
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSERS nanostructures with engineered active peptides against an immune checkpoint protein†
Marina
Gobbo
 a,
Isabella
Caligiuri
b,
Micaela
Giannetti
c,
Lucio
Litti
a,
Claudia
Mazzuca
c,
Flavio
Rizzolio
bd,
Antonio
Palleschi
c and
Moreno
Meneghetti
*a
a,
Isabella
Caligiuri
b,
Micaela
Giannetti
c,
Lucio
Litti
a,
Claudia
Mazzuca
c,
Flavio
Rizzolio
bd,
Antonio
Palleschi
c and
Moreno
Meneghetti
*a
aDepartment of Chemical Sciences, University of Padova, via F. Marzolo 1, 35131 Padova, Italy. E-mail: moreno.meneghetti@unipd.it
bPathology Unit, Centro di Riferimento Oncologico di Aviano (CRO) IRCCS, via F. Gallini 2, 33081 Aviano, PN, Italy
cDepartment of Chemical Science and Technologies, University of Rome “Tor Vergata”, and CSGI unit of Rome, Via della Ricerca Scientifica, 00133 Rome, Italy
dDepartment of Molecular Sciences and Nanosystems, Ca’ Foscari University of Venice, via Torino 155, 30172 Venice, Italy
First published on 5th February 2024
Abstract
The immune checkpoint programmed death ligand 1 (PD-L1) protein is expressed by tumor cells and it suppresses the killer activity of CD8+ T-lymphocyte cells binding to the programmed death 1 (PD-1) protein of these immune cells. Binding to either PD-L1 or PD1 is used for avoiding the inactivation of CD8+ T-lymphocyte cells. We report, for the first time, Au plasmonic nanostructures with surface-enhanced Raman scattering (SERS) properties (SERS nanostructures) and functionalized with an engineered peptide (CLP002: Trp-His-Arg-Ser-Tyr-Tyr-Thr-Trp-Asn-Leu-Asn-Thr), which targets PD-L1. Molecular dynamics calculations are used to describe the interaction of the targeting peptide with PD-L1 in the region where the interaction with PD-1 occurs, showing also the poor targeting activity of a peptide with the same amino acids, but a scrambled sequence. The results are confirmed experimentally since a very good targeting activity is observed against the MDA-MB-231 breast adenocarcinoma cancer cell line, which overexpresses PD-L1. A good activity is observed, in particular, for SERS nanostructures where the CLP002-engineered peptide is linked to the nanostructure surface with a short charged amino acid sequence and a long PEG chain. The results show that the functionalized SERS nanostructures show very good targeting of the immune checkpoint PD-L1.
1. Introduction
Immunotherapy approaches include advanced strategies for activating immune responses against tumors. Immune actions are complex cascades of processes, which start with the recognition of tumor-associated antigens (TAAs) overexpressed on tumor cells.1 TAAs are present in the tumor environment and are taken by dendritic cells (DCs) for their recognition by CD4+ helper T-lymphocyte cells (Th). Activated Th release cytokines, such as tumor necrosis factor-α, interleukin-12 and interferon-γ, induce the suppression of tumor growth and increase the presentation of TAAs on the tumor cell surface. This process favors the activation of CD8+ T-lymphocyte cells for the recognition of TAAs on the tumor cells, which follows the triggering of the programmed death of tumor cells. Th also interact with B cells and induce the differentiation of memory B-lymphocyte cells that produce specific antibodies for TAAs for long-term surveillance.However, different mechanisms can also be activated by the tumor for suppressing the immune response. Among them, some derive from surface proteins called immune checkpoints that can inactivate CD8+ T-cells. The programmed death ligand 1 (PD-L1, CD274 or B7-H1) is a checkpoint expressed by tumor cells because it can link to the programmed death 1 (PD-1, CD279) expressed by the CD8+ T-cells, suppressing their activity.2,3 This is one of the important processes which inactivate the immune response against a tumor. Therefore, a strategy for preserving the immune response activity of CD8+ T-cells is to avoid the interaction between PD-1 and PD-L1. This is usually obtained with monoclonal antibodies (mAbs), which bind to PD-1 or PD-L1 and therefore avoid their interaction. However, the administration of efficacious concentrations of these mABs is usually associated with adverse related events, such as diarrhea, colitis or more serious effects like diabetes, which cause discontinuation of the treatment.4
Using nanostructures is a valid approach to solve these problems because they are platforms that can be engineered to give theragnostic responses, namely diagnostic and therapeutic responses, exploiting only one type of nanostructure.5–7
Surface-enhanced Raman scattering (SERS) nanostructures are plasmonic nanostructures with strong SERS signals derived from the presence of the so-called hot spots, in which molecules show an enhancement of the Raman signals of about one billion or more with respect to normal signals.8 These types of nanostructures were not previously considered for targeting PD-L1/PD-1.5–7 The narrowness of SERS signals, which are vibrational in nature, with respect to the fluorescence ones, and their huge intensities, comparable with those of fluorescence, make these nanostructures useful in a novel efficacious approach for imaging and diagnosis.9
Au nanostructures are among the most used plasmonic nanostructures because of their inertness in a biological environment. They can also be used for a therapeutic approach like photothermal therapy (PTT), since they are strong absorbers in the near-IR spectral region and can easily induce, when irradiated, an increase of the local temperature, at the level of a single cell, by as much as ten degrees, triggering cell death processes.10 Nanostructures constructed with Au nanoparticles are easily functionalized with thiols, exploiting the formation of stable sulfur–gold bonds. Using laser-ablated nanoparticles, which can be produced without any stabilizing ligands to obtain stable colloidal solutions,11 the functionalization is quickly completed and there are no problems of toxicity derived from the ligands.
