
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Self-brushing for nanopatterning: achieving perpendicular domain orientation in block copolymer thin films†
Hongbo
Feng
 *a,
Wen
Chen
a,
Gordon S. W.
Craig
a,
Stuart J.
Rowan
abc and
Paul F.
Nealey
a
*a,
Wen
Chen
a,
Gordon S. W.
Craig
a,
Stuart J.
Rowan
abc and
Paul F.
Nealey
a
aPritzker School of Molecular Engineering, University of Chicago, 5640 S. Ellis Avenue, Chicago, Illinois 60637, USA. E-mail: hfeng9@alum.utk.edu
bDepartment of Chemistry, University of Chicago, 5735 S. Ellis Avenue, Chicago, Illinois 60637, USA
cCenter for Molecular Engineering, Argonne National Laboratory, 9700 S. Cass Avenue, Lemont, Illinois 60439, USA
First published on 11th April 2024
Abstract
The self-assembly of thin films of block copolymers (BCPs) with perpendicular domain orientation offers a promising approach for nanopatterning on a variety of substrates, which is required by advanced applications such as ultrasmall transistors in integrated circuits, nanopatterned materials for tissue engineering, and electrocatalysts for fuel cell applications. In this study, we created BCPs with an A-b-(B-r-C) architecture that have blocks with equal surface energy (γair) and that can bind to the substrate, effectively creating a non-preferential substrate coating via self-brushing that enables the formation of through-film perpendicular domains in thin films of BCPs. We employed a thiol–epoxy click reaction to functionalize polystyrene-block-poly(glycidyl methacrylate) with a pair of thiols to generate an A-b-(B-r-C) BCP and tune γair of the B-r-C block. The secondary hydroxyl and thiol ether functionality generated by the click reaction was utilized to bind the BCP to the substrates. Scanning electron microscopy revealed that perpendicular orientation was achieved by simply annealing a thin film of the BCP on the bare substrate without the usual extra step of coating a random copolymer brush on the substrate. The self-brushing capability of the BCP was also examined on gold, platinum, titanium, aluminum nitride, and silicon nitride surfaces. These results demonstrate that self-brushing is a promising approach for achieving perpendicular domain orientation in thin films of BCP for nanopatterning on a variety of useful surfaces.
Introduction
The ability to manifest the unique properties of finely patterned functional inorganic materials is tremendously important in advanced applications.1,2 One example is the formation of ultrasmall transistors, which enhance computation power while simultaneously providing greater energy efficiency.3 In another example, thin films of silicon nitride (SiNx) have been used as diffusion barriers, capping layers, and etch stops in the manufacturing of integrated circuits.4,5 While semiconductor applications provide common examples of patterned inorganic materials, there are applications of these materials that are far removed from semiconductor applications. For example, nanopatterned gold (Au) has been utilized to control cell behavior in tissue engineering.6–8 Nanopatterned platinum (Pt) in the form of a Pt nanowire array has been proposed for high-performance electrocatalysts for fuel cell applications.9 Titanium (Ti), which is strong, light, and biocompatible, has been actively investigated for promising orthopedic implants, where the tissue adhesion can be significantly boosted by functionalizing the Ti surface at the nanoscale.10 Thin films of aluminum nitride (AlNx) are widely used in acoustic wave devices, optoelectronics, and energy harvesters because of the high band gap and high thermoelectric conductivity of AlNx.11 To continue to apply these inorganic materials for advanced applications requires nanopatterning at finer and finer scales. The creation of such features at the nanoscale remains a grand challenge.Self-assembly of block copolymers (BCPs) in thin films offers a promising approach to generate patterns at the nanoscale.12–14 Directed self-assembly (DSA) of BCPs has been implemented in semiconductor manufacturing and non-semiconductor applications such as the fabrication of bit patterned media, nanoporous membranes, and phononic crystals.14–17 For self-assembly to form well-defined features with minimal defects in the assembled structures, the BCP must have a suitable value of χN, where χ is the Flory–Huggins interaction parameter and N is the degree of polymerization of the BCP. The ideal BCP morphology for pattern transfer consists of through-film domains that are perpendicular to both the substrate and the free surface. While the development of approaches such as solvent annealing or the use of a top-coat to achieve perpendicular domains at the free surface has been impressive, these treatments add complexity to the overall processes, and simple thermal annealing is preferred. Perpendicular domain orientation via thermal annealing with a free surface requires that each block has the same surface energy with air (γair) at the annealing temperature, such that the difference in γair between the two blocks (Δγair) = 0. Similarly, the difference of the surface energies of each block with the substrate (Δγsub) must also equal zero. To manage these often-conflicting properties, we recently reported a series of high-throughput BCP platforms with an A-block-(B-random-C) (A-b-(B-r-C)) polymer architecture that can achieve both small feature sizes (<10 nm) and Δγair = 0, and then coated the substrate with a non-preferential copolymer brush to achieve Δγsub = 0.18–22
Copolymer coatings are commonly used to modulate the block–substrate interaction.23–25 The coating is typically a random copolymer brush consisting of the constituent monomers of the BCP and additional functionality that can either crosslink or form a covalent bond with the surface of the substrate. Recently, we reported a B-r-C random copolymer that could enable Δγsub = 0 between an A-b-(B-r-C) BCP and a silicon substrate.26 Surprisingly, in some instances more than one composition ratio of B and C (φ; φ = nC/(nB + nC), where ni is the number of repeat units of type i) of the coating could enable Δγsub = 0. Previous work reported using an amphiphilic BCP to create Langmuir–Blodgett films and used poly(2,2,6,6-tetramethyl-4-piperidyl methacrylate)-block-poly(glycidyl methacrylate) to deposit coatings on the surface of indium-doped tin oxide, in which the glycidyl methacrylate block could covalently bond to the surface oxides.27–29 These studies suggested that incorporating into the BCP domain functionalities that can form stable interactions with the substrate offers a pathway to form a monolayer coating, which may effectively serve as a self-brushing layer (SBL) and obviate the extra step of coating a random copolymer brush on the substrate. This approach could enable perpendicular assembly of domains by only spin-coating and annealing the functionalized BCP.
