
Cell membrane coated nanoparticles as a biomimetic drug delivery platform for enhancing cancer immunotherapy
Zichen
Zhong†
,
Wen
Deng†
,
Jian
Wu
,
Haojie
Shang
,
Yonghua
Tong
,
Yu
He
,
Qiu
Huang
,
Xiaozhuo
Ba
,
Zhiqiang
Chen
* and
Kun
Tang
 *
*
Department of Urology, Tongji Hospital, Tongji medical college, Huazhong University of Science and Technology, Wuhan 430030, Hubei, China. E-mail: zhqchen8366@163.com; tangsk1990@163.com
First published on 15th April 2024
Abstract
Cancer immunotherapy, a burgeoning modality for cancer treatment, operates by activating the autoimmune system to impede the growth of malignant cells. Although numerous immunotherapy strategies have been employed in clinical cancer therapy, the resistance of cancer cells to immunotherapeutic medications and other apprehensions impede the attainment of sustained advantages for most patients. Recent advancements in nanotechnology for drug delivery hold promise in augmenting the efficacy of immunotherapy. However, the efficacy is currently constrained by the inadequate specificity of delivery, low rate of response, and the intricate immunosuppressive tumor microenvironment. In this context, the investigation of cell membrane coated nanoparticles (CMNPs) has revealed their ability to perform targeted delivery, immune evasion, controlled release, and immunomodulation. By combining the advantageous features of natural cell membranes and nanoparticles, CMNPs have demonstrated their unique potential in the realm of cancer immunotherapy. This review aims to emphasize recent research progress and elucidate the underlying mechanisms of CMNPs as an innovative drug delivery platform for enhancing cancer immunotherapy. Additionally, it provides a comprehensive overview of the current immunotherapeutic strategies involving different cell membrane types of CMNPs, with the intention of further exploration and optimization.
1. Introduction
Immunotherapy, a promising cancer treatment, activates the host immune system to combat solid tumors and circulating tumor cells.1 In recent years, immunotherapy has been used clinically as an adjuvant treatment after surgery, radiotherapy, and chemotherapy.2,3 Current cancer immunotherapies include cancer vaccines, immune checkpoint blockade (ICB) therapy, cytokine therapy, and adoptive immunotherapy.4 Despite their potential, issues like low tumor antigen expression, abnormal cytokine secretion, inadequate immune cell activation, and the complex immunosuppressive tumor microenvironment (TME) lead to drug resistance and adverse reactions.5,6 Together with the accompanying toxic and inflammation reactions during therapy, the clinical efficacy is limited.7 Hence, the imperative challenges lie in precisely targeting drug delivery to tumor sites, ameliorating the immunosuppressive TME, augmenting immune responsiveness, and mitigating systemic immunotoxicity.8Nanotechnology has introduced innovative approaches to cancer immunotherapy.9 Nanoparticles (NPs) have demonstrated considerable utility in delivering singular or multiple drugs to tumor sites, significantly impacting diagnostic imaging, photoimmunotherapy, chemotherapy, and gene therapy.10–13 Their systemic delivery leverages the enhanced penetration and retention (EPR) effect,14 and their controlled properties and low toxicity ensure prolonged drug retention and compatibility, mitigating delivery challenges in cancer immunotherapy.15 However, NPs face challenges in immunotherapy due to recognition and clearance by the reticuloendothelial system (RES), leading to low effectiveness.16,17 Prior attempts to enhance NP circulation and targeting with modifications like polyethylene glycol (PEG) or antigenic peptides have limitations due to immune response and complex synthesis process.18–20 The ideal NPs should possess immune evasion, extended retention, targeted delivery, biosafety, and scalability.21,22
Cell membrane coating technology presents a solution by encapsulating natural cell membranes on NPs. These synthesized CMNPs are endowed with both NPs’ properties and those of the coated cell membranes.23–25 Various cell membrane types, such as erythrocyte, leukocyte, platelet, cancer cell, immune cell, and hybrid cell membranes, have been utilized, resulting in CMNPs with immune evasion, targeted delivery, controlled release, and immunomodulation. These advances have found applications in diagnostic imaging,26,27 infection,28,29 inflammation,30–32 cancer chemotherapy,33 and immunotherapy. This review provides an overview of the applications of CMNPs in enhancing cancer immunotherapy (Fig. 1). It delineates the process of cancer immunotherapy and the synthesis of CMNPs, while emphasizing the latest research progress and the underlying mechanisms of CMNPs concerning different cell membrane types and delivery components. Furthermore, it discusses the challenges and prospects of this technology.
 | ||
| Fig. 1 Graphical abstract of CMNPs delivering different therapeutic agents for enhancing cancer immunotherapy. NP: nanoparticle; NK cell: natural killer cell; DC: dendritic cell; ICD: immunogenic cell death; DAMP: damage associated molecular pattern; CTL: cytotoxic T lymphocytes; mDC: mature dendritic cell; TAM: tumor associated macrophage; CAF: cancer associated fibroblasts; PTT: photothermal therapy; PDT: photodynamic therapy; ICIs: immune checkpoint inhibitors. | ||
2. Cancer immune cycle and cancer immunotherapy
The immune system recognizes cancer cells as foreign entities, triggering innate and adaptive immune responses to eliminate them. This process, termed the “cancer immune cycle”, was conceptualized by Chen and Mellman in 2013.34 Dendritic cells (DCs) act as specialized antigen-presenting cells (APCs) to capture antigens released by tumors (step 1), and then express the captured antigens as major histocompatibility complex (MHC)-antigen peptide complexes on cell membranes (step 2). After DCs deliver the complexes to the secondary lymphoid organs (step 3), T cells are activated by the interactions between T cell receptors (TCRs) and the MHC-antigen peptide complexes along with other ligands and receptors. Activated tumor-specific cytotoxic T cells (CTLs) migrate to the tumor sites, guided by chemokines in TME (step 4), and gradually infiltrate the tumor tissues (step 5). Specific recognition and binding of TCRs to MHC-antigen peptide complex on associated tumor cells (step 6) lead to tumor cell destruction, releasing more tumor-associated antigens (TAAs), and perpetuating the cycle (step 7). Each step has checkpoints or inhibitors to finely regulate tumor immunity.35,36 Consequently, cancer immunotherapy aims to enhance and amplify the cancer immune cycle to achieve a heightened immune response without inducing autoimmune-associated inflammation (Fig. 2).37,38 | ||
| Fig. 2 Schematic of the cancer immunity cycle and mechanisms of CMNPs loaded with different therapeutic agents and common immunotherapies for enhancing the anti-cancer immunity. | ||
Cancer immunotherapy has made substantial advancements, representing a pivotal development in the realm of cancer treatment. Immune checkpoint inhibitors (ICIs) are notable for restoring immune cells’ capacity to counter cancer cells by blocking inhibitory molecules (e.g., PD-1, PD-L1, CTLA-4) on immune cells.39 These inhibitors have gained FDA approval for melanoma, lung cancer, renal cancer, and other conditions, yielding noteworthy therapeutic outcomes.40–44 Adoptive cell therapy, such as CAR-T cell therapy, primarily targets hematological malignancies (like leukemia and lymphoma). It involves genetically modifying T cells with specific antigen receptor genes, enabling them to release substances like perforin and granzyme B to directly eliminate identified tumor cells.45,46 Cancer vaccines, in contrast, aim to activate the patient's immune system against tumor cells. Notable vaccine types include TAA vaccines, individualized cancer vaccines, viral vector vaccines, and combination vaccines with immunostimulants or antitumor drugs.47–49 Cytokine therapy regulates the growth, differentiation, and function of immune cells within TME, enhancing immune cell activity and alleviating immunosuppression.50 Several cytokines, including IL-2, IL-7, IL-12, IL-15, interferon (IFN), and tumor necrosis factor (TNF), are partially utilized in cancer immunotherapy.51–55 Each of these immunotherapeutic approaches reinitiates the cancer immune cycle by acting on one or more stages, effectively controlling tumor progression, metastasis, and recurrence, thereby representing significant progress for cancer patients.
Despite being at the forefront of cancer treatment, immunotherapy's clinical efficacy remains suboptimal, primarily due to patient resistance to immunotherapeutic drugs. Various mechanisms contribute to cancer cells developing resistance after immunotherapy.6,56 Tumor cells evade immune cell recognition by down-regulating TAA expression, diminish immune clearance by expressing immunosuppressive molecules (PD-L1 and CTLA-4), and develop resistance to immunotherapy, as seen in tumor cells treated with photo-immunotherapy that up-regulate heat shock proteins (HSPs) to counteract thermal ablation effects.57,58 Additionally, multiple factors in the intricate TME, such as immune-suppressing cells (Tregs, M2-type macrophages, and CAFs), immunosuppressive cytokines (TGF-β, IL-10), physical barriers like dense extracellular matrix, and signaling pathways promoting anti-apoptotic and anti-inflammatory responses, hinder immune cell function.59 Moreover, high-frequency gene mutations in tumor cells during proliferation can result in the expression of neoantigens or mutant tumor antigens, leading to a low or no response to immunotherapeutic drugs.60 A sustained anti-tumor immune response also leads to impaired function, inactivation, or death of immune cells.5,61–63 These intricate and interconnected mechanisms collectively contribute to the development of drug resistance. Besides, the limitations of cancer immunotherapy, including low response rates, limited durability, and immune-related adverse events (irAEs), pose challenges for its rapid development.64,65 Consequently, it has become a challenge to develop new immunotherapeutic strategies to provide patients with safe, durable, and specific cancer immunotherapy.
3. Applications of nanoparticles in cancer immunotherapy
The emergence of nanoparticles has provided new ideas to address these challenges of cancer immunotherapy approaches described above. Nanotechnology has significantly improved drug delivery by utilizing drug-loaded NPs that enhance penetration, retention, and accumulation of immunotherapy drugs in cancer tissues, leading to safer and more efficient drug delivery.14,15 For instance, Xu et al. have loaded melanoma TAAs and adjuvants into lipid calcium phosphate (LCP) NPs to develop a cancer vaccine. This approach stimulated an effective anti-melanoma T cell response by enhancing phagocytosis and presentation of DCs.66 Another study involved combining mRNA with lipid-based NPs to create an mRNA vaccine. The nanoparticles protected mRNA from degradation and clearance, thereby increasing its accumulation in DCs and enhancing the expression of specific tumor antigens, resulting in effective T cell responses in models of melanoma and lung cancer.67Certain nanomaterials with photosensitive properties, such as gold nanocages and copper sulfide, can enhance the effectiveness of immunotherapy. They achieve this by inducing immunogenic cell death (ICD) in tumor cells through photodynamic or photothermal therapy, releasing pro-inflammatory factors to promote APC maturation, and enhancing the depth of the cancer immune cycle.68,69 For instance, Wang et al. demonstrated that polyethanolated gold nanostructures, when exposed to near-infrared (NIR) light, prolonged blood circulation time, accumulated at tumor sites, and exhibited significant therapeutic effects in a breast cancer mouse model by converting light into thermal energy.70 Another study utilized mesoporous silica-coated up-conversion NPs loaded with photosensitizers, resulting in the generation of a large amount of reactive oxygen species (ROS) upon 980 nm laser irradiation, showcasing the photodynamic effect in a colon cancer mouse model.71
Additionally, novel nano-delivery systems can enhance immunotherapy effectiveness by targeting neovascularization, fibroblasts, and immunosuppressive factors in TME to reshape its immunosuppressive nature. Ferumoxytol, an iron oxide NP, has been found to induce the repolarization of M2-type macrophages to M1-type, initiating a pro-inflammatory immune response that inhibits tumor growth.72 Lipid NPs loaded with anti-angiogenic drugs and other chemotherapeutics have been designed to normalize tumor blood vessels, bolster endothelial resistance, and promote vascular normalization.73
However, despite the promising future of NPs in cancer immunotherapy, there exist limitations that hinder their application. While NPs accumulate in tumor tissues based on their specific properties, they can also permeate normal tissues, potentially leading to toxic reactions.74 Furthermore, NPs are often recognized as foreign entities by the immune system, resulting in expedited clearance and reduced efficacy. Despite some advances in delivery, the issue of tumor resistance to immunotherapeutic agents remains unresolved.75 Consequently, the development of safe, controlled, and targeted NPs has become a paramount challenge for advancing cancer immunotherapy.
To address the limitations of individual nanoparticles in cancer therapy, different types of CMNPs have been investigated and applied across various fields. Undoubtedly, CMNPs, leveraging the biological functionalities inherent in cell membranes, have significantly enhanced the versatility of nanoparticles in cancer immunotherapy, catalyzing a spectrum of innovative advancements. Primarily, CMNPs leverage natural cells as encapsulating agents, endowing them with surface characteristics reminiscent of native cells, thereby facilitating enhanced interactions with the immune system.76 By modulating the surface proteins or ligands of nanoparticles, CMNPs can precisely target specific tumor cells, facilitating accurate delivery and controlled release of therapeutic agents.77 Moreover, CMNPs exhibit the capability to not only transport chemical drugs but also incorporate immunomodulators, nucleic acid drugs, and other therapeutic agents, thereby creating a multifunctional therapeutic platform.78 This multifaceted approach enables simultaneous engagement of multiple immunotherapeutic mechanisms, consequently augmenting the overall efficacy of tumor treatment strategies. In fact, CMNPs are not independent of the current rapidly evolving cancer immunotherapy modalities, but rather a complementary form of drug delivery that undoubtedly provides a theoretically safer and more efficient platform for the applications of for ICIs, CAR-T cell therapy, cytokine therapy, cancer vaccines, and other immunotherapeutic approaches.79,80
4. Characterization and synthesis of CMNPs
Since Hu et al. formed CMNPs by co-extruding erythrocyte membranes with PLGA nanoparticles in 2011, the biomimetic strategy of CMNPs began to flourish and provided a perfect platform for cancer therapy.23 The combinations of different cell membranes with nanoparticle cores have given CMNPs the functions of membrane-derived cells and the unique advantages of nanoparticles in cancer immunotherapy. Multiple cell membranes are currently used in this biomimetic strategy, including the membranes of erythrocytes, platelets, cancer cells, immune cells, stem cells, bacteria, and even hybrid cell membranes.81,82The complex functions of cell membranes enable CMNPs to adapt to the intricate tumor microenvironment. After wrapping membranes on NPs as carriers, and loading with drugs or engineering on the surface, CMNPs exhibit diverse characteristics, particularly in targeted drug delivery and immune evasion that conventional therapies lack, presenting a novel concept for cancer immunotherapy.24
4.1. Unique functions of CMNPs from different cell membrane sources
Following the coating of cell membranes, the proteins, signaling molecules, and antigens on the cell membrane surface remain intact, thereby ensuring the seamless transfer of these biologically functional constituents to CMNPs. The specialized functions that CMNPs derive from original cells are outlined next, based on the different cell types.Macrophages are versatile innate immune cells widely distributed in tissues. In adult individuals, they predominantly originate from hematopoietic stem cells/monocytes in the bone marrow. Macrophages could assume diverse functional states depending on their local microenvironment. M1-type macrophages are activated by interferon, bacterial lipopolysaccharide (LPS), or granulocyte-macrophage colony-stimulating factor (CM-CSF), and promote inflammatory processes and adaptive immunity. Conversely, M2-type macrophages are primarily activated by elevated levels of cytokines such as IL-4, and IL-13, and are involved in tissue repair, angiogenesis, and the suppression of inflammation.94 As a natural immune barrier, macrophages express integrins on the membranes to contact with vascular cell adhesion molecules (VCAM-1) on tumor cells, thus enabling them to target tumor sites and realize prolonged circulation time.95 Macrophages in TME can transform into TAM under the influence of chemokines.96 As the most abundant immune cell population in the TME, M1-type TAMs secrete pro-inflammatory cytokines and enhance their ability to combat tumors. Conversely, most TAMs exhibit M2-type characteristics, which can inhibit the function of CD4+ and CD8+ T cells, recruit Tregs, suppress the anti-tumor immune response, and facilitate tumor cell proliferation, infiltration, and metastasis.97,98 Thus, modulating macrophage polarization is an ideal approach to reverse immunosuppression of TME.99
Dendritic cells, being the primary APCs in the body, play a pivotal role in recognizing, internalizing, and presenting antigens, which are essential for activating specific T cell immune responses. Since mature DCs expressing MHC-antigen peptide complexes on the surfaces are key players in the tumor immune process, DC membrane coated NPs are poised to amplify the cancer immune cycle by inheriting their antigen-presenting and T cell activation abilities, thereby offering novel possibilities for CMNPs-based immunotherapy.100
NK cells can generate non-specific anti-tumor responses without prior antigen sensitization or antibody involvement. This is mainly accomplished by perforin and tumor necrosis factor secreted by activated NK cells. Additionally, NK cells release various cytokines to play immunomodulatory roles, such as promoting the maturation of APCs and inducing M1-type macrophage polarization. Moreover, NK cells can target tumor cells through surface activation receptors CD16 and NKG2D. Consequently, NK CMNPs hold promise for enhancing targeted drug delivery and regulating the immune microenvironment.101
