
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Probing scaffold size effects on multivalent lectin–glycan binding affinity, thermodynamics and antiviral properties using polyvalent glycan-gold nanoparticles†
Rahman
Basaran
a,
Darshita
Budhadev
a,
Amy
Kempf
b,
Inga
Nehlmeier
b,
Nicole
Hondow
c,
Stefan
Pöhlmann
bd,
Yuan
Guo
 e and
Dejian
Zhou
*a
e and
Dejian
Zhou
*a
aSchool of Chemistry and Astbury Centre for Structural Molecular Biology, University of Leeds, Leeds LS2 9JT, UK. E-mail: d.zhou@leeds.ac.uk
bInfection Biology Unit, German Primate Centre - Leibniz Institute for Primate Research, 37077 Göttingen, Germany
cSchool of Chemical and Process Engineering, University of Leeds, Leeds LS2 9JT, UK
dFaculty of Biology and Psychology, Georg-August-University Göttingen, 37073 Göttingen, Germany
eSchool of Food Science and Nutrition, and Astbury Centre for Structural Molecular Biology, University of Leeds, Leeds LS2 9JT, UK
First published on 17th June 2024
Abstract
Multivalent lectin–glycan interactions (MLGIs) are pivotal for viral infections and immune regulation. Their structural and biophysical data are thus highly valuable, not only for understanding their basic mechanisms but also for designing potent glycoconjugate therapeutics against target MLGIs. However, such information for some important MGLIs remains poorly understood, greatly limiting research progress. We have recently developed densely glycosylated nanoparticles, e.g., ∼4 nm quantum dots (QDs) or ∼5 nm gold nanoparticles (GNPs), as mechanistic probes for MLGIs. Using two important model lectin viral receptors, DC-SIGN and DC-SIGNR, we have shown that these probes can not only offer sensitive fluorescence assays for quantifying MLGI affinities, but also reveal key structural information (e.g., binding site orientation and binding mode) useful for MLGI targeting. However, the small sizes of the previous scaffolds may not be optimal for maximising MLGI affinity and targeting specificity. Herein, using α-manno-α-1,2-biose (DiMan) functionalised GNP (GNP-DiMan) probes, we have systematically studied how GNP scaffold size (e.g., 5, 13, and 27 nm) and glycan density (e.g., 100, 75, 50 and 25%) determine their MLGI affinities, thermodynamics, and antiviral properties. We have developed a new GNP fluorescence quenching assay format to minimise the possible interference of GNP's strong inner filter effect in MLGI affinity quantification, revealing that increasing the GNP size is highly beneficial for enhancing MLGI affinity. We have further determined the MLGI thermodynamics by combining temperature-dependent affinity and Van't Hoff analyses, revealing that GNP-DiMan–DC-SIGN/R binding is enthalpy driven with favourable binding Gibbs free energy changes (ΔG°) being enhanced with increasing GNP size. Finally, we show that increasing the GNP size significantly enhances their antiviral potency. Notably, the DiMan coated 27 nm GNP potently and robustly blocks both DC-SIGN and DC-SIGNR mediated pseudo-Ebola virus cellular entry with an EC50 of ∼23 and ∼49 pM, respectively, making it the most potent glycoconjugate inhibitor against DC-SIGN/R-mediated Ebola cellular infections. Our results have established GNP-glycans as a new tool for quantifying MLGI biophysical parameters and revealed that increasing the GNP scaffold size significantly enhances their MLGI affinities and antiviral potencies.
Introduction
Multivalent lectin–glycan interactions (MLGIs) are widespread and vital for pathogen infection and immune regulation.1–5 As monovalent lectin–glycan interactions are often too weak to be bio-active, lectins mostly form multimeric structures with clustered carbohydrate-binding-domains (CRDs), allowing them to bind multivalently with multivalent glycan ligands to enhance affinity and define specificity via spatial matching. The overall MLGI affinity is not only defined by the monovalent affinity, but also by the binding mode and glycan valency.3 In general, a pair of spatially matched MLGI partners may bind simultaneously with each other to form small individual lectin–ligand complexes, giving rise to a great MLGI affinity enhancement due to both favourable binding enthalpy and entropy terms.6–8 While those without perfect spatial and orientational matches may crosslink each other to maximize the binding enthalpies, this often forms large-scale protein–ligand assemblies with relatively low MLGI affinity enhancement due to large entropic penalties.9 Therefore, understanding the structural and molecular mechanisms of target MLGIs is of great importance, allowing us to design spatially-matched glycoconjugates to target specific MLGIs potently for therapeutic interventions. Notably, the development of glycoconjugates as potent entry inhibitors to block virus binding and infecting host cells can be advantageous over other anti-viral strategies because this can effectively prevent virus mutation and develop resistance.4,10,11 This can be particularly beneficial for controlling infections induced by unstable RNA viruses, including SARS-CoV-2, where rapid mutations and variations can lead to effective evasion of host antibody responses induced by past infections and/or vaccines.A large number of glycoconjugates, built upon various scaffolds, have been widely employed to study their MLGI properties and are being exploited as potential therapeutics.3–5,7,11–21 Some of these have exhibited potent inhibition against pathogen infections.6,7,11,15,16,18–20 In most cases, their MLGI biophysical data were obtained by conventional techniques, such as surface plasmon resonance (SPR),4,22 isothermal titration calorimetry (ITC),4,23 and/or fluorescence competition assays.24,25 While ITC and SPR are widely used biophysical techniques that can provide quantitative binding affinity, thermodynamic, and/or kinetic data, they cannot provide key structural information, such as lectin binding site orientation, inter-binding site distance, and binding mode, which are very useful for designing potent, specific glycoconjugate inhibitors. Moreover, these methods can also suffer from limitations, such as low sensitivity (requiring large sample amounts) and unreliable binding affinity measurement for very strong interactions (with equilibrium dissociation constants, Kds, of low nM or below) for ITC,23,26 while the affinities measured by SPR can be strongly affected by the density and orientation of the immobilized binding partners.22 Furthermore, most previous MLGI studies have been based on “passive” scaffolds, where nanomaterials are used as scaffolds only to create a polyvalent display of glycans to enhance their MLGI affinity, specificity, and/or therapeutic potency. The unique size-dependent optical/electrical properties of nano-scaffolds, the cornerstones of many nanomaterials, have not been explored as readouts for probing MLGI biophysical and structural data.
Recently, we have developed the “active” nano-scaffold strategy for MLGI research, where we have not only exploited the nano-scaffolds’ unique optical properties as new MLGI affinity assays but also their nanoscale size and high TEM contrast to reveal key MLGI structural information.11,19,27–29 We have employed the dendritic cell tetrameric lectin, DC-SIGN,30,31 and a closely-related endothelial cell lectin, DC-SIGNR,32 (collectively abbreviated as DC-SIGN/R hereafter) as model lectins. These lectins play a key role in mediating or facilitating the infection of many viruses, e.g., HIV, HCV, Ebola, Zika, and SARS-CoV-2.20,21,30,32,33 Despite displaying identical CRD-mannose monovalent binding motifs,34 and almost identical tetrameric architectures,31,35 these two lectins actually exhibit distinct virus transmitting properties. For example, DC-SIGN is more effective in transmitting the HIV infection than DC-SIGNR,36 while only DC-SIGNR, but not DC-SIGN, is able to transmit the West Nile virus infection.37 The structural mechanisms underlying such differences in DC-SIGN/R remain not fully understood. By displaying glycans polyvalently onto a CdSe/ZnS quantum dot (QD)19,27,28 or a small gold nanoparticle (GNP) scaffold11 as multifunctional probes, we have developed a new ratiometric QD-FRET (Förster resonance energy transfer)19,27,28 or GNP-fluorescence quenching affinity assay for MLGIs.11 We have found that glycan nanoparticles exhibit greatly enhanced MLGI affinities compared to the corresponding monovalent binding (up to 1.8 million fold)29 and the enhancement with DC-SIGN is significantly greater than that with DC-SIGNR (by ∼20–200 fold), arising from their distinct binding modes: DC-SIGN binds tetravalently with all 4 CRDs to one glycan nanoparticle, while DC-SIGNR crosslinks with multiple glycan nanoparticles.11,19,28 These glycan nanoparticles were found to potently block DC-SIGN-, but not DC-SIGNR-, mediated pseudo-Ebola virus infections with sub-nM EC50 (concentration giving 50% apparent inhibition) values.11,19 Despite success, our previous studies were all built upon small nanoparticle scaffolds (e.g., ∼4 nm QD or 5 nm GNP), which may not be optimal for maximising the MLGI affinity, specificity, and antiviral potency.
In this paper, we have systematically varied the GNP scaffold size (e.g., ∼5, ∼13 and ∼27 nm, denoted as G5, G13 and G27, respectively) and their surface glycan densities (e.g., 100, 75, 50 and 25%, by diluting the active glycan ligand, lipoic acid-tetra(ethylene glycol)-α-manno-α-1,2-biose (LA-EG4-DiMan), using an inert spacer ligand, LA-EG4-OH; see Fig. 1 for their chemical structures) to investigate how these factors control their MLGI properties with DC-SIGN/R. We have developed a new format of the GNP-fluorescence quenching assay to minimize the interference arising from GNPs’ strong inner filter effect, allowing us to robustly quantify MLGI affinities for both small and large GNP-glycans. We found that the MLGI affinities between DC-SIGN/R and GNP-glycans are enhanced significantly by increasing the GNP size, while reducing the glycan density from 100% to 50% does not markedly affect their affinity, although reducing the glycan density further to 25% weakens their affinity. By quantifying temperature-dependent MLGI affinities via GNP fluorescence quenching and applying Van't Hoff analysis, we have quantified their MLGI biophysical parameters, revealing that DC-SIGN/R bindings with GNP-glycans are enthalpy driven, and the negative binding enthalpy changes (ΔHs) for DC-SIGN are ∼4 times that of the monovalent binding, while that for DC-SIGNR is about twice that of the monovalent binding, under our assay conditions. Finally, we report that glycan-GNPs potently block DC-SIGN/R mediated pseudo-Ebola viral infection of host cells, with potencies being enhanced with increasing GNP size. In particular, a 27 nm GNP capped with a pure LA-EG4-DiMan ligand (G27-DiMan) potently and robustly blocks both DC-SIGN and DC-SIGNR mediated virus infections with EC50 values of ∼23 and ∼49 pM, respectively, making it the most potent glycoconjugate inhibitor against DC-SIGN/R mediated Ebola cellular infection. Together, our results have not only established GNP-glycans as a new platform tool for quantifying important MLGI biophysical data (e.g., affinity, thermodynamics, binding mode and binding site orientation, etc.) but also revealed that increasing the GNP scaffold size is strongly beneficial for GNP-glycan's MLGI affinities and antiviral potencies and therefore have successfully addressed an important knowledge gap in current MLGI research.