Here, we employed a thiol–epoxy click reaction to functionalize polystyrene-block-poly(glycidyl methacrylate) (S-b-G) with pairs of thiols to generate an A-b-(B-r-C) BCP with Δγair = 0. The thiol–epoxy reaction generated a secondary hydroxy group at each epoxy repeat unit that could form a covalent bond with silanol groups on the surface of the silicon substrate. This A-b-(B-r-C) BCP served as an exemplar BCP to explore the self-brushing behavior. This concept was extended to substrates that are essential in semiconductor manufacturing, such as Si, Au, Pt, Ti, AlNx, and SiNx. BCP domain orientation was examined with scanning electron microscopy (SEM). The chain conformation of the BCP self-brushing layer was also estimated using film thickness (h) measurements.
Experimental
Materials and syntheses
Styrene (99%, Aldrich), glycidyl methacrylate (GMA, 99%, Aldrich), 2-cyano-2-propyl benzodithioate (CPDB, 97%, Strem Chemicals), 2-mercaptopyridine (2MP, 99%, Aldrich), methyl thioglycolate (MTG, 95%, Aldrich, volatile and with very unpleasant smell!), 2,2,2-trifluoroethanethiol (TFET, 95%, Aldrich, volatile and with very unpleasant smell!), 2,2′-azobis(2-methylpropionitrile) (AIBN, 98% Aldrich), lithium hydroxide (LiOH, 99.99%, Aldrich), tetrahydrofuran (THF, 99.9%, Fisher Chemical), N,N-dimethylformamide (DMF, anhydrous, 99.8%, Aldrich), diiodomethane (CH2I2, 99%, Aldrich) and propylene glycol methyl ether acetate (PGMEA, >99.5%, Aldrich) were purchased and used as received unless otherwise noted. LiOH aqueous solutions were freshly made before each thiol–epoxy reaction at a concentration of approximately 20 mg mL−1 in deionized water. Inhibitor in the styrene and GMA monomers was removed by passing the monomers through a basic alumina column prior to use. AIBN was recrystallized from methanol twice prior to use. Cross-linkable polystyrene (x-S) was purchased from polymer source and used as received.Material characterization
1H NMR was performed on a Bruker AVANCE II+ 500. The NMR samples were dissolved in a deuterated solvent (CDCl3) at a concentration of approximately 15 mg mL−1. Mn and Đ were characterized with SEC on a Shimadzu gel permeation chromatography system equipped with a Wyatt DAWN HELEOS II multi-angle light scattering detector, a Wyatt ViscoStar III differential viscometer, a Wyatt Optilab T-rEX differential refractive index detector, and a Shimadzu SPD-M30A photodiode array detector (200–800 nm). The pump used was a Shimadzu HPLC LC20-AD. THF with 250 ppm of BHT was used as the eluent solvent and the columns sets were 2 Agilent PLgel 5 μm MIXED-D plus guard.SEM images were acquired on a Zeiss Merlin high-resolution field-emission SEM with a 1–1.5 keV accelerating voltage at a working distance <4 mm using the in-lens secondary electron detector. Image brightness and contrast were adjusted for presentation. Atomic force microscopy (AFM) was performed on a Bruker Nanoscope IIIa Multimode 5 using tapping mode in air. Small-angle X-ray scattering (SAXS) data was collected on a SAXSLAB (Xenocs)'s Ganesha with a rotating anode (Cu Kα) providing a focused X-ray beam with λ = 0.154 nm. The detector used was a Gabriel-type multi-wire area detector (1024 × 1024 pixels).
Grazing-incidence small-angle X-ray scattering (GISAXS) images were collected with the Beamline 8-ID-E at the Advanced Photon Source, Argonne National Lab. Diffraction patterns were collected using a photon energy of 10.9 keV. An incidence angle of 0.14° was chosen for all reported measurements. This angle was chosen because it is above the typical critical angle of organic thin film but below the critical angle of the silicon substrate. The exposure time for the measurements was 3–10 s. The images were collected at a sample-to-detector distance of 2185 mm using a Pilatus MF pixel array detector. To minimize beam damage and background air scatter, measurements were performed in a vacuum chamber at ∼10−3 Torr.
The h of each film was measured using a J. A. Woollam Alpha SE ellipsometer and fitted using a Si–SiOx–Cauchy model where the native oxide thickness was preset at 1.5 nm. For films with h < 10 nm, the optical constants were first fit to thick films, and then locked to determine h more accurately.