Here SERS nanostructures will be functionalized with an engineered peptide to target the immune checkpoint PD-L1. These types of nanostructures were not previously synthesized.5–7 We will verify the activity of the nanostructures with in vitro tests using MDA-MB-231 breast adenocarcinoma epithelial cells, which overexpress PD-L1. The results will be rationalized with molecular dynamics calculations12 of the interaction of the peptide with the region of PD-L1 which binds to PD-1.
In principle, peptides present some drawbacks which, however, can be solved by using nanostructures. Peptides do not bind to antigens as strongly as antibodies. However, since the number of peptides on a nanoparticle is of the order of several thousand, the avidity of such structures rivals that of antibodies.13 Another problem with peptides, which are small parts of proteins, is that they can be easily degraded by enzymes in a biological environment. We have shown that when they are clustered on the nanoparticles, they are not degraded by enzymes that are unable to interact properly with them.14 Therefore, the targeting activity of peptide-functionalized nanostructures is preserved for a long time also in the presence of degrading enzymes.14
Peptides have important properties such as the absence of immunogenicity, a critical aspect of antibodies, and lower manufacturing costs with well standardized and reproducible procedures.
The results show that the SERS nanostructures functionalized with engineered peptides are an important tool for targeting specific antigens on the cell surface and are therefore very useful as a platform for immunotherapeutic approaches.
2. Results and discussion
2.1. Design and synthesis of the engineered peptides for PD-L1 targeting
Peptides have advantages over both small molecules and antibodies. In addition to their low toxicity and high fidelity of synthesis, their sizes allow them to bind more easily to a relatively flat surface of the protein–protein interface and cover a much wider portion of the interacting area, which translates into more potent and selective inhibition.Among the peptide and peptidomimetic inhibitors targeting PD-L1 with high affinity,15,16 we selected the dodecapeptide CLP002 (Table 1) identified by bacterial surface display methods.17 This peptide exhibits high affinity and specificity to the human PD-L1 protein. It was also shown to block the CD80/PD-L1 interaction and inhibit tumor growth, increasing the survival of CT26 tumor-bearing mice.17
| Peptide/ligand | Sequencea |
|---|---|
| a Standard abbreviation for natural amino acids; Ac: acetyl group; O2oc: –NH(CH2CH2O)2CH2–CO–. | |
| CLP002 | Trp-His-Arg-Ser-Tyr-Tyr-Thr-Trp-Asn-Leu-Asn-Thr |
| sCLP002 | Thr-Arg-Trp-Ser-His-Tyr-Asn-Thr-Leu-Trp-Tyr-Asn |
| L1 | Ac-Cys-(O2oc)2-CLP002-NH2 |
| L2 | Ac-CLP002-(O2oc)2-Cys-NH2 |
| L3 | HS-PEG-Lys3-Gly2-CLP002-NH2 |
| L4 | HS-PEG-Lys3-Gly2-sCLP002-NH2 |
As a targeting unit, CLP002 exhibits several advantages over other peptide inhibitors of PD-L1 such as the unbranched structure and the lack of unnatural amino acids or internal cysteine residues, which could be involved in binding the gold nanoparticles used as nanostructure scaffolds (see below). Adding an extra cysteine to the N- (L1 in Table 1) or C- (L2 in Table 1) terminal end of the peptide sequence allows control of the orientation of the peptide on the gold nanostructures, which can affect the targeting efficiency.13 Ligands L1 and L2 were also designed with a short oligoethylene linker between the CPL002 sequence and the last cysteine residue to enhance the mobility of the targeting peptide bound to the nanoparticles.