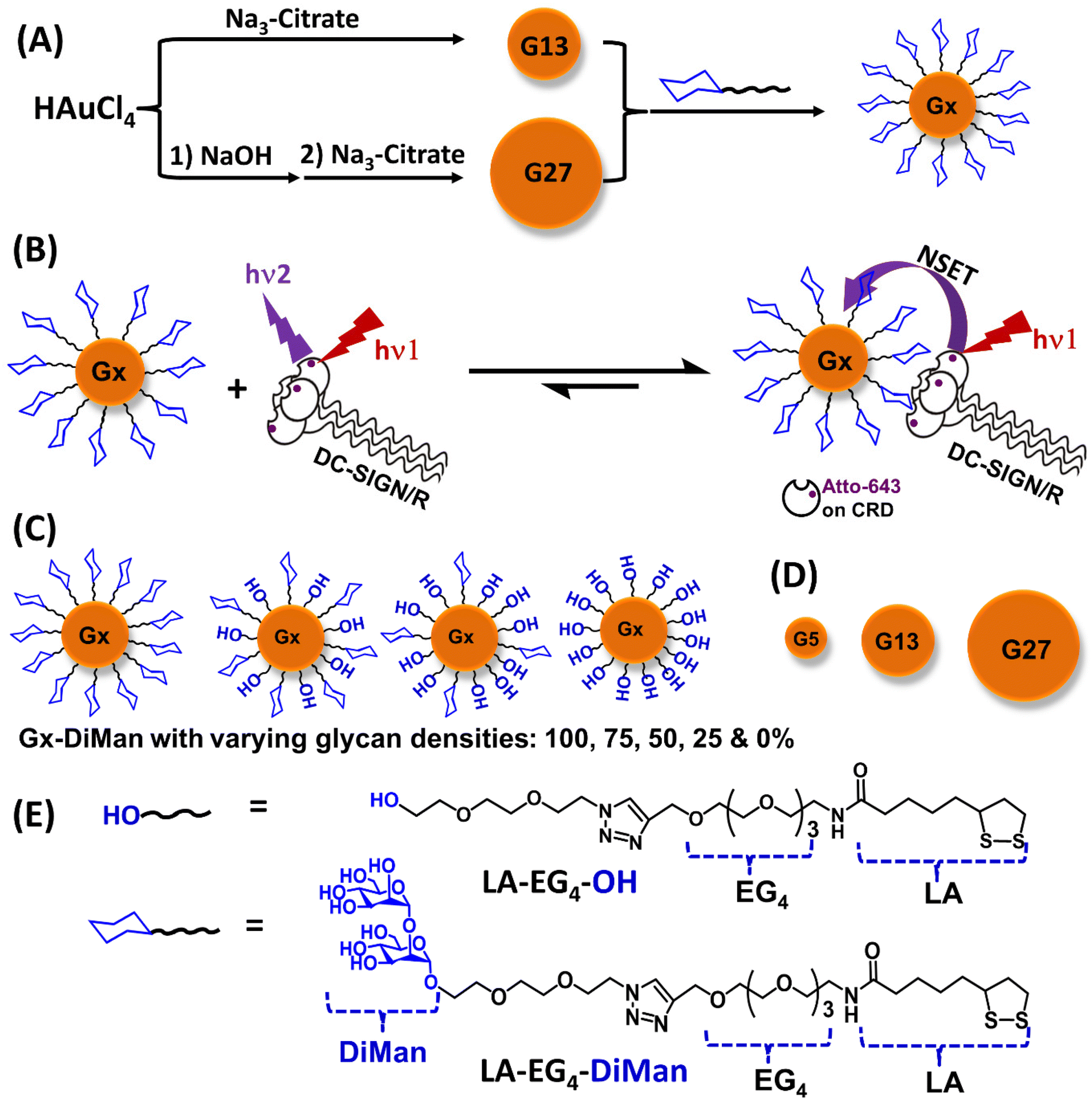 | ||
| Fig. 1 (A) A schematic showing the procedure of synthesising G13 (via citrate reduction of HAuCl4) and G27 (via NaOH treatment of HAuCl4 followed by citrate reduction) by further glycosylation to make Gx-glycans. (B) A schematic showing the principle of the GNP-fluorescence quenching assay for MLGI. Prior to binding with Gx-DiMan, excitation of the Atto-647 label on DC-SIGN/R CRDs gives strong fluorescence, while upon binding, the Atto-643 labels on DC-SIGN/R CRDs is efficiently and completely quenched by Gx-DiMan at proximity via a nano-surface energy transfer (NSET) mechanism; thus the quenching efficiency represents the proportion of DC-SIGN/R that have bound to Gx-DiMan at equilibrium. (C) Schematic representation of Gx-DiMan coated with varying glycan densities ranging from 100% (coated with 100% of LA-EG4-DiMan), 75%, 50%, 25% to 0% (coated with 100% of LA-EG4-OH) diluted by the LA-EG4-OH negative control ligand. (D) Schematic showing the different sizes of GNPs (Gx; x = 5, 13, and 27 nm), and (E) the chemical structures of LA-EG4-DiMan and LA-EG4-OH ligands used in this study. | ||
Results and discussion
Ligand design and synthesis
A lipoic acid-tetra(ethylene glycol)-α-manno-α-1,2-biose (LA-EG4-DiMan) based multifunctional ligand was synthesised via our established procedures (see Fig. 1E for its chemical structure).11 Each ligand was designed to contain three unique functional domains:19,38 an LA anchoring domain to chelate on the GNP surface by forming two strong Au–S bonds;11,38 a hydrophilic flexible EG4 linker domain to offer high water solubility, excellent biocompatibility and resistance against non-specific interactions;39–42 and a terminal DiMan domain to promote strong and specific binding with DC-SIGN/R.14,19 A hydroxyl group (OH) terminating ligand having the same LA anchoring and EG4 linker domains, LA-EG4-OH (Fig. 1E), was also synthesised and employed as an inert spacer ligand to tune the GNP surface glycan density. It also served as a negative control to correct the fluorescence quenching background (in the absence of specific MLGIs) caused by GNP's inner filter effect. We showed previously that displaying LA-EGx-DiMan (x is the number of repeating EG units between the LA and DiMan groups) based glycan ligands polyvalently onto QDs (x = 3 and 11)19,28 or G5 (x = 2)11 yielded strong MLGI affinities with DC-SIGN,11,19 while those coated with the equivalent -OH terminating (no terminal DiMan group) ligands gave no measurable binding,28 suggesting that LA-EG4-OH is a suitable negative control ligand for this study.The synthetic route to the LA-EG4-DiMan and LA-EG4-OH spacer ligands is shown in Scheme 1. First, an LA-EG4-acetylene linker was synthesised using the standard dicyclohexylcarbodiimide/N,N-dimethyl aminopyridine (DCC/DMAP) mediated amide coupling between lipoic acid and H2N-EG4-acetylene.11 Then, α-1-manno-α-1,2-biose appending an EG2-azide linker (N3-EG2-DiMan) to offer some flexibility to the terminal DiMan group was synthesised using our established route as described previously.19 Finally, the LA-EG4-acetylene linker was efficiently coupled to N3-EG2-DiMan or N3-EG2-OH (commercial) via the Cu-catalyzed click chemistry to give the desired LA-EG4-DiMan or LA-EG4-OH ligand in good yields, respectively.11,43 Their chemical structures were confirmed by 1H/13C NMR and LC-MS spectra (see ESI section 3† for details).
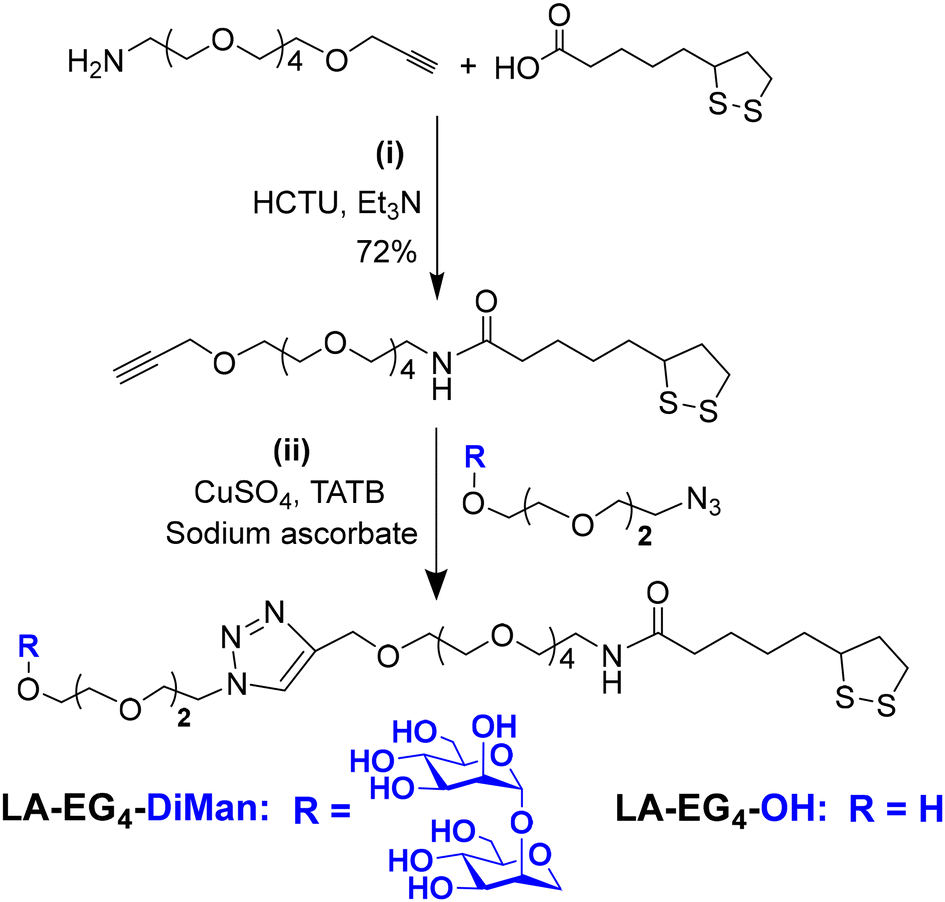 | ||
| Scheme 1 The synthetic route to the LA-EG4-DiMan and LA-EG4-OH spacer ligands. | ||
GNP synthesis and characterisation
Three different-sized GNPs with average diameters of ∼5, ∼13 and ∼27 nm (see ESI Fig. S1 and S2† for their TEM images), denoted as G5, G13 and G27, respectively, were employed in this study. Our motivation here was that tuning the GNP scaffold size may lead to an optimal glycan-GNP with excellent spatial and topological match to those of the four carbohydrate-recognition domains (CRDs) in each DC-SIGN/R molecules, thereby greatly enhancing their MLGI affinity and selectivity.5 The GNPs were either purchased commercially (for G5)11 or synthesised in house using the standard citrate reduction of HAuCl4 (for G13) or NaOH treatment of HAuCl4 followed by citrate reduction (for G27, Fig. 1A) via the literature protocols with minor modifications.44,45 Their detailed synthesis procedures are described in the Experimental section. Interestingly, the LA-EG4-DiMan ligand coated G5 (denoted as G5-DiMan hereafter) has a similar size and surface glycan coating to a gp160 trimer, the HIV surface densely glycosylated spike protein, which is responsible for mediating HIV–DC-SIGN interactions and viral infections.46,47 Thus, G5-DiMan may serve as a good mimic of gp160 for probing its interaction with DC-SIGN. The larger G13 and G27 were employed to investigate how the GNP scaffold size impacts their MLGIs with DC-SIGN/R. The citrate stabilised G13 and G27 as prepared gave single plasmonic absorption bands at ∼520 nm and ∼522 nm, respectively. They also displayed a single Gaussian distribution species with hydrodynamic diameters (Dh, volume population) of ∼16 and ∼29 nm for G13 and G27, respectively, consistent with that expected for isolated single G13 and G27 particles coated with a thin layer of citrate ligands (ESI, Fig. S1 and S2†).Preparation of GNP–glycan conjugates
Gx-glycan conjugates (Gx particles capped with the corresponding LA-EG4-DiMan and/or LA-EG4-OH ligands) were prepared by incubation of citrate-stabilised GNPs overnight with the pure LA-EG4-DiMan or mixed LA-EG4-DiMan/LA-EG4-OH ligands (with the glycan ligand content varying from 100%, 75%, 50%, and 25% to 0% to tune the GNP surface glycan ligand density) in water under a fixed total ligand: a GNP molar ratio of 1000![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 for G5, 3000 for G13, or 10000 for G27. We have found previously that GNP-glycans prepared using the LA-glycan ligands and their reduced dihydrolipoic acid (DHLA)-forms have an identical Dh value and stability.11 Therefore, the air-stable LA-glycan ligands were directly used to make the GNP-glycans without reduction. Since the LA-EG4-DiMan and LA-EG4-OH spacer ligands have the same GNP anchoring and EG4-linker groups, they should have the same GNP binding properties. Therefore, the GNP surface ligand contents should match those used in GNP conjugation which can be readily tuned by varying the solution glycan and spacer ligand ratio. The resulting G5-glycan conjugates were purified by ultra-filtration using 30 K MWCO filter tubes and washing with pure water as described previously.11 The G13- and G27-glycan conjugates were purified by centrifugation and washing with pure water. The unbound free ligands in the supernatant and washings were collected and used to determine the Gx surface glycan valency as described previously.11 The Gx-glycan conjugates were found to completely resist NaCl (250 mM) induced aggregation, suggesting that their surfaces were successfully functionalised with the desired ligands (the citrate stabilised GNPs readily aggregate in a moderate NaCl content, due to NaCl screening of the electrostatic repulsions among negatively charged GNPs). The Gx-glycans were found to be uniform and monodisperse both in pure water and a binding buffer (20 mM HEPES, 100 mM NaCl, 10 mM CaCl2, pH 7.8) with Dhs of ∼13, ∼22, and ∼32 nm for G5-, G13- and G27-DiMan, respectively (ESI, Fig. S5–S7†). These Dhs were a few nm larger than those of their corresponding citrate stabilised Gxs, matching what was expected for single Gx particles coated with a monolayer of LA-EG4-DiMan ligands which is slightly longer than the native citrate ligands. The Gx-glycan solutions were highly stable; no changes of physical appearance or precipitation were observed after prolonged storage (>12 months) in a fridge at 4 °C. The numbers of glycan ligands on each GNP (i.e., glycan valency) were estimated from the ligand amount difference between that added and that remained unbound in the supernatant after GNP conjugation via a phenol-sulphuric acid carbohydrate quantification method as described previously,11,19,48 giving a glycan valency per Gx particle of ∼730, ∼2200 and ∼6290 for G5-, G13- and G27-DiMan100%, respectively (ESI, Table 1). The average inter-glycan distance (d) was estimated from their Dh value and glycan valency using the method reported previously,11,49 giving d values of ∼0.95, ∼0.93 and ∼0.80 nm for G5-, G13- and G27-DiMan100%, respectively (Table 1). These d values match well with the majority of inter-glycan sequon spaces (e.g. 0.7–1.3 nm) found on the HIV surface glycoprotein gp160 trimer.46 Therefore, our Gx-glycans are good mimics for probing gp160-DC-SIGN interactions which are responsible for mediating HIV infections.
:1 for G5, 3000 for G13, or 10000 for G27. We have found previously that GNP-glycans prepared using the LA-glycan ligands and their reduced dihydrolipoic acid (DHLA)-forms have an identical Dh value and stability.11 Therefore, the air-stable LA-glycan ligands were directly used to make the GNP-glycans without reduction. Since the LA-EG4-DiMan and LA-EG4-OH spacer ligands have the same GNP anchoring and EG4-linker groups, they should have the same GNP binding properties. Therefore, the GNP surface ligand contents should match those used in GNP conjugation which can be readily tuned by varying the solution glycan and spacer ligand ratio. The resulting G5-glycan conjugates were purified by ultra-filtration using 30 K MWCO filter tubes and washing with pure water as described previously.11 The G13- and G27-glycan conjugates were purified by centrifugation and washing with pure water. The unbound free ligands in the supernatant and washings were collected and used to determine the Gx surface glycan valency as described previously.11 The Gx-glycan conjugates were found to completely resist NaCl (250 mM) induced aggregation, suggesting that their surfaces were successfully functionalised with the desired ligands (the citrate stabilised GNPs readily aggregate in a moderate NaCl content, due to NaCl screening of the electrostatic repulsions among negatively charged GNPs). The Gx-glycans were found to be uniform and monodisperse both in pure water and a binding buffer (20 mM HEPES, 100 mM NaCl, 10 mM CaCl2, pH 7.8) with Dhs of ∼13, ∼22, and ∼32 nm for G5-, G13- and G27-DiMan, respectively (ESI, Fig. S5–S7†). These Dhs were a few nm larger than those of their corresponding citrate stabilised Gxs, matching what was expected for single Gx particles coated with a monolayer of LA-EG4-DiMan ligands which is slightly longer than the native citrate ligands. The Gx-glycan solutions were highly stable; no changes of physical appearance or precipitation were observed after prolonged storage (>12 months) in a fridge at 4 °C. The numbers of glycan ligands on each GNP (i.e., glycan valency) were estimated from the ligand amount difference between that added and that remained unbound in the supernatant after GNP conjugation via a phenol-sulphuric acid carbohydrate quantification method as described previously,11,19,48 giving a glycan valency per Gx particle of ∼730, ∼2200 and ∼6290 for G5-, G13- and G27-DiMan100%, respectively (ESI, Table 1). The average inter-glycan distance (d) was estimated from their Dh value and glycan valency using the method reported previously,11,49 giving d values of ∼0.95, ∼0.93 and ∼0.80 nm for G5-, G13- and G27-DiMan100%, respectively (Table 1). These d values match well with the majority of inter-glycan sequon spaces (e.g. 0.7–1.3 nm) found on the HIV surface glycoprotein gp160 trimer.46 Therefore, our Gx-glycans are good mimics for probing gp160-DC-SIGN interactions which are responsible for mediating HIV infections.
