in the context of biomaterials, J. Biomed. Mater. Res., Part A, 2011, 96(1), 239–260 CrossRef PubMed.
- V. R. Shinde, et al., Enhanced permeability and retention effect: A key facilitator for solid tumor targeting by nanoparticles, Photodiagn. Photodyn. Ther., 2022, 39, 102915 CrossRef CAS PubMed.
- E. J. Wherry, T cell exhaustion, Nat. Immunol., 2011, 12(6), 492–499 CrossRef CAS PubMed.
- L. Liu, et al., Single-cell analysis reveals HBV-specific PD-1(+)CD8(+) TRM cells in tumor borders are associated with HBV-related hepatic damage and fibrosis in HCC patients, J. Exp. Clin. Cancer Res., 2023, 42(1), 152 CrossRef CAS PubMed.
- S. Jin, et al., Inference and analysis of cell-cell communication using CellChat, Nat. Commun., 2021, 12(1), 1088 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr00554f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |