
Solvent-induced structural transformation in a one-dimensional coordination polymer†
 *b
Jinhee
Park
*a
*b
Jinhee
Park
*a
Abstract
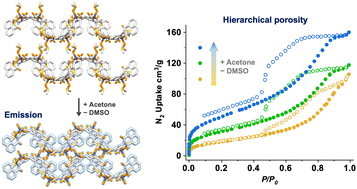
We have rationally designed a one-dimensional coordination polymer (1D CP), termed 1D-DGIST-18, that exhibits intrinsic structural flexibility. This 1D CP enables its expansion into a three-dimensional network through supramolecular interactions involving coordinated solvents and/or ligands. The strategic selection of solvents for solvent exchange, prior to drying, significantly influences the structures of 1D-DGIST-18 by removing certain coordinating solvents and modulating π–π stacking. Consequently, a hierarchical porosity emerges, ranging from micro- to meso- to macroporous structures, which is attributed to its inherent structural dynamics. Additionally, the formation of excimers endows 1D-DGIST-18, when immersed in acetone, with ‘turn-on’ fluorescence, as evidenced by fluorescence decay profiles. These structural transitions within 1D-DGIST-18 are further elucidated using single-crystal X-ray diffractometry. The insights from this study provide a foundation for the design of materials with structural dynamics and tunable properties.
- This article is part of the themed collections: 2025 Lunar New Year Collection and Nanoscale 2024 Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