γ air values were determined from contact angle measurements, which were performed with a KRÜSS drop shape analyzer DSA100 (KRÜSS GmbH). Solutions of the nanocoating in PGMEA or THF were spin-coated on a Piranha-cleaned silicon wafer to form thin films with h = ∼30 nm. The thin films were then annealed on a hot plate at 150 °C for 1 h inside a N2 glovebox. Before each contact angle measurement, dry N2 was blown over the film surface to remove particle contaminants. The contact angles of two probing liquids, deionized H2O and CH2I2, were recorded using the sessile drop method with a drop volume of 1 μL for each measurement. The left and right contact angles of each drop were averaged, and 10 sessile drops were deposited for each sample. γair was then calculated using the OWRK method.30,31
Substrate surface preparation
Silicon wafers (4′, N-type, 〈100〉 orientation) were purchased from Pure Wafer. First, an O2 plasma was used to clean the wafer surface. Then 50 nm of Au, Ti, and Pt were deposited on silicon wafer with a 1 Å s−1 deposition rate and a 10 rpm rotation speed using an Angstrom EvoVac electron beam evaporator. Double-sided SiNx wafers were purchased from Pure Wafer. A 100 nm nitride layer with a tensile stress <250 ± 50 MPa was deposited on both sides of the silicon wafer using low-pressure chemical vapor deposition (LPCVD). AlNx coatings (∼30 nm) were deposited on silicon wafers at 250 °C with 0.64 Å/cycle deposition rate using an Ultratech Fiji G2 Plasma-Enhanced atomic layer deposition system.Deposition of BCP onto the substrate
Thin films of the model BCP were prepared by spin-coating the BCP solution in THF onto the substrate of interest. Silicon wafers were pre-cleaned using a hot piranha solution (a mixture of 30% H2O2 and 70% (v/v) concentrated H2SO4, Danger!) and rinsed with deionized H2O. Other substrates including Au, Pt, Ti, AlNx, and SiNx were fabricated as described above. The BCP-coated wafer was then annealed in a glove box filled with N2 at 150 °C for 1 h.Directed self-assembly (DSA) with self-brushing
To conduct the self-brushing DSA process, a 15 nm thick film of x-S was spin-coated onto a clean silicon wafer and then thermally cross-linked. The x-S was then patterned using e-beam lithography with a CSAR-Neu 7520 from ALLRESIST GmbH. Then the resist was developed using MIF 726 for 1 min, followed by a quick water rinse. The pattern was trim etched into the x-S substrate underneath it to achieve ∼1.5L0 wide guide stripes on different pitches. After stripping any remaining resist, the BCP was spin-coated on top of the x-S pattern to a thickness of 2L0 and annealed at 175 °C for 10 min in a nitrogen glovebox. Our previous study determined that self-brushing of the BCP would selectively occur only on the silicon wafer when the film was annealed at 175 °C for 10 min.18 This determination was critical because formation of the brush on the guide stripe would prohibit the chemoepitaxial guiding effect.Results and discussion
The chemical structure of the exemplar BCP and film construction is depicted in Fig. 1a. First, S-b-G(TFET-r-2MP) (Mn = 19.8 kg mol−1, Đ = 1.13) with a value of φ that provided Δγair = 0 was synthesized following a procedure reported previously.18 The matched surface energy was confirmed by an island-hole test, where a 0.5L0 topography was formed after thermal annealing the BCP thin films on a preferential S-coated silicon wafer.18 The BCP thin film was then deposited and annealed on a substrate that can bind to the functionality in the BCP, such as secondary –OH or the thiol ether. Such a binding interaction leads to the formation of an SBL, which can be used to modulate the substrate–BCP interactions. Fig. 1b–d shows representative top-down SEM images of S-b-G(TFET-r-2MP) films with different thickness annealed on piranha-cleaned silicon substrates. | ||
| Fig. 1 (a) The chemical structure of S-b-G(TFET-r-2MP) with Δγair = 0 at φ of 0.35 and L0 of 14.3 nm when annealed at 150 °C. This BCP is used to form a self-brushing layer (SBL) through the reactive moiety, either the secondary hydroxyl group or the thiol ether group, with the substrates as indicated by the dashed box. The proposed SBL formation is depicted on the right. (b–d) Top-down SEM micrographs of S-b-G(TFET-r-2MP) with film thicknesses of (b) 26.5 nm, (c) 9.2 nm, and (d) 5.6 nm, on a piranha-treated Si surface. (e) Left: SEM image of chemoepitaxy DSA with 5× density multiplication (n = 5) of S-b-G(TFET-r-2MP) using crosslinked S (x-S) as the guide. Right: SEM image of the DSA pattern post rinse. To enhance the contrast of the nanostructures, the pattern was stained with Al2O3 using the sequential infiltration synthesis process. The bright white stripes are the guide pattern of x-S. Partial alignment of the SBL is indicated by the red arrows. | ||
A through-film perpendicular orientation of the domains can be achieved with thermal annealing when Δγair = 0 and Δγsub = 0. Thus, it was initially surprising when the thin films of the BCPs spontaneously formed perpendicular domains after thermal annealing on a piranha-cleaned silicon wafer, which is a high surface energy that is not likely to provide Δγsub = 0 for the BCP. The surface tensions of substrates used in this work in summarized in Table S1.† This formation of perpendicular domains suggested the presence of a coating layer formed in situ that balanced the interaction of each block with the substrate. A series of rinses of the BCP-coated silicon wafer in THF and THF/DMF revealed a residual thin film layer that functioned as an SBL. The SBL was formed through a condensation reaction between the secondary hydroxyl groups of the BCP and silanol groups on the surface of the silicon substrate. The secondary hydroxy group could also undergo a transesterification reaction resulting in crosslinking. However, this reaction was less pronounced than the condensation reaction due to steric hinderance and the absence of a catalyst.32 To mimic the SBL, a thinner BCP film with only 5.6 nm (h/L0 = 0.40) was deposited on a piranha-cleaned wafer, yielding the fingerprint feature shown in Fig. 1d. A high-resolution SEM micrograph of a S-b-G(TFET-r-2MP) thin film after annealing on a piranha-treated Si surface is shown in Fig. S1a.† With the encouraging results showing the perpendicular assembly of BCP domains with SBL, a chemoepitaxy DSA was then performed as a proof-of-concept to demonstrate the capability of SBLs for nanopatterning. x-S was used as the guide (Fig. S2a†). Representative SEM images of DSA with 5× density multiplication are shown in Fig. S2b.† Although defects are present after DSA, the images shows that self-brushing has significant potential for nanopatterning applications. In addition, a rinsing test was performed on a film of BCP after DSA with 5× density multiplication, which revealed features of the SBL aligned to the guide stripes, as shown in Fig. 1e. This alignment, albeit poor, implies that the SBL can be guided by a chemical pattern and could potentially mitigate the etch and pattern transfer related issues that occur when using a random copolymer brush in the background regions of the chemical pattern.
GISAXS was performed to probe the self-assembled structures quantitively over a large scale, as shown in Fig. 2a. First, the perpendicular domain orientation (out-of-plane) was observed in the two-dimensional profile, which agrees well with the SEM results. Fig. 2b shows the full width at half maximum (FWHM) of the principal diffraction peak, which is inversely proportional to the grain size (ζ) of the self-assembled in-plane structure, as a function of normalized h. ζ of the self-assembled feature decreased monotonically as h decreased, following a power law fit of ζ ∼ (h/L0)−0.602, which agreed well with the SEM results. The reduced grain sizes were likely due to the thickness confinement effect, which prevented polymer chain compression and extension such that they could not efficiently move across another chain to form more uniform structures, similar to previously reported studies.33–35 The formation of fingerprint patterns in both the thick and thin films demonstrated the capability of the SBL to balance the domain–substrate interaction of the two different blocks of the BCP.
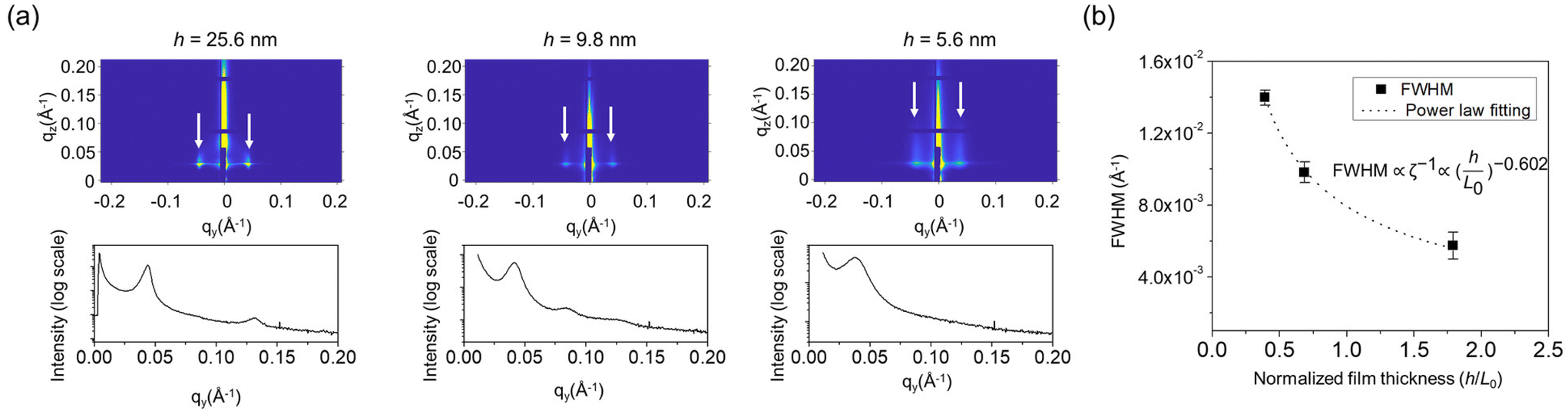 | ||
| Fig. 2 (a) 2D profile of grazing incidence small angle X-ray scattering of S-b-G(TFET-r-2MP) with film thicknesses (h) of 25.6, 9.8, and 5.6 nm, annealed on a piranha-treated Si surface. Below each 2D profile is the corresponding 1D profile. The white arrows indicate the presence of perpendicular assembly. (b) The full width at half-maximum (FWHM) of the principal peak of the 1D profile as a function of normalized film thickness. The FWHM, which is inversely proportional to the grain size (ζ), shows a good power law fit to the normalized film thickness, as shown by the dashed line. The FWHM and its standard deviation were obtained from a Gaussian peak fitting function using OriginPro v8.1. | ||
To investigate the conformation of the SBL, several scenarios of chain conformation of self-assembled BCP monolayer on silicon substrate were considered, as shown in Fig. 3a. In the first scenario, the silicon surface is exclusively occupied by G(TFET-r-2MP), causing the S block to reside above the SBL–BCP interface. This is analogous to submerging a substrate coated with a thin film of BCP into a selective solvent that is preferential to one of the blocks. By varying the solvent quality and grafting density, the number density of the reactive moiety, and the chain length, the conformation of the extended S chain can be in the brush or the mushroom regime. In the second case, only part of the –OH groups in the G block can react with the surface silanols, such that both the S and part of the G block reside above the SBL–BCP interface. As the number density of the reactive moiety decreases, more G block chains can extend into the SBL–BCP interface along with S. Following this analysis, the number density of grafting sites also has a direct impact on the fraction of the two blocks