The shielding effect of polymers like polyethylene glycol (PEG) on the nanoparticles is well known and has been exploited to reduce non-specific interactions in complex biological media, providing better pharmacokinetic profiles.18 Moreover, we have recently shown that the targeting peptide linked to PEG on the nanoparticles is protected against proteolytic enzymes and does not undergo rapid degradation common to natural peptides.14 Therefore, to obtain biocompatible nanoparticles, CLP002 was also linked to a thiolated PEG3000 chain (L3 in Table 1) through a cationic pentapeptide spacer (KKKGG) that fosters the exposition of the targeting peptide on the nanoparticle surface.12 To assess the specificity of the CLP002 sequence in PD-L1 recognition, a ligand (L4 in Table 1) similar to L3 but with a scrambled sequence of the targeting peptide (sCLP002 in Table 1) was also prepared.
All ligands were synthesized by the solid phase peptide synthesis method. Their characterization by analytical high performance liquid chromatography and mass spectrometry (HPLC-MS) is reported in the ESI.†
2.2. Construction of the SERS nanostructures and their functionalization
The SERS nanostructures used in the present work are plasmonic nanostructures of aggregated gold nanoparticles synthesized by laser ablation in solution.Nanoparticles were obtained by the ablation of bulk gold under a micromolar water solution of NaCl using 8 ns laser pulses at 1064 nm, the fundamental frequency of a Nd:YAG laser.11 As previously shown, ablation using a focused beam with a fluence of 2 J cm−2 allows easy synthesis in a solution (LASiS) of spherical nanoparticles with an average dimension of 20 nm and a negative surface charge due to the partial oxidation of surface gold atoms.19 The colloidal solution is very stable without the presence of stabilizing molecules due to the Coulomb interaction. The functionalization of these nanoparticles is very easy because without the presence of a stabilizing coating, it is a direct process that does not need the exchange of stabilizing molecules with those of interest.
The SERS effect produces a huge enhancement of the vibrational Raman signal of molecules in proximity to the surface of metallic nanoparticles due to the excitation of their localized plasmon resonance.20 One can calculate the enhancement, which is very large, in particular, near the contact space between the aggregated nanoparticles (hot spots) where enhancement factors also of one billion are found.8 Aggregation of the nanoparticles can be obtained with a Raman-active thiolated molecule, which remains trapped within the hot spots and can be used as a SERS reporter. This is called electromagnetic enhancement, the main effect for SERS with respect to the chemical enhancement.20 Further enhancement derives from the resonance with the electronic transitions of the SERS reporter. That is why a tiny quantity of a thiol derivative of Texas Red (TR-SH) was used as a SERS reporter.
The functionalization of the nanostructures is easy because the surface of the nanoparticles is clean. In the first step, we functionalized the nanoparticles obtained by LASiS with a very small amount of TR-SH (see the Experimental section) to induce aggregation and produce the SERS nanostructures. In the second step, the nanostructures were functionalized with the engineered peptides exploiting their thiol function for binding to the gold surface.
We evaluated the average number of peptides per nanoparticle from the difference of the UV absorption spectra at 280 nm of the incubation solution of the ligands and that of the supernatant solution recovered after the functionalization and centrifugation of the nanostructures. We estimated that, on average, several thousand engineered peptides functionalized the nanoparticles: 6500 peptides/nanoparticles for L1@SERS-NS, 5700 for L2@SERS-NS, 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 for L3@SERS-NS and 2600 for L4@SERS-NS, where Ln@SERS-NS indicates the SERS nanostructure functionalized with the Ln peptide (see Table 1).
500 for L3@SERS-NS and 2600 for L4@SERS-NS, where Ln@SERS-NS indicates the SERS nanostructure functionalized with the Ln peptide (see Table 1).
Fig. 1 shows, as an example, the UV-vis-NIR spectra of the Au nanoparticle starting solution and that of the final solution for L3@SERS-NS. This spectrum is representative of all the other Ln@SERS-NS samples because all these spectra are dominated by the extinction spectra of the similar aggregation of Au nanoparticles.
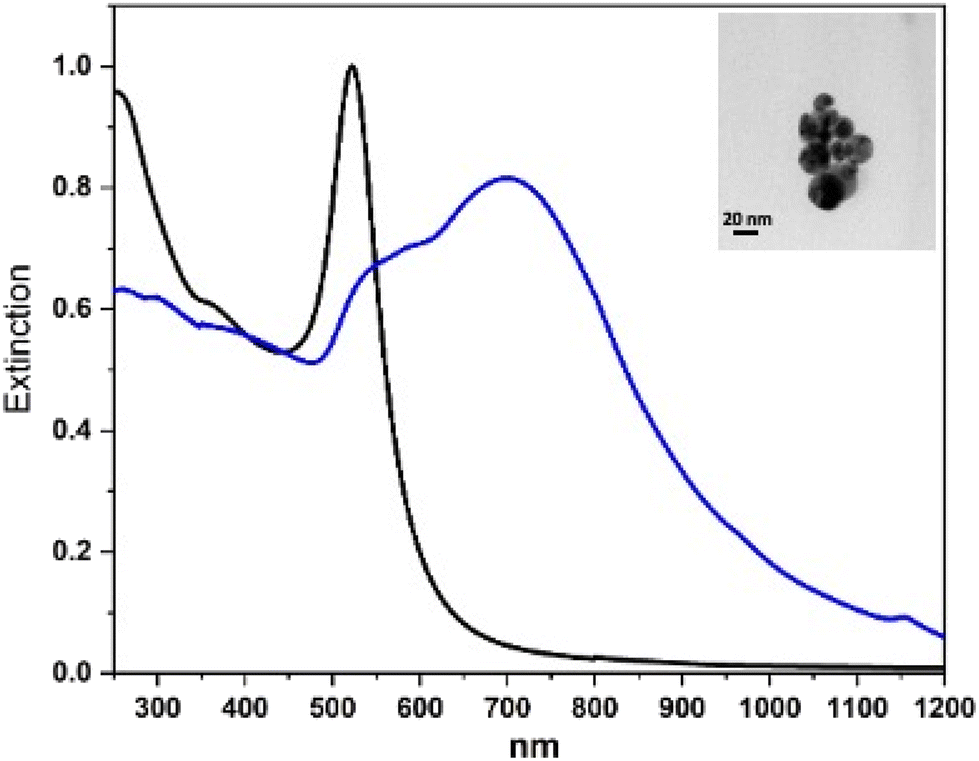 | ||
| Fig. 1 UV-vis-NIR spectra of LASiS nanoparticles (black line, normalized at 522 nm) and L3@SERS-NS after the functionalization of SERS-NS with the engineered peptide L3 (blue line, normalized in the gold interband transition region with that of the LASiS nanoparticles). Inset: TEM image of a SERS nanoaggregate. | ||
One notes that the sharp extinction peak at 522 nm of the nanoparticles of the ablated solution transforms into a broader structure for the aggregated nanoparticles centered at about 700 nm with a shoulder at about 530 nm and a fast decreasing extinction toward 1200 nm. The sharp peak of the nanoparticles of the ablated solution can be fitted with the calculated spectrum of isolated nanoparticles with an average diameter of 20 nm and this makes it possible to evaluate their concentration, whereas the broader one can be fitted with that of an ensemble of aggregated nanostructures with up to 10 nanoparticles.8 For the SERS nanostructures, one can also predict8 a huge enhancement of the SERS signals up to 109–1010. The nanoparticles usually aggregate into more or less rounded nanostructures with the largest dimension of the order of 100 nm.12,13
The SERS spectrum of L3@SERS-NS is presented in Fig. 2 as a representative of all Ln@SERS-NS spectra, which are always dominated by the SERS spectrum of the SERS reporter, TR-SH, characterized by two bands at 1505 and 1649 cm−1 originating from the normal mode of vibrations with important contributions of C–N and C–C stretching vibrations, respectively. These signals are used to identify the presence of the nanostructures in the incubated cells, which appear when the peptides have targeted the antigen on the cell surface and, therefore, are not washed out in the washing steps of the protocol.