4.2. Engineering strategies of cell membranes
There are inevitable limitations in using natural cell membranes as the biological function source for CMNPs. With the increasing comprehensiveness and stringency of CMNP requirements, there's a gradual shift in the coating materials from natural to engineered cell membranes (Fig. 3).113 To achieve multifunctional modification, engineering strategies are gaining attention to modify the cell membrane by inserting specific ligands, functional proteins, or directly at the cellular level.114,115 Hybrid cell membranes, discussed earlier, represent a mature engineering technique for physical fusion, overcoming the inherent limitations of single-cell membranes and achieving functional compatibility.103 | ||
| Fig. 3 Engineering strategies of cell membranes via membrane hybridization, chemical conjugation, and biological modification. aDEC205: anti-DEC205; aSIRPα: signal regulatory protein alpha; OVA: ovalbumin; AUNP12-Mal-PEG-FAB: AUNP12(anti-PD1 peptide)-maleimide-polyethylene glycol-formyl benzoic acid; CBP-12: 12-mer Clec9a binding peptide; cRGD: cyclo (Arg-Gly-AspD-Tyr-Lys) peptide; YSA: ephrin-A2 receptor-specific peptide. | ||
Chemical modification is widely employed for engineering cell membranes. The phospholipid bilayer structure allows the embedding amphiphiles containing diverse functional groups into the cell membrane via a non-covalent manner by hydrophobic interactions. For instance, DSPE-PEG-Man, inserted into the phospholipid bilayers, increased the ability to target APCs in lymph nodes or macrophages in tumor sites through mannose introduction.116,117 Recognizing the low stability of non-covalent binding, researchers prefer chemical bonding for the stable attachment of active molecules like peptides or proteins to membrane surfaces.118
Biological modification of cell membranes is a favorable and promising approach. One study employed DOX pretreatment to obtain cell membranes overexpressing CRT for promoting the active uptake of nanovaccines by DCs.119 Genetic engineering techniques enable precise control of cellular biosynthesis at the gene level, resulting in cell membranes that stably express the desired functional molecules. For instance, Rao and co-workers realized overexpression of the SIRPα variants on cancer cell membranes by lentiviral transfection, heightening the affinity for CD47.120 Similarly, Tang et al. genetically engineered a HEK293T cell line stably overexpressing M-αPD-L1 to encapsulate NPs to enhance immunotherapy.121
4.3. The synthesis of CMNPs
The process of acquiring cell membrane materials involves two key stages: cell membrane lysis and subsequent purification (Fig. 4). Current methodologies for cell membrane lysis encompass hypotonic solution, freeze–thaw cycling, and ultrasound techniques. Hypotonic lysis necessitates the preparation of a suitable hypotonic lysis buffer (pH 7.4), typically composed of Tris, MgCl2, among others, with the addition of the protease inhibitor PMSF (excluding EDTA) on ice to prevent the degradation of crucial functional proteins on the cell membrane surface. Subsequently, cell pellets are harvested via centrifugation and subjected to washing for further processing.122 Freeze–thaw cycling involves the iterative freezing of cell suspensions in liquid nitrogen for about 20 minutes, succeeded by thawing in a water bath (37 °C) for 5 minutes, until substantial disruption of cell morphology is achieved, accompanied by the disappearance of intact nuclei and perinuclear structures, as visualized under a microscope. Subsequent centrifugation is performed to eliminate dense cell nuclei and a fraction of undamaged cells.123 Although ultrasound-assisted cell lysis is a convenient and rapid method, the potential for membrane protein damage and protein denaturation due to ultrasound-induced heating underscores the necessity for meticulous control over ultrasound parameters. Presently, researchers commonly employ the former two methods for cell lysis, adapting protocols as needed to preserve the integrity of cell membrane surface proteins and ion channel structures and functionalities.105 Following lysis, further processing of the obtained cell suspension is required to obtain purified cell membrane fragments. For nucleus-free cells such as red blood cells and platelets, purification of cell membranes is relatively straightforward through high-speed centrifugation. However, for nucleated cells like tumor cells and immune cells, the presence of intracellular organelles and complex protein and enzyme components presents challenges to membrane material acquisition.124 Typically, a discontinuous sucrose gradient centrifugation method is employed to segregate and purify cell membranes. This is followed by low-speed centrifugation to eliminate residual cell nuclei and organelles, prior to subjecting the resulting supernatant to high-speed centrifugation for the precipitation of cell membrane fractions.125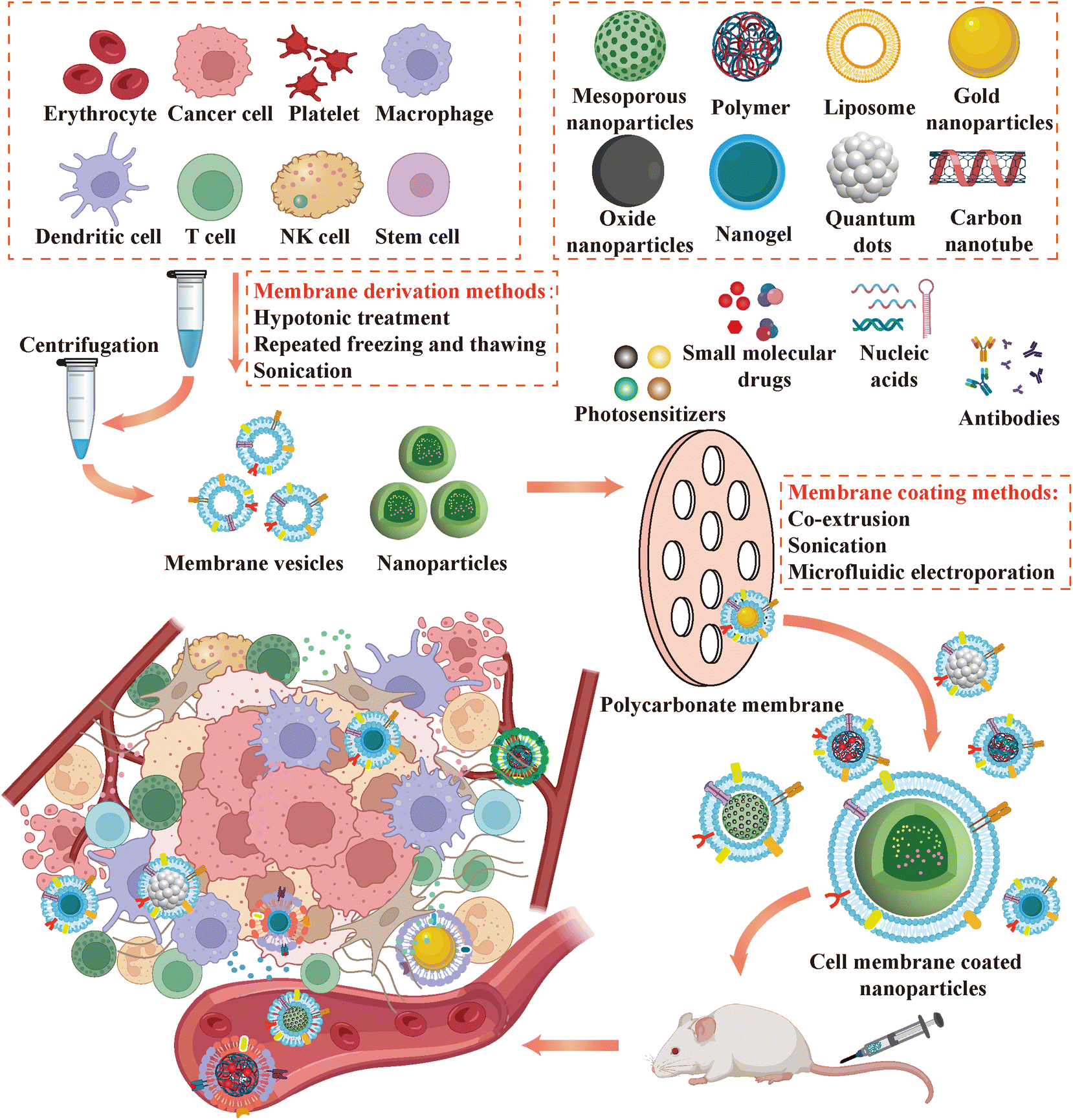 | ||
| Fig. 4 Schematic of sources and synthesis process of cell membrane coated nanoparticles for cancer immunotherapy. | ||
The selection of a coating technique for CMNPs significantly influences their functionality. Physical extrusion emerges as the preferred method in most investigations, entailing the repeated extrusion of purified cell membranes and nanoparticles through porous polycarbonate membranes with diameters in the hundreds of nanometers.126 Mechanical force transiently disrupts membrane fluidic structures, facilitating the reassembly of protein-laden membrane fragments around nanoparticle cores. This approach enables the production of uniformly sized and coated CMNPs by adjusting membrane pore sizes, thereby ensuring consistency with the surface proteins of the source cells.25 Nonetheless, this method is primarily suitable for laboratory-scale production and entails considerable time and labor. The inherent asymmetric charge interactions between nanoparticle cores and the inner and outer layers of cell membranes culminate in the formation of shell–core structures with specific right-side-out membrane orientation subsequent to coating.127 This phenomenon is crucial for preserving the integrity of various ion channels and functional protein components on the cell membrane surface. Ultrasound, based on acoustic principles, is another coating method where ultrasound energy can disrupt membrane stability, leading to self-assembly of membranes and nanoparticles into shell–core structures, facilitating large-scale production.128 However, careful consideration is warranted regarding potential alterations in biological membrane component functionality attributable to excessive local ultrasound energy and drug deformation and leakage. Microfluidic electroporation represents an innovative coating approach wherein membranes and nanoparticles traverse distinct channels within a microfluidic chip, achieving thorough mixing before fusing at the electroporation region between two electrodes under the influence of electrical pulses.129 Various parameters such as mixing velocity, voltage, and pulse intensity can be finely tuned within the device, thereby advancing membrane coating technology towards heightened efficiency and precision. The attainment of comprehensive coverage of cell membranes onto nanoparticle surfaces holds pivotal importance in constructing proficient drug delivery systems.130
Ensuring complete coverage of cell membranes on the surface of nanoparticles is crucial for constructing drug delivery systems. Many studies characterize the physicochemical properties and biological characteristics of CMNPs to verify the successful encapsulation. The encapsulation process, owing to the negative charge of cell membranes, elicits alterations in nanoparticle surface charge and particle size. Consequently, dynamic light scattering (DLS) analyzers serve as prevalent tools for quantifying and contrasting the particle size and zeta potential of nanoparticles pre- and post-coating. Moreover, transmission electron microscopy (TEM) enables direct visualization of the shell–core architecture of CMNPs, with discernible density disparities between lipid bilayers and nanoparticles evident upon staining, manifesting as a halo encircling effectively coated nanoparticles.131 Spectroscopic techniques are employed to scrutinize the optical characteristics and surface chemistry of CMNPs. For instance, UV-visible absorption spectroscopy enables the discrimination between absorption spectra originating from cell membranes and nanoparticle cores. This differentiation facilitates the characterization and authentication of CMNPs by discerning between individual nanoparticles exhibiting either dual or single absorption modes.132,133 Furthermore, X-ray diffraction, Fourier transform infrared spectroscopy, and nuclear magnetic resonance spectroscopy are utilized to probe the structural configuration and compositional makeup of CMNPs.134
An indispensable facet of characterization experiments involves the analysis of surface proteomics and lipidomics of synthesized CMNPs. Conventionally, sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) is deployed to confirm distinct membrane protein profiles, while western blot analysis evaluates the retention of coated proteins. Notably, for engineered cell membrane coated nanoparticles, Coomassie Brilliant Blue staining and western blotting targeting coupled or overexpressed proteins assume critical importance.135 Mass spectrometry emerges as a robust tool for identifying surface biological functional constituents. Liquid chromatography-tandem mass spectrometry (LC-MS/MS) facilitates the analysis of surface protein constituents and their amino acid sequences, along with the identification and quantification of lipid species and contents on the membrane surface.136 Immunological detection methodologies, such as immunoblotting and immunofluorescence staining, are instrumental in examining the presence, spatial distribution, and relative abundance of specific components on CMNP surfaces, especially for overexpressed engineered cell membranes, where localization of overexpressed proteins by spotlight confocal microscopy is essential.77
In summary, recent years have witnessed significant advancements in the preparation techniques for CMNPs. Alongside conventional blood cells and tumor cells, diverse cell membrane sources such as T cells, stem cells, and hybrid membranes have emerged as promising coating materials owing to their distinct advantages.137 The rapid progress in engineered cell membrane technologies, encompassing chemical conjugation and genetic engineering, has introduced diverse strategies to enhance the functionalities of coated membranes. Given the reliance of CMNPs on straightforward top-down fabrication methods, stringent requirements for core nanomaterials are not imperative.25 Various materials including polymers, metallic substances, and oxides can serve as drug carriers, greatly expanding the repertoire of shell–core structures.138,139 On the synthesis front, besides conventional physical extrusion and sonication methods, innovative techniques such as microfluidic electroporation have been developed to yield more stable and homogeneous CMNPs. Collectively, these advancements contribute to the ongoing progress of CMNPs in the realm of cancer immunotherapy, and ongoing optimization of synthesis processes promises to expand its applicability for the treatment of cancer and other diseases.
5. Classification of CMNPs delivery components
CMNPs, as a nascent drug delivery platform, possess distinctive drug-loading capabilities. Encasing NPs in cell membranes provides a biocompatible shield, reducing toxicity and immunogenicity, preventing premature drug clearance and degradation, and enhancing drug bioavailability and patient adherence. Cell membranes derived from specific cell types enable targeted delivery through protein binding and surface modification, augmenting their biological functions. This biocompatible, targeted, stable, and versatile platform opens new avenues for precise drug delivery and therapy. These delivered therapeutic agents can be broadly classified into the following groups shown in Table 1.88,107,116,121,140–162| Type | Therapeutic agents | Core nanoparticles | Membrane types | Ref. |
|---|---|---|---|---|
| Photosensitizers | Gold nanoparticles | Au@Carbon | Human oral adenosquamous carcinoma cell | 140 |
| Black phosphorus | Black phosphorus quantum dots | 4T1 and B16F10 cancer cell | 141 | |
| Polydopamine | Polydopamine | 4T1 cancer cell | 142 | |
| Indocyanine green | Fe3O4 | Hybrid membrane (erythrocyte and ID8 cell membrane) | 143 | |
| Protoporphyrin IX | Copper peroxide | Erythrocyte | 144 | |
| Chlorin e6 | DGL-G3 | Platelet | 145 | |
| TCPP | Metal–organic frameworks | Hybrid membrane (DC and 4T1 cell membrane) | 146 | |
| Small molecule compounds | ||||
| Targeting immune cells | R837 | PLGA | B16-OVA cell | 116 |
| R848 | CuS | 4T1 cell | 147 | |
| Monophosphoryl lipid A | PLGA | B16F10 cell | 148 | |
| cGMP-AMP | PLGA | B16-OVA cell | 149 | |
| IL-15 | PLGA | NK cell | 150 | |
| Acetaminophen | Nanogel | Panc02 cell | 151 | |
| Magnetic nanoparticles | Fe3O4 | M1 macrophage | 152 | |
| Targeting cancer cells | Doxorubicin | Mesoporous organosilica nanoparticles | 4T1 cell | 153 |
| Paclitaxel | Hollow manganese dioxide nanoparticles | Mesenchymal stem cell | 154 | |
| Sorafenib | Hollow mesoporous silica nanoparticles | HepG2 cell | 88 | |
| Gemcitabine | PEG-PDPA | 4T1 cell | 155 | |
| Docetaxel | PLGA | 4T1 cell | 156 | |
| Targeting cancer metabolic mechanisms | Glucose oxidase | Zeolitic imidazolate framework | 4T1 cell | 157 |
| Indoximode | Gold nanocage | M1 macrophage | 158 | |
| Catalase | Zeolitic imidazolate framework | B16F10 cell | 159 | |
| Nucleic acid | Fibrinogen-like protein 1 siRNA | PLGA | Hybrid membrane (macrophage and 4T1 cell membrane) | 107 |
| IL-1α siRNA | Black phosphorus | Erythrocyte | 160 | |
| Protein tyrosine phosphatase non-receptor type 2 shRNA | HA-DOX | M1 macrophage | 161 | |
| IL-12 mRNA | Calcium carbonate | GL261 cell | 162 | |
| Antibody | aPD-L1 | Barium titanate nanoparticle | 293T cell | 121 |
| aPD-L1 | Hollow mesoporous silica nanoparticles | Platelet | 88 |
5.1. Photosensitizers
Photothermal therapy (PTT) is a novel, non-invasive tumor thermal ablation technology. After the materials with high photothermal conversion efficiency are injected into the body, they can convert external near-infrared light into heat, inducing tumor cell ICD. The apoptotic tumor cells release massive new antigens and damage-associated molecular patterns (DAMPs), initiating the first step of the tumor immune cycle, recruiting APCs, and mobilizing T cells to amplify the immunotherapy.163 Currently, various photothermal agents are being incorporated into CMNPs to address issues like inadequate accumulation at the target site, limited circulation time, and potential toxic reactions.Nanoparticles with inherent photothermal properties, such as gold NPs utilizing localized surface plasmon resonance (LSPR), have been employed in the treatment of prostate cancer. In one study, Au@C NPs were synthesized through one-step hydrothermal method using gold perchlorate (HAuCl4·3H2O) and glucose as the carbon source, while Au@C-CCM was produced by coating CAL-27 cancer cell membranes. The encapsulation provided stable photothermal properties to gold NPs, achieving 44.2% photothermal conversion efficiency, potentially improving PTT efficacy coupled with cancer cell membrane-based targeting.140 Black phosphorus (BP), a 2D material with superior photothermal conversion efficiency, is a preferred photothermal agent in PTT applications. In a study, black phosphorus Quantum Dots (BPQDs) were prepared using a modified liquid stripping technique, forming BPQD-CCNVs after coating them with cancer cell membranes. These NPs exhibited rapid surface temperature increase under near-infrared light within 2 min.141 Polydopamine (PDA) is a melanin-like substance with excellent physicochemical properties. CPCaNPs were pH-responsive and imaging-guided PTT platforms, which were synthesized by dopamine-mediated biomineralization and 4T1 cell membrane coating (Fig. 5). Upon targeting the acidic microenvironment, CPCaNPs decomposed to produce CO2 bubbles that significantly enhance the ultrasound signal for imaging. These NPs exhibited concentration-dependent heating ability with a 36.7% photothermal conversion efficiency and enabled photoacoustic (PA) imaging.142
 | ||
| Fig. 5 CMNPs for cancer multimodal imaging and photothermal immunotherapy.142 (A) Schematic of the synthesis of CPCaNPs. (B) Thermal infrared images at different concentrations. (C) Temperature change curve at different concentrations. (D) Temperature variation curve under different power densities of the 808 nm laser. (E) In vivo distribution and tumor targeting examination. (F) Tissue distribution in the removed organs and tumors. (G) PA imaging of the tumor after injection. (H) Photothermal images of laser-irradiated tumors after injection. (I) CEUS imaging of the tumor after injection. Copyright 2022 American Chemical Society. | ||
Many other photosensitizers have been incorporated into CMNPs to exert photothermal effects, including indocyanine green (ICG), IR-797, etc. For instance, ICG was loaded into Fe3O4 coated with hybrid cell membranes (erythrocyte and ID8 cell membranes), achieving a desirable photothermal effect. This combination reached a temperature of 55.5 °C when exposed to 808 nm near-infrared light, demonstrating promising potential for tumor ablation.143,164 Moreover, organic NPs, such as porphyrin compounds and specific organic polymers, can regulate the photothermal effects through molecular structure and functional unit adjustments.165
In the realm of photodynamic therapy (PDT), a range of photosensitizers can be selected to generate ROS or other active substances upon excitation by laser light at specific wavelengths, enabling selective damage to tumor cells. ICG, as mentioned earlier in PTT, serves as a notable representative and an FDA-approved imaging reagent for near-infrared PDT.12 Commonly used photosensitizers include protoporphyrin IX (PpIX), methylene blue (MB), chlorin e6 (Ce6), and low-wavelength photosensitizers.144,166,167 Despite its potential for highly effective and non-invasive cancer therapy, PDT's progress remains hindered by the limited availability of suitable photosensitizers and delivery challenges.