exposed to the SBL–BCP interface, which can differ from the volume fraction of the blocks in bulk. The presence of both S and G(TFET-r-2MP) in the SBL–BCP interface creates a nonpreferential substrate coating, similar to the case of a nonpreferential poly(styrene-co-methyl methacrylate) (S-r-M) brush coating for polystyrene-block-poly(methyl methacrylate) (S-b-M) BCPs in previous reports.23Fig. 3b shows a comparison of γ of the SBL (γcoat) to γ of S (γS), G(TFET) (γG(TFET)), and G(2MP) (γG(2MP)), which are determined with contact angle measurements using H2O and CH2I2. Γcoat is close to γS and a φ-average of γG(TFET) and γG(2MP), suggesting that both S and G(TFET-r-2MP) were exposed to the free surface, effectively forming the SBL–BCP interface in the assembled structures. To estimate the grafting density and evaluate the chain conformation in the SBL, h is measured. For a similar Mn, the h of the SBL is approximately half of the h of an OH-terminated polymer brush grafted on a piranha-cleaned silicon wafer. Table S2† summarizes the literature values of h and Mn of the polymer brushes. Both the γ and h results indicate that only a fraction of the hydroxyl groups react with the surface where multiple anchoring points are possible, which agrees well with previous studies on side-chain grafted polymer coatings.36,37
 | ||
| Fig. 3 (a) Schematics of the possible interactions of the self-assembled monolayer and the silicon substrate. With a substrate that is preferential to the G(TFET-r-2MP) block, many bonds form with each chain, which fully occupies the silicon surface, such that the number and density of BCPs adsorbed on the surface is low. In this situation, the S chain extends away from the substrate, making the self-assembled monolayer preferential to the S block. When the substrate is preferential to both blocks, single or multiple anchoring points are possible, but a greater density of BCPs can connect with the surface. In this configuration, both S and G(TFET-r-2MP) blocks extend away from the substrate, making the self-assembled monolayer non-preferential to the S and G(TFET-r-2MP) blocks. (b) The comparison of surface energies of the residue layer. | ||
Depositing thin films of a BCP on an Au surface eliminates the condensation reaction between the hydroxy groups in the BCP and the surface silanols but allows for the formation of S–Au bonds through chemisorption.38–41 The binding affinity is sufficiently high to leave a residue layer after multiple rinses with DMF.42 The chain conformation, similar to the case of secondary –OH on a silicon surface, depends on the number density of the thiol–ether grafting sites. Upon annealing a 26 nm thin film of S-b-G(TFET-r-2MP) on an Au surface, a fingerprint pattern is apparent in the SEM images, as shown in Fig. 4a, indicating Δγsub = 0. For comparison, annealing a thin film of lamellae-forming S-b-M, which does not contain a moiety that is reactive with Au, on an Au surface yields a parallel assembly of domains at a temperature known to have Δγair = 0 for S-b-M. Previous work has determined that Au is preferential to the S domain over the M domain.43,44 Thus, the distinct perpendicular domain orientation of this work must be attributed to the SBL formed by the thiol–ether functionality in G(TFET-r-2MP). The chain conformation should be similar to the case of the polymer on Si. A fingerprint pattern is also observed after annealing the S-b-G(TFET-r-2MP) film on a Pt surface, as shown in Fig. 4b. One noticeable difference is that a grain-like structure, indicated by the dashed circle in Fig. 4a, is observed in the film on the Au surface but not on the Pt surface. One possible reason is that the Au–S interaction (418 kJ mol−1) is stronger than the Pt–S interaction (234 kJ mol−1),45 which can reduce chain relaxation and prevent the formation of more uniform structures. Because the Ti surface is prone to oxidation, a thin layer of titanium oxide (TiO2) can be formed when exposed to air.46 Like the case of the S-b-G(TFET-r-2MP) thin film annealed on Si, perpendicular assembly is obtained on the Ti surface, as shown in Fig. 4c. The SBL likely forms through the TiO2–OH, TiO2–S and Ti–S interactions.47,48
 | ||
| Fig. 4 (a–e) Top-down SEM images of a 26 nm thick film of S-b-G(TFET-r-2MP) annealed on different surfaces. (a) Au. (b) Pt. (c) Ti. (d) AlNx. (e) SiNx. In (a), the yellow dashed circles indicate the grain-like structure. A thin layer of Ti was also used as an adhesion layer when creating the Au and Pt substrates. (a) and (b) show a thin film of Ti between the metal and the Si, which was used as an adhesion promoter. The image in (e) is featureless because the assembled film lacks perpendicular domains. (f) Large-area SEM image of a self-assembled thin film of 26 nm thick S-b-G(TFET-r-2MP) on SiNx that exhibits island and hole topographic features, indicative of a parallel domain orientation at the free surface. | ||