| Gx-DiMan | Glycan valency, N | D h (nm) | Glycan footprint (nm2) | Deflection angle, θ (°) | Inter-glycan distance, d (nm) |
|---|---|---|---|---|---|
| G5-DiMan | 730 ± 52 | 12.9 | 0.72 ± 0.05 | 8.48 ± 0.30 | 0.95 ± 0.04 |
| G13-DiMan | 2200 ± 172 | 21.9 | 0.68 ± 0.05 | 4.88 ± 0.19 | 0.93 ± 0.03 |
| G27-DiMan | 6290 ± 440 | 32.4 | 0.52 ± 0.04 | 2.89 ± 0.10 | 0.80 ± 0.04 |
Quantifying GNP-glycan-DC-SIGN/R binding affinity
GNPs are well-known for their strong and universal quenching properties for a range of different fluorophores.50 Moreover, its quenching efficiency (QE) – distance dependence has been shown to follow the nano-surface energy transfer (NSET, where QE is proportional to the inverse 4th power dependence on distance),51–53 rather than the Förster resonance energy transfer (FRET, where QE is proportional to the inverse 6th power dependence on distance) mechanism exhibited by organic quenchers. As a result, its quenching is far more efficient and able to cover a longer distance range than those relying on the FRET mechanism.50,51 Indeed, the GNP's superior quenching ability has been widely exploited for biosensing, bioimaging and biodiagnostic applications.54,55 Recently, we have demonstrated that the outstanding fluorescence quenching properties of GNPs can be harnessed as a new readout method for quantifying MLGI affinities using a small G5-glycan.11 In that case, we first introduced a site-specific cysteine mutation on the CRD of the DC-SIGN/R extracellular segment. The mutation site lies close to, but outside of, its glycan binding pocket, and therefore does not affect its glycan binding property. The mutant proteins have been shown to retain faithfully the tetrameric structure and glycan-binding properties of the full-length lectins.11,19,27 We then labelled the lectins using a maleimide-modified Atto-643 dye with a labelling efficiency of >80% per monomer (ESI, Fig. S11†). Then, we measured the fluorescence spectra of the labelled lectins alone and their 1:1 molar mixture with G5-glycans over a range of concentrations (e.g., 0.2–64 nM) and fitted the obtained fluorescence quenching efficiency (QE)–concentration (C) relationships using Hill's equation to derive their apparent binding Kds.11 While this method worked well with the small G5-glycans, due to its relatively low absorption extinction coefficient (ε = 1.10 × 107 M−1 cm−1), unfortunately, it cannot work for the larger GNP-glycans. This is because the absorption extinction coefficient of GNPs roughly increases linearly with volume,11,56 where the very strong absorption (hence inner filter effect) of the large GNPs (e.g., ε = 1.10 × 107, 2.32 × 108 and 2.39 × 109 M−1 cm−1 for G5, G13, and G27, respectively) severely interferes with the fluorescence quenching measurement, even at moderately high Cs (e.g., >10 nM, the highest C should be greater than Kds to ensure good Kd measurement accuracy), making it inaccurate to measure affinities even for strong MLGIs (e.g., Kds at nM levels).
To overcome this problem, a fixed C value of 10, 4, or 0.5 nM for G5, G13, or G27, respectively, was employed while the lectin concentration was varied in this study. In this case, all lectin-Gx-DiMan binding samples had the same background absorption, which can be easily corrected by using their respective Gx-OH (Gx coated with pure LA-EG4-OH ligand) negative controls at that concentration. The fluorescence spectra of the lectins (varying Cs) without and with a fixed concentration of Gx-glycans were recorded at an excitation wavelength, λex, of 630 nm, where Gxs have minimal absorption, to minimise any possible interference from GNP's inner filter effect. All binding studies were carried out in a binding buffer containing 1 mg mL−1 bovine serum albumin (BSA) as described earlier,11 to make the binding conditions closely resemble real biological situations. Moreover, this also greatly reduced any non-specific interactions and adsorption of proteins and GNPs to surfaces, which can be a major source of experiment errors at low Cs, e.g. < 10 nM.57
The fluorescence spectra showing the binding between DC-SIGN/R and Gx-DiMan are given in ESI Fig. S12–S17† and their corresponding (QE × C) vs. C plots are shown in ESI Fig. S18.† In the absence of Gx-DiMan, the fluorescence intensities of labelled DC-SIGN/R both increased linearly with C, but their fluorescence was greatly reduced in the presence of Gx-DiMan (ESI, Fig. S12†), consistent with binding-induced fluorescence quenching by the GNPs in proximity.11,50,51,55 Moreover, free mannose effectively competed with Gx-DiMan binding with DC-SIGN/R, giving rise to a significant, dose-dependent recovery of DC-SIGN/R fluorescence (ESI, Fig. S19 and 20†). Furthermore, mixing DC-SIGN/R with the control Gx-OH produced negligible fluorescence quenching, confirming that the fluorescence quenching observed with Gx-DiMan is due to the specific MLGIs between Gx-DiMan and DC-SIGN/R (ESI, Fig. S21†).
To analyse the binding data quantitatively, we first calculated the quenching efficiency (QE) for each (Gx-DiMan + lectin) sample at each protein concentration viaeqn. (1):11
| QE = (IF0 − IF)/IF0 | (1) |
Given that a GNP has been shown to quench fluorophores in close proximity by up to 99.97%,50 it is safe to assume that the observed QE represents the fraction of the added lectins that have bound to Gx-DiMan.11 Previously, we measured the QEs for 1:1 mixed lectin + G5-glycan samples at a variety of Cs, and then fitted the QE – C relationships using the Hill equation to derive their apparent Kds.11 While this worked well for small G5-glycans (due to its relatively low ε), it did not work for large G13- and G27-glycans, owing to their much stronger inner filter effect (e.g., ε for G13 and G27 being ∼21 and ∼220 fold that of G5, respectively).11
Interestingly, we found that the QE remained almost constant for a fixed amount of Gx-DiMan after mixing with DC-SIGN over a certain protein: Gx molar ratio (PGR) range, although the QE was found to decrease as PGR was further increased, possibly due to surface saturation (see ESI Fig. S12†). This result suggests that before surface saturation, Gx-DiMan surface-bound DC-SIGN molecules do not hinder further binding of other DC-SIGN molecules on the same Gx-DiMan, and a Gx-DiMan with a few bound DC-SIGN molecules still produces the same level of QE as a free Gx-DiMan. Therefore, the equilibrium for multiple DC-SIGN molecules binding to one Gx-DiMan can be approximated as multiple copies of one-to-one DC-SIGN-Gx-DiMan binding interactions as given in eqn (2) below.
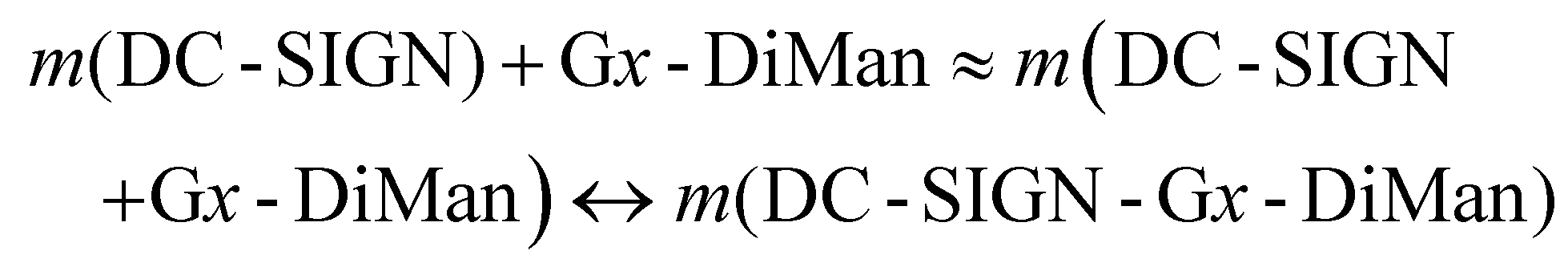 | (2) |
Note here that each Gx-DiMan particle has been cycled m times in binding to m copies of DC-SIGN molecules (where m is less than the lectin saturation number on each Gx-DiMan). As a result, the calculation of Kd can be greatly simplified by using the 1:1 binding equilibrium:
| DC-SIGN + Gx-DiMan ↔ DC-SIGN-Gx-DiMan | (3) |
| Kd = [DC-SIGN] [Gx-DiMan]/[DC-SIGN-Gx-DiMan] | (4) |
For a 1:1 interaction with equal starting C for both components, [DC-SIGN]0 = [Gx-DiMan]0 = C0. Since the QE represents the portion of lectin bound to Gx-DiMan, [DC-SIGN-Gx-DiMan] = C0 × QE, and thus the equilibrium C of free [DC-SIGN] = [Gx-DiMan] = C0 × (1 − QE). Substituting these numbers into eqn (4) allows us to calculate their binding Kd using eqn (5):
| Kd = [C0 × (1 − QE)]2/(C0 × QE) = C0 × (1 − QE)2/QE | (5) |
To measure QE more accurately, a plot of (QE × C) vs. C relationship over a lectin concentration range below that required to saturate the Gx-DiMan surface was employed to determine the average QE from linear fitting, where the slope obtained from the fit represents the average QE (see ESI Fig. S12E & S18†). The fitting parameters and calculated Kds using eqn (5) for DC-SIGN/R binding with Gx-DiMan at a variety of glycan densities are summarised in Table 2.