The emergence of CMNPs offers a new strategy for photosensitizer delivery. Metal–organic frameworks (MOFs) containing photosensitizers can overcome the self-combustion disadvantage and enhance the diffusion of the generated ROS. For example, a study employed TCPP to synthesize porphyrin-based Zr-MOF (PCN-224) and coated it with hybrid membrane composed of 4T1 cell and DC membranes via ultrasonication in an ice bath. This assembly was then irradiated with 660 nm light to generate ROS.146 Ce6, similar to most photosensitizers, exhibits hydrophobicity and tends to aggregate in solution. To address this, non-covalent interactions were harnessed to load Ce6 into OVA NPs encapsulated with cancer cell membranes, enhancing stability and efficient ROS generation under laser irradiation.145
Additionally, photosensitizers can be embedded into the surface of biological membranes to mediate PDT. For instance, through the chimeric peptide with electrostatic interaction and hydrophobic effect, PpIX was inserted into the erythrocyte membranes tightly, forming a multifunctional nanoplatform CP@mRBC-PpIX after coating copper peroxide (CP) NPs.144
Given the variability in materials with distinct absorption spectra and phototherapeutic properties, the selection of photosensitizers should be customized based on disease characteristics, patient conditions, and research or clinical treatment objectives.
5.2. Small molecule compounds
In the realm of cancer immunotherapy, immune-modulating drugs have been widely researched and applied. Since cytokines such as IL-2 were applied in immunotherapy, the development of various small molecule immunotherapy drugs such as cell growth inhibitors, cytokines, chemokines, and metabolism modulators has been spurred.50 Despite tremendous progress, small molecule drugs often encounter resistance and adverse reactions in clinical applications, prompting innovations in drug delivery strategies. CMNPs, with superior delivery properties, are extensively employed in research to overcome drug resistance and enhance targeted drug delivery efficiency. The following section will provide an overview of several widely used immunotherapeutic drugs loaded into CMNPs.Alongside TLR agonists, interferon gene-stimulating factor (STING) agonists can also activate innate immunity as adjuvants.149 DNA fragments from lysed tumor cells activate STING via cGMP-AMP synthase (cGAS) within APCs and T cells, leading to downstream IFN-dependent antitumor immunity. Combining PC7A (activating STING pathway) with TLR-9 agonist CpG has been investigated for its vaccine adjuvant properties. The PH-responsive PC7A multimer formed cores with CpG loaded and coated with Mycobacterium smegmatis (MS) membranes containing pathogen-associated molecular patterns. This synthesized structure, known as BNP, was further modified with imine moieties (Mal) to enhance antigen uptake.112
Additionally, cytokine therapy, a classical form of immunotherapy, can also benefit from improved drug delivery through CMNPs. In a recent study, IL-15, known for its activation of T cell and NK cell functions, was loaded into PLGAs and enveloped by cRGD peptide-modified NK cell membranes (referred to as R-NKm@NPs). This approach improved cytokine delivery and enabled pH-responsive drug release (Fig. 6). NK cell membrane components, NKG2D and DNAM1, facilitated the NPs in crossing the blood–brain barrier (BBB) to target GBM cells. Their ability to target glioma TME was further amplified after modification with the brain tumor-targeting ligand cGRD, which exhibited a high affinity with αvβ3 on GBM cell surfaces.101
 | ||
| Fig. 6 NK cell membrane coated NPs delivered cytokine and chemotherapeutics for targeted GBM chemo-immunotherapy.150 (A) Schematic of the synthesis of R-NKm@NPs. (B) SDS-PAGE and western blotting analysis. (C) Drug releasing profile. (D) Confocal images of the permeability in the in vitro BBB model. (E) In vivo bioluminescence imaging and ex vivo fluorescence imaging of the major organs. (F) Ex vivo fluorescence imaging of the brain and the semiquantitative biodistribution of DiD. Copyright 2023 Wiley. | ||
Conventional anti-inflammatory drugs have also demonstrated a modulatory effect on immune cells. In one study, Panc02 cell membranes (PCM) were employed to coat pH/oxidation-responsive nanogels for delivering the cyclooxygenase inhibitor Acetaminophen (APAP) to modulate NK cell function.151 Beyond drug loading, nanoparticles themselves can serve as immune cell modulators. For example, Rao et al. encapsulate magnetic nanoparticles (MNs) with membranes derived from genetically engineered cells that overexpress SIRPα, effectively reprogramming TAMs.120 Commonly used nanoparticles, such as iron oxide NPs (Fe3O4), have been encapsulated with various cell membranes, including M1 membranes and hybrid cell membranes, to participate in the regulation of immune processes.143,152 Furthermore, there is a growing trend of combining traditional immunotherapeutic drugs with NPs, offering new avenues for cancer immunotherapy.
For instance, doxorubicin (DOX), a non-specific anticancer agent, which inhibits DNA and RNA synthesis, has been incorporated into various CMNPs. In one study, DOX was loaded into mesoporous organosilica nanoparticles (MONs) coated with 4T1 cell membranes, allowing for controlled release through ray-responsive diselenide bonding.153 Another study loaded DOX with IND and Ce6 into M1 membrane coated bilirubin NPs (BPs) through stirring and centrifugation to exert synergistic effects.158
Paclitaxel (PTX), the first discovered drug that inhibits cancer cell mitosis by interacting with microtubules, can be precisely delivered using hollow manganese dioxide NPs coated with human umbilical cord-derived mesenchymal stem cell membranes, as demonstrated by Xie et al.154 Various other classic anticancer drugs, including docetaxel, gemcitabine, and sorafenib, have also been integrated into nanoplatforms for cancer immunotherapy.88,155,156
The robust proliferative capacity of tumor cells relies on the high-energy supply, leading to enhanced glycolysis and glutaminolysis for energy and biosynthesis. To address this, CMNP with glucose oxidase (Gox) as the major body was developed. Gox solution was centrifuged by mixing and stirring with 2-MI solution so that Gox was co-loaded with EPI and hemin on zeolitic imidazolate framework (ZIF-8). Ultrasound encapsulation in 4T1 membrane enabled the simultaneous reduction of glucose and glutathione (GSH) levels at the tumor site.157
Tryptophan metabolism in tumors, regulated by enzymes tryptophan 2,3-dioxygenase (TDO) and indoleamine 2,3-dioxygenase (IDO), generates immunosuppressive metabolites such as kynurenine. Tryptophan deficiency and metabolite accumulation inhibit effector T cell function, promoting the development of Treg and myeloid-derived suppressor cells (MDSCs). To disrupt tumor cell tryptophan metabolism, IDO1 inhibitor indoximode (IND) was loaded into various CMNPs, such as M1 macrophage coated gold nanocages and GL261 cell membrane coated Cu2−xSe NPs.158,170
Hypoxia, another significant feature of TME, results from excessive oxygen consumption by proliferating tumors and insufficient oxygen supply from abnormal blood vessels. Researchers have utilized CMNPs to alleviate tumor hypoxia to reverse immunosuppressive TME. For example, they incorporated catalase (CAT) for hydrogen peroxide breakdown into ZIF-8 with B16F10 cell membranes to create mZCD.159 Another study developed manganese dioxide multifunctional biomimetic nanoparticles, HM-BPT NPs, with hybrid cell membrane encapsulation (MSC membranes and pH-sensitive liposomes) to leverage manganese oxides’ catalase-like function through Mn2+-mediated Fenton-like reactions.171 Moreover, drugs targeting the arginine and adenosine metabolic pathways are also expected to amplify their effects based on CMNPs.
5.3. Nucleic acid agents
Nucleic acid therapy, a rapidly evolving biotechnology, is widely applied in addressing infectious diseases, cancer, autoimmune disorders, and related fields through regulating intracellular gene expression.172 Distinct from traditional small molecule and antibody drugs, nucleic acid drugs primarily function through base complementary pairing, enabling specific targeting of intracellular and extracellular proteins by modulating protein-coding genes. Nucleic acids, which carry a negative charge, require binding to auxiliary agents to facilitate cellular uptake. However, their clinical translation faces challenges, including the risk of degradation by nucleases, transmembrane transport difficulties, immunogenicity, and instability. Consequently, innovation in nucleic acid drugs concentrates on improving their delivery systems. Traditional viral vector delivery strategies, due to their limitations, are being replaced by alternative methods like GalNac (N-acetylgalactosamine) modification and liposomal delivery technologies.173 Several notable drugs have been introduced using these approaches. Similarly, CMNPs, characterized by multiple advantageous features, have garnered attention as drug delivery platforms for acid-based drugs to enhance cancer immunotherapy.Small interfering RNAs, a prominent class of small nucleic acid drugs, are favored by researchers for their precise gene silencing capabilities. For example, Gong et al. employed a double emulsion technique to co-encapsulate fibrinogen-like protein 1 siRNA (siFGL1) and Met-CO2 into PLGA and coated with a hybrid membrane derived from RAW264.7 and 4T1 cells using ultrasound. siFGL1 effectively downregulated FGL1 expression in tumor cells, reducing its binding to lymphocyte activation gene 3.107 In another study, IL-1α siRNA disrupted inflammation-associated IL-1α expression and regulated Treg cells through BP NPs coated with erythrocyte membranes.160 Yang et al. used CMNPs to deliver short hairpin RNAs (shRNAs) targeting protein tyrosine phosphatase non-receptor type 2 (Ptpn2) for gene therapy. They co-assembled Ptpn2-targeted shRNA plasmids with iRGD and DOX via electrostatic adsorption and co-extruded them with M1 macrophage membranes to downregulate Ptpn2 gene expression.161
Furthermore, the delivery of mRNAs can also be improved through CMNPs. For example, IL-12 is a kind of cytokine with significant activation of anti-tumor immunity but difficult to achieve by intravenous administration. Zhao et al. utilized reverse microemulsion to load IL-12 mRNA onto calcium carbonate (CaCO3) and synthesized mRNA@CMCaCO3 NPs by co-extrusion with GL261 cell membranes in 200 nm polycarbonate membranes, enabling BBB penetration and cytoplasmic transfection of IL-12 mRNA (Fig. 7).162
 | ||
| Fig. 7 Cancer cell membrane coated NPs delivered IL-12 mRNA for targeted GBM immunotherapy.162 (A) Schematic of the synthesis of IL-12 mRNA@cRGD-CM-CaCO3 NPs. (B) Proposed mechanism of NPs for BBB penetration, TME navigation and sono-immunotherapy synergistic anti-tumor effects in GBM. (C) Homotypic targeting through fluorescence measurement. (D) Luminescence intensity of NPs. (E) In vivo luminescence imaging of brain tumor model. (F) Quantitative bioluminescence signal intensity. Copyright 2022 BioMed Central. | ||
As research advances, nucleic acid delivery strategies like exosomes and peptide nanomaterials are under development. Combining these approaches with cell membrane coating technology holds promise for enhancing nucleic acid drug delivery efficiency in future research.
5.4. Antibody agents
Since the first introduction of therapeutic anti-CD3 antibodies in clinical practice in 1986, antibody-based drugs have rapidly proliferated and found widespread applications in areas like chronic inflammation, autoimmune disorders, and cancer treatment. In cancer therapy, these antibodies fall into three categories: monoclonal antibodies (mAbs), bispecific antibodies, and antibody–drug conjugates (ADCs).174 They function by modulating various signaling pathways within cancer cells. Notably, ICIs, predominantly targeting PD-1/PD-L1 and CTLA-4 with monoclonal antibodies, have gained prominence as classical immunotherapeutic agents in cancer treatment.Despite the benefits of ICIs, such as high specificity, relatively low side effects, and extended half-lives, there is ongoing research focused on enhancing their therapeutic effectiveness using CMNPs. For instance, to enhance the precision of anti-PD-L1 therapy, a genetically engineered aPD-L1 with MMP2 activation properties was expressed on the surface of HEK 293T cells. The membranes were obtained through low-permeability lysis and cyclic freezing and thawing, which were then coated onto ultra-small barium titanate (BTO) NPs (Fig. 8). The N-terminal binding domain of aPD-L1 was concealed by the MMP2 substrate peptide, allowing its inhibitory effect to be exerted specifically within MMP2-rich tumor tissues.121 Additionally, research has explored improving the delivery of aPD-L1 with CMNPs, such as by mixing aPD-L1 with synthesized platelet membrane-encapsulated hollow mesoporous silica NPs at room temperature and subjecting them to high-speed centrifugation to facilitate the co-delivery of aPD-L1 and antitumor drugs.88 In fact, ICIs are more frequently used as a combination immunotherapy regimen, co-administered with CMNPs to achieve synergistic therapeutic effects.
 | ||
| Fig. 8 Genetically engineering CMNPs for MMP2-activated immunotherapy. (A) Schematic of synthesis of M@BTO.121 (B) Schematic of mechanism of MMP2-activated piezocatalysis-immunotherapy. (C) Fluorescence images of genetically engineered cells. (D) Fluorescence images of DCFH-DA in B16F10 cells. (E) Fluorescence images of oxygen probe in B16F10 cells. (F) Fluorescence imaging of organs collected after different injections. Copyright 2023 Wiley. | ||
In parallel with CMNPs, numerous drug delivery systems have undergone extensive investigation. Among these, liposomes represent an early-developed system characterized by one or more phospholipid bilayers forming spherical vesicles. Typically, liposomes consist of cationic lipids, helper lipids, cholesterol, and PEGylated lipids. They excel in encapsulating hydrophobic drugs within their lipid bilayer, thereby enhancing drug solubility and prolonging residence time.175 Lipid nanoparticle formulations have been extensively employed in clinical trials for delivering various therapeutic agents, including anticancer, anti-inflammatory, antibiotic, antifungal, and gene therapies, notably in nucleic acid drug delivery, such as mRNA vaccine development.176,177 Nevertheless, liposomes encounter challenges such as reduced stability and limited biocompatibility compared to CMNPs, especially in scenarios where effective penetration of biological barriers like the blood–brain barrier remains elusive. The majority of nanoparticles available commercially are simplistic liposomal structures, underscoring the need for more sophisticated designs to enhance patient outcomes via nanodelivery systems.178
Extracellular vesicles (EVs) represent another class of membranous vesicles secreted by cells into the extracellular matrix, playing crucial roles in intercellular communication.179 Similar in size and structure to liposomes, EVs harbor various bioactive molecules such as proteins and lipids on their surface, along with internal nucleic acids. This feature endows EVs with the potential for complex drug delivery functionalities. EVs are commonly categorized into exosomes, microvesicles, and apoptotic bodies based on their cellular origin.180 Their low immunogenicity and distinctive advantages in intercellular signaling render them promising candidates as drug carriers in cancer therapy.181 In addition, EVs can efficiently load proteins, nucleic acids, cytokines, and other substances via endogenous expression within cells, utilizing the cell's inherent expression machinery.182 Nonetheless, despite these advantages, EVs exhibit certain limitations compared to CMNPs. For example, in terms of pharmacokinetics, externally introduced EVs may undergo rapid hepatic clearance, resulting in a shortened half-life.183 Furthermore, achieving clinical translation of extracellular vesicle delivery necessitates elevated standards of isolation and characterization. Moreover, the purification of extracellular vesicles typically involves intricate methodologies and specialized equipment, and is susceptible to contamination, thereby posing challenges in large-scale production and purification processes.184
Consequently, in comparison to analogous drug delivery systems, CMNPs, with their intricate surface biomolecular composition, heightened biocompatibility and stability, and more sophisticated design strategies, demonstrate superior potential for advancement in the realm of tumor immunotherapy.