AlNx and SiNx are two essential coating materials in nanofabrication and in numerous applications. The surface properties of AlNx depend heavily on the deposition process. A report in the literature states that the pristine AlNx surface contains a significant amount of O in addition to Al and N, suggesting the presence of Al–OH groups on the surface.49Fig. 4d shows the fingerprint structure that is formed after depositing and annealing a 26 nm film of S-b-G(TFET-r-2MP) on an AlNx coating on Si. The SBL is formed through the condensation reaction of the hydroxy groups in G(TFET-r-2MP) with the surface Al–OH groups. The analysis done in the case of Si should be applicable. Surprisingly, a parallel assembly was observed in the case of SiNx as shown in Fig. 4e and f. Carduner et al. and Okada et al. reported the chemical composition of the surface layer of SiNx was SiOxNy, which is composed of Si–NH2, Si–OH, Si2–NH, and Si–H.50,51 Comparing the AlNx and SiNx surfaces, the BCP film that formed on SiNx did not lead to Δγsub = 0. The preference for one of the blocks in the BCP is presumably attributed to the higher grafting density of the SBL, similar to the first scenario in Fig. 3a. The high grafting density is a result of a greater number of reactive sites for G(TFET-r-2MP). For example, the hydroxyl group can react with Si–H through deprotonation,52 and the ester group can react with Si–NH2 to form an amide bond.53 The surface functionality is known to be process-dependent, which could be leveraged to control BCP domain orientation for nanopatterning applications.46 Topographic features such as surface roughness have been shown to induce perpendicular assembly, however, it is not applicable to the case study in this work because the substrates were atomically smooth. Nevertheless, the study here utilizing the spontaneous perpendicular assembly of BCP films simplifies the process for the fabrication of nanopatterned features on different surfaces. We anticipate this concept can be extended to other BCPs with Δγair = 0 and with blocks containing functionalities that can bind to the substrate of interest. Future work should focus on the fundamentals of the interaction between the substrate and the BCP and utilization of the created nanostructures for downstream applications.
Conclusions
A model BCP, S-b-G(TFET-r-2MP) with an A-b-(B-r-C) architecture, where A and B-r-C have equal γair and the B-r-C block contains functionality that can bind to the substrate, was deposited onto Si, Au, Pt, Ti, SiNx, and AlNx surfaces. Examination of the thin films that remained after rinsing determined that S-b-G(TFET-r-2MP) could form an SBL either through a condensation reaction or chemisorption with the surface functionality of the substrates. An SBL formed on the Si, Au, and Pt substrates that balanced the BCP domain–substrate interaction for the two blocks, as evidenced by the perpendicular domains that spontaneously self-assembled via thermal annealing. The chain conformation and surface energies of the self-brushing layer were evaluated, where multiple grafting sites on the substrates caused both S and G(TFET-r-2MP) blocks to extend through the SBL–BCP interface, making the SBL nonpreferential to S-b-G(TFET-r-2MP). Parallel domains were observed when a SiNx layer coated the substrate. This research presents self-brushing as a new approach to realize perpendicular domain orientation without the use of separate coatings. Future work should focus on investigating the capability of self-brushing for BCPs with other morphologies. From a technical point of view, with subsequent selective etching, we anticipate that self-brushing can simplify the fabrication of useful nanofeatures for advanced applications.Author contributions
The manuscript was written through the contributions of all authors. All authors have approved the final version of the manuscript. H. F. designed and conducted the experiments and analyzed the data under the supervision of S. J. R. and P. F. N. W. C. prepared the substrate surfaces. H. F., W. C., G. S. W. C., S. J. R., and P. F. N. wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the U.S. Department of Commerce, National Institute of Standards and Technology, as part of the Center for Hierarchical Materials Design (CHiMaD). This work made use of the shared facilities at the University of Chicago Materials Research Science and Engineering Center, supported by the National Science Foundation under award number DMR-2011854. It also made use of the Pritzker Nanofabrication Facility of the Pritzker School of Molecular Engineering at the University of Chicago, which receives support from Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF ECCS-1542205), a node of the National Science Foundation's National Nanotechnology Coordinated Infrastructure. Part of this work was carried out at the Soft Matter Characterization Facility of the University of Chicago.References
- C. Kinnear, T. L. Moore, L. Rodriguez-Lorenzo, B. Rothen-Rutishauser and A. Petri-Fink, Chem. Rev., 2017, 117, 11476–11521 CrossRef CAS PubMed.
- J. W. Steed and J. L. Atwood, Supramolecular chemistry, John Wiley & Sons, 2022 Search PubMed.