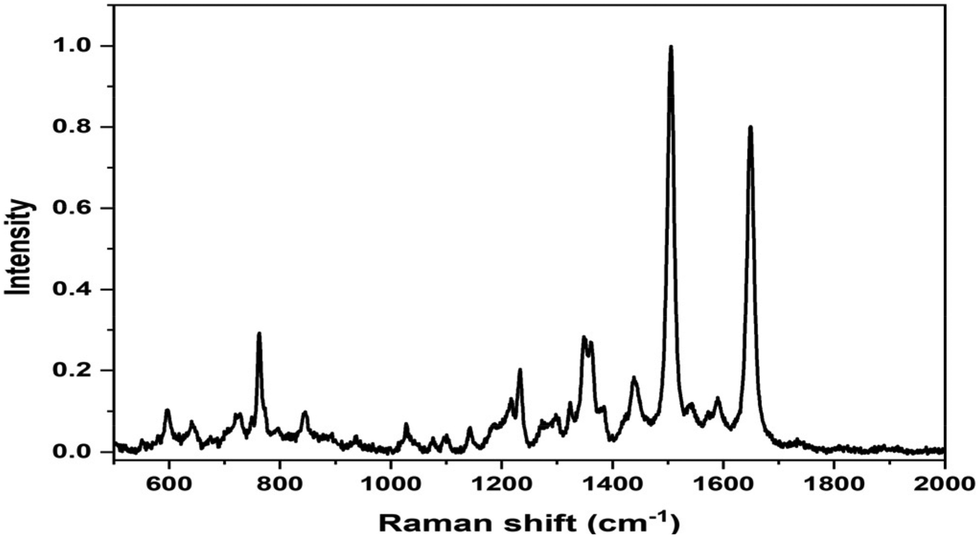 | ||
| Fig. 2 SERS spectrum of L3@SERS-NS where the dominant features are those of the SERS spectrum of TR-SH at 1505 and 1649 cm−1. | ||
2.3. Molecular dynamics calculations of the interaction of the targeting peptide with PD-L1
Detailed information on the interaction of CLP002 with PD-L1 was obtained by molecular dynamics (MD) simulations. A study of the interaction of the scrambled peptide (sCLP002) with PD-L1 was also performed. In both cases, the starting peptide structure was the most stable conformation obtained by MD simulations of a single peptide in water solution. For CLP002, the MD simulations showed the presence of a main ensemble of calculated structures with 90% of all the obtained conformations. The mean conformation (Fig. 3a) shows an elongated shape stabilized by one intramolecular hydrogen bond between Asn9 and Asn11 and many π–π interactions between the side chains Trp1, His2, and Tyr5, as well as between Tyr6 and Trp8. In the case of the sCLP002 peptide, only one ensemble (98% of total conformations) was obtained, characterized by a hairpin-like structure stabilized by four hydrogen bonds (Fig. 3b). Differences in the adopted secondary structure were confirmed from both the CD and FTIR spectra (Fig. S3 in the ESI†). Scrambling the primary sequence of peptides leads to interesting changes in the conformations adopted by the peptide.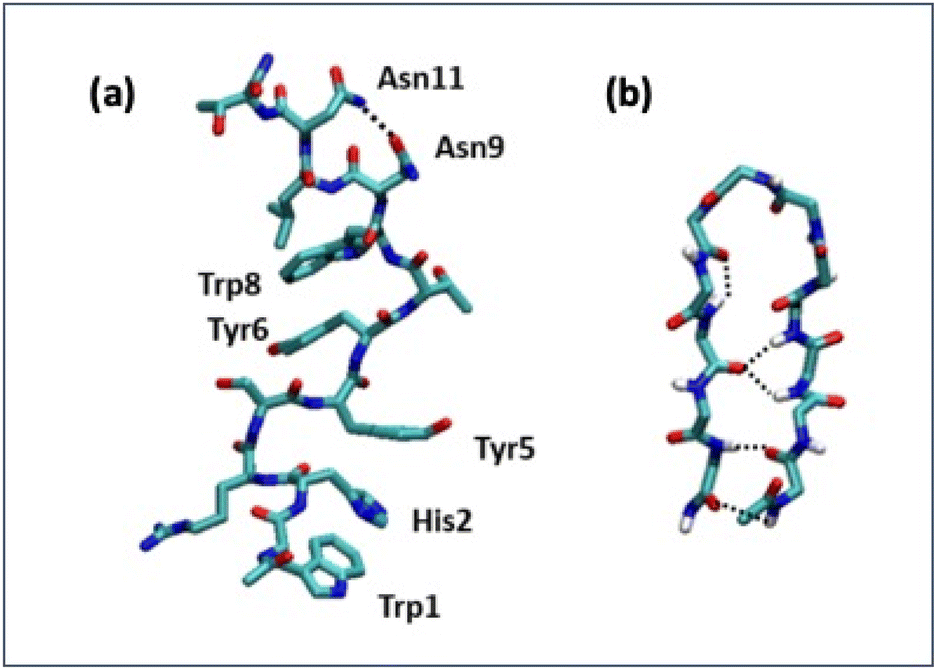 | ||
| Fig. 3 Stable conformers of CLP002 (a) and sCLP002 (b) in water. | ||
To perform the MD simulations of the PD-L1/peptide system, the X-ray structure of PD-L121 was used as the starting conformation of the protein, whereas the conformation reported in Fig. 3 was adopted for the peptides. For each peptide, three replicas of 100 ns were performed; in all cases, the starting distance between the peptide and the protein was greater than 1.4 nm and non-bonded interaction cutoffs were used.