| Gx-DiMan | DC-SIGN | DC-SIGNR | ||
|---|---|---|---|---|
| QE | K d (nM) | QE | K d (nM) | |
| G5-DiMan100% | 0.473 ± 0.012 | 5.8 ± 0.3 | 0.314 ± 0.006 | 14.9 ± 0.5 |
| G13-DiMan100% | 0.603 ± 0.003 | 1.00 ± 0.03 | 0.445 ± 0.001 | 2.77 ± 0.02 |
| G13-DiMan75% | 0.535 ± 0.004 | 1.6 ± 0.1 | 0.397 ± 0.008 | 3.66 ± 0.16 |
| G13-DiMan50% | 0.462 ± 0.008 | 2.5 ± 0.1 | 0.349 ± 0.006 | 4.86 ± 0.17 |
| G13-DiMan25% | 0.391 ± 0.014 | 3.8 ± 0.2 | 0.348 ± 0.004 | 4.89 ± 0.12 |
| G27-DiMan100% | 0.523 ± 0.025 | 0.22 ± 0.03 | 0.367 ± 0.006 | 0.54 ± 0.02 |
| G27-DiMan75% | 0.468 ± 0.017 | 0.30 ± 0.03 | 0.313 ± 0.011 | 0.75 ± 0.05 |
| G27-DiMan50% | 0.490 ± 0.016 | 0.26 ± 0.03 | 0.314 ± 0.011 | 0.75 ± 0.05 |
| G27-DiMan25% | 0.400 ± 0.025 | 0.45 ± 0.06 | 0.296 ± 0.017 | 0.83 ± 0.08 |
Based on the calculated Kds (Table 2), four notable conclusions can be drawn: (1) DC-SIGN bound more strongly to all Gx-DiMans than DC-SIGNR did, irrespective of the GNP size and glycan density. As G5-DiMan here presents a good mimic for the HIV's trimeric gp120 spike, its stronger affinity with DC-SIGN over DC-SIGNR thus may help explain why DC-SIGN is more effective than DC-SIGNR in transmitting HIV infections.36 This result agreed well with our earlier results obtained with QD-DiMan19 and G5-EG2-DiMan (G5 coated with the same LA-DiMan based ligand, except for its EGx linker being 2 EG unit shorter than that used in this study) conjugates.11 Given that the CRDs in DC-SIGN/R have the same mannose-binding motifs,34 the different affinities here may indicate that the DC-SIGN/R might adopt different modes in binding to Gx-DiMan, similar to those observed previously with QD-DiMan19 and G5-EG2-DiMan.11 (2) The MLGI affinity between Gx-DiMan and DC-SIGN/R increased significantly with the increasing GNP scaffold size. For example, the Kds for Gx-DiMan (x = 5, 13, and 27 nm) were found to be ∼5.8, ∼1.0, and ∼0.2 nM for DC-SIGN, and ∼14.9, ∼2.7, and ∼0.54 nM for DC-SIGNR, respectively, suggesting that a larger GNP scaffold provided a more favourable glycan display to enhance their DC-SIGN/R binding. This is likely due to the lower surface curvature of larger GNP scaffolds which improves their surface glycan accessibility to DC-SIGN/R CRDs for multivalent binding.29 (3) Decreasing the Gx surface glycan density gradually weakened their MLGI affinities for both lectins. This result was also consistent with the literature that glycoconjugates of larger sizes and higher glycan valency generally offered higher viral inhibition potencies.4,5,58 (4) G27-DiMan (100%) exhibited the strongest DC-SIGN affinity among all Gx-DiMan conjugates studied here, with an apparent Kd of ∼0.2 nM, its affinity was ∼29 and ∼5 fold stronger than its G5- and G13-DiMan(100%) counterparts. Interestingly, the same trend of GNP size dependent affinity enhancement was also observed for DC-SIGNR binding with Gx-DiMan. Together, our results indicate that increasing the GNP scaffold size is strongly beneficial for enhancing their MLGI affinities with both DC-SIGN and DC-SIGNR, likely due to the reduced surface curvature of the larger GNP scaffold which affords the glycan ligands a more suitable spatial topology for forming strong MLGIs with both lectins.29
Probing the DC-SIGN/R-Gx-DiMan binding mode by dynamic light scattering (DLS)
Previously, we studied the binding mode between wild-type (WT)-DC-SIGN/R (no cysteine mutation) and G5-EG2-DiMan by monitoring the hydrodynamic diameters (Dhs) of the resulting lectin-G5-EG2-DiMan assemblies by DLS.11 We found that binding of multiple DC-SIGN molecules to each G5-EG2-DiMan gave small assemblies (i.e., saturate Dh ∼ 45 nm), matching that expected for single G5-EG2-DiMan particles coated with a monolayer of DC-SIGN molecules, implying that DC-SIGN binds to a single G5-EG2-DiMan with all four of its CRDs. In contrast, binding of multiple DC-SIGNR molecules to G5-EG2-DiMan yielded extensive, large-scale assemblies (Dhs ∼ 2 μm) which were too big to be individual lectin-G5-EG2-DiMan assemblies, suggesting that DC-SIGN and G5-EG2-DiMan crosslink with each other to form large assemblies.11 To investigate whether the GNP scaffold size will affect the binding mode between Gx-DiMan and DC-SIGN/R, we further studied the binding between G13/G27-DiMan(100%) and WT-DC-SIGN/R using DLS.11,29 First, we monitored the Dhs of binding-induced Gx-DiMan-lectin assemblies under a variety of PGRs using a fixed C of 5 nM for G13-DiMan or 1 nM for G27-DiMan. Both WT-DC-SIGN/R alone displayed a single narrow Gaussian species with a Dh of ∼12 nm (full width at half-maximum, FWHM, of ∼3.7 nm, see ESI Fig. S10†). The Dh histograms (volume population) of the resulting Gx-DiMan–DC-SIGN/R complexes under a variety of PGRs are shown in ESI Fig. S22–25.† Their Dh – PGR relationships are shown in Fig. 3A. In general, the Dhs of the Gx-DiMan–DC-SIGN complexes increased gradually with an increase in the PGR before reaching saturation, where the Dh values remained roughly constant. This behaviour closely resembled that observed for DC-SIGN binding with G5-EG2-DiMan previously.11 The saturated Dhs for DC-SIGN binding with G13-DiMan and G27-DiMan were found to be ∼60 and ∼75 nm, respectively, which roughly matched those expected for single Gx-DiMan particles coated with a monolayer of DC-SIGN molecules.31,34 The saturated Dhs here are ∼15 and ∼30 nm larger than that observed previously with G5-EG2-DiMan (saturate Dh∼ 45 nm), respectively. This result is not unexpected considering their larger GNP scaffold size and more DC-SIGN molecules being able to bind to each Gx-DiMan particle. However, the trend of Dh – PGR relationship for DC-SIGNR binding with Gx-DiMan was found to be significantly different from that of DC-SIGN; with the former consistently giving larger Dhs than the latter. In fact, the Dhs of the formed Gx-DiMan–DC-SIGNR complexes (e.g., ∼200 and ∼500 nm for x = 13 and 27, respectively) were too big to be isolated single particles, and thus indicating the formation of aggregates or clusters induced by DC-SIGNR-G13/G27-DiMan crosslinking. This result is also similar to that observed with G5-EG2-DiMan–DC-SIGNR binding reported previously.11 Using a surface binding footprint of ∼35 nm2 per DC-SIGN molecule,19 and the glycan surface areas of Gx-DiMan calculated from their Dhs (e.g., ∼1500 and ∼3300 nm2), a PGR of ∼43 or ∼94 with DC-SIGN was estimated to be able to fully saturate the surface of G13-DiMan or G27-DiMan, respectively.The evolution of Dh – time dependence of binding-induced Gx-DiMan–DC-SIGN/R complexes (under a fixed PGR of 32 for G13-DiMan and 80 for G27-DiMan, both below their respective surface saturation PGRs) was further monitored by DLS and the results are shown in Fig. 2B. The corresponding time-dependent Dh distribution (volume population) histograms are given in ESI Fig. S26–S29.† Binding of DC-SIGN with G13/27-DiMan gave only one single Gaussian species with Dh values of ∼60 nm and ∼77 nm, respectively. Such species formed very rapidly (<20 min) and showed no further changes in Dh over the next 320 min. These results are consistent with DC-SIGN binding simultaneously to one Gx-DiMan via all four of its CRDs, thereby forming a monolayer of DC-SIGN molecules on each Gx-DiMan particle to give small, isolated Gx-DiMan-lectin assemblies.11,19 In contrast, binding of DC-SIGNR gave considerably larger Dhs values at 20 min (e.g., >200 nm for G13-DiMan and >400 nm for G27-DiMan, respectively), which also increased significantly with time to >600 nm for G13-DiMan or >900 nm for G27-DiMan at 160 min and finally both stabilised at ∼700 nm at 320 min (Fig. 2B). Such Dh values were far too big to be individual Gx-DiMan particles coated with a monolayer of proteins, a strong indication of DC-SIGNR and G13/G27-DiMan crosslinking to form large Gx-DiMan-lectin assemblies. Such crosslinking reactions occurred over a relatively long period, leading to gradually increased sizes for lectin-Gx-DiMan assemblies which eventually precipitated out of the solution after 5 h (Fig. 2C). In contrast, DC-SIGN-Gx-DiMan assemblies were highly stable and showed no signs of colour change or precipitation after 5 h (Fig. 2C). Overall, these results indicate that the binding mode of DC-SIGN/R with Gx-DiMan conjugates are very different: DC-SIGN binds simultaneously to a single Gx-DiMan via all four CRDs and forms small assemblies made of single Gx-DiMan particles coated with a monolayer of DC-SIGN molecules, whereas DC-SIGNR and Gx-DiMan crosslink each other to form extensive, large-scale assemblies. These results were very similar to those observed with G5-EG2-DiMan previously,11 indicating that increasing the GNP scaffold size from 5 to 27 nm did not change the binding modes between Gx-DiMan and DC-SIGN/R.
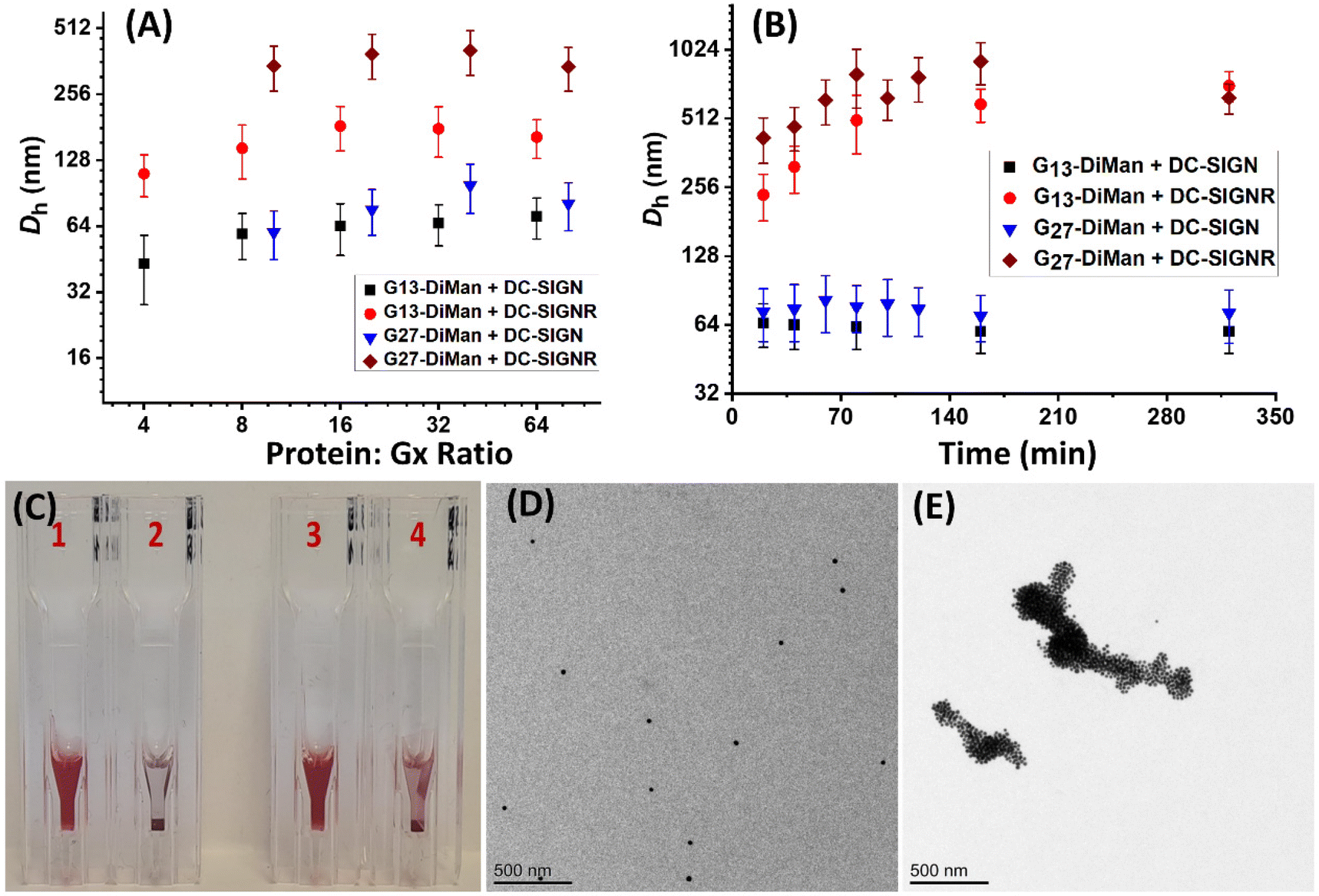 | ||
| Fig. 2 (A) Hydrodynamic diameter (Dh, volume population) – PGR relationship for G13/27-DiMan binding with DC-SIGN/R, and (B) Dh–time relationship for G13/27-DiMan binding with DC-SIGN/R under a fixed PGR of 32 or 80 for G13-DiMan or G27-DiMan, respectively. Dh values (volume population) are displayed as mean Dh ± ½ FWHM. For distributions containing two different Dh species, mean Dh = (xc1·A1%) + (xc2·A2%), and mean FWHM = (w1·A1%) + (w2·A2%). (C) Optical photographs of representative DLS samples at 5 h after mixing Gx-DiMan and lectins: (1) G27-DiMan + DC-SIGN (PGR: 80); (2) G27-DiMan + DC-SIGNR (PGR: 80); (3) G13-DiMan + DC-SIGN (PGR: 32); and (4) G13-DiMan + DC-SIGNR (PGR: 32). (D and E) TEM images of cryo-prepared G27-DiMan + DC-SIGN (D) or G27-DiMan + DC-SIGNR (E) samples after 40 min of incubation (PGR = 80) in a binding buffer. Scale bar = 500 nm. | ||
The different binding modes of Gx-DiMan with DC-SIGN/R were further verified by “cryo-snapshot” TEM imaging, which has been shown to be able to capture nanoparticles in their native dispersion or assembly states in solution.59 This was achieved by rapid plunge-freezing of the sample into liquid ethane, followed by drying under vacuum before being loaded onto the TEM grids for TEM imaging.11,59 Here, G27-DiMan was employed in the investigation. G27-DiMan was first mixed with DC-SIGN or DC-SIGNR at a PGR of 80 and incubated for 40 min, then they were plunge-frozen for sample preparation, and finally applied for TEM imaging. The resulting TEM images (Fig. 2D and E) clearly revealed that binding of DC-SIGN with G27-DiMan gave completely isolated single particles, whereas binding of DC-SIGNR produced extensive, large-scale clustered GNP assemblies. These results were similar to those observed for G5-EG2-DiMan binding with DC-SIGN/R reported previously.11 These results also fully agreed with their Dh size measurement described in the previous section. The combined TEM and DLS data thus reaffirmed the distinct modes for DC-SIGN/R in binding to G27-DiMan, where DC-SIGN binds simultaneously with all four binding sites to one G27-DiMan to form small, isolated GNP/protein core/shell particles, while DC-SIGNR cross-links with different G27-DiMan particles to form large scale lectin-Gx-DiMan assemblies. These results completely agree with those observed previously between DC-SIGN/R and G5-DiMan or (4 nm) QD-DiMan.11,19 Therefore, increasing the GNP scaffold size in Gx-DiMan from 5 to 27 nm did not impact their binding mode with DC-SIGN/R molecules in solution, although this did enhance their MLGI affinities significantly.