- I. L. Markov, Nature, 2014, 512, 147–154 CrossRef CAS PubMed.
- F. L. Riley, J. Am. Ceram. Soc., 2000, 83, 245–265 CrossRef CAS.
- J. X. J. Zhang and K. Hoshino, in Molecular Sensors and Nanodevices, ed. J. X. J. Zhang and K. Hoshino, Academic Press, 2nd edn, 2019, pp. 43–111. DOI:10.1016/B978-0-12-814862-4.00002-8.
- G. Chauhan, A. L. Ángeles, E. Gonzalez-González, M. M. Kulkarni, B. Cardenas-Benitez, M. F. Jiménez, G. Trujillo-de Santiago, M. M. Alvarez, M. Madou and S. O. Martinez-Chapa, Adv. Mater. Interfaces, 2020, 7, 2000238 CrossRef CAS.
- K. S. Kwok, Y. Zuo, S. J. Choi, G. J. Pahapale, L. Gu and D. H. Gracias, Nano Lett., 2023, 23, 7477–7484 CrossRef CAS PubMed.
- J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Chem. Rev., 2005, 105, 1103–1170 CrossRef CAS PubMed.
- H. Li, X. Wu, X. Tao, Y. Lu and Y. Wang, Small, 2020, 16, 2001135 CrossRef CAS PubMed.
- A. I. M. Greer, V. Goriainov, J. Kanczler, C. R. M. Black, L.-A. Turner, R. M. D. Meek, K. Burgess, I. MacLaren, M. J. Dalby, R. O. C. Oreffo and N. Gadegaard, ACS Appl. Mater. Interfaces, 2020, 12, 33541–33549 CrossRef CAS PubMed.
- R. M. R. Pinto, V. Gund, C. Calaza, K. K. Nagaraja and K. B. Vinayakumar, Microelectron. Eng., 2022, 257, 111753 CrossRef CAS.
- H. Feng, X. Lu, W. Wang, N.-G. Kang and J. W. Mays, Polymers, 2017, 9, 494 CrossRef PubMed.
- S. O. Kim, H. H. Solak, M. P. Stoykovich, N. J. Ferrier, J. J. De Pablo and P. F. Nealey, Nature, 2003, 424, 411–414 CrossRef CAS PubMed.
- C.-C. Liu, E. Franke, Y. Mignot, R. Xie, C. W. Yeung, J. Zhang, C. Chi, C. Zhang, R. Farrell, K. Lai, H. Tsai, N. Felix and D. Corliss, Nat. Electron., 2018, 1, 562–569 CrossRef CAS.
- K. Hono, Y. K. Takahashi, G. Ju, J.-U. Thiele, A. Ajan, X. Yang, R. Ruiz and L. Wan, MRS Bull., 2018, 43, 93–99 CrossRef CAS.
- C. Zhou, T. Segal-Peretz, M. E. Oruc, H. S. Suh, G. Wu and P. F. Nealey, Adv. Funct. Mater., 2017, 27, 1701756 CrossRef.
- C. Zhou, N. Tambo, E. M. Ashley, Y. Liao, J. Shiomi, K. Takahashi, G. S. W. Craig and P. F. Nealey, ACS Nano, 2020, 14, 6980–6989 CrossRef CAS PubMed.
- H. Feng, M. Dolejsi, N. Zhu, S. Yim, W. Loo, P. Ma, C. Zhou, G. S. W. Craig, W. Chen, L. Wan, R. Ruiz, J. J. de Pablo, S. J. Rowan and P. F. Nealey, Nat. Mater., 2022, 21, 1426–1433 CrossRef CAS PubMed.
- H. Feng, M. Dolejsi, N. Zhu, P. J. Griffin, G. S. W. Craig, W. Chen, S. J. Rowan and P. F. Nealey, Adv. Funct. Mater., 2022, 32, 2206836 CrossRef CAS.
- H. Feng, C.-G. Chae, C. Eom, G. S. W. Craig, S. J. Rowan and P. F. Nealey, Macromolecules, 2023, 56, 2675–2685 CrossRef CAS.
- W. S. Loo, H. Feng, T. J. Ferron, R. Ruiz, D. F. Sunday and P. F. Nealey, ACS Macro Lett., 2023, 12, 118–124 CrossRef CAS PubMed.
- H. Feng, L. Schneider, W. Loo, G. S. W. Craig, C. Eom, Z. Wang, R. Ruiz, J. J. de Pablo, S. J. Rowan and P. F. Nealey, Macromolecules, 2023, 56, 4591–4601 CrossRef CAS.
- P. Mansky, Y. Liu, E. Huang, T. P. Russell and C. Hawker, Science, 1997, 275, 1458–1460 CrossRef CAS.
- C. C. Liu, A. Ramirez-Hernandez, E. Han, G. S. W. Craig, Y. Tada, H. Yoshida, H. M. Kang, S. X. Ji, P. Gopalan, J. J. de Pablo and P. F. Nealey, Macromolecules, 2013, 46, 1415–1424 CrossRef CAS.
- E. Huang, L. Rockford, T. P. Russell and C. J. Hawker, Nature, 1998, 395, 757–758 CrossRef CAS.