The binding between PD-L1 and peptides was studied, in particular, in the region of contact between PD-L1 and PD1, as obtained from the X-ray structure present in the protein data bank.21 The contact region shows both a hydrophobic part and a hydrophilic part (Fig. S4 in the ESI†).
To estimate the binding of peptides to PD-L1, the solvent accessible surface (SAS) of PD-L1 in the presence of the peptide was calculated during the simulation time (Fig. 4a).
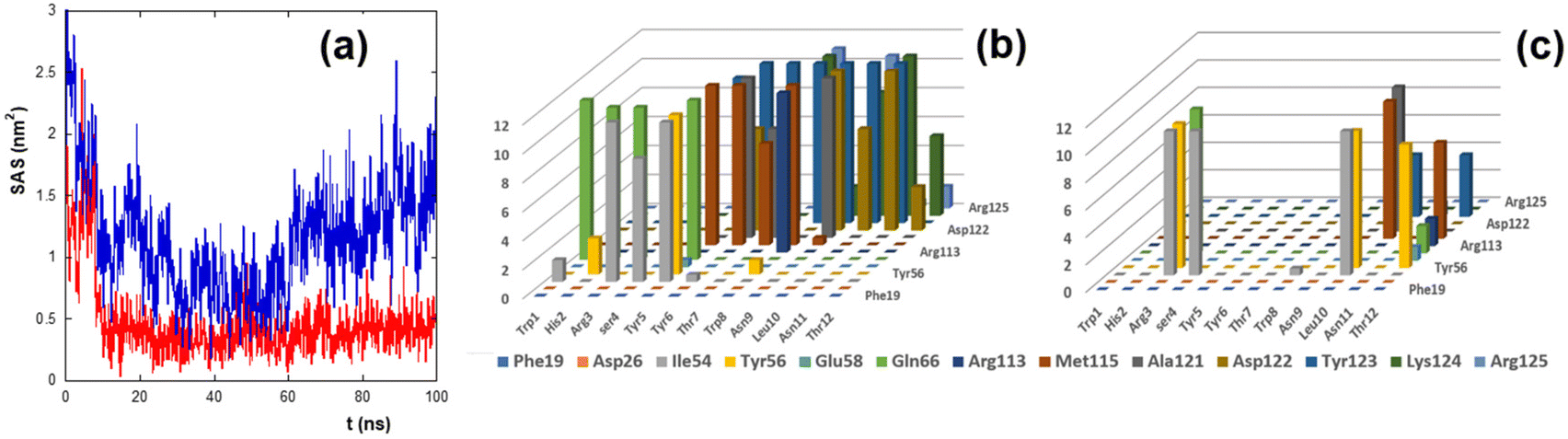 | ||
| Fig. 4 (a) Time evolution of the solvent accessible surface (SAS) of the hydrophobic residues of the PD-L1 contact region. The red and blue lines refer to the SASs of CLP002 and sCLP002, respectively. (b, c) Persistence time (ns, z-axis) of the residues of the contact region between PD-L1 and CLP002 (b) or sCLP002 (c). The peptide and PD-L1 residues are presented on the x-axis and y-axis, respectively. | ||
The SAS of the hydrophobic residues of the PD-L1 contact region decreases strongly after 100 ns in the presence of CLP002 (red line) but not in the presence of sCLP002 (blue line), indicating that the former realizes a hydrophobic core similar to that of the PD-L1/PD-1 adduct. This result strongly suggests that CLP002 is more efficient than sCLP002 in preventing the binding of PD-1 to PD-L1.
The interaction of the PD-L1/peptide adduct was also analyzed in terms of the persistence time of the contacts (distance less than 5 Å) between the protein and the peptide residues in the last 10 ns of the simulations. As shown in Fig. 4b and c, all residues in the contact region of PD-L1 interact with those of CLP002 (Fig. 4b), whereas only some of them interact with sCLP002 (Fig. 4c).