6. Applications of CMNPs in cancer immunotherapy
This review article surveys various types of CMNPs aimed at augmenting the efficacy of tumor immunotherapy, as depicted in Table 2.80,88,100,101,105,107,116,117,120,128,139,141,143–149,151–153,155,157–162,164,166,170,185–211 The predominant mechanisms underlying most CMNPs are twofold: (1) delivering photosensitizers or capitalizing on the optical properties of NPs, facilitating photoimmunotherapy under near-infrared light irradiation. This process directly induces ICD through PTT or PDT for tumor ablation and the release of a significant quantity of tumor-associated antigens to reinitiate the cancer immune cycle. (2) Delivering small molecule immunotherapeutic drugs, immune modulators, chemical agents, or signaling molecules that induce tumor cell apoptosis, enhancing the immune system's response to tumor tissue. These compounds also act either on immune cells, immunosuppressive factors in TME, or both, thereby reshaping the immunosuppressive TME and bolstering anti-tumor immunity.| Types of membrane | Core nanoparticles | Modification of CMNPs | Delivered drugs | Animal tumor models | Applications | Ref. |
|---|---|---|---|---|---|---|
| PLGA: poly(lactic-co-glycolic acid); PLB: plumbagin; DIH: dihydrotanshinone I; Hb: hemoglobin; YSA: ephrin-A2 receptor-specific peptide; BP: black phosphorus; BPQDs: black phosphorus quantum dots; PTX: paclitaxel; CP: copper peroxide; PpIX: protoporphyrin IX; PLA: polylactic acid; SAS: sulfasalazine; DGL-G3: third-generation poly-L-lysine dendrimer; Ce6: chlorin e6; DTX: docetaxel; MOF: metal organic framework; Lox: lactate oxidase; Oxa: oxaliplatin; HMSNs: hollow mesoporous silica nanoparticles; SO: sorafenib; MNs: magnetic nanoparticles; OVA: ovalbumin; ZIF-8: zeolitic imidazolate framework; CAT: catalase; DOX: doxorubicin; MSNs: mesoporous silica nanoparticles; Pro: propranolol; CBP-12: 12-mer Clec9a binding peptide; 1G3-Cu: phosphorus dendrimer–copper(II) complexes; Toy: toyocamycin; MPDA: mesoporous polydopamine; EPI: epirubicin; Gox: glucose oxidase; DPPA-1: D-peptide antagonist; AFT: afatinib; 2-BP: 2-bromopalmitate; DLMSNs: dendritic large-pore mesoporous silica nanoparticles; AUNP12: anti-PD1 peptide; GM-CSF: granulocyte-macrophage colony-stimulating factor; LPS: lipopolysaccharide; ALD: alendronate; APAP: acetaminophen; TCPP: 4,4′,4′′,4′′′-(porphine5,10,15,20-tetrayl) tetrakis (benzoic acid); cRGD: cyclo (Arg-Gly-AspD-Tyr-Lys) peptide; AMNPs: allomelanin nanoparticles; IND: indoximod; RAPA: rapamycin; TMZ: temozolomide; Gem: gemcitabine; AuNCs: gold nanoclusters; UCNP: upconversion-nanoparticle; HA: hyaluronic acid; NS: nanosuspensions; PCN-224: porphyrin-based Zr-MOF; SPN: semiconducting polymer nanoparticles; Met: metformin; siFGL1: fibrinogen-like protein 1 siRNA. | ||||||
| Erythrocyte | PLGA | DSPE-PEG-Mannose inserted | hgp100 peptides, MPLA | Breast cancer | • Biomimetic vaccine: co-delivered antigen peptide and adjuvant to APCs | 185 |
| PLGA | DSPE-PEG-Mannose inserted | PLB, DIH, NH4HCO3 | Hepatoma | • Delivered PLB and DIH to induce ICD to reverse the immunosuppressive TME | 186 | |
| PLGA | DSPE-PEG-Mannose inserted | hgp peptides, MPLA | Melanoma | • Biomimetic vaccine: co-delivered antigen peptide and MPLA to APCs | 187 | |
| • Combined with DOX induced ICD | ||||||
| Mesoporous TiO2 | Hb on the NP surface | RRx-001 | Breast cancer | • Reactive nitrogen species (RNS) induced ICD | 188 | |
| BP | YSA anchored onto membrane | IL-1α siRNA, PTX | Colorectal cancer | • Deliver siRNA to block IL1 expression and restrict Treg cell accumulation | 160 | |
| BPQDs | Breast cancer | • PTT induced ICD and synergized with aPD-1 therapy | 189 | |||
| CP | PpIX inserted | Melanoma | • PDT induced ICD and CP induced reverse of immunosuppressive TME | 144 | ||
| Platelet | PLA | R848 | Colorectal cancer and breast cancer | • Delivered immunostimulatory R848 to TME | 128 | |
| Fe3O4 | SAS | Breast cancer | • Delivered SAS to induce ferroptosis and M1 macrophage polarization | 190 | ||
| DGL-G3 | Ce6, DTX | Melanoma | • PDT and delivery of DTX induced ICD | 166 | ||
| MOF | Lox, Oxa | Breast cancer | • Delivered Lox to consume lactate and Oxa to amplify ICD-induced immunotherapy | 191 | ||
| HMSNs | aPD-L1 coupled to membrane | SO | Hepatoma | • Delivered aPD-L1 and SO to CTCs effectively | 88 | |
| Cancer cell | ||||||
| MNs | SIRPα variant overexpressed on membrane | Melanoma and breast cancer | • Blocked CD47-SIRPα pathway and repolarized TAMs to M1-type to enhance macrophage immune response | 120 | ||
| B16F10 | PLGA | OVA and CD80 overexpressed on the membrane | Melanoma | • Delivered antigen and costimulatory signals to stimulate T cells directly | 192 | |
| PEI25k | CRT induced and CD47 knockout | CpG | Melanoma | • Biomimetic vaccine: co-delivered antigen and CpG to stimulate DCs and combined with ICIs | 193 | |
| PLGA | MPLA | Melanoma | • Biomimetic vaccine: co-delivered antigen and MPLA to APCs effectively | 148 | ||
| ZIF-8 | CAT, DOX | Melanoma | • Delivered CAT to relieve hypoxia in TME and enhance the aPD1 therapy | 159 | ||
| MSN | CpG, Pro | Melanoma | • Delivered Pro to block β-AR to reverse suppressive effects on T cells | 139 | ||
| • Delivered CpG to promote DC maturation | ||||||
| B16-OVA | OVA NPs | Ce6 | Melanoma | • PDT induced ICD | 145 | |
| PLGA | CBP-12 peptide inserted | 2′3′-cGAMP | Melanoma and breast cancer | • Delivered antigen and STING agonist to DCs to enhance IFN-stimulated expression of genes and antigen cross-presentation | 149 | |
| PLGA | DSPE-PEG-Mannose inserted | R837 | Melanoma | • Biomimetic vaccine: co-delivered antigen and R837 to stimulate DCs and combined with aPD1 therapy | 116 | |
| 1G3-Cu | Toy | Melanoma | • Delivered 1G3-Cu and Toy to induce ICD through amplifying ER stress and mitochondrial dysfunction and combined with aPD-L1 therapy | 194 | ||
| 4T1 | MPDA | R848 | Breast cancer | • Biomimetic vaccine: co-delivered antigen and R848 to activate APCs and combined with aPD-L1 therapy | 195 | |
| MONs | DOX | Breast cancer | • Delivered DOX to induce ICD and combined with aPD-L1 therapy | 153 | ||
| ZIF-8 | Calreticulin over-expressed on the membrane | EPI, Gox, hemin | Breast cancer | • Delivered EPI, Gox, and hemin to induce ICD and enhance ICI therapy | 157 | |
| PLGA | DPPA-1 anchored to the membrane | AFT, 2-BP | Breast cancer | • Delivered AFT and 2-BP to induce apoptosis and proliferation inhibition | 196 | |
| • Delivered DPPA-1 for PD-1/PD-L1 blockade | ||||||
| DLMSNs | Thiol functional groups; AUNP-12 | CuS, R848 | Breast cancer | • PTT induced ICD and R848 induced T lymphocyte activation | 147 | |
| DMSNs and Au NPs | Membrane mixed with PEOz liposome | R837 | Breast cancer | • PTT induced ICD | 197 | |
| • Delivered R837 to enhance immunotherapy | ||||||
| BPQDs | Loaded into thermosensitive hydrogel with GM-CSF and LPS | Melanoma and breast cancer | • PTT enhanced recruitment and migration of DCs and combined with aPD1 therapy | 141 | ||
| AML | PLGA | CpG | Acute myeloid leukemia | • Biomimetic vaccine: co-delivered AML-associated antigen and CpG to APCs effectively | 80 | |
| K7M2 | Hollow MnO2 | ALD | Ginsenosides Rh2 | Osteosarcoma | • Delivered ginsenosides Rh2 and Mn2+ to induce immuno-chemo-dynamic therapy | 198 |
| Panc02 | pH/oxidation nanogels | APAP | Pancreatic cancer | • Delivered APAP to enhance the activation of NK cells and NK cells-dependent DCs recruitment, combined with ICIs | 151 | |
| GL261 | CaCO3 | cRGD inserted | IL-12 mRNA | Glioma | • CO2 induced necroptosis of GBM cells under US irradiation | 162 |
| • Delivered IL-12 mRNA to stimulate the proliferation and activation of CTLs | ||||||
| AMNPs | CLP002 | Glioma | • PTT induced ICD | 199 | ||
| • Delivered CLP002 to amplify ICB therapy | ||||||
| Cu2−xSe | IND and JQ1 | Glioma | • PDT induced ICD | 170 | ||
| • Delivered IND, JQ1 to remodel immunosuppressive TME | ||||||
| LLC | Iron(II)-CpG | Cell membrane pre-treated with DOX to over-express ICD-related proteins | CpG | Lewis lung carcinoma | • Delivered ICD-related proteins and CpG to promote DC maturation | 200 |
| Dendritic cell | PLGA | DC membrane was pre-pulsed with CCM-coated NPs | Melanoma | • Cross-primed T cells directly for immunotherapy and combined with ICIs | 201 | |
| PLGA | DC membrane was preactivated to present tumor antigens | RAPA | Glioblastoma | • Delivered tumor antigen and RAPA to promote DC maturation | 100 | |
| DSPE-PEG | IR-797 | Breast cancer | • PTT induced ICD and enhanced T cell activation | 164 | ||
| NK cell | PLGA | TCPP | Breast cancer | • PDT induced ICD and M1 macrophage polarization | 202 | |
| PLGA | cRGD inserted | TMZ, IL-15 | Glioma | • Delivered TMZ and IL-15 to stimulate NK cell proliferation and activation | 101 | |
| Macrophage | PLGA and Fe3O4 | Lipopolysaccharide treated | R837 | Breast cancer | • Enhanced M1 phenotype polarization | 152 |
| PLGA | R837, DOX, lansoprazole | Breast cancer | • Delivered DOX to induce ICD and R837 to induce macrophage repolarization | 203 | ||
| PEG-PDPA | Gem | Breast cancer | • Delivered Gem to promote lymphocyte infiltration | 155 | ||
| AuNCs | DOX, Ce6, IND | Breast cancer and melanoma | • PDT and delivery of DOX and IND induced ICD and reversed immunosuppressive TME | 158 | ||
| UCNP | CSF1R overexpressed on the membrane and PMMA modified | RB | Breast cancer | • PDT induced ICD | 204 | |
| • Blocked CSF1–CSF1R axis | ||||||
| FFVLK-PEG | Ce6, PTX, IND | Breast cancer | • PDT and delivery of PTX induced ICD | 205 | ||
| • Delivered IND to induce inhibition of IDO pathway to reverse the immunosuppressive TME | ||||||
| SiO2 | DOX, R848, CAT | Hepatoma and colorectal cancer | • Delivered DOX and R848 to induce ICD and promote DC maturation | 206 | ||
| • Delivered Cat to alleviate A2AR pathway to reverse immunosuppressive TME | ||||||
| SiO2 | DSPE-PEG-Mannose inserted | PFC, Ce6, PTX | Breast cancer | • PDT induced ICD | 117 | |
| • Delivered PTX to induce the M2 to M1 reprogramming | ||||||
| HA-DOX | iRGD inserted and PEI condensed shRNA-Ptpn2 | shRNA, DOX | Melanoma | • Delivered DOX to induce ICD and shRNA for M1 macrophage polarization and DC maturation | 161 | |
| T cell | MSNs | CAR-T cell | IR780 | Hepatoma | • PTT induced ICD | 207 |
| BSA | PD-1 overexpressed on membrane | ORY-1001 | Breast cancer and colorectal cancer | • Delivered ORY-1001 to upregulate IFNs and downstream MHC I and PDL1 | 208 | |
| • Delivered PD-1 to block PD-L1 | ||||||
| Hybrid membrane | ||||||
| DC and cancer cell | PLGA | CpG-ODN | Colorectal cancer and glioma | • Delivered antigens, costimulatory signals, and CpG-ODN to T cells | 105 | |
| PLGA | CpG-ODN | Ovarian cancer | • Delivered antigens and CpG-ODN to stimulate T cells | 209 | ||
| NS | DTX | Glioblastoma | • Delivered antigen to stimulate T cell and DTX to induce apoptosis | 210 | ||
| PCN-224 | Breast cancer | • Delivered antigen and co-stimulating factors to DCs and T cells | 146 | |||
| • PDT induced ICD | ||||||
| SPN | Breast cancer | • Delivered antigen and T cell stimulating factors to DCs and T cells | 211 | |||
| • PTT induced ICD | ||||||
| RBC and cancer cell | Fe3O4 | ICG | Ovarian cancer | • PTT induced ICD | 143 | |
| Macrophage and cancer cell | PLGA | Met-CO2, siFGL1 | Breast cancer | • Delivered Met and siRNA to induce PD-L1 blockade and FGL1 gene silencing | 107 | |
Many types of CMNPs exhibit a multifaceted approach to tumor clearance, combining various immunotherapy techniques and even traditional cancer treatments like chemotherapy and radiotherapy. The following sections categorize CMNP applications based on key immunotherapeutic agents and their impact on anti-tumor immunotherapy.