Probing Gx-DiMan binding thermodynamics with DC-SIGN/R
The binding thermodynamics between Gx-DiMan and DC-SIGN/R were derived by measuring their apparent Kds via GNP fluorescence quenching at three different temperatures (e.g., 25, 30, and 35 °C). The temperature was controlled using a dry bath for samples, and the cuvette temperature was maintained by using a temperature-controlled water pump system.28 The resulting fluorescence spectra and the (QE × C) versus C plots are given in ESI Fig. S30–S34.† The apparent Kds and fitting parameters are given in ESI Table S4.† The calculated Kds at different temperatures were then combined with Van't Hoff analysis to derive the binding thermodynamic data. By integrating the two Gibbs free energy equations (eqn (6) and (7)), the resulting changes in the MLGI enthalpy (ΔH) and entropy (ΔS) terms can be obtained from the linear fits of the In(Kd) vs. (1/T) plots using eqn (8).28| ΔG = −RTln(Ka) = RTln(Kd) | (6) |
| ΔG = ΔH − TΔS | (7) |
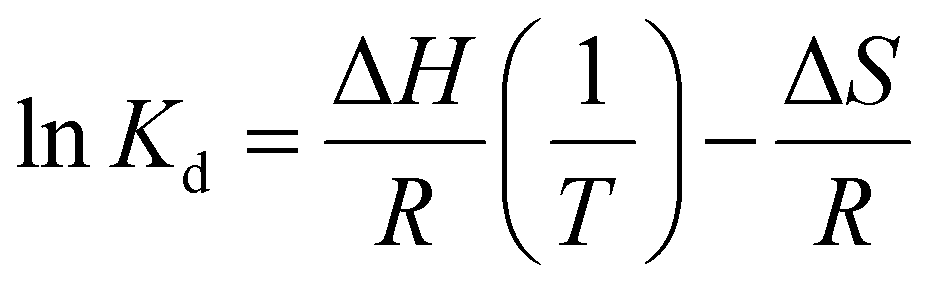 | (8) |
Fig. 3A and B show the Van't Hoff plots and linear fits of the ln(Kd) – (1/T) relationships for the Gx-DiMan – DC-SIGN/R binding data. The slope and intercept obtained from the linear fits correspond to the (ΔH/R) and (–ΔS/R) terms, respectively, allowing us the derive the ΔH and ΔS values of the MLGIs. The resulting MLGI thermodynamic parameters obtained for Gx-DiMan–DC-SIGN/R binding are shown in Fig. 3C, and the detailed thermodynamic parameters are given in Table 3.
 | ||
| Fig. 3 Van't Hoff analyses of the ln(Kd) − 1/T relationships for Gx-DiMan binding with Atto643 labelled- (A) DC-SIGN and (B) DC-SIGNR. All data have been corrected for Gx's inner filter effects obtained with their respective Gx-OH negative control. (C) Comparison of the standard (T = 298 K) binding ΔH° (red), TΔS° (blue), and ΔG° (green) for Gx-DiMan binding with DC-SIGN/R. Error bars represent the fitting errors. | ||
| Gx-DiMan–Lectin | ΔH° (kJ mol−1) | ΔS° (J mol−1 K−1) | −TΔS° (kJ mol−1) | ΔG° (kJ mol−1) |
|---|---|---|---|---|
| G5-DiMan–DC-SIGN | −132 ± 32 | −288 ± 105 | 86 ± 31 | −47 ± 44 |
| G13-DiMan–DC-SIGN | −111 ± 10 | −207 ± 33 | 62 ± 9 | −50 ± 14 |
| G27-DiMan–DC-SIGN | −100 ± 14 | −138 ± 46 | 41 ± 14 | −59 ± 19 |
| G5-DiMan–DC-SIGNR | −38.2 ± 2.0 | 17.6 ± 6.0 | −5.2 ± 2.0 | −43.5 ± 3.0 |
| G13-DiMan–DC-SIGNR | −45.5 ± 4.0 | 14.3 ± 11.0 | −4.2 ± 4.0 | −49.7 ± 5.0 |
| G27-DiMan–DC-SIGNR | −43.8 ± 2.0 | 34.9 ± 7.0 | −10.4 ± 2.0 | −54.2 ± 3.0 |
Two notable conclusions can be drawn from the results shown in Table 3. (1) DC-SIGN binding interactions with all three Gx-DiMan (x = 5, 13 and 27) are enthalpy-driven with strongly favourable (negative) ΔH° terms. Their binding ΔH° values were found to be similar (after accounting for the experimental errors) at −132 ± 32, −111 ± 10 and −100 ± 14 kJ mol−1, for G5, G13, and G27, respectively. These ΔH° values are roughly 4 times that of the monovalent binding between DC-SIGN CRD and DiMan measured by ITC (−25.8 kJ mol−1),60 suggesting that all four CRDs in each DC-SIGN have participated in Gx-DiMan binding. This result is fully consistent with the mode of simultaneous tetravalent binding, via all four CRDs in each DC-SIGN molecule, to one Gx-DiMan described in the previous section. The good agreement between the ΔH° values obtained here and that measured by ITC suggests that our GNP fluorescence quenching can act as a reliable new method for quantifying DC-SIGN MLGI thermodynamics. (2) The ΔS° values for DC-SIGN binding with all three Gx-DiMan conjugates were found to be negative (unfavourable), and their absolute values were reduced with an increase in the GNP scaffold size. This result implies that the unfavourable entropy changes for DC-SIGN-Gx-DiMan binding are reduced with an increase in the GNP size, leading to enlarged negative ΔG° values. Thus, the enhancement of DC-SIGN-Gx-DiMan MLGI affinity with an increase in the GNP scaffold size observed in the previous section mainly comes from the reduced binding entropy penalty, and not from the enhanced binding enthalpy. The total MLGI entropy changes reflect the net contributions of the binding induced changes in translational, rotational, and conformational entropies of lectins and Gx-DiMan ligands as well as their surrounding media (e.g. binding buffer).28 Therefore, a likely reason for the reduced entropic penalty for the bigger GNPs observed here could be due to the presence of a higher proportion of unbound surface DiMan ligands (as all measurements were performed at PGRs below surface saturation) which may have largely retained their conformational and rotational degrees of freedom. Taking together, the thermodynamic data of DC-SIGN-Gx-DiMan binding obtained here are fully consistent with that expected for DC-SIGN's simultaneous tetravalent binding mode with a single Gx-DiMan described in the earlier sections.
In contrast, the binding thermodynamics of DC-SIGNR with Gx-DiMan was found to differ significantly from those of DC-SIGN. The highly favourable ΔH° values observed in DC-SIGN were significantly reduced in DC-SIGNR-Gx-DiMan binding. Interestingly, the ΔH°s of DC-SIGNR binding with all three Gx-DiMans were similar, ∼half of that observed with DC-SIGN. This result may indicate that only the binding or unbinding of two CRDs in the DC-SIGNR based MLGIs with Gx-DiMan were captured under our experimental conditions. This result is consistent with their Dh studies where DC-SIGNR cross-links with different Gx-DiMans, presumably first by using 2 CRDs to form partially bound Gx-DiMan–DC-SIGNR structural units, which then crosslink each other to form extended large assemblies.28 The later step may happen over a relatively long period and hence may not be captured in the current measurement. Another possible reason could be that the Kd method employed herein was too simplified to provide accurate measurement for DC-SIGNR based crosslinking MLGIs, although the trend of enhanced MLGI affinity as a function of the increasing GNP scaffold size measured here did match well with their enhanced antiviral potencies (see antiviral results in the next section). Interestingly, the large entropic penalties observed in DC-SIGN binding with Gx-DiMan appeared to have completely disappeared and were replaced by a small favourable entropic term in DC-SIGNR binding. The net result here is a gradually enhanced favourable binding ΔG° (hence affinity) with an increase in the GNP scaffold size. Overall, these results indicate that DC-SIGNR binding exhibits a smaller favourable enthalpy term than DC-SIGN, but this is partially compensated by a small favourable entropic term,61 giving rise to only a slightly smaller negative binding ΔG° value than that of its DC-SIGN counterpart.
Inhibition of DC-SIGN/R-promoted EBOVpp entry into cells
Given the strong affinities (low apparent Kds) of Gx-DiMan in binding to DC-SIGN/R, we anticipated that they should be strong inhibitors in blocking DC-SIGN/R-mediated viral infections, exemplified using the pseudo-Ebola virus. Therefore, Gx-DiMans were tested for their ability to inhibit cell surface DC-SIGN/R promoted entry of the pseudo-Ebola virus using a model cellular infection assay.11,19 Here, single cycle vesicular stomatitis virus (VSV) particles pseudotyped with the Ebola virus glycoprotein (EBOV-GP) and encoding the luciferase gene (abbreviated as EBOVpp) were used.11,19 We employed the human embryonic kidney cells, 293T, transfected to express DC-SIGN and DC-SIGNR as the host cells as described previously.11,19 This well-established cellular assay provides a robust readout (luciferase activity indicating infectious entry) for evaluating the antiviral properties of glycoconjugates targeting DC-SIGN/R based viral receptors.11,19 Binding of Gx-DiMan to cell surface DC-SIGN/R will block these lectin receptors from binding to EBOV-GP spikes on the virus particle surface, reducing virus cellular entry and hence luciferase production as shown schematically in Fig. 4A1 and A2. VSV particles bearing the vesicular stomatitis virus glycoprotein (VSV-G) that does not employ DC-SIGN/R for cell entry were employed as a specificity control, while VSV particles encoding the luciferase gene but bearing no viral glycoproteins were used as a negative control.11 All experiments assessing the antiviral activity of Gx-DiMan (positive controls) and Gx-OH (negative controls) were performed in DMEM cell culture medium supplemented with 10% fetal bovine serum (FBS) at 37 °C as described previously.11 The unprocessed luciferase activities of 293T cells treated with Gx-DiMan and Gx-OH controls are given in ESI Fig. S35–S37.† After Gx-DiMan treatment, luciferase activities upon EBOVpp infection were significantly and dose-dependently reduced as demonstrated by statistical analysis (ESI, Fig. S35†), while those measured upon VSV-G-driven infections were almost unaffected (ESI, Fig. S36†). Moreover, treatment with the Gx-OH negative controls lacking the terminal DiMan showed no apparent inhibition (ESI, Fig. S37†). These results clearly indicated that the observed inhibitions were specific, due to the specific binding of Gx-DiMan to cell surface DC-SIGN/R receptors which blocked their subsequent binding to EBOV-GP to augment viral infection. The normalized viral inhibition data were fitted by a modified inhibition model described in eqn (9):11,43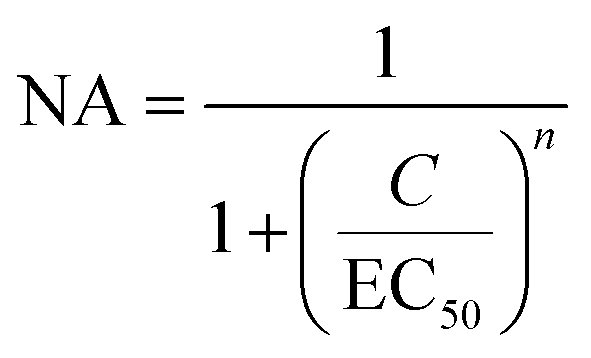 | (9) |
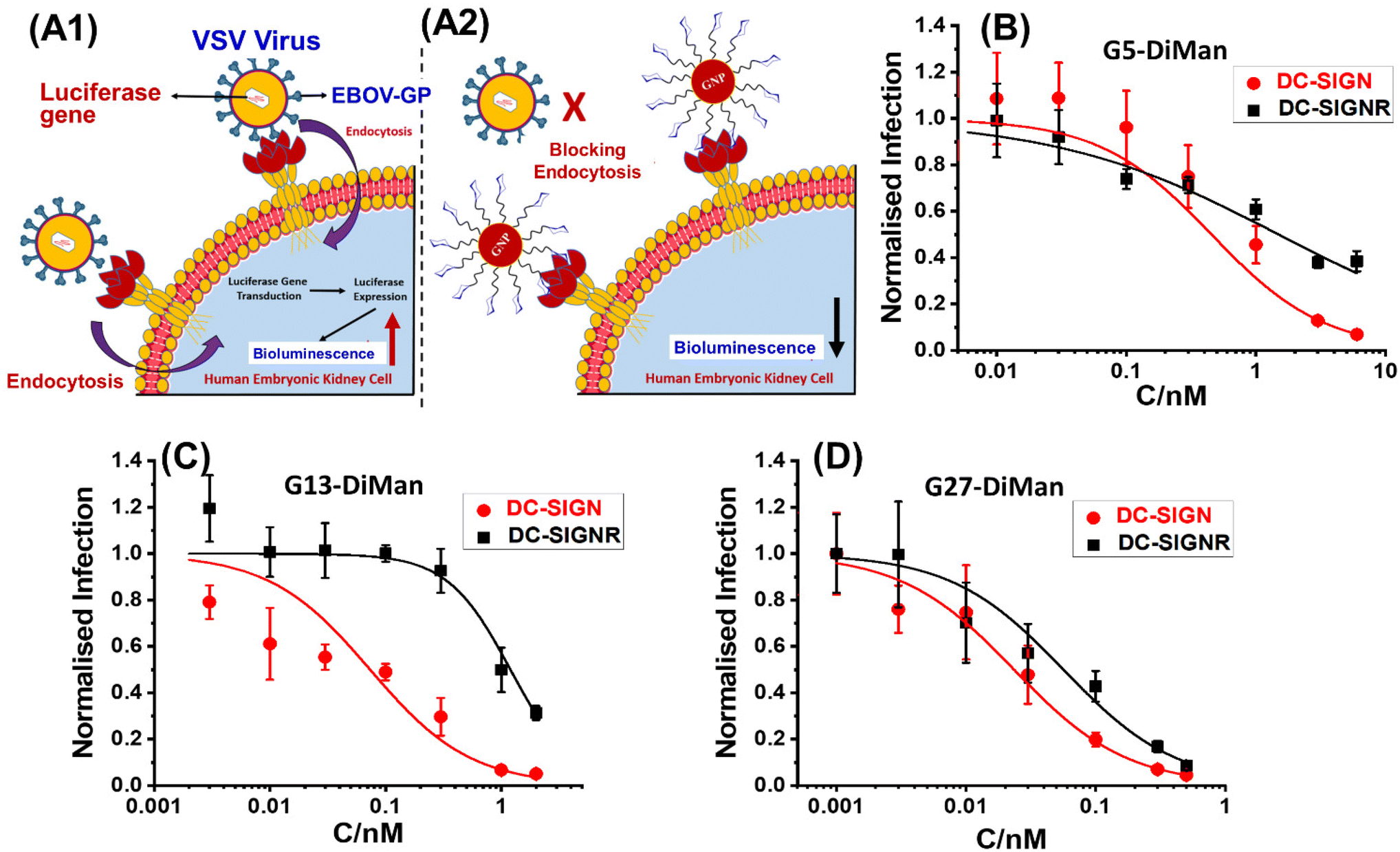 | ||
| Fig. 4 A schematic illustration of our cellular infection assays with the pseudo-Ebola virus model. (A1) In the absence of GNP-DiMan, vesicular stomatitis virus (VSV) particles bind efficiently to cell surface DC-SIGN/R lectin receptors via their surface Ebola glycoproteins (EBOV-GPs), leading to efficient endocytosis and luciferase gene production in host cells. (A2) Binding of high affinity GNP-DiMan particles on cell surface DC-SIGN/R receptors efficiently blocks these lectin receptors from binding to virus particles, thereby reducing the transduction of the luciferase gene. (B–D) Plots of normalised luciferase activity (NA) against the Gx-DiMan concentration for the 293T cells after treatment with varying doses of (B) G5-DiMan, (C) G13-DiMan or (D) G27-DiMan. The data were fitted using eqn (9). The luciferase activities of all treatment samples were subtracted by their corresponding pcDNA control backgrounds and then normalised by those obtained in the absence of Gx-DiMan. | ||
| Gx-DiMan | Lectin receptor | n | EC50 (nM) | R 2 |
|---|---|---|---|---|
| G5-DiMan | DC-SIGN | 1 | 0.45 ± 0.02 | 0.959 |
| G13-DiMan | DC-SIGN | 1 | 0.073 ± 0.007 | 0.948 |
| G27-DiMan | DC-SIGN | 1 | 0.023 ± 0.001 | 0.980 |
| G5-DiMan | DC-SIGNR | 0.50 ± 0.06 | 1.49 ± 0.23 | 0.942 |
| G13-DiMan | DC-SIGNR | 1.59 ± 0.25 | 1.19 ± 0.10 | 0.987 |
| G27-DiMan | DC-SIGNR | 1 | 0.049 ± 0.002 | 0.976 |
It was exciting that all three Gx-DiMan (x = 5, 13, and 27) conjugates potently and non-cooperatively blocked DC-SIGN-promoted EBOVpp entry into 293T cells, with impressively low EC50 values of 0.45 ± 0.02, 0.073 ± 0.007, and 0.023 ± 0.001 nM, respectively. Thus, their antiviral potency was found to have enhanced significantly with an increase in the GNP scaffold size. This was fully consistent with their enhanced DC-SIGN affinity measured by the GNP fluorescence quenching assay (Table 2). Moreover, all three Gx-DiMan inhibitors were found to act in a non-cooperative fashion (n = 1), indicating that Gx-DiMan could serve as a viable, potent inhibitor against DC-SIGN-mediated viral infections at the cellular level. Notably, G27-DiMan, with its impressively low EC50 of 23 ± 1 pM, is considerably more potent than some of the most potent glycoconjugate inhibitors against DC-SIGN-mediated viral infections reported in the literature (e.g., the giant globular multivalent glycofullerenes, EC50: ∼0.67 nM,15 the virus-like glycodendrinanoparticles, EC50: ∼0.91 nM,18 and our previous QD-EG3-DiMan, EC50: ∼0.70 nM,19 and G5-EG2-DiMan, EC50: ∼0.095 nM (ref. 11)). Interestingly, compared to G5-EG2-DiMan (G5 coated by the same LA-EGx-DiMan ligand except for the EGx linker being two EG units shorter), G5-DiMan here was found to be less potent (EC50: ∼0.45 nM vs. ∼0.095 nM (ref. 11)), although this is consistent with its weaker DC-SIGN affinity than the former (Kd: ∼5.8 nM vs. ∼3.8 nM (ref. 11)). Thus, increasing the EGx linker length has weakened the DC-SIGN binding affinity and hence the antiviral potency of Gx-DiMan, similar to that observed previously with the QD-EGx-DiMan conjugates.19 Nonetheless, a suitable EG linker length is required to promote high stability and resistance against non-specific interactions41 for glycan-nanoparticles, which are essential for potential applications under challenging in vivo conditions.