The picture of PD-L1 interacting with CLP002, shown in red, after 100 ns is presented in Fig. 5 and shows that the elongated form of CLP002 fits to the PD-L1 contact region, giving the reason for the result reported in Fig. 4a.
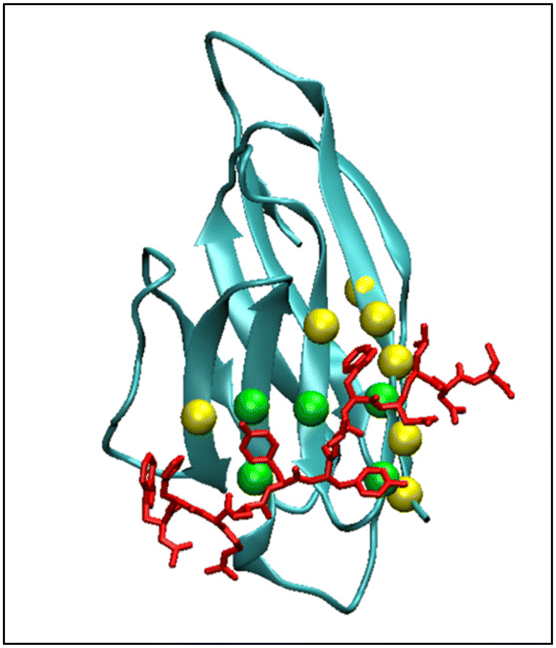 | ||
| Fig. 5 MD simulation frame at 100 ns showing the binding of PD-L1 to CLP002. The spheres indicate the residues of PD-L1 that bind to PD-1. The residues involved in the hydrophobic core of the PD-L1/PD-1 adduct are colored green. | ||
The other two replicas of PD-L1/CLP002 (see Fig. S5, ESI†) allowed results like those reported in Fig. 5 to be found, demonstrating the good targeting of PD-L1 by CLP002.
These results predict that a strong targeting activity difference between L3@SERS-NS and L4@SERS-NS should be observed.
2.4. Targeting of the PD-L1 immune checkpoint
MDA-MB-231 breast adenocarcinoma cancer cells express a high level of PD-L1 and were used as a model for verifying the targeting activity of the prepared SERS-NS. The cells were treated in a well with different concentrations of Ln@SERS-NS for 2 h. Subsequently, the cells were fixed and washed before recording the SERS signals of single cells. The SERS signals indicate the presence of the nanostructures and therefore that a targeting activity occurred. The SERS signals were recorded with a micro-Raman instrument (Renishaw InVia), which allows recording the SERS signals of single cells. 100 or more cells were considered for each experiment. An excitation at 633 nm with a He–Ne laser was used because the SERS spectrum of TR-SH is strong with such an excitation. All the spectra were analyzed by finding the Pearson correlation coefficient for each spectrum using a reference SERS spectrum of TR-SH. A plot of the intensity of the main peak of the SERS spectrum at 1505 cm−1 against the Pearson correlation coefficient (see Fig. 6) allowed a Pearson correlation coefficient threshold of 0.8 to be chosen as an indication of an efficient targeting.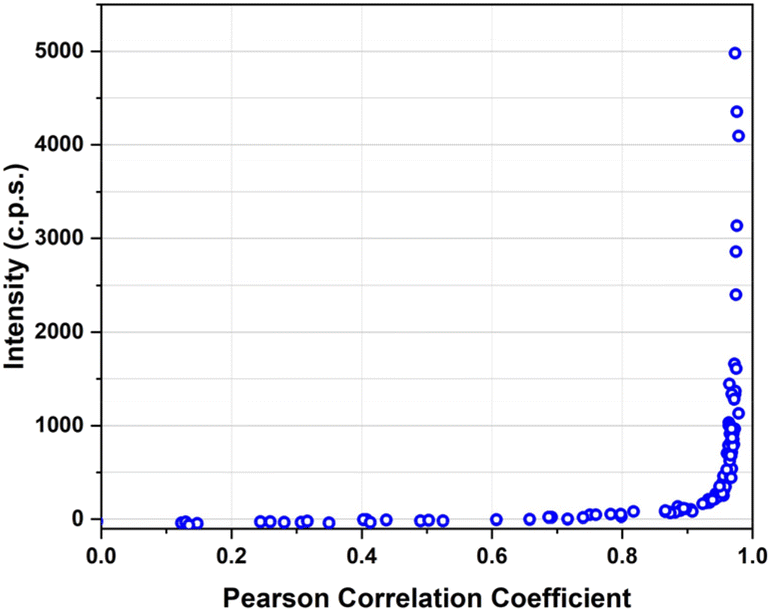 | ||
| Fig. 6 Intensity of the main peak of TR-SH at 1505 cm−1 against the Pearson correlation coefficient of the spectrum for all the spectra recorded in single cells of MDA-MB-231 incubated with a solution of L3@SERS-NS at a concentration of 100 pM. | ||
The incubation solutions were obtained with decreasing concentrations of Ln@SERS-NS and the percentage of cells with a SERS spectrum above the threshold was determined.