6.1. Photoimmunotherapy based on photosensitizer delivery
Photoimmunotherapy involves the accumulation of photosensitizers at the tumor site, achieved through active or passive targeting strategies. By irradiating with specific light wavelengths, the phototherapeutic agent transitions from an inactive to an active state, inducing ICD in tumor cells and releasing DAMPs including CRTs, high-mobility group box 1 (HMGB1), and adenosine triphosphate, which trigger an enhanced anti-tumor immune response.212The effectiveness of photothermal therapy relies on selecting NPs with high photothermal conversion efficiency and facilitating their localized aggregation at the tumor site. To boost the efficacy of photoimmunotherapy, researchers employ strategies such as the application of BPQDs with photothermal properties. For instance, Liang et al. designed BPQD-RM, comprising BPQDs at its core and encapsulated with erythrocyte membranes (Fig. 9). BPQDs exhibit a high photothermal conversion efficiency of 28.4% and excellent loading capabilities. Additionally, the presence of erythrocyte membranes reduces in vivo degradation and prolongs circulation time, with a half-life of 23.91 ± 0.2 h. A 10 min exposure to 808 nm NIR irradiation during BPQD-RM treatment raised 4T1 tumor temperatures to 52.5 °C, resulting in PTT-induced tumor cell apoptosis, DCs aggregation, and significant infiltration of CD4+ and CD8+ T cells at the tumor sites. However, tumor cells upregulated PD-1 expression, depleting infiltrating T cells. To address this, the study employed a combination of BPQD-RMNVs, NIR, and aPD-1, which enhanced T cell function, showing promise in treating triple-negative breast cancer.189
 | ||
| Fig. 9 Cancer cell membrane coated BPQDs for cancer photothermal immunotherapy. (A) Schematic of synthesis of Gel-BPQD-CCNVs.189 (B) Schematic of mechanism for cancer immunotherapy. (C) TEM image of empty CCNV, single BPQD-CCN, and BPQD-CCNVs. (D) Confocal images indicating the membrane coated successfully. (E) IR thermographic maps and in vitro collective release of GM-CSF and BPQD-CCNVs. (F) Quantitative analysis of cells in draining lymph nodes; PBS (#1), Gel-BPQD-CCNVs + NIR (GM-CSF free, #2), Gel-BPQD-CCNVs (#3), Gel-BPQD-CCNVs + NIR (#4). (G) Tumor growth curves, weights of tumors, and survival curves; (#1) PBS, (#2) CCNV, (#3) a-PD1, (#4) Gel-BPQD-CCNVs, (#5) Gel-BPQD-CCNVs + NIR, (#6) Gel-BPQD-CCNVs + NIR + a-PD1. Copyright 2019 American Chemical Society. | ||
Efficient delivery of photothermal materials to the tumor site is crucial. To achieve this, researchers synthesized Gel-BPQD-CCNV by encapsulating BPQD with cancer cell membranes and loading them into a heat-sensitive hydrogel containing GM-CSF and LPS. This facilitated the sustained release of GM-CSF and LPS upon heating, effectively recruiting DCs to promote antigen cross-presentation. Similarly, this platform significantly enhanced CD8+ and CD4+ T cell infiltration and remarkably increased IFN-γ and TNF-α levels in a recurrent tumor model when combining PTT and aPD-1, offering a novel idea for treating residual and metastatic tumors following primary tumor resection.141
Moreover, small-molecule photosensitizers, similar to photothermal NPs, can be used for comprehensive photoimmunotherapy based on CMNPs. However, the method of enhancing photothermal effects by increasing the photothermal conversion rate may inadvertently harm immune cells while improving tumor ablation. In one study, DCs were activated in a controlled environment through co-cultivation with 4T1 tumor antigen and a TLR-3 agonist. These activated DCs exhibited a heightened expression of key co-stimulatory signals on membranes, notably MHC molecules, CD80, and CD86. Subsequently, an intelligent dendritic cell (iDC) was synthesized by encapsulating DC membrane with NPs carrying IR-797. The administered iDC effectively migrated to the lymph nodes, where it stimulated the proliferation of T cells and the secretion of pro-inflammatory cytokines, such as TNF-α, IL-2, and IFN-γ. This, in turn, led to the suppression of HSP family expression in cancer cells and diminished their ability to adapt to high temperatures. Under the increased sensitivity to heat stress, maintaining a temperature range of 42–45 °C through 808 nm laser induced ICD in 4T1 cells, further increasing the activation percentage of CD8+ and CD4+ T cells.164
Additionally, another study involved the fusion of ID8 cell and erythrocyte membranes at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, achieved through sonication at 37 °C for 10 min. This composite was then coated onto Fe3O4 NPs loaded with ICG to enhance its targeting ability toward ovarian cancer tissues and reduce RES phagocytosis. The resulting Fe3O4-ICG@IRM composite displayed tumor-inhibiting properties, even in the absence of NIR treatment. Upon applying NIR irradiation at 2.0 W cm−2 for 10 min, the tumor site's temperature rose significantly, reaching 58.7 °C. Sufficient PTT led to an increased proportion of CD4+ and CD8+ T cells, accompanied by a reduction in immunosuppressive Foxp3+ T cells.143
:1 ratio, achieved through sonication at 37 °C for 10 min. This composite was then coated onto Fe3O4 NPs loaded with ICG to enhance its targeting ability toward ovarian cancer tissues and reduce RES phagocytosis. The resulting Fe3O4-ICG@IRM composite displayed tumor-inhibiting properties, even in the absence of NIR treatment. Upon applying NIR irradiation at 2.0 W cm−2 for 10 min, the tumor site's temperature rose significantly, reaching 58.7 °C. Sufficient PTT led to an increased proportion of CD4+ and CD8+ T cells, accompanied by a reduction in immunosuppressive Foxp3+ T cells.143
In the context of enhancing PDT-mediated photoimmunotherapy, researchers have developed some multifunctional nanoplatforms such as PCN@FM. This platform exhibited targeted specificity for 4T1 tumors and inherited DC-like characteristics, including the ability to migrate to lymph nodes and express co-stimulatory molecules required for T cell activation. Under laser irradiation, PCN@FM triggered the release of ROS, inducing ICD. As a result, the PCN@FM group achieved enhanced activation of DCs and CTLs compared to other experimental groups.146
Since the mentioned strategy involves delivering antigens and stimulation signals to T cells, which can be challenging to prepare and apply, some studies have explored the use of cell membranes capable of modulating immune cells without prior sensitization to deliver photosensitizers. Deng et al. reported NK cell membrane-based biomimetic systems (NK-NPs) loaded with TCPP for PDT against 4T1 tumors. NK-NPs also triggered the polarization of M1-type macrophages through surface-bound proteins like IRGM1 and RAB 10.202
The cell membrane's phospholipid bilayer structure facilitates the integration of functional molecules, offering novel possibilities for photosensitizer applications. For instance, CP@mRBC-PpIX, embedded within the erythrocyte membrane, generated substantial ROS upon 630 nm laser irradiation, leading to pronounced cytotoxicity in just two min. Additionally, copper peroxide reacted with hydrogen ions to raise the pH at the tumor site, mitigating the acidic and hypoxic TME. The results demonstrated that CP@mRBC-PpIX enhanced the expression of STING, CRT, and HMGB1 proteins, promoted macrophage polarization toward the M1-type, and effectively recruited CD8+ T cells through the immunomodulatory effects of PDT and CP.144
6.2. Immunotherapy based on small molecular drug delivery
In recent years, CMNPs undoubtedly provide a new platform for enhancing biomimetic cancer vaccines by delivering specific tumor antigens on cell membranes to improve the body's immune response. The introduction of immune adjuvants further strengthens the function of immune cells such as DCs. Yang et al. created a melanoma tumor vaccine called NP-R@M-M using PLGA NPs loaded with the R837 and mannose-modified B16-OVA cell membranes. Similarly, this combination promoted DC maturation and induced CTL to control tumor progression and metastasis. Combining this approach with ICIs further enhanced its therapeutic potential.116
In addition to cancer vaccines comprising tumor antigens and adjuvants, various pharmaceuticals and NPs are available for direct modulation of immune cells within TME based on CMNPs. Deng and colleagues introduced a nanogel coated with Panc02 cell membranes (PCM@RNGs). Given the high expression of prostaglandin E2 (PGE2) in malignant tumors, which directly inhibits DC maturation and NK cell function, they incorporated the cyclooxygenase inhibitor APAP (PCM@APAP@RNG) to counteract PGE2's suppressive effects. The PCM@APAP@RNG promptly released APAP in the acidic, glutathione-rich TME, promoting NK cell activation. Activated NK cells produced IFN-γ to induce cancer cell apoptosis, released CCL5 and XCL1 to influence the migration of BMDCs, and in combination with aPD-L1 therapy, enhanced CD8+ T cell infiltration, significantly suppressing pancreatic tumor growth and postoperative tumor recurrence.151
Considering that TAMs can drive tumor invasion and metastasis by adopting an immunosuppressive M2-type, and due to the plasticity of TAMs under the influence of inflammatory factors, their polarization towards the M1-type has become a prominent immunotherapeutic strategy. A study introduced gene-edited NPs (gCM-MNs) where SIRPα variants overexpressed on the cell membrane exhibited a 50000-fold increase in affinity for CD47, substantially enhancing macrophage uptake of gCM-MNs through competitive binding. Meanwhile, ROS generated by MNs via the Fenton reaction induced TAM repolarization towards an anti-tumor M1-type. The results demonstrated an increase in CD8+ T cell infiltration at the tumor site, accompanied by elevated secretion of IFN-γ and TNF-α, resulting in a noteworthy inhibitory effect on melanoma.120
Similarly, HMnO2-MSC-TAT@PTX further optimized the delivery of lung cancer-associated chemotherapeutic agents by facilitating the accumulation of Mn2+ and PTX in tumor cell nucleus via TAT peptides (Fig. 10). This led to enhanced PTX-mediated DNA damage, coupled with Mn2+-mediated activation of cyclic adenosine monophosphate synthase (cCAS) and STING, promoting DC maturation and T cell infiltration for a synergistic therapeutic effect on non-small cell lung cancer.154 Additionally, studies also explored the combined effects of chemotherapeutic agents with photosensitizers and nucleic acid drugs.167
 | ||
| Fig. 10 Mesenchymal stem cell membrane coated MnO2 delivered PTX for nucleus-targeted combination cancer therapy.154 (A) Schematic of synthesis of HMnO2-MSC-TAT NPs and schematic illustration of chemo-immunotherapy. (B) CLSM images of A549 cells incubated with different NPs. (C) Amount of cellular and nuclear uptake of NPs. (D) In vivo imaging of A549 tumor-bearing mice. (E) Amount of released HMGB1. (F) LLC tumor growth curves in mice. (G) Percentages of surviving mice treated with different formulations. Copyright 2023 American Chemical Society. | ||
Another study aimed to reverse the immunosuppressive effects of adenosine accumulation in TME, often caused by hypoxia. Adenosine binds to the G-protein-coupled A2A receptor (A2AR) and suppresses immune cell function, including the recruitment of Tregs. To address this, Wen et al. co-encapsulated Cat, Dox, and R848 in mesoporous silica (Cat@SiO2-M) to promote the uptake of NPs by hepatocellular carcinoma cells through macrophage membrane encapsulation (Fig. 11). The encapsulated Cat converted H2O2 into oxygen, relieving hypoxia and alleviating immunosuppression associated with the adenosine-A2AR pathway. Additionally, the ICD and DC maturation effects of DOX and R848 reduced the population of Tregs by 90.21% while increasing CD8+ T cells, effectively counteracting immune cell dysfunction caused by adenosine accumulation.206
 | ||
| Fig. 11 Macrophage membrane coated mesoporous silica nanoplatform inhibiting adenosine A2AR via in situ oxygen supply for immunotherapy.206 (A) Schematic of synthesis of D/R/C@SiO2-M. (B) The mechanism of D/R/C@SiO2-M mediated immunotherapy. (C) Fluorescence images and the MFI of tumor tissue. (D) In vivo bioluminescence images of tumor-bearing mice and in vitro tissues. (E) Tumor growth curves and percentages of surviving mice treated with different formulations. (F) Western blot analysis and average gray value in mice tumor tissue 2 days after the last treatment. Copyright 2023 Elsevier. | ||
6.3. Immunotherapy based on nucleic acid delivery
The inherent stability of nucleic acid drugs necessitates innovative delivery systems, such as CMNPs, widely employed in studies for enhancing cancer immunotherapy through nucleic acid-based drug delivery. In a study, Ou et al. utilized poly-L-histidine (H)-grafted BP to carry PTX and IL-1α siRNA (ILsi) for combined cancer therapy (Fig. 12). The BP-H-ILsi-X@EM-YSA system, synthesizing an erythrocyte membrane shell embedded with Ephrin-A2 receptor-specific peptide (YSA), selectively targeted MC38 tumor tissues. Upon NIR exposure, ROS production degraded the erythrocyte membrane shell, releasing ILsi and PTX into the cytoplasm. ILsi notably silenced IL-1α expression, reducing CCL22 secretion, hindering Treg migration to the tumor site, and in combination with PTX, enhancing CD8+ T cell infiltration.160 | ||
| Fig. 12 Erythrocyte membrane coated NPs delivered IL-1α siRNA and paclitaxel for targeted and combination cancer therapy.160 (A) Schematic of synthesis of BP-H-ILsi-X@EM-YSA and schematic illustration of immunotherapy. (B) Confocal images and flow cytometry data of cellular uptake levels. (C) Confocal analysis of endosomal escape of ILsi from MC-38 cells. (D) ELISA analysis of IL levels in MC-38 tumors. (E) ELISA analysis of intratumor CCL22 levels. (F) Flow cytometry analysis of the percentages of DCs and T cells and ratio between CD8+ and Treg in the treated tumor. Copyright 2019 Theranostics. | ||
In another study, nanopolymers encapsulated in M1 macrophage membranes were used to deliver shRNA-Ptpn2 with DOX. Polarized M1 macrophage membranes facilitated DOX accumulation and targeted intranuclear delivery of shRNA, significantly down-regulating Ptpn2 mRNA expression, and enhancing T cell responsiveness to IFN-γ. The combination with DOX increased CD8+ T cell ratios in the TME, augmenting the immunotherapeutic impact on melanoma.161
6.4. Immunotherapy based on antibody delivery
ICIs, following extensive research, have been successfully integrated into clinical practice and demonstrate remarkable tumor suppression capabilities. With the introduction of CMNPs in cancer immunotherapy, ICIs are frequently combined with CMNPs loaded with immunotherapeutic agents. Additionally, ICIs can also realize more effective therapeutic outcomes through CMNPs.For instance, in the context of treating intracerebral glioblastoma, one study employed CLP002, a low molecular weight PD-L1 inhibitor, loaded into allomelanin NPs (AMNPs) – a melanin analogue with photothermal properties (Fig. 13). AMNPs were encapsulated with GL261 membranes (AMNP@CLP@CCM) to enhance BBB and targeting precision. Subsequent laser irradiation at 808 nm and 0.5 W cm−2 allowed for effective photothermal therapy in glioblastoma tissues. The combined action of in situ PTT and ICIs led to DC maturation, CD8+ T cell infiltration, and long-term immune memory T cell generation, achieving therapeutic effects against intracranial tumors.199
 | ||
| Fig. 13 Cancer cell membrane coated AMNPs delivered ICIs for targeted photothermal immunotherapy.199 (A) Schematic of synthesis and therapeutic mechanism of AMNP@CLP@CCM for immunotherapy. (B) Flow histogram and corresponding quantification analysis of the blocking effect of PD-L1 on the surface of GL261 cells. (C) Representative fluorescence images of frozen brain sections. (D) Bio-TME images of the tumor tissue after being administrated for 12 h. (E) In vivo infrared thermal imaging in orthotopic GBM-bearing mice. (F) Flow cytometric quantification of activated CTL from the tumor. (G) Immunofluorescence images of CD8+ CD44+ T cells in glioma tumor sections. Copyright 2023 Elsevier. | ||
The non-specific distribution of ICIs via injection contributes to their limited efficacy and adverse effects. Alongside CMNP-mediated targeted delivery, research has harnessed MMP2 found in TME to enhance the precision of ICI therapy. For instance, BTO was encapsulated in genetically engineered membranes with aPD-L1 overexpressed. The helical peptides on aPD-L1 were specifically cleaved by MMP2 to realize specific anti-PD-L1 therapy. The synergistic effects of BTO and ROS-induced ICD not only boosted intracellular oxygen levels but also enhanced the aggregation of immune-suppressive cells in the hypoxic TME, facilitating the transformation of “cold” tumors into “hot” tumors.121
Various strategies have also been devised for ICI delivery through CMNPs, including lipid nanoparticles loaded with RNA editing enzyme ADAR1 siRNA (siAdar) enclosed by PD1 overexpressed membranes. This approach achieved immune checkpoint inhibition and promoted interferon production via gene silencing.213 As CMNP technology advances, new avenues for effectively regulating immune checkpoint axis will continue to emerge.
6.5. Combination therapy
To enhance therapeutic outcomes and overcome limitations in immunotherapy, immunotherapeutic approaches based on CMNPs are typically combined with other modalities in oncology. For instance, ICIs, as a matured immunotherapeutic, often administered systemically, are used in conjunction with CMNP-mediated photoimmunotherapy or cancer nanovaccines.189,193,195 This combined therapeutic approach has the potential for comprehensive immune system activation, displaying promising clinical translational prospects. Surgical intervention remains an effective treatment for many early-stage solid tumors. However, residual lesions or metastasized tumor cells significantly impact surgical efficacy. Studies have explored utilizing extracted cell membranes from excised tumor tissues to encapsulate NPs loaded with immune adjuvants, compensating for surgical limitations.141 Moreover, traditional systemic radiotherapy and chemotherapy can synergize or amplify CMNP-mediated immunotherapy by directly killing tumor cells.214,215 Ongoing research and advancement of these combinational therapeutic strategies hold the promise of offering more effective treatment options for cancer patients.7. Conclusions and future perspectives
This review highlights the application of different CMNPs in cancer immunotherapy. Recent advances in cancer therapy have led to the increasing popularity of immunotherapy, which activates the immune system to combat cancer cells. However, the effectiveness of immunotherapy varies depending on factors like cancer cell resistance. The introduction of new nanotechnology in cancer treatment has opened up avenues for enhancing cancer immunotherapy depending on its excellent drug delivery capacity or photothermal property. The arrival of biomimetic nano-platforms allows a wider application of NPs in cancer immunotherapy. Due to the inheritance of unique characteristics of both cell membranes and NPs, CMNPs enable prolonged circulation in the bloodstream, precise drug delivery, and interaction with components in TME, making them a superior platform for immunotherapy. Consequently, CMNPs can enhance PTT or PDT by delivering photosensitizers to tumor tissues. They can also stimulate the innate and adaptive immune system against tumors and reverse the immunosuppressive TME by delivering immunomodulatory agents.While CMNPs show promise in cancer immunotherapy and demonstrate significant anti-tumor effectiveness in studies, their clinical application faces numerous challenges and limitations. The synthesis process of CMNPs is critical for their anti-tumor effectiveness. Extracting intact and functional cell membranes poses a pressing issue, as the loss and inactivation of proteins during membrane extraction compromise CMNPs’ therapeutic effectiveness.216 Heterogeneity in cell membranes expanded in vitro can trigger inflammatory responses, necessitating the isolation of blood cells or autologous tumor cells, increasing operational complexity. Current purification methods, mainly using differential centrifugation, can lead to immune reactions and self-damage due to the retention of complex components like nucleic acids and cytoplasm, potentially causing cell carcinogenesis. Improving the cell membrane extraction process is essential to maintain CMNPs’ quality. Furthermore, the challenge of achieving mass production hinders the clinical adoption of CMNPs. The current methods, such as physical extrusion and ultrasonication, suffer from inefficiency, complexity, and uneven coating. Although production methods are improving, they are still in the early stages of research, posing challenges for scaling up to clinical needs.217
As a novel anti-tumor strategy, CMNPs must address biosafety concerns to determine their clinical viability. The stability of the membrane coating impacts in vivo metabolism and is critical for achieving favorable pharmacokinetics in the case of CMNPs. The evolving field of NPs provides diverse core options for CMNPs, raising considerations about potential immune responses, organism toxicity, degradation kinetics, and metabolite toxicity associated with NPs. Moreover, the intricate protein composition on CMNPs’ surfaces can lead to hypersensitivity reactions in allergic individuals or adverse interactions with normal cells.