The inhibition of DC-SIGNR-promoted EBOVpp entry by Gx-DiMan was also enhanced with an increase in the GNP scaffold size (e.g., with EC50 values of 1.5 ± 0.2, 1.2 ± 0.1, and 0.049 ± 0.002 nM for G5-, G13-, and G27-DiMan, respectively), similar to the trend observed against DC-SIGN-mediated viral infections. However, their inhibition cooperativity showed a clear scaffold size dependence: it changed from negative (n = ∼0.5) to positive (n = ∼1.6) and finally non-cooperative (n = 1) as the GNP scaffold size increased from 5 to 27 nm. This may indicate a change in binding behaviour (or mode) for Gx-DiMan with cell surface DC-SIGNR receptors as the scaffold size changes. While the smallest G5-DiMan may retain its crosslinking mode in binding to DC-SIGNR on the cell surface, which would make it difficult to achieve complete inhibition (n < 1) due to the cell membrane restrictions, the largest G27-DiMan may be able to bind simultaneously to a single DC-SIGNR receptor and completely block its binding to EBOV-GP, similar to that occurring with DC-SIGN on the cell membrane, giving rise to a non-cooperative inhibition (n = 1) behaviour. Together with an impressively low EC50 of ∼49 pM, these results indicate that G27-DiMan can serve as a potent, viable inhibitor against DC-SIGNR-mediated viral infections. Given that both DC-SIGN and DC-SIGNR have shown to play an important role in facilitating SARS-CoV-2 infections,33 the excellent potency and non-cooperative inhibition behaviour observed with G27-DiMan may make it a highly promising, viable entry inhibitor against SARS-CoV-2 infections.
Interestingly, a ∼2.2-fold higher potency for G27-DiMan against DC-SIGN- over DC-SIGNR-mediated viral infections also agreed well with their relative MLGI affinity differences measured by our GNP fluorescence quenching assay (i.e., apparent Kd: ∼0.20 vs. ∼0.54 nM). A comparison of the MLGI affinities and viral inhibition data for Gx-DiMan revealed that their Kd and EC50 values do not match directly, possibly due to the different binding environments used in these studies (e.g., in solution vs. on the cell membrane),11 as well as the potential inaccuracies associated with the over-simplified Kd calculation method here. However, there appeared to be a clear positive correlation between the Kd and EC50 values (i.e., the lower the Kd, the lower the EC50, signifying more potent inhibition). This result suggests that the GNP fluorescence quenching based affinity method developed here could act as a rapid method for estimating the relative antiviral potentials for GNP-glycan based entry inhibitors.
Conclusion
In summary, we have developed a new format of GNP fluorescence quenching assay which has successfully addressed the potential interference arising from GNPs’ strong inner filter effect, making it suitable for measuring the MLGI affinities and thermodynamics for both small and large GNP-based glycoconjugates. Using this assay, we have revealed that the MLGI affinities of both DC-SIGN and DC-SIGNR binding are enhanced significantly (∼30 fold) as the GNP size is increased from 5 to 27 nm. Moreover, we have shown that binding of DC-SIGN with Gx-DiMan is enthalpy driven, with strongly negative binding ΔH° values of ∼ 4 times that of the corresponding monovalent binding, suggesting that all four CRDs in each DC-SIGN molecule are engaged in Gx-DiMan binding. This result is fully consistent with DC-SIGN's binding mode with Gx-DiMan (via all four CRDs in binding to a single Gx-DiMan) derived from the combined Dh and TEM analyses of binding-induced lectin-GNP assemblies. We have further revealed that the enhanced MLGI affinity of DC-SIGN in binding to the larger GNP-DiMan over its smaller GNP counterparts is due to reduced entropic penalty and not increased binding enthalpy, whereas the enhanced affinity for DC-SIGNR in the same situation is due to a combined contribution of both favourable enthalpic and entropic terms. Despite different thermodynamic mechanisms, the overall binding ΔG° values for both DC-SIGN/R are increased with an increase in the GNP scaffold size, leading to enhanced MLGI affinity. Finally, we have demonstrated that Gx-DiMans are highly potent entry inhibitors against DC-SIGN/R-mediated pseudo-Ebola virus cellular infection. Their antiviral potencies are enhanced significantly with an increase in the GNP scaffold size, matching well to their enhanced DC-SIGN/R affinities (reduced Kd values) and more favourable binding thermodynamics (larger negative ΔG° values). Interestingly, the scaffold size of Gx-DiMan has been found to exhibit a significant, but very different impact against the simultaneously binding DC-SIGN-mediated viral infections from the crosslinking DC-SIGNR-mediated viral infections. Against DC-SIGN-mediated viral infections, increasing the scaffold size can enhance the antiviral potency without affecting inhibition cooperativity (n = 1), whereas against DC-SIGNR-mediated viral infections, increasing the scaffold size can not only enhance the antiviral potency but also impact the inhibition cooperativity, shifting from negative- (for G5), to positive- (for G13), and finally to non- (for G27) cooperative inhibition. In particular, G27-DiMan can potently and robustly (n = 1) block both DC-SIGN- and DC-SIGNR-mediated virus infection with impressively low EC50 values of ∼23 and ∼49 pM, respectively, making it the most potent glycoconjugate inhibitor against DC-SIGN/R-mediated Ebola cellular infections reported to date. Our work thus demonstrates the great potential of G27-DiMan as a highly potent entry inhibitor against a wide range of DC-SIGN/R-promoted viral infections. A potential limitation of the current study is that all MLGI binding affinity and thermodynamic studies were performed in solution using isolated lectin molecules which have the freedom to move in all three dimensions. This represents a very different binding environment from their native environment on cell membranes (in two dimensions with in-plane mobility). As a result, the MLGI biophysical data obtained herein may not be directly transferable to that happening on cell membranes. Therefore, further studies under conditions that mimic more closely those of DC-SIGN/R immobilised on cell membranes are still needed, which will be addressed in a follow-on study.Experimental section
Materials
Gold(III) chloride trihydrate, sodium hydroxide, trisodium citrate, copper sulphate, sodium sulphate, calcium chloride, HEPES, lipoic acid (LA), sodium ascorbate, tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl] amine (TBTA), methanol, ethanol, chloroform, phenol, bovine serum albumin, tetrahydrofuran, tris-(hydroxymethyl)aminomethane (Tris base), hydrochloric acid, sodium chloride, ethylenediamine tetraacetic acid (EDTA), and guanidine hydrochloride were purchased from Sigma-Aldrich, Alfa Aesar, Fluorochem, and Thermo Scientific with >99% impurity and used as-received without further purification unless specified elsewhere. 2-[2-(2-Azido-ethoxy) ethoxy]ethanol and 5 nm GNPs suspended in a citrate buffer were purchased from Sigma-Aldrich. NH2-EG4-C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH was purchased from PurePEG LLC. Maleimide-modified Atto-643 dye was commercially purchased from ATTO-Tech GmbH. Ultrapure water (resistance >18.2 MΩ cm) purified using an ELGA Purelab classic UVF system was used for all experiments and making all buffers. These include the binding buffer (20 mM HEPES, 100 mM NaCl, 10 mM CaCl2, pH 7.8); lectin elution buffer (20 mM HEPES, 100 mM NaCl, 2.5 mM EDTA, pH 7.8); lectin labelling buffer (20 mM HEPES, 100 mM NaCl, 10 mM CaCl2, pH 7.2); and lectin elution buffer post dye-labelling (20 mM HEPES, 100 mM NaCl, 2.5 mM EDTA, pH 7.2).
CH was purchased from PurePEG LLC. Maleimide-modified Atto-643 dye was commercially purchased from ATTO-Tech GmbH. Ultrapure water (resistance >18.2 MΩ cm) purified using an ELGA Purelab classic UVF system was used for all experiments and making all buffers. These include the binding buffer (20 mM HEPES, 100 mM NaCl, 10 mM CaCl2, pH 7.8); lectin elution buffer (20 mM HEPES, 100 mM NaCl, 2.5 mM EDTA, pH 7.8); lectin labelling buffer (20 mM HEPES, 100 mM NaCl, 10 mM CaCl2, pH 7.2); and lectin elution buffer post dye-labelling (20 mM HEPES, 100 mM NaCl, 2.5 mM EDTA, pH 7.2).
Synthesis of 13 nm gold nanoparticles (G13s)45,62
A freshly prepared aqueous solution of gold(III) chloride trihydrate (1 mM, 400 mL) was placed in a 500 mL three-necked round-bottomed flask, and the solution was then heated to reflux in an oil bath (130 °C) under stirring. When the solution began to reflux, trisodium citrate solution (38 mM, 40 mL) was quickly added. The solution colour quickly turned from yellow to wine red in ∼1 min, indicating the formation of GNPs. The reaction was further refluxed under magnetic stirring for another 1 h to ensure that the reaction was complete. The GNP solution was then removed from the oil bath and was allowed to cool down to room temperature (RT) naturally under stirring. This produced citrate-stabilized GNPs with a core diameter of ∼13 nm as confirmed by TEM imaging (ESI, Fig. S1†). The resulting G13 stock was transferred to a clean glass container and stored at RT until use.Synthesis of 27 nm gold nanoparticles (G27s)45,62
A freshly prepared aqueous solution of gold(III) chloride trihydrate (0.25 mM, 400 mL) was placed in a 500 mL two-necked round-bottomed flask, and NaOH (1 mM, 50 mL) was then added directly into the solution. The mixture was stirred for 30 min and then heated to reflux in a 130 °C oil bath under magnetic stirring. After the solution started to reflux, trisodium citrate solution (166 mM, 6 mL) was then quickly added. The solution colour gradually changed from yellow to light red in 15 min. The reaction was refluxed for another 1 h to complete the synthesis. The solution was then taken out of the oil bath and kept stirring for 1 h until it was cooled down to RT. This produced citrate stabilized GNPs with a mean diameter of ∼27 nm (G27) as confirmed by TEM images (ESI, Fig. S2†). The G27 stock was transferred to a clean glass container and stored at RT until use.Synthesis of LA-EG4-DiMan11
LA-EG4-CCH was synthesised by amide coupling between lipoic acid (LA) and commercial NH2-EG4-CCH as described previously.43 1-Azido-3,6-dioxaoct-8-yl-α-d-mannopyranosyl-(1 → 2)-α-d-mannopyranoside (N3-EG2-DiMan) was synthesised via our established protocols as described previously.19 LA-EG4-CCH (50 mg, 0.120 mmol), N3-EG2-DiMan (66 mg, 0.132 mmol), CuSO4·5H2O (1.07 mg, 0.0043 mmol), TBTA (4.01 mg, 0.0075 mmol), and sodium ascorbate (3.21 mg, 0.0162 mmol) were dissolved in 2 mL of THF/H2O (1:1, vol/vol) to allow for an efficient click reaction between LA-EG4-CCH and N3-EG2-DiMan.11 The resulting solution was stirred overnight at RT in the dark. The next day, the consumption of all starting compounds was confirmed by TLC. The solvent was then evaporated, and the desired ligand was purified by size exclusion chromatography using a Biogel P2 column using ammonium formate as an eluent to afford the desired product, LA-EG4-DiMan, in 77% yield.11 TLC: (CHCl3/MeOH 3:1) Rf 0.57; 1H NMR (400 MHz, D2O) δ (ppm): 8.10 (s, 1H), 5.12 (s, 1H), 5.03 (s, 1H), 4.73–4.60 (m, 3H), 4.08 (s, 1H), 3.99 (dd, 3H, J = 10.2, 5.1 Hz), 3.94–3.82 (m, 5H), 3.69 (dt, 31H, J = 12.8, 7.1, 6.7 Hz), 3.45–3.30 (m, 2H), 3.30–2.33 (m, 2H), 2.26 (t, 2H, J = 7.3 Hz), 1.99 (dt, 1H, J = 12.9, 6.9 Hz), 1.78–1.54 (m, 4H), 1.42 (q, 1H, J = 7.6 Hz); 13C NMR (100 MHz, D2O) δ (ppm): 176.7, 144.1, 125.5, 102.2, 98.3, 78.6, 73.2, 72.7, 70.2, 70.1, 69.9, 69.6, 69.6, 69.5, 69.5, 69.4, 69.2, 68.9, 68.8, 68.7, 66.9, 66.8, 66.5, 66.5, 63.2, 63.1, 61.4, 61.1, 60.8, 59.3, 56.5, 50.1, 50.0, 46.6, 40.2, 38.9, 38.1; LC-MS: calculated m/z for C37H66N4O18S2 (M + H)+ 919.38, found 919.78 (see ESI Fig. S3†).