We used the same procedure for all the Ln@SERS-NS samples.
The results of all the incubation solutions with all the Ln@SERS-NS samples are reported in Fig. 7.
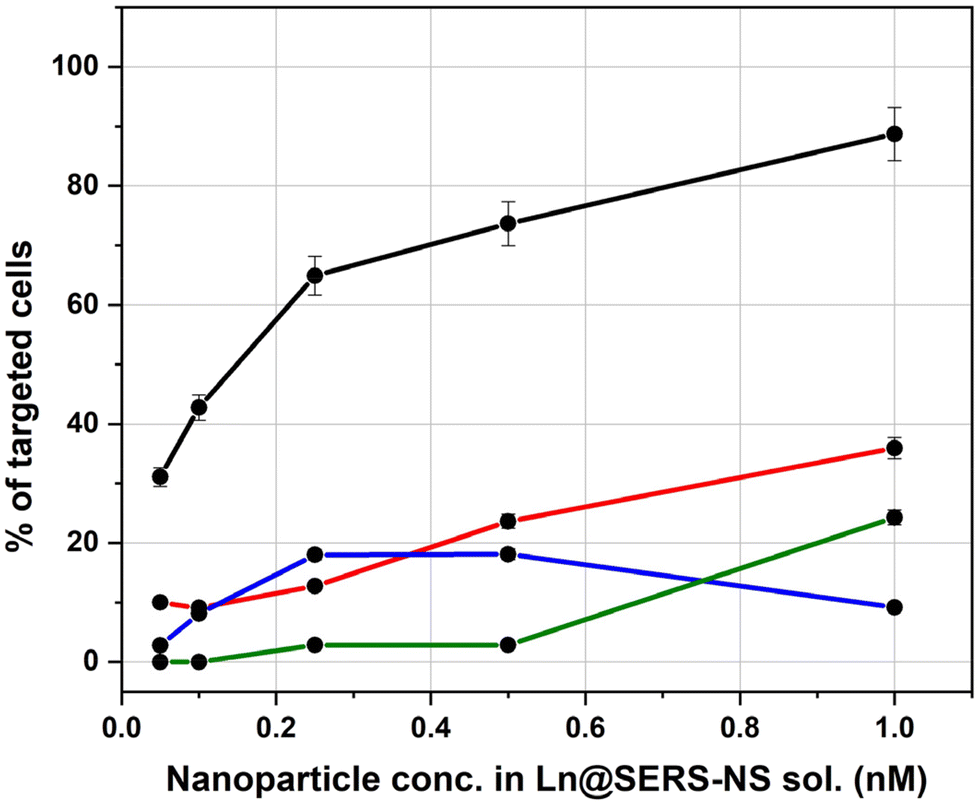 | ||
| Fig. 7 Activity of Ln@SERS-NS for the targeting of MDA-MB-231 overexpressing PD-L1. The % of the cell with a positive targeting, namely with a Pearson correlation coefficient above the threshold of 0.8, is plotted against the concentration of Ln@SERS-NS used for incubation (L1@SERS-NS: red line, L2@SERS-NS: blue line, L3@SERS-NS: black line, and L4@SERS-NS: green line). Concentrations refer to the nanoparticles present in the solutions. Error bars refer to the variability observed by recording the SERS signals within single cells. | ||
It is clear that L3@SERS-NS has a good targeting activity with respect to the other Ln@SERS-NS samples.
These results show different important characteristics of the engineered peptides. Comparison of the curves for L1@SERS-NS and L2@SERS-NS with that for L3@SERS-NS shows the importance of the presence of the PEG chain and the charged amino acid sequence made from three lysines and two glycines (KKKGG). The peptides present in L1@SERS-NS and