In current research, various cell membranes, including organelle membranes, have been explored to emulate the biomimetic function of CMNPs. Future investigations into novel cell membrane types, like B cell membranes, hold promise for improving CMNPs. Targeted drug delivery is a primary focus in CMNP research. While existing studies mention the use of different cell membrane coatings for drug targeting toward tumor or immune cells, most approaches are essentially “passive” targeting, lacking specificity. Engineered cell membranes have enabled precise drug delivery to specific components in TME, such as targeting macrophages with mannose or crossing BBB for homologous targeting of brain tumor cells through cRGD modification.162 Genetically engineered cell membranes are emerging as a promising strategy for active targeting in cancer therapy. For instance, NPs enclosed within the membranes of T cells with PD-1 overexpressed allow for specific interactions with tumor cells exhibiting high levels of PD-L1.208 In general, engineered CMNPs represent a burgeoning area of interest for active targeting and enhancing the efficacy of cancer immunotherapy.
The amalgamation of biofilm functions and nanoparticle properties in CMNPs presents significant potential in cancer therapy. Efforts in future research should focus on developing bionanosystems with enhanced multifunctionality and precise targeting capabilities, while prioritizing biosafety. This will enable the full utilization of their unique advantages in clinical cancer immunotherapy.
Abbreviations
| CMNPs | Cell membrane coated nanoparticles |
| ICB | Immune checkpoint blockade |
| TME | Tumor microenvironment |
| NPs | Nanoparticles |
| RES | Reticuloendothelial system |
| PEG | Polyethylene glycol |
| DC | Dendritic cell |
| APC | Antigen-presenting cell |
| CTL | Cytotoxic T cell |
| TAA | Tumor-associated antigen |
| ICI | Immune checkpoint inhibitor |
| IFN | Interferon |
| TNF | Tumor necrosis factor |
| PD-1 | Programmed cell death 1 |
| PD-L1 | Programmed cell death 1 ligand 1 |
| CTLA-4 | Cytotoxic T lymphocyte-associated antigen-4 |
| Treg | Regulatory T cell |
| CAF | Cancer associated fibroblast |
| ICD | Immunogenic cell death |
| NIR | Near-infrared |
| ROS | Reactive oxygen species |
| CAR-T | Chimeric antigen receptor T-cell immunotherapy |
| PLGA | Poly(lactic-co-glycolic acid) |
| SIRPα | Signal-regulated protein alpha |
| NK cell | Natural killer cell |
| CRT | Calreticulin |
| PTT | Photothermal therapy |
| DAMPs | Damage-associated molecular patterns |
| BP | Black phosphorus |
| PDA | Polydopamine |
| PDT | Photodynamic therapy |
| PpIX | Protoporphyrin IX |
| MOF | Metal organic framework |
| Ce6 | Chlorin e6 |
| TLR | Toll-like receptor |
| STING | Interferon gene-stimulating factor |
| BBB | Blood–brain barrier |
| MNs | Magnetic nanoparticles |
| DOX | Doxorubicin |
| MONs | Mesoporous organosilica nanoparticles |
| PTX | Paclitaxel |
| ZIF | Zeolitic imidazolate framework |
| GSH | Glutathione |
| IDO | Indoleamine 2,3-dioxygenase |
| MDSC | Myeloid-derived suppressor cells |
| siFGL1 | Fibrinogen-like protein 1 siRNA |
| shRNA | Short hairpin RNA |
| MMP | Matrix metalloproteinases |
| EV | Extracellular vesicle |
| HMGB1 | High-mobility group box 1 |
Author contributions
Zichen Zhong and Wen Deng wrote the original draft. Jian Wu, Haojie Shang and Yonghua Tong completed the recombination and arrangement of figures. Yu He, Qiu Huang and Xiaozhuo Ba collected information and organized materials. Zhiqiang Chen and Kun Tang came up with the overall structure of the manuscript. All authors reviewed and edited this manuscript.Data availability
No data was used in the research and all related researches were referenced.Conflicts of interest
There exist no conflicts in the review.Acknowledgements
Our research was funded by the National Natural Science Foundation of China (No. 82170779, 82270804), the Natural Science Foundation of Hubei Province (No. 2023AFB867), Young Elite Scientists Sponsorship Program by CAST (No. YESS20220646), the Natural Science Foundation of Hainan Province (No. 823MS177) and 2019 Wuhan Yellow Crane Talent Program (Outstanding Young Talents).References
- Y. Zhang and Z. Zhang, Cell. Mol. Immunol., 2020, 17, 807–821 CrossRef CAS PubMed.
- J. Isaacs and T. E. Stinchcombe, Drugs, 2022, 82, 855–863 CrossRef CAS PubMed.
- A. Arina, S. I. Gutiontov and R. R. Weichselbaum, Clin. Cancer Res., 2020, 26, 2777–2782 CrossRef CAS PubMed.
- C. Pan, H. Liu, E. Robins, W. Song, D. Liu, Z. Li and L. Zheng, J. Hematol. Oncol., 2020, 13, 29 CrossRef PubMed.
- J. S. O'Donnell, M. W. L. Teng and M. J. Smyth, Nat. Rev. Clin Oncol., 2019, 16, 151–167 CrossRef PubMed.
- Y. Zheng, S. Li, H. Tang, X. Meng and Q. Zheng, Front. Immunol., 2023, 14, 1153990 CrossRef CAS PubMed.
- M. Dougan, A. M. Luoma, S. K. Dougan and K. W. Wucherpfennig, Cell, 2021, 184, 1575–1588 CrossRef CAS PubMed.
- A. Finck, S. I. Gill and C. H. June, Nat. Commun., 2020, 11, 3325 CrossRef CAS PubMed.
- N. Gong, N. C. Sheppard, M. M. Billingsley, C. H. June and M. J. Mitchell, Nat. Nanotechnol., 2021, 16, 25–36 CrossRef CAS PubMed.
- V. Gowd, A. Ahmad, M. Tarique, M. Suhail, T. A. Zughaibi, S. Tabrez and R. Khan, Semin. Cancer Biol., 2022, 86, 624–644 CrossRef CAS PubMed.
- E. Pavitra, B. Dariya, G. Srivani, S. M. Kang, A. Alam, P. R. Sudhir, M. A. Kamal, G. S. R. Raju, Y. K. Han, B. Lakkakula, G. P. Nagaraju and Y. S. Huh, Semin. Cancer Biol., 2021, 69, 293–306 CrossRef CAS PubMed.
- B. Ji, M. Wei and B. Yang, Theranostics, 2022, 12, 434–458 CrossRef CAS PubMed.
- R. A. Revia, Z. R. Stephen and M. Zhang, Acc. Chem. Res., 2019, 52, 1496–1506 CrossRef CAS PubMed.
- Y. Shi, R. van der Meel, X. Chen and T. Lammers, Theranostics, 2020, 10, 7921–7924 CrossRef PubMed.
- C. Xu, C. Lei, Y. Wang and C. Yu, Angew. Chem., Int. Ed., 2022, 61, e202112752 CrossRef CAS PubMed.
- M. Yu and J. Zheng, ACS Nano, 2015, 9, 6655–6674 CrossRef CAS PubMed.
- Y. N. Zhang, W. Poon, A. J. Tavares, I. D. McGilvray and W. C. W. Chan, J. Controlled Release, 2016, 240, 332–348 CrossRef CAS PubMed.
- J. S. Suk, Q. Xu, N. Kim, J. Hanes and L. M. Ensign, Adv. Drug Delivery Rev., 2016, 99, 28–51 CrossRef CAS PubMed.
- F. Wang, M. Hu, N. Li, X. Sun, G. Xing, G. Zheng, Q. Jin, Y. Liu, C. Cui and G. Zhang, ACS Appl. Mater. Interfaces, 2022, 14, 39843–39857 CrossRef CAS PubMed.
- W. Qin, J. Chandra, M. A. S. Abourehab, N. Gupta, Z. S. Chen, P. Kesharwani and H. L. Cao, Mol. Cancer, 2023, 22, 87 CrossRef CAS PubMed.
- M. J. Mitchell, M. M. Billingsley, R. M. Haley, M. E. Wechsler, N. A. Peppas and R. Langer, Nat. Rev. Drug Discovery, 2021, 20, 101–124 CrossRef CAS PubMed.
- M. E. Aikins, C. Xu and J. J. Moon, Acc. Chem. Res., 2020, 53, 2094–2105 CrossRef CAS PubMed.
- C. M. Hu, L. Zhang, S. Aryal, C. Cheung, R. H. Fang and L. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 10980–10985 CrossRef CAS PubMed.
- R. H. Fang, W. Gao and L. Zhang, Nat. Rev. Clin Oncol., 2023, 20, 33–48 CrossRef PubMed.
- R. H. Fang, A. V. Kroll, W. Gao and L. Zhang, Adv. Mater., 2018, 30, e1706759 CrossRef PubMed.
- M. Li, H. Fang, Q. Liu, Y. Gai, L. Yuan, S. Wang, H. Li, Y. Hou, M. Gao and X. Lan, Biomater. Sci., 2020, 8, 1802–1814 RSC.
- Z. Wang, M. Zhang, S. Chi, M. Zhu, C. Wang and Z. Liu, Adv. Healthc. Mater., 2022, 11, e2200521 CrossRef PubMed.
- X. Wang, Z. Xia, H. Wang, D. Wang, T. Sun, E. Hossain, X. Pang and Y. Liu, Theranostics, 2023, 13, 3224–3244 CrossRef CAS PubMed.
- J. Ma, L. Jiang and G. Liu, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2022, 14, e1825 CAS.
- J. H. Park, Y. Jiang, J. Zhou, H. Gong, A. Mohapatra, J. Heo, W. Gao, R. H. Fang and L. Zhang, Sci. Adv., 2021, 7, eabf7820 CrossRef CAS PubMed.
- Q. Zhang, D. Dehaini, Y. Zhang, J. Zhou, X. Chen, L. Zhang, R. H. Fang, W. Gao and L. Zhang, Nat. Nanotechnol., 2018, 13, 1182–1190 CrossRef CAS PubMed.
- C. Gao, Q. Huang, C. Liu, C. H. T. Kwong, L. Yue, J. B. Wan, S. M. Y. Lee and R. Wang, Nat. Commun., 2020, 11, 2622 CrossRef CAS PubMed.
- Y. Miao, Y. Yang, L. Guo, M. Chen, X. Zhou, Y. Zhao, D. Nie, Y. Gan and X. Zhang, ACS Nano, 2022, 16, 6527–6540 CrossRef CAS PubMed.
- D. S. Chen and I. Mellman, Immunity, 2013, 39, 1–10 CrossRef CAS PubMed.
- Y. Zhu, X. An, X. Zhang, Y. Qiao, T. Zheng and X. Li, Mol. Cancer, 2019, 18, 152 CrossRef PubMed.
- R. Pio, D. Ajona, S. Ortiz-Espinosa, A. Mantovani and J. D. Lambris, Front. Immunol., 2019, 10, 774 CrossRef CAS PubMed.
- S. Zuo, J. Song, J. Zhang, Z. He, B. Sun and J. Sun, Theranostics, 2021, 11, 7471–7487 CrossRef CAS PubMed.
- X. Fan, K. Wang, Q. Lu, Y. Lu and J. Sun, Small, 2023, 19, e2205166 CrossRef PubMed.
- S. Bagchi, R. Yuan and E. G. Engleman, Annu. Rev. Pathol., 2021, 16, 223–249 CrossRef CAS PubMed.
- G. Chen, A. C. Huang, W. Zhang, G. Zhang, M. Wu, W. Xu, Z. Yu, J. Yang, B. Wang, H. Sun, H. Xia, Q. Man, W. Zhong, L. F. Antelo, B. Wu, X. Xiong, X. Liu, L. Guan, T. Li, S. Liu, R. Yang, Y. Lu, L. Dong, S. McGettigan, R. Somasundaram, R. Radhakrishnan, G. Mills, Y. Lu, J. Kim, Y. H. Chen, H. Dong, Y. Zhao, G. C. Karakousis, T. C. Mitchell, L. M. Schuchter, M. Herlyn, E. J. Wherry, X. Xu and W. Guo, Nature, 2018, 560, 382–386 CrossRef CAS PubMed.
- M. S. Carlino, J. Larkin and G. V. Long, Lancet, 2021, 398, 1002–1014 CrossRef CAS PubMed.
- A. Lahiri, A. Maji, P. D. Potdar, N. Singh, P. Parikh, B. Bisht, A. Mukherjee and M. K. Paul, Mol. Cancer, 2023, 22, 40 CrossRef PubMed.
- M. Reda, W. Ngamcherdtrakul, M. A. Nelson, N. Siriwon, R. Wang, H. Y. Zaidan, D. S. Bejan, S. Reda, N. H. Hoang, N. A. Crumrine, J. P. C. Rehwaldt, A. Bindal, G. B. Mills, J. W. Gray and W. Yantasee, Nat. Commun., 2022, 13, 4261 CrossRef CAS PubMed.
- L. Au, E. Hatipoglu, M. Robert de Massy, K. Litchfield, G. Beattie, A. Rowan, D. Schnidrig, R. Thompson, F. Byrne, S. Horswell, N. Fotiadis, S. Hazell, D. Nicol, S. T. C. Shepherd, A. Fendler, R. Mason, L. Del Rosario, K. Edmonds, K. Lingard, S. Sarker, M. Mangwende, E. Carlyle, J. Attig, K. Joshi, I. Uddin, P. D. Becker, M. W. Sunderland, A. Akarca, I. Puccio, W. W. Yang, T. Lund, K. Dhillon, M. D. Vasquez, E. Ghorani, H. Xu, C. Spencer, J. I. Lopez, A. Green, U. Mahadeva, E. Borg, M. Mitchison, D. A. Moore, I. Proctor, M. Falzon, L. Pickering, A. J. S. Furness, J. L. Reading, R. Salgado, T. Marafioti, M. Jamal-Hanjani, P. Consortium, G. Kassiotis, B. Chain, J. Larkin, C. Swanton, S. A. Quezada, S. Turajlic and T. R. R. Consortium, Cancer Cell, 2021, 39, 1497–1518 CrossRef CAS PubMed.
- J. Lu and G. Jiang, Mol. Cancer, 2022, 21, 194 CrossRef PubMed.
- Y. H. Yang, J. W. Liu, C. Lu and J. F. Wei, Int. J. Biol. Sci., 2022, 18, 2609–2626 CrossRef CAS PubMed.
- U. Sahin and O. Tureci, Science, 2018, 359, 1355–1360 CrossRef CAS PubMed.
- A. Harari, M. Graciotti, M. Bassani-Sternberg and L. E. Kandalaft, Nat. Rev. Drug Discovery, 2020, 19, 635–652 CrossRef CAS PubMed.
- Z. Liu, J. Lv, Q. Dang, L. Liu, S. Weng, L. Wang, Z. Zhou, Y. Kong, H. Li, Y. Han and X. Han, Int. J. Biol. Sci., 2022, 18, 5607–5623 CrossRef CAS PubMed.
- D. J. Propper and F. R. Balkwill, Nat. Rev. Clin Oncol., 2022, 19, 237–253 CrossRef CAS PubMed.
- R. Hernandez, J. Poder, K. M. LaPorte and T. R. Malek, Nat. Rev. Immunol., 2022, 22, 614–628 CrossRef CAS PubMed.
- S. Nakao, Y. Arai, M. Tasaki, M. Yamashita, R. Murakami, T. Kawase, N. Amino, M. Nakatake, H. Kurosaki, M. Mori, M. Takeuchi and T. Nakamura, Sci. Transl. Med., 2020, 12, eaax7992 CrossRef CAS PubMed.
- C. S. Garris, S. P. Arlauckas, R. H. Kohler, M. P. Trefny, S. Garren, C. Piot, C. Engblom, C. Pfirschke, M. Siwicki, J. Gungabeesoon, G. J. Freeman, S. E. Warren, S. Ong, E. Browning, C. G. Twitty, R. H. Pierce, M. H. Le, A. P. Algazi, A. I. Daud, S. I. Pai, A. Zippelius, R. Weissleder and M. J. Pittet, Immunity, 2018, 49, 1148–1161 CrossRef CAS PubMed.
- S. Ma, M. A. Caligiuri and J. Yu, Trends Immunol., 2022, 43, 833–847 CrossRef CAS PubMed.
- M. Jiang, P. Chen, L. Wang, W. Li, B. Chen, Y. Liu, H. Wang, S. Zhao, L. Ye, Y. He and C. Zhou, J. Hematol. Oncol., 2020, 13, 81 CrossRef CAS PubMed.
- M. D. Vesely, T. Zhang and L. Chen, Annu. Rev. Immunol., 2022, 40, 45–74 CrossRef CAS PubMed.
- A. Kalbasi and A. Ribas, Nat. Rev. Immunol., 2020, 20, 25–39 CrossRef CAS PubMed.
- C. Wang, Y. Zhang, K. Guo, N. Wang, H. Jin, Y. Liu and W. Qin, Int. J. Cancer, 2016, 138, 1824–1834 CrossRef CAS PubMed.
- A. Goenka, F. Khan, B. Verma, P. Sinha, C. C. Dmello, M. P. Jogalekar, P. Gangadaran and B. C. Ahn, Cancer Commun., 2023, 43, 525–561 CrossRef PubMed.
- T. N. Schumacher and R. D. Schreiber, Science, 2015, 348, 69–74 CrossRef CAS PubMed.