Synthesis of LA-EG4-OH11
LA-EG4-CCH (50 mg, 0.120 mmol), 2-[2-(2-Azido-ethoxy) ethoxy] ethanol, N3-EG2-OH (23.1 mg, 0.132 mmol), CuSO4·5H2O (1.07 mg, 0.0043 mmol), TBTA (4.01 mg, 0.0075 mmol), and sodium ascorbate (3.21 mg, 0.0162 mmol) were dissolved in 2 mL of THF/H2O (1:1, vol/vol) to allow for an efficient click reaction to occur between LA-EG4-CCH and N3-EG2-OH. The resulting solution was stirred overnight at RT in the dark. The next day, the consumption of all starting compounds was confirmed by TLC. The solvent was then evaporated, and the desired ligand was purified by size exclusion chromatography using a Biogel P2 column using ammonium formate as an eluent to obtain the desired product in 75% yield.11 TLC: (CHCl3/MeOH 10:1) Rf 0.45; 1H NMR (500 MHz, D2O) δ (ppm): 8.01 (s, 1H), 3.93–3.87 (m, 2H), 3.66–3.59 (m, 17H), 3.58–3.56 (m, 4H), 3.55–3.51 (m, 5H), 3.48–3.45 (m, 2H), 3.29 (t, 2H, J = 5.2 Hz), 3.15–3.07 (m, 1H), 2.39 (dq, 1H, J = 12.4, 6.1 Hz), 2.16 (t, 2H, J = 7.3 Hz), 1.88 (dq, 1H, J = 13.8, 6.9 Hz), 1.70–1.60 (m, 1H), 1.53 (ddd, 3H, J = 14.9, 7.6, 5.3 Hz), 1.31 (p, 2H, J = 7.6 Hz); 13C NMR (125 MHz, D2O) δ (ppm): 183.1, 142.9, 125.4, 71.6, 69.6, 69.4, 68.9, 68.7, 65.9, 61.5, 60.3, 56.4, 49.9, 48.4, 43.9, 43.7, 43.2, 41.7, 38.0, 36.3, 33.6, 33.4, 24.9, 17.3; LC-MS: calculated m/z for C25H46N4O8S2 (M + H)+ 595.28, found 595.57 (see ESI Fig. S4†).
Preparation of Gx-DiMan/OH conjugates
G5-DiMans were prepared by mixing G5 and the LA-EG4-DiMan ligand via self-assembly in an aqueous solution.11 Commercially G5s (6 mL, 91 nM) suspended in citrate solution were concentrated to 250 μL using a 30 K MWCO spin column and washed with H2O (3 × 250 μL) to remove any unbound impurities. The resulting concentrated G5 solution was then directly mixed with the LA-EG4-DiMan ligand at a G5:ligand molar ratio of 1:1 000. The resulting mixture was left stirring at RT in the dark overnight to form G5-DiMan conjugates. After that, the mixture was transferred to a 30 K MWCO centrifugal filter and centrifuged at 4000 rpm for 20 min, and the G5-DiMan residues were washed with H2O (3 × 250 μL) to remove any unbound free ligands, and then dispersed in pure water to make the G5-DiMan stock. Its concentration was determined by the Beer–Lambert law from the plasmon peak absorbance at ∼515 nm and a molar extinction coefficient of 1.10 × 107 M−1 cm−1 for G5 (see ESI Fig. S5† for the Dh histogram and the UV-vis spectrum).
For the preparation of G13/27-DiMan, 20 mL each of the citrate stabilized G13 or G27 stock solution was directly added with the required amount of the LA-EG4-DiMan ligand stock solution in water at a GNP: ligand molar ratio of 1:3000 for G13 or 1:10000 for G27. The resulting solution was stirred at RT in the dark overnight to form Gx-DiMan conjugates via gold–thiol self-assembly. After that, the resulting mixtures were divided into 1.5 mL portions, added into Eppendorf tubes and centrifuged at 17000g for 30 min for G13-DiMan and 6 000g for 15 min for G27-DiMan conjugates to remove any unbound free ligands. After careful withdrawal of the supernatant, the Gx-DiMan residues were washed with pure water (3 × 500 μL), followed by centrifugation and washing with water three times to remove any unbound free ligands. For G27, the Eppendorf tubes were pre-washed with 0.025% Tween-20 aqueous solution before being used for Gx-DiMan purification to prevent nanoparticles from sticking to the Eppendorf tube walls.
To prepare Gx-DiMan conjugates with different surface glycan densities (e.g., 75%, 50% and 25%), LA-EG4-OH and LA-EG4-DiMan were mixed in the desired percentages first before being used in GNP conjugation at the same total ligand: Gx molar ratios and purified using the same method as Gx-DiMan. The Dh histograms and UV-vis spectra for G13/27-DiMan/-OH are given in Fig. S6 and S7 (ESI).† The concentrations of Gx-DiMan/OH conjugates were determined using the Beer–Lambert law with the peak absorbance at ∼520 nm and molar extinction coefficient of 2.32 × 108 M−1 cm−1 for G13 and 2.39 × 109 M−1 cm−1 for G27, respectively (see ESI Fig. S8†).
All the filtrate and washing-through liquids were collected, combined, freeze-dried, and re-dissolved in 1.40 mL of pure water to determine the amount of unbound LA-EG4-DiMan ligand using the phenol-sulphuric acid method described previously.11,19 25 μL of each solution was diluted with water to a final volume of 125 μL. This solution was then mixed with 125 μL of 5% phenol and 625 μL of H2SO4, and then allowed to incubate at RT for 30 min. The absorbance of the solution was recorded at 490 nm, and the dilution factors were then corrected to calculate the total amount of unconjugated glycan ligand against a standard calibration curve obtained with the pure LA-EG4-DiMan ligand. The difference in the LA-EG4-DiMan ligand amount between that added and that remained in the supernatant was conserved to have conjugated onto the GNP surface.11
Protein production and labelling11,19
The soluble extracellular segments of DC-SIGN and DC-SIGNR, which faithfully replicated the tetrameric structure and glycan binding properties of full-length lectins,11,19 were expressed as inclusion bodies in E. coli and purified by mannose-Sepharose affinity column chromatography as reported earlier.27 The mutant proteins, DC-SIGN Q-274C and DC-SIGNR R278C, were constructed by site-directed mutagenesis and labelled with Atto-643 maleimide as described previously.19,28 The labelled proteins were purified by mannose-Sepharose affinity columns. All the proteins were characterised by high-resolution mass spectroscopy (HRMS, see ESI, Fig. S9 and S11†). The dye labelling efficiency (per protein monomer) was determined to be ∼82% and ∼90% for DC-SIGN and DC-SIGNR, respectively, based on the relative peak areas of the labelled and unlabelled protein peaks measured by HR-MS (see ESI Fig. S11†).Fluorescence spectra11
All fluorescence spectra were recorded with a Horiba FluoroMax-4 spectrofluorometer using a 0.70 mL quartz cuvette at a fixed excitation wavelength (λex) of 630 nm. Emission spectra over 650–800 nm were collected with excitation and emission slit widths of 5 nm at a slow scan speed. All measurements were carried out in a binding buffer (20 mM HEPES, pH 7.8, 100 mM NaCl, 10 mM CaCl2) containing 1 mg mL−1 BSA to reduce any non-specific interactions and absorption to cuvette walls. The required amounts of Gx-DiMan and DC-SIGN/R were mixed and then incubated at RT for 20 min before recording their fluorescence spectra. The fluorescence spectra from 650 to 800 nm were integrated and used to calculate the quenching efficiency (QE).Dynamic light scattering (DLS)11
All DLS measurements were performed using a Malvern Zetasizer NanoZS DLS system using a sample volume of 400 μL in 1 cm disposable polystyrene cuvettes. The hydrodynamic diameters (Dh, all volume populations) of wild-type DC-SIGN/R, Gx-DiMan and Gx-DiMan + DC-SIGN or DC-SIGNR samples were measured in a binding buffer (20 mM HEPES, 100 mM NaCl, 10 mM CaCl2, pH 7.8) at RT with ten consecutive runs, and each lasting 120 seconds, and the averages of the ten runs were used to determine size distributions. Each sample was analysed in triplicate to obtain their average Dh and standard deviations (SDs).Virus inhibition studies
The inhibition effects of Gx-DiMan (100% glycan density) were evaluated using vesicular stomatitis virus (VSV) vector particles pseudotyped with the Ebola virus glycoprotein (EBOV-GP) and 293T cells via our established procedures.11,19 Briefly, 293T cells seeded in 96-well plates were transfected with plasmids encoding DC-SIGN/R or control transfected with empty plasmid (pcDNA). The cells were washed at 16 h post transfection and further cultivated at 37 °C, 5% CO2 in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS). At 48 h post-transfection, the cells were exposed to twice the final concentration of Gx-DiMan inhibitor in OptiMEM-medium for 30 min in a total volume of 50 μL. After that, the resulting cells were inoculated with 50 μL of preparations of the luciferase gene encoding VSV vector particles bearing either EBOV-GP which can use DC-SIGN/R for the augmentation of host cell entry or the vesicular stomatitis virus glycoprotein (VSV-G). The latter cannot use DC-SIGN/R to enhance host cell entry, and thus was employed as a specificity control. Under these conditions, binding of Gx-DiMan particles to 293T cell surface DC-SIGN/R receptors can block EBOV-GP interactions with such lectin receptors, reducing the virus particle transduction efficiency and producing reduced cellular luciferase activity. At 16–20 h post infection luciferase activities in cell lysates were determined using a commercially available kit (PJK), following the manufacturer's instructions, as described in our previous publications.11,19Data analysis and fitting11
All fluorescence and DLS data were analyzed using Origin software (version 2022b). The fluorescence spectra of lectins alone and lectin + Gx-DiMan samples were integrated and used to calculate the QEs and presented as mean ± standard errors (SEs). The (QE × C) vs. C plots were fitted by the linear function, accounting for the SEs of each data point, to give the best fits (highest R2 values). The DLS histograms were fitted by the standard Gaussian function (single or multiple, depending on the data distribution) to obtain the Dh (shown as Xc in the fitting parameters), full-width at half-maximum (FWHM, shown as W), and area abundance (shown as A) and are depicted in each DLS graph. For samples containing two species, a linear average of the two based on their volume abundances was used, e.g., mean Dh = Dh1 × A1/(A1 + A2) + Dh2 × A2/(A1 + A2); mean FWHM = FWHM1 × A1/(A1 + A2) + FWHM2 × A2/(A1 + A2).11 The results obtained from the best fits were listed in the relevant tables with the standard fitting errors. Statistical analyses of viral inhibition data were analysed using the Microsoft Excel software with a two-tailed T-test. The unprocessed luciferase activity data (indicative of viral infection) of samples after treatment with varying doses of Gx-DiMan were compared with their respective control sample in the absence of Gx-DiMan inhibitors. Samples showing no significant statistical differences (p > 0.05) from their controls were unmarked, while those showing significant statistical differences were marked as *: p ≤ 0.05; **: p ≤ 0.01; and ***: p ≤ 0.001 (see ESI Fig. S35†).Data availability
All the spectra and plots supporting this paper have been included in the ESI.† The original data of this study are available from the corresponding authors upon request. For the purpose of open access, the authors have applied a Creative Commons Attribution (CC BY) license to any Author Accepted Manuscript version arising from this submission.Conflicts of interest
There is no conflict of interest to declare.Acknowledgements
This project was supported by a UK Biotechnology and Biological Science Research Council grant (grant number: BB/R007829/1 to D. Z., N. H., & Y. G.). R. B. thanks the Turkish Ministry of National Education for providing a PhD scholarship to support his study at the University of Leeds, United Kingdom.References
- G. D. Brown, J. A. Willment and L. Whitehead, Nat. Rev. Immunol., 2018, 18, 374–389 CrossRef CAS PubMed.
- T. B. H. Geijtenbeek and S. I. Gringhuis, Nat. Rev. Immunol., 2009, 9, 465–479 CrossRef CAS PubMed.
- M. Mammen, S. K. Choi and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 2754–2794 CrossRef PubMed.
- A. Bernardi, J. Jiménez-Barbero, A. Casnati, C. De Castro, T. Darbre, F. Fieschi, J. Finne, H. Funken, K. E. Jaeger, M. Lahmann, T. K. Lindhorst, M. Marradi, P. Messner, A. Molinaro, P. V. Murphy, C. Nativi, S. Oscarson, S. Penadés, F. Peri, R. J. Pieters, O. Renaudet, J. L. Reymond, B. Richichi, J. Rojo, F. Sansone, C. Schäffer, W. B. Turnbull, T. Velasco-Torrijos, S. Vidal, S. Vincent, T. Wennekes, H. Zuilhof and A. Imberty, Chem. Soc. Rev., 2013, 42, 4709–4727 RSC.