- H. Feng, B. Kash, S. Yim, K. Bagchi, G. S. W. Craig, W. Chen, S. J. Rowan and P. F. Nealey, Langmuir, 2023, 39, 14688–14698 CrossRef CAS PubMed.
- C.-S. Cho, T. Kotaka and T. Akaike, J. Biomed. Mater. Res., 1993, 27, 199–206 CrossRef CAS PubMed.
- J. Kumaki, Polym. J., 2016, 48, 3–14 CrossRef CAS.
- K. Takahashi, K. Korolev, K. Tsuji, K. Oyaizu, H. Nishide, E. Bryuzgin, A. Navrotskiy and I. Novakov, Polymer, 2015, 68, 310–314 CrossRef CAS.
- D. K. Owens and R. Wendt, J. Appl. Polym. Sci., 1969, 13, 1741–1747 CrossRef CAS.
- D. Kaelble, J. Adhes., 1970, 2, 66–81 CrossRef CAS.
- J. Otera, Chem. Rev., 1993, 93, 1449–1470 CrossRef CAS.
- A. M. Welander, G. S. W. Craig, Y. Tada, H. Yoshida and P. F. Nealey, Macromolecules, 2013, 46, 3915–3921 CrossRef CAS.
- N. Mahadevapuram, I. Mitra, A. Bozhchenko, J. Strzalka and G. E. Stein, J. Polym. Sci., Part B: Polym. Phys., 2016, 54, 339–352 CrossRef CAS.
- T. P. Russell, G. Coulon, V. R. Deline and D. C. Miller, Macromolecules, 1989, 22, 4600–4606 CrossRef CAS.
- Y. Yu, G. J. Vancso and S. de Beer, Eur. Polym. J., 2017, 89, 221–229 CrossRef CAS.
- B. K. H. Feng, S. Yim, K. Bagchi, G. S. W. Craig, W. Chen, S. J. Rowan and P. F. Nealey, Langmuir, 2023, 39, 14688–14698 CrossRef PubMed.
- A. Ulman, S. D. Evans, Y. Shnidman, R. Sharma, J. E. Eilers and J. C. Chang, J. Am. Chem. Soc., 1991, 113, 1499–1506 CrossRef CAS.
- R. L. Garrell, J. E. Chadwick, D. L. Severance, N. A. McDonald and D. C. Myles, J. Am. Chem. Soc., 1995, 117, 11563–11571 CrossRef CAS.
- R. G. Nuzzo, B. R. Zegarski and L. H. Dubois, J. Am. Chem. Soc., 1987, 109, 733–740 CrossRef CAS.
- Y. Xue, X. Li, H. Li and W. Zhang, Nat. Commun., 2014, 5, 4348 CrossRef CAS PubMed.
- S. Slavin, A. H. Soeriyadi, L. Voorhaar, M. R. Whittaker, C. R. Becer, C. Boyer, T. P. Davis and D. M. Haddleton, Soft Matter, 2012, 8, 118–128 RSC.
- S.-J. Jeong, J. E. Kim, H.-S. Moon, B. H. Kim, S. M. Kim, J. B. Kim and S. O. Kim, Nano Lett., 2009, 9, 2300–2305 CrossRef CAS PubMed.
- S.-M. Park, M. P. Stoykovich, R. Ruiz, Y. Zhang, C. T. Black and P. F. Nealey, Adv. Mater., 2007, 19, 607–611 CrossRef CAS.
- Z. Li, S.-C. Chang and R. S. Williams, Langmuir, 2003, 19, 6744–6749 CrossRef CAS.
- W. Boonrawd, K. R. Awad, V. Varanasi and E. I. Meletis, Surf. Coat. Technol., 2021, 414, 127119 CrossRef CAS PubMed.
- G. Mani, D. M. Johnson, D. Marton, V. L. Dougherty, M. D. Feldman, D. Patel, A. A. Ayon and C. M. Agrawal, Langmuir, 2008, 24, 6774–6784 CrossRef CAS PubMed.
- X. Tao, J. Wang, Z. Ying, Q. Cai, G. Zheng, Y. Gan, H. Huang, Y. Xia, C. Liang, W. Zhang and Y. Cui, Nano Lett., 2014, 14, 5288–5294 CrossRef CAS PubMed.
- J. Kaaos, G. Ross and M. Paulasto-Kröckel, ACS Appl. Mater. Interfaces, 2021, 13, 38857–38865 CrossRef CAS PubMed.
- K. R. Carduner, R. O. Carter, M. E. Milberg and G. M. Crosbie, Anal. Chem., 1987, 59, 2794–2797 CrossRef CAS.
- K. Okada, K. Fukuyama and Y. Kameshima, J. Am. Ceram. Soc., 1995, 78, 2021–2026 CrossRef CAS.
- L.-H. Liu, D. J. Michalak, T. P. Chopra, S. P. Pujari, W. Cabrera, D. Dick, J.-F. Veyan, R. Hourani, M. D. Halls, H. Zuilhof and Y. J. Chabal, J. Phys.: Condens. Matter, 2016, 28, 094014 CrossRef PubMed.
- B. Gnanaprakasam and D. Milstein, J. Am. Chem. Soc., 2011, 133, 1682–1685 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr00223g |
| This journal is © The Royal Society of Chemistry 2024 |