- R. Saleh and E. Elkord, Semin. Cancer Biol., 2020, 65, 13–27 CrossRef CAS PubMed.
- G. Morad, B. A. Helmink, P. Sharma and J. A. Wargo, Cell, 2022, 185, 576 CrossRef CAS PubMed.
- A. S. Bear, R. H. Vonderheide and M. H. O'Hara, Cancer Cell, 2020, 38, 788–802 CrossRef CAS PubMed.
- S. Baxi, A. Yang, R. L. Gennarelli, N. Khan, Z. Wang, L. Boyce and D. Korenstein, Br. Med. J., 2018, 360, k793 CrossRef PubMed.
- M. Ramos-Casals, A. Flores-Chavez, P. Brito-Zeron, O. Lambotte and X. Mariette, Pharmacol. Ther., 2022, 237, 108250 CrossRef CAS PubMed.
- Z. Xu, S. Ramishetti, Y. C. Tseng, S. Guo, Y. Wang and L. Huang, J. Controlled Release, 2013, 172, 259–265 CrossRef CAS PubMed.
- L. M. Kranz, M. Diken, H. Haas, S. Kreiter, C. Loquai, K. C. Reuter, M. Meng, D. Fritz, F. Vascotto, H. Hefesha, C. Grunwitz, M. Vormehr, Y. Husemann, A. Selmi, A. N. Kuhn, J. Buck, E. Derhovanessian, R. Rae, S. Attig, J. Diekmann, R. A. Jabulowsky, S. Heesch, J. Hassel, P. Langguth, S. Grabbe, C. Huber, O. Tureci and U. Sahin, Nature, 2016, 534, 396–401 CrossRef PubMed.
- W. Song, J. J. Hu, S. J. Song, Y. Xu, H. Yang, F. Yang, Y. Zhou, T. Yu and W. X. Qiu, ACS Appl. Mater. Interfaces, 2022, 14, 42931–42939 CrossRef CAS PubMed.
- S. Niu, X. Zhang, G. R. Williams, J. Wu, F. Gao, Z. Fu, X. Chen, S. Lu and L. M. Zhu, Acta Biomater., 2021, 126, 408–420 CrossRef CAS PubMed.
- Y. Wang, K. C. Black, H. Luehmann, W. Li, Y. Zhang, X. Cai, D. Wan, S. Y. Liu, M. Li, P. Kim, Z. Y. Li, L. V. Wang, Y. Liu and Y. Xia, ACS Nano, 2013, 7, 2068–2077 CrossRef CAS PubMed.
- B. Ding, S. Shao, C. Yu, B. Teng, M. Wang, Z. Cheng, K. L. Wong, P. Ma and J. Lin, Adv. Mater., 2018, 30, e1802479 CrossRef PubMed.
- S. Zanganeh, G. Hutter, R. Spitler, O. Lenkov, M. Mahmoudi, A. Shaw, J. S. Pajarinen, H. Nejadnik, S. Goodman, M. Moseley, L. M. Coussens and H. E. Daldrup-Link, Nat. Nanotechnol., 2016, 11, 986–994 CrossRef CAS PubMed.
- S. Du, H. Xiong, C. Xu, Y. Lu and J. Yao, Biomater. Sci., 2019, 7, 1147–1160 RSC.
- Y. Luo, H. Yang, Y. F. Zhou and B. Hu, J. Controlled Release, 2020, 317, 195–215 CrossRef CAS PubMed.
- Z. Mirza and S. Karim, Semin. Cancer Biol., 2021, 69, 226–237 CrossRef CAS PubMed.
- M. Sushnitha, M. Evangelopoulos, E. Tasciotti and F. Taraballi, Front. Bioeng. Biotechnol., 2020, 8, 627 CrossRef PubMed.
- N. Krishnan, Y. Jiang, J. Zhou, A. Mohapatra, F. X. Peng, Y. Duan, M. Holay, S. Chekuri, Z. Guo, W. Gao, R. H. Fang and L. Zhang, Nat. Nanotechnol., 2024, 19, 345–353 CrossRef CAS PubMed.
- H. Liu, Y. Y. Su, X. C. Jiang and J. Q. Gao, Drug Delivery Transl. Res., 2023, 13, 716–737 CrossRef CAS PubMed.
- S. Yaman, H. Ramachandramoorthy, P. Iyer, U. Chintapula, T. Nguyen, M. Sabnani, T. Kotadia, S. Ghaffari, L. M. Pop, R. Hannan, J. A. Weidanz and K. T. Nguyen, Bioact. Mater., 2024, 34, 422–435 CAS.
- D. T. Johnson, J. Zhou, A. V. Kroll, R. H. Fang, M. Yan, C. Xiao, X. Chen, J. Kline, L. Zhang and D. E. Zhang, Leukemia, 2022, 36, 994–1005 CrossRef CAS PubMed.
- H. Wu, T. Zhang, N. Li and J. Gao, J. Controlled Release, 2023, 360, 169–184 CrossRef CAS PubMed.
- S. Li, X. Meng, B. Peng, J. Huang, J. Liu, H. Xiao, L. Ma, Y. Liu and J. Tang, Acta Biomater., 2024, 174, 26–48 CrossRef CAS PubMed.
- S. Han, W. Wang, S. Wang, S. Wang, R. Ju, Z. Pan, T. Yang, G. Zhang, H. Wang and L. Wang, Nanoscale, 2019, 11, 20206–20220 RSC.
- A. A. Sousa-Junior, F. Mello-Andrade, J. V. R. Rocha, T. G. Hayasaki, J. S. de Curcio, L. D. C. Silva, R. C. de Santana, E. Martins Lima, C. G. Cardoso, E. P. Silveira-Lacerda, S. A. Mendanha and A. F. Bakuzis, Pharmaceutics, 2023, 15, 943 CrossRef CAS PubMed.
- M. Koupenova, A. C. Livada and C. N. Morrell, Circ. Res., 2022, 130, 288–308 CrossRef CAS PubMed.
- O. Eriksson, C. Mohlin, B. Nilsson and K. N. Ekdahl, Front. Immunol., 2019, 10, 1590 CrossRef CAS PubMed.
- H. Han, R. Bartolo, J. Li, M. A. Shahbazi and H. A. Santos, Eur. J. Pharm. Biopharm., 2022, 172, 1–15 CrossRef CAS PubMed.
- X. Da, J. Mo, Q. Li, B. Cao, J. Huang, Y. Lu, L. Lu, M. Fan and H. Lu, Biochem. Biophys. Res. Commun., 2023, 671, 335–342 CrossRef CAS PubMed.
- A. S. Lonsdorf, B. F. Kramer, M. Fahrleitner, T. Schonberger, S. Gnerlich, S. Ring, S. Gehring, S. W. Schneider, M. J. Kruhlak, S. G. Meuth, B. Nieswandt, M. Gawaz, A. H. Enk and H. F. Langer, J. Biol. Chem., 2012, 287, 2168–2178 CrossRef CAS PubMed.
- F. Huang, Q. Zhang, J. Xiao, X. Zhang, X. Han, X. Shi, J. Hu, L. Li and X. Qian, Int. J. Nanomed., 2023, 18, 2261–2273 CrossRef CAS PubMed.
- S. Li, S. Dong, J. Wu, X. Lv, N. Yang, Q. Wei, C. Wang and J. Chen, ACS Appl. Mater. Interfaces, 2023, 15, 7878–7886 CrossRef CAS PubMed.
- A. Ocana, C. Nieto-Jimenez, A. Pandiella and A. J. Templeton, Mol. Cancer, 2017, 16, 137 CrossRef PubMed.
- F. Oroojalian, M. Beygi, B. Baradaran, A. Mokhtarzadeh and M. A. Shahbazi, Small, 2021, 17, e2006484 CrossRef PubMed.
- M. Orecchioni, Y. Ghosheh, A. B. Pramod and K. Ley, Front. Immunol., 2020, 11, 234 CrossRef PubMed.
- F. Xie, Z. Liu, P. Wang, M. Cai, Y. Li, J. Yan, Q. Lin and F. Luo, Adv. Healthc. Mater., 2022, 11, e2202115 CrossRef PubMed.
- K. Wu, K. Lin, X. Li, X. Yuan, P. Xu, P. Ni and D. Xu, Front. Immunol., 2020, 11, 1731 CrossRef CAS PubMed.
- Q. Zhang and M. Sioud, Int. J. Mol. Sci., 2023, 24, 7493 CrossRef CAS PubMed.
- J. Gao, Y. Liang and L. Wang, Front. Immunol., 2022, 13, 888713 CrossRef CAS PubMed.
- C. Kerneur, C. E. Cano and D. Olive, Front. Immunol., 2022, 13, 1026954 CrossRef CAS PubMed.
- X. Ma, L. Kuang, Y. Yin, L. Tang, Y. Zhang, Q. Fan, B. Wang, Z. Dong, W. Wang, T. Yin and Y. Wang, ACS Nano, 2023, 17, 2341–2355 CrossRef CAS PubMed.
- L. Zhang, Y. Zhang, X. Wang, Y. Zhou, J. Qi, L. Gu, Q. Zhao, R. Yu and X. Zhou, Small, 2023, e2301439, DOI:10.1002/smll.202301439.
- H. Y. Chen, J. Deng, Y. Wang, C. Q. Wu, X. Li and H. W. Dai, Acta Biomater., 2020, 112, 1–13 CrossRef CAS PubMed.
- Y. Zhao, A. Li, L. Jiang, Y. Gu and J. Liu, Biomacromolecules, 2021, 22, 3149–3167 CrossRef CAS PubMed.
- D. Wang, H. Dong, M. Li, Y. Cao, F. Yang, K. Zhang, W. Dai, C. Wang and X. Zhang, ACS Nano, 2018, 12, 5241–5252 CrossRef CAS PubMed.
- J. Ma, F. Liu, W. C. Sheu, Z. Meng, Y. Xie, H. Xu, M. Li, A. T. Chen, J. Liu, Y. Bao, X. Zhang, S. Zhang, L. Zhang, Z. Zou, H. Wu, H. Wang, Y. Zhu and J. Zhou, Nano Lett., 2020, 20, 4084–4094 CrossRef CAS PubMed.
- L. L. Bu, L. Rao, G. T. Yu, L. Chen, W. W. Deng, J. F. Liu, H. Wu, Q. F. Meng, S. S. Guo, X. Z. Zhao, W. F. Zhang, G. J. Chen, Z. Gu, W. Liu and Z. J. Sun, Adv. Funct. Mater., 2019, 29, 1807733 CrossRef.
- C. Gong, X. Yu, W. Zhang, L. Han, R. Wang, Y. Wang, S. Gao and Y. Yuan, J. Nanobiotechnol., 2021, 19, 58 CrossRef CAS PubMed.
- S. Zang, K. Huang, J. Li, K. Ren, T. Li, X. He, Y. Tao, J. He, Z. Dong, M. Li and Q. He, Acta Biomater., 2022, 148, 181–193 CrossRef CAS PubMed.
- L. Rao, Q. F. Meng, Q. Q. Huang, Z. X. Wang, G. T. Yu, A. Li, W. J. Ma, N. G. Zhang, S. S. Guo, X. Z. Zhao, K. Liu, Y. F. Yuan and W. Liu, Adv. Funct. Mater., 2018, 28, 1803531 CrossRef.
- H. H. Wu, Y. Zhou, Y. Tabata and J. Q. Gao, J. Controlled Release, 2019, 294, 102–113 CrossRef CAS PubMed.
- Y. Zou, Y. Sun, Y. Wang, D. Zhang, H. Yang, X. Wang, M. Zheng and B. Shi, Nat. Commun., 2023, 14, 4557 CrossRef CAS PubMed.
- R. B. Patel, M. Ye, P. M. Carlson, A. Jaquish, L. Zangl, B. Ma, Y. Wang, I. Arthur, R. Xie, R. J. Brown, X. Wang, R. Sriramaneni, K. Kim, S. Gong and Z. S. Morris, Adv. Mater., 2019, 31, e1902626 CrossRef PubMed.
- Y. Yang, K. Wang, Y. Pan, L. Rao and G. Luo, Adv. Sci., 2021, 8, e2102330 CrossRef PubMed.
- M. Zhang, S. Cheng, Y. Jin, N. Zhang and Y. Wang, Clin. Transl. Med., 2021, 11, e292 CrossRef PubMed.
- W. Song, P. Jia, Y. Ren, J. Xue, B. Zhou, X. Xu, Y. Shan, J. Deng and Q. Zhou, Bioact. Mater., 2023, 23, 80–100 Search PubMed.
- R. Yang, J. Xu, L. Xu, X. Sun, Q. Chen, Y. Zhao, R. Peng and Z. Liu, ACS Nano, 2018, 12, 5121–5129 CrossRef CAS PubMed.
- J. Yoon, X. T. Le, J. Kim, H. Lee, N. T. Nguyen, W. T. Lee, E. S. Lee, K. T. Oh, H. G. Choi and Y. S. Youn, J. Controlled Release, 2023, 360, 482–495 CrossRef CAS PubMed.
- C. M. Csizmar, J. R. Petersburg and C. R. Wagner, Cell Chem. Biol., 2018, 25, 931–940 CrossRef CAS PubMed.
- X. Xiong, J. Zhao, J. Pan, C. Liu, X. Guo and S. Zhou, Nano Lett., 2021, 21, 8418–8425 CrossRef CAS PubMed.
- L. Rao, S. K. Zhao, C. Wen, R. Tian, L. Lin, B. Cai, Y. Sun, F. Kang, Z. Yang, L. He, J. Mu, Q. F. Meng, G. Yao, N. Xie and X. Chen, Adv. Mater., 2020, 32, e2004853 CrossRef PubMed.
- Q. Tang, S. Sun, P. Wang, L. Sun, Y. Wang, L. Zhang, M. Xu, J. Chen, R. Wu, J. Zhang, M. Gong, Q. Chen and X. Liang, Adv. Mater., 2023, 35, e2300964 CrossRef PubMed.
- L. Zou, Y. Zhang, N. Cheraga, O. D. Abodunrin, K. Y. Qu, L. Qiao, Y. Q. Ma, Y. Hang, N. P. Huang and L. J. Chen, Small, 2024, 20, e2304110 CrossRef PubMed.
- X. Sun, G. He, C. Xiong, C. Wang, X. Lian, L. Hu, Z. Li, S. J. Dalgarno, Y. W. Yang and J. Tian, ACS Appl. Mater. Interfaces, 2021, 13, 3679–3693 CrossRef CAS PubMed.
- Z. Cao, X. Liu, W. Zhang, K. Zhang, L. Pan, M. Zhu, H. Qin, C. Zou, W. Wang, C. Zhang, Y. He, W. Lin, Y. Zhang, D. Han, M. Li and J. Gu, ACS Nano, 2023, 17, 23746–23760 CrossRef CAS PubMed.
- C. Wang and S. Wu, Front. Bioeng. Biotechnol., 2022, 10, 944518 CrossRef PubMed.
- A. Fernandez-Borbolla, L. Garcia-Hevia and M. L. Fanarraga, Int. J. Mol. Sci., 2024, 25, 2071 CrossRef CAS PubMed.
- C. M. Hu, R. H. Fang, B. T. Luk, K. N. Chen, C. Carpenter, W. Gao, K. Zhang and L. Zhang, Nanoscale, 2013, 5, 2664–2668 RSC.
- B. Bahmani, H. Gong, B. T. Luk, K. J. Haushalter, E. DeTeresa, M. Previti, J. Zhou, W. Gao, J. D. Bui, L. Zhang, R. H. Fang and J. Zhang, Nat. Commun., 2021, 12, 1999 CrossRef CAS PubMed.
- F. Wang, S. Lin, Z. Yu, Y. Wang, D. Zhang, C. Cao, Z. Wang, D. Cui and D. Chen, Lab Chip, 2022, 22, 2624–2646 RSC.
- L. Rao, B. Cai, L. L. Bu, Q. Q. Liao, S. S. Guo, X. Z. Zhao, W. F. Dong and W. Liu, ACS Nano, 2017, 11, 3496–3505 CrossRef CAS PubMed.
- V. Bommakanti, M. Banerjee, D. Shah, K. Manisha, K. Sri and S. Banerjee, Environ. Res., 2022, 214, 113919 CrossRef CAS PubMed.
- J. C. Potts, A. Jain, D. B. Amabilino, F. J. Rawson and L. Perez-Garcia, Anal. Chem., 2023, 95, 12998–13002 CrossRef CAS PubMed.
- G. R. Nirmal, Z. C. Lin, C. H. Lin, C. T. Sung, C. C. Liao and J. Y. Fang, Biomater. Sci., 2022, 10, 6172–6189 RSC.
- Y. Fan, M. Marioli and K. Zhang, J. Pharm. Biomed. Anal., 2021, 192, 113642 CrossRef CAS PubMed.
- T. Liu, Z. Zhou, M. Zhang, P. Lang, J. Li, Z. Liu, Z. Zhang, L. Li and L. Zhang, J. Controlled Release, 2023, 362, 502–512 CrossRef CAS PubMed.
- A. Kulyyassov, M. Fresnais and R. Longuespee, Proteomics, 2021, 21, e2100153 CrossRef PubMed.
- M. Wang, Y. Xin, H. Cao, W. Li, Y. Hua, T. J. Webster, C. Zhang, W. Tang and Z. Liu, Biomater. Sci., 2021, 9, 1088–1103 RSC.
- J. Xiong, H. Tang, L. Sun, J. Zhu, S. Tao, J. Luo, J. Li, J. Li, H. Wu and J. Yang, Acta Biomater., 2024, 175, 293–306 CrossRef CAS PubMed.