- S. Bhatia, L. C. Camacho and R. Haag, J. Am. Chem. Soc., 2016, 138, 8654–8666 CrossRef CAS PubMed.
- T. R. Branson, T. E. McAllister, J. Garcia-Hartjes, M. A. Fascione, J. F. Ross, S. L. Warriner, T. Wennekes, H. Zuilhof and W. B. Turnbull, Angew. Chem., Int. Ed., 2014, 53, 8323–8327 CrossRef CAS PubMed.
- P. I. Kitov, J. M. Sadowska, G. Mulvey, G. D. Armstrong, H. Ling, N. S. Pannu, R. J. Read and D. R. Bundle, Nature, 2000, 403, 669–672 CrossRef CAS PubMed.
- J. Huskens, L. J. Prins, R. Haag and B. J. Ravoo, Multivalency: concepts, research and applications, John Wiley & Sons, 2018 Search PubMed.
- H. Feinberg, R. Castelli, K. Drickamer, P. H. Seeberger and W. I. Weis, J. Biol. Chem., 2007, 282, 4202–4209 CrossRef CAS PubMed.
- B. M. Illescas, J. Rojo, R. Delgado and N. Martin, J. Am. Chem. Soc., 2017, 139, 6018–6025 CrossRef CAS PubMed.
- D. Budhadev, E. Poole, I. Nehlmeier, Y. Liu, J. Hooper, E. Kalverda, U. S. Akshath, N. Hondow, W. B. Turnbull, S. Pöhlmann, Y. Guo and D. Zhou, J. Am. Chem. Soc., 2020, 142, 18022–18034 CrossRef CAS PubMed.
- X. Chen, O. Ramström and M. Yan, Nano Res., 2014, 7, 1381–1403 CrossRef CAS PubMed.
- F. Lasala, E. Arce, J. R. Otero, J. Rojo and R. Delgado, Antimicrob. Agents Chemother., 2003, 47, 3970–3972 CrossRef CAS PubMed.
- O. Martínez-Ávila, K. Hijazi, M. Marradi, C. Clavel, C. Campion, C. Kelly and S. Penadés, Chem. – Eur. J., 2009, 15, 9874–9888 CrossRef PubMed.
- A. Muñoz, D. Sigwalt, B. M. Illescas, J. Luczkowiak, L. Rodríguez-Pérez, I. Nierengarten, M. Holler, J. S. Remy, K. Buffet, S. P. Vincent, J. Rojo, R. Delgado, J. F. Nierengarten and N. Martín, Nat. Chem., 2016, 8, 50–57 CrossRef PubMed.
- J. Ramos-Soriano, J. J. Reina, B. M. Illescas, N. De La Cruz, L. Rodriguez-Perez, F. Lasala, J. Rojo, R. Delgado and N. Martin, J. Am. Chem. Soc., 2019, 141, 15403–15412 CrossRef CAS PubMed.
- N. C. Reichardt, M. Martín-Lomas and S. Penadés, Chem. Soc. Rev., 2013, 42, 4358–4376 RSC.
- R. Ribeiro-Viana, M. Sánchez-Navarro, J. Luczkowiak, J. R. Koeppe, R. Delgado, J. Rojo and B. G. Davis, Nat. Commun., 2012, 3, 1303 CrossRef PubMed.
- Y. Guo, I. Nehlmeier, E. Poole, C. Sakonsinsiri, N. Hondow, A. Brown, Q. Li, S. Li, J. Whitworth, Z. Li, A. Yu, R. Brydson, W. B. Turnbull, S. Pöhlmann and D. Zhou, J. Am. Chem. Soc., 2017, 139, 11833–11844 CrossRef CAS PubMed.
- B. M. Illescas, J. Rojo, R. Delgado and N. Martín, J. Am. Chem. Soc., 2017, 139, 6018–6025 CrossRef CAS PubMed.
- V. Porkolab, M. Lepsík, S. Ordanini, A. St John, A. Le Roy, M. Thépaut, E. Paci, C. Ebel, A. Bernardi and F. Fieschi, ACS Cent. Sci., 2023, 9, 709–718 CrossRef CAS PubMed.
- V. Porkolab, C. Pifferi, I. Sutkeviciute, S. Ordanini, M. Taouai, M. Thépaut, C. Vivès, M. Benazza, A. Bernardi, O. Renaudet and F. Fieschi, Org. Biomol. Chem., 2020, 18, 4763–4772 RSC.
- A. Velazquez-Campoy and E. Freire, Nat. Protoc., 2006, 1, 186–191 CrossRef CAS PubMed.
- X. Wang, O. Ramström and M. D. Yan, Adv. Mater., 2010, 22, 1946–1953 CrossRef CAS PubMed.
- S. H. Liyanage and M. D. Yan, Chem. Commun., 2020, 56, 13491–13505 RSC.
- J. H. Rao, J. Lahiri, L. Isaacs, R. M. Weis and G. M. Whitesides, Science, 1998, 280, 708–711 CrossRef CAS PubMed.
- Y. Guo, C. Sakonsinsiri, I. Nehlmeier, M. A. Fascione, H. Zhang, W. Wang, S. Pöhlmann, W. B. Turnbull and D. Zhou, Angew. Chem., Int. Ed., 2016, 55, 4738–4742 CrossRef CAS PubMed.
- J. Hooper, Y. Y. Liu, D. Budhadev, D. F. Ainaga, N. Hondow, D. J. Zhou and Y. Guo, ACS Appl. Mater. Interfaces, 2022, 14, 47385–47396 CrossRef CAS PubMed.
- J. Hooper, D. Budhadev, D. L. F. Ainaga, N. Hondow, D. J. Zhou and Y. Guo, ACS Appl. Nano Mater., 2023, 6, 4201–4213 CrossRef CAS PubMed.
- T. B. H. Geijtenbeek, D. S. Kwon, R. Torensma, S. J. van Vliet, G. C. F. van Duijnhoven, J. Middel, I. Cornelissen, H. Nottet, V. N. KewalRamani, D. R. Littman, C. G. Figdor and Y. van Kooyk, Cell, 2000, 100, 587–597 CrossRef CAS PubMed.
- Y. Guo, H. Feinberg, E. Conroy, D. A. Mitchell, R. Alvarez, O. Blixt, M. E. Taylor, W. I. Weis and K. Drickamer, Nat. Struct. Mol. Biol., 2004, 11, 591–598 CrossRef CAS PubMed.
- S. Pöhlmann, E. J. Soilleux, F. Baribaud, G. J. Leslie, L. S. Morris, J. Trowsdale, B. Lee, N. Coleman and R. W. Doms, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 2670–2675 CrossRef PubMed.
- R. Amraei, W. Yin, M. A. Napoleon, E. L. Suder, J. Berrigan, Q. Zhao, J. Olejnik, K. B. Chandler, C. Xia and J. Feldman, ACS Cent. Sci., 2021, 7, 1156–1165 CrossRef CAS PubMed.
- H. Feinberg, D. A. Mitchell, K. Drickamer and W. I. Weis, Science, 2001, 294, 2163–2166 CrossRef CAS PubMed.
- Y. Guo, C. E. Atkinson, M. E. Taylor and K. Drickamer, J. Biol. Chem., 2006, 281, 16794–16798 CrossRef CAS PubMed.
- N. P. Y. Chung, S. K. J. Breun, A. Bashirova, J. G. Baumann, T. D. Martin, J. M. Karamchandani, J. W. Rausch, S. F. J. Le Grice, L. Wu, M. Carrington and V. N. KewalRamani, J. Biol. Chem., 2010, 285, 2100–2112 CrossRef CAS PubMed.
- C. W. Davis, H. Y. Nguyen, S. L. Hanna, M. D. Sánchez, R. W. Doms and T. C. Pierson, J. Virol., 2006, 80, 1290–1301 CrossRef CAS PubMed.
- K. Susumu, H. T. Uyeda, I. L. Medintz, T. Pons, J. B. Delehanty and H. Mattoussi, J. Am. Chem. Soc., 2007, 129, 13987–13996 CrossRef CAS PubMed.
- D. J. Zhou, A. Bruckbauer, C. Abell, D. Klenerman and D. J. Kang, Adv. Mater., 2005, 17, 1243–1248 CrossRef CAS.
- K. L. Prime and G. M. Whitesides, Science, 1991, 252, 1164–1167 CrossRef CAS PubMed.
- K. L. Prime and G. M. Whitesides, J. Am. Chem. Soc., 1993, 115, 10714–10721 CrossRef CAS.
- D. Zhou, A. Bruckbauer, L. M. Ying, C. Abell and D. Klenerman, Nano Lett., 2003, 3, 1517–1520 CrossRef CAS.
- D. Budhadev, J. Hooper, C. Rocha, I. Nehlmeier, A. M. Kempf, M. Hoffmann, N. Krüger, D. J. Zhou, S. Pöhlmann and Y. Guo, JACS Au, 2023, 3, 1755–1766 CrossRef CAS PubMed.
- X. Ji, X. Song, J. Li, Y. Bai, W. Yang and X. Peng, J. Am. Chem. Soc., 2007, 129, 13939–13948 CrossRef CAS PubMed.
- L. Song, V. H. B. Ho, C. Chen, Z. Q. Yang, D. S. Liu, R. J. Chen and D. J. Zhou, Adv. Healthcare Mater., 2013, 2, 275–280 CrossRef CAS PubMed.
- G. B. E. Stewart-Jones, C. Soto, T. Lemmin, G.-Y. Chuang, A. Druz, R. Kong, P. V. Thomas, K. Wagh, T. Zhou, A.-J. Behrens, T. Bylund, C. W. Choi, J. R. Davison, I. S. Georgiev, M. G. Joyce, Y. D. Kwon, M. Pancera, J. Taft, Y. Yang, B. Zhang, S. S. Shivatare, V. S. Shivatare, C.-C. D. Lee, C.-Y. Wu, C. A. Bewley, D. R. Burton, W. C. Koff, M. Connors, M. Crispin, U. Baxa, B. T. Korber, C.-H. Wong, J. R. Mascola and P. D. Kwong, Cell, 2016, 165, 813–826 CrossRef CAS PubMed.
- J. H. Lee, G. Ozorowski and A. B. Ward, Science, 2016, 351, 1043–1048 CrossRef CAS PubMed.
- S. K. Saha and C. F. Brewer, Carbohydr. Res., 1994, 254, 157–167 CrossRef CAS PubMed.
- H. D. Hill, J. E. Millstone, M. J. Banholzer and C. A. Mirkin, ACS Nano, 2009, 3, 418–424 CrossRef CAS PubMed.
- B. Dubertret, M. Calame and A. J. Libchaber, Nat. Biotechnol., 2001, 19, 365–370 CrossRef CAS PubMed.
- T. L. Jennings, M. P. Singh and G. F. Strouse, J. Am. Chem. Soc., 2006, 128, 5462–5467 CrossRef CAS PubMed.
- E. Dulkeith, M. Ringler, T. A. Klar, J. Feldmann, A. Muñoz Javier and W. J. Parak, Nano Lett., 2005, 5, 585–589 CrossRef CAS PubMed.
- T. Pons, I. L. Medintz, K. E. Sapsford, S. Higashiya, A. F. Grimes, D. S. English and H. Mattoussi, Nano Lett., 2007, 7, 3157–3164 CrossRef CAS PubMed.
- D. S. Seferos, D. A. Giljohann, H. D. Hill, A. E. Prigodich and C. A. Mirkin, J. Am. Chem. Soc., 2007, 129, 15477–15479 CrossRef CAS PubMed.
- S. P. Song, Z. Q. Liang, J. Zhang, L. H. Wang, G. X. Li and C. H. Fan, Angew. Chem., Int. Ed., 2009, 48, 8670–8674 CrossRef CAS PubMed.
- X. Liu, M. Atwater, J. Wang and Q. Huo, Colloids Surf., B, 2007, 58, 3–7 CrossRef CAS PubMed.
- H. Y. Zhang, G. Q. Feng, Y. Guo and D. J. Zhou, Nanoscale, 2013, 5, 10307–10315 RSC.
- E. Boisselier and D. Astruc, Chem. Soc. Rev., 2009, 38, 1759–1782 RSC.
- N. Hondow, R. Brydson, P. Y. Wang, M. D. Holton, M. R. Brown, P. Rees, H. D. Summers and A. Brown, J. Nanopart. Res., 2012, 14, 977 CrossRef.
- A. Holla and A. Skerra, Protein Eng., Des. Sel., 2011, 24, 659–669 CrossRef CAS PubMed.
- J. D. Chodera and D. L. Mobley, in Annual Rev. Biophy, ed. K. A. Dill, 2013, vol. 42, pp. 121–142 Search PubMed.
- L. Song, Y. Guo, D. Roebuck, C. Chen, M. Yang, Z. Q. Yang, S. Sreedharan, C. Glover, J. A. Thomas, D. S. Liu, S. R. Guo, R. J. Chen and D. J. Zhout, ACS Appl. Mater. Interfaces, 2015, 7, 18707–18716 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials, instruments, and methods showing the synthesis and characterization of LA-EG4-DiMan and LA-EG4-OH ligands by 1H/13C NMR and MS spectra, the production, characterization and labelling of DC-SIGN/R by HR-MS; UV-vis, DLS histograms and TEM images of citrate stabilised G13 and G27; DLS histograms of Gx-DiMan and Gx-DiMan-lectin complexes at different protein to GNP ratios and their time-dependent evolution; the fluorescence spectra of lectins alone and lectin + Gx-DiMan samples at different protein to GNP molar ratios and at three different temperatures, and the resulting (QE × C) vs. C plots with linear fits; and the unprocessed virus inhibition data showing relevant cellular luciferase activities after treatment with different Gx-DiMan inhibitors and Gx-OH controls. See DOI: https://doi.org/10.1039/d4nr00484a |
| This journal is © The Royal Society of Chemistry 2024 |