- C. Yang, Y. He, F. Chen, F. Zhang, D. Shao and Z. Wang, Small, 2023, 19, e2207029 CrossRef PubMed.
- Q. Wu, L. Chen, X. Huang, J. Lin, J. Gao, G. Yang, Y. Wu, C. Wang, X. Kang, Y. Yao, Y. Wang, M. Xue, X. Luan, X. Chen, Z. Zhang and S. Sun, Int. J. Oral Sci., 2023, 15, 9 CrossRef CAS PubMed.
- X. Ye, X. Liang, Q. Chen, Q. Miao, X. Chen, X. Zhang and L. Mei, ACS Nano, 2019, 13, 2956–2968 CrossRef CAS PubMed.
- L. Wan, Y. Cao, C. Cheng, R. Tang, N. Wu, Y. Zhou, X. Xiong, H. He, X. Lin, Q. Jiang, X. Wang, X. Guo, D. Wang, H. Ran, J. Ren, Y. Zhou, Z. Hu and P. Li, ACS Appl. Mater. Interfaces, 2023, 15, 1784–1797 CrossRef CAS PubMed.
- J. Xiong, M. Wu, J. Chen, Y. Liu, Y. Chen, G. Fan, Y. Liu, J. Cheng, Z. Wang, S. Wang, Y. Liu and W. Zhang, ACS Nano, 2021, 15, 19756–19770 CrossRef CAS PubMed.
- A. Chen, F. Yang, J. Kuang, Y. Xiong, B. B. Mi, Y. Zhou, J. J. Hu, S. J. Song, T. Wan, Z. Z. Wan, H. Y. Huang, X. R. Li, W. Song and W. X. Qiu, ACS Appl. Mater. Interfaces, 2021, 13, 45335–45345 CrossRef CAS PubMed.
- H. Wang, K. Wang, L. He, Y. Liu, H. Dong and Y. Li, Biomaterials, 2020, 244, 119964 CrossRef CAS PubMed.
- W. L. Liu, M. Z. Zou, T. Liu, J. Y. Zeng, X. Li, W. Y. Yu, C. X. Li, J. J. Ye, W. Song, J. Feng and X. Z. Zhang, Adv. Mater., 2019, 31, e1900499 CrossRef PubMed.
- Y. Cheng, Q. Chen, Z. Guo, M. Li, X. Yang, G. Wan, H. Chen, Q. Zhang and Y. Wang, ACS Nano, 2020, 14, 15161–15181 CrossRef CAS PubMed.
- R. H. Fang, C. M. Hu, B. T. Luk, W. Gao, J. A. Copp, Y. Tai, D. E. O'Connor and L. Zhang, Nano Lett., 2014, 14, 2181–2188 CrossRef CAS PubMed.
- S. Gou, W. Liu, S. Wang, G. Chen, Z. Chen, L. Qiu, X. Zhou, Y. Wu, Y. Qi and Y. Gao, Nano Lett., 2021, 21, 9939–9950 CrossRef CAS PubMed.
- L. Zhang, Y. Zhang, X. Wang, Y. Zhou, J. Qi, L. Gu, Q. Zhao, R. Yu and X. Zhou, Small, 2023, 19, e2301439 CrossRef PubMed.
- J. Deng, W. Xu, S. Lei, W. Li, Q. Li, K. Li, J. Lyu, J. Wang and Z. Wang, Small, 2022, 18, e2203114 CrossRef PubMed.
- L. Liu, Y. Wang, X. Guo, J. Zhao and S. Zhou, Small, 2020, 16, e2003543 CrossRef PubMed.
- D. Shao, F. Zhang, F. Chen, X. Zheng, H. Hu, C. Yang, Z. Tu, Z. Wang, Z. Chang, J. Lu, T. Li, Y. Zhang, L. Chen, K. W. Leong and W. F. Dong, Adv. Mater., 2020, 32, e2004385 CrossRef PubMed.
- L. Xie, C. Zhang, M. Liu, J. Huang, X. Jin, C. Zhu, M. Lv, N. Yang, S. Chen, M. Shao, X. Du and G. Feng, ACS Appl. Mater. Interfaces, 2023, 15, 10541–10553 CrossRef CAS PubMed.
- J. Li, Y. Wu, J. Wang, X. Xu, A. Zhang, Y. Li and Z. Zhang, ACS Nano, 2023, 17, 322–336 CrossRef CAS PubMed.
- Q. Chen, L. Zhang, L. Li, M. Tan, W. Liu, S. Liu, Z. Xie, W. Zhang, Z. Wang, Y. Cao, T. Shang and H. Ran, J. Nanobiotechnol., 2021, 19, 449 CrossRef CAS PubMed.
- Z. Li, H. Cai, Z. Li, L. Ren, X. Ma, H. Zhu, Q. Gong, H. Zhang, Z. Gu and K. Luo, Bioact. Mater., 2023, 21, 299–312 CrossRef CAS PubMed.
- C. Hu, T. Lei, Y. Wang, J. Cao, X. Yang, L. Qin, R. Liu, Y. Zhou, F. Tong, C. S. Umeshappa and H. Gao, Biomaterials, 2020, 255, 120159 CrossRef CAS PubMed.
- M. Z. Zou, W. L. Liu, C. X. Li, D. W. Zheng, J. Y. Zeng, F. Gao, J. J. Ye and X. Z. Zhang, Small, 2018, 14, e1801120 CrossRef PubMed.
- W. Ou, J. H. Byeon, Z. C. Soe, B. K. Kim, R. K. Thapa, B. Gupta, B. K. Poudel, S. K. Ku, C. S. Yong and J. O. Kim, Theranostics, 2019, 9, 6780–6796 CrossRef CAS.
- C. Yang, Y. Ming, K. Zhou, Y. Hao, D. Hu, B. Chu, X. He, Y. Yang and Z. Qian, Research, 2022, 2022, 9768687 CAS.
- P. Zhao, Y. Tian, Y. Lu, J. Zhang, A. Tao, G. Xiang and Y. Liu, J. Nanobiotechnol., 2022, 20, 525 CrossRef CAS PubMed.
- X. Huang, Y. Lu, M. Guo, S. Du and N. Han, Theranostics, 2021, 11, 7546–7569 CrossRef CAS PubMed.
- Z. Sun, G. Deng, X. Peng, X. Xu, L. Liu, J. Peng, Y. Ma, P. Zhang, A. Wen, Y. Wang, Z. Yang, P. Gong, W. Jiang and L. Cai, Biomaterials, 2021, 279, 121228 CrossRef CAS PubMed.
- H. Montaseri, C. A. Kruger and H. Abrahamse, Int. J. Mol. Sci., 2020, 21, 3358 CrossRef CAS.
- H. Yan, Y. Zhang, Y. Zhang, Y. Li, X. Kong, D. Liu, J. Li, Y. Xi, J. Ji, L. Ye and G. Zhai, Biomater. Sci., 2022, 10, 6583–6600 RSC.
- J. Ni, J. Song, B. Wang, H. Hua, H. Zhu, X. Guo, S. Xiong and Y. Zhao, Biomed. Pharmacother., 2020, 126, 110046 CrossRef CAS PubMed.
- Y. Xun, H. Yang, B. Kaminska and H. You, J. Hematol. Oncol., 2021, 14, 176 CrossRef CAS PubMed.
- X. Wei, L. Liu, X. Li, Y. Wang, X. Guo, J. Zhao and S. Zhou, J. Controlled Release, 2019, 313, 42–53 CrossRef CAS PubMed.
- T. Wang, H. Zhang, W. Qiu, Y. Han, H. Liu and Z. Li, Bioact. Mater., 2022, 16, 418–432 CAS.
- J. Zhang, L. Wei, X. Ma, J. Wang, S. Liang, K. Chen, M. Wu, L. Niu and Y. Zhang, Acta Biomater., 2023, 166, 470–484 CrossRef CAS PubMed.
- J. A. Kulkarni, D. Witzigmann, S. B. Thomson, S. Chen, B. R. Leavitt, P. R. Cullis and R. van der Meel, Nat. Nanotechnol., 2021, 16, 630–643 CrossRef CAS PubMed.
- E. Samaridou, J. Heyes and P. Lutwyche, Adv. Drug Delivery Rev., 2020, 154–155, 37–63 CrossRef CAS PubMed.
- S. Jin, Y. Sun, X. Liang, X. Gu, J. Ning, Y. Xu, S. Chen and L. Pan, Signal Transduction Targeted Ther., 2022, 7, 39 CrossRef CAS.
- T. M. Allen and P. R. Cullis, Adv. Drug Delivery Rev., 2013, 65, 36–48 CrossRef CAS PubMed.
- S. A. Dilliard, Q. Cheng and D. J. Siegwart, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2109256118 CrossRef CAS.
- S. H. Kiaie, N. Majidi Zolbanin, A. Ahmadi, R. Bagherifar, H. Valizadeh, F. Kashanchi and R. Jafari, J. Nanobiotechnol., 2022, 20, 276 CrossRef CAS PubMed.
- F. Zahednezhad, M. Saadat, H. Valizadeh, P. Zakeri-Milani and B. Baradaran, J. Controlled Release, 2019, 305, 194–209 CrossRef CAS PubMed.
- A. Becker, B. K. Thakur, J. M. Weiss, H. S. Kim, H. Peinado and D. Lyden, Cancer Cell, 2016, 30, 836–848 CrossRef CAS PubMed.
- V. R. Minciacchi, M. R. Freeman and D. Di Vizio, Semin. Cell Dev. Biol., 2015, 40, 41–51 CrossRef CAS PubMed.
- H. Chen, T. Sun and C. Jiang, J. Controlled Release, 2022, 348, 572–589 CrossRef CAS PubMed.
- X. Zhang, H. Zhang, J. Gu, J. Zhang, H. Shi, H. Qian, D. Wang, W. Xu, J. Pan and H. A. Santos, Adv. Mater., 2021, 33, e2005709 CrossRef PubMed.
- K. W. Witwer and J. Wolfram, Nat. Rev. Mater., 2021, 6, 103–106 CrossRef CAS PubMed.
- R. Xu, D. W. Greening, H. J. Zhu, N. Takahashi and R. J. Simpson, J. Clin. Invest., 2016, 126, 1152–1162 CrossRef PubMed.
- Y. Guo, D. Wang, Q. Song, T. Wu, X. Zhuang, Y. Bao, M. Kong, Y. Qi, S. Tan and Z. Zhang, ACS Nano, 2015, 9, 6918–6933 CrossRef CAS PubMed.
- S. Han, S. Bi, T. Guo, D. Sun, Y. Zou, L. Wang, L. Song, D. Chu, A. Liao, X. Song, Z. Yu and J. Guo, J. Controlled Release, 2022, 348, 250–263 CrossRef CAS PubMed.
- Y. Bao, Q. Hu, X. Wang, X. Feng, Y. He, Y. Guo and D. Fu, Biomed. Pharmacother., 2020, 129, 110377 CrossRef CAS.
- Q. Feng, Y. Li, N. Wang, Y. Hao, J. Chang, Z. Wang, X. Zhang, Z. Zhang and L. Wang, Small, 2020, 16, e2002138 CrossRef.
- X. Liang, X. Ye, C. Wang, C. Xing, Q. Miao, Z. Xie, X. Chen, X. Zhang, H. Zhang and L. Mei, J. Controlled Release, 2019, 296, 150–161 CrossRef CAS PubMed.
- Q. Jiang, K. Wang, X. Zhang, B. Ouyang, H. Liu, Z. Pang and W. Yang, Small, 2020, 16, e2001704 CrossRef PubMed.
- H. Wang, C. Wu, X. Tong and S. Chen, J. Controlled Release, 2023, 353, 727–737 CrossRef CAS PubMed.
- Y. Jiang, N. Krishnan, J. Zhou, S. Chekuri, X. Wei, A. V. Kroll, C. L. Yu, Y. Duan, W. Gao, R. H. Fang and L. Zhang, Adv. Mater., 2020, 32, e2001808 CrossRef PubMed.
- S. Liu, J. Wu, Y. Feng, X. Guo, T. Li, M. Meng, J. Chen, D. Chen and H. Tian, Bioact. Mater., 2023, 22, 211–224 CAS.
- Y. Guo, Y. Fan, Z. Wang, G. Li, M. Zhan, J. Gong, J. P. Majoral, X. Shi and M. Shen, Adv. Mater., 2022, 34, e2206861 CrossRef.
- T. Li, G. Chen, Z. Xiao, B. Li, H. Zhong, M. Lin, Y. Cai, J. Huang, X. Xie and X. Shuai, Nano Lett., 2022, 22, 3095–3103 CrossRef CAS.
- X. Wang, X. Zhu, B. Li, X. Wei, Y. Chen, Y. Zhang, Y. Wang, W. Zhang, S. Liu, Z. Liu, W. Zhai, P. Zhu, Y. Gao and Z. Chen, ACS Appl. Mater. Interfaces, 2022, 14(20), 23152–23163 CrossRef CAS PubMed.
- Z. Li and L. Rong, ACS Appl. Mater. Interfaces, 2021, 13, 23469–23480 CrossRef CAS PubMed.
- L. Fu, W. Zhang, X. Zhou, J. Fu and C. He, Bioact. Mater., 2022, 17, 221–233 CAS.
- M. Sun, Y. Li, W. Zhang, X. Gu, R. Wen, K. Zhang, J. Mao, C. Huang, X. Zhang, M. Nie, Z. Zhang, C. Qi, K. Cai and G. Liu, Acta Biomater., 2023, 166, 552–566 CrossRef CAS PubMed.
- S. Li, S. Jiang, M. S. U. Rahman, J. Mei, X. Wang, J. Jiang, Y. Chen, S. Xu and Y. Liu, Small Methods, 2023, 7, e2201569 CrossRef PubMed.
- F. Chen, Z. Geng, L. Wang, Y. Zhou and J. Liu, Small, 2022, 18, e2203066 CrossRef PubMed.
- G. Deng, Z. Sun, S. Li, X. Peng, W. Li, L. Zhou, Y. Ma, P. Gong and L. Cai, ACS Nano, 2018, 12, 12096–12108 CrossRef CAS PubMed.
- Y. Li, R. Yuan, Y. Luo, X. Guo, G. Yang, X. Li and S. Zhou, Adv. Mater., 2023, 35, e2300216 CrossRef PubMed.
- C. Chen, M. Song, Y. Du, Y. Yu, C. Li, Y. Han, F. Yan, Z. Shi and S. Feng, Nano Lett., 2021, 21, 5522–5531 CrossRef CAS PubMed.
- R. Liu, Y. An, W. Jia, Y. Wang, Y. Wu, Y. Zhen, J. Cao and H. Gao, J. Controlled Release, 2020, 321, 589–601 CrossRef CAS PubMed.
- X. Wen, X. Xiong, G. Yang, W. Xiao, J. Hou, T. Pan, Y. Hu and S. Zhou, J. Controlled Release, 2023, 353, 535–548 CrossRef CAS PubMed.
- W. Ma, D. Zhu, J. Li, X. Chen, W. Xie, X. Jiang, L. Wu, G. Wang, Y. Xiao, Z. Liu, F. Wang, A. Li, D. Shao, W. Dong, W. Liu and Y. Yuan, Theranostics, 2020, 10, 1281–1295 CrossRef CAS.
- Y. Zhai, J. Wang, T. Lang, Y. Kong, R. Rong, Y. Cai, W. Ran, F. Xiong, C. Zheng, Y. Wang, Y. Yu, H. H. Zhu, P. Zhang and Y. Li, Nat. Nanotechnol., 2021, 16, 1271–1280 CrossRef CAS.
- L. Zhang, W. Zhao, J. Huang, F. Li, J. Sheng, H. Song and Y. Chen, Front. Immunol., 2022, 13, 828263 CrossRef CAS PubMed.
- W. Hao, Y. Cui, Y. Fan, M. Chen, G. Yang, Y. Wang, M. Yang, Z. Li, W. Gong, Y. Yang and C. Gao, J. Nanobiotechnol., 2021, 19, 378 CrossRef CAS PubMed.
- C. Xu, Y. Jiang, Y. Han, K. Pu and R. Zhang, Adv. Mater., 2021, 33, e2008061 CrossRef PubMed.
- H. Kobayashi and P. L. Choyke, Acc. Chem. Res., 2019, 52, 2332–2339 CrossRef CAS PubMed.
- L. Ding, X. Zhang, P. Yu, F. Peng, Y. Sun, Y. Wu, Z. Luo, H. Li, Y. Zeng, M. Wu and X. Liu, Mol. Ther., 2023, 31, 2489–2506 CrossRef CAS PubMed.
- J. Deng, S. Xu, W. Hu, X. Xun, L. Zheng and M. Su, Biomaterials, 2018, 154, 24–33 CrossRef CAS PubMed.
- T. Liu, C. Shi, L. Duan, Z. Zhang, L. Luo, S. Goel, W. Cai and T. Chen, J. Mater. Chem. B, 2018, 6, 4756–4764 RSC.
- X. Zhen, P. Cheng and K. Pu, Small, 2019, 15, e1804105 CrossRef PubMed.
- L. Liu, D. Pan, S. Chen, M. V. Martikainen, A. Karlund, J. Ke, H. Pulkkinen, H. Ruhanen, M. Roponen, R. Kakela, W. Xu, J. Wang and V. P. Lehto, Nat. Commun., 2022, 13, 6181 CrossRef CAS PubMed.
Footnote |
| † Zichen Zhong and Wen Deng are joint first authors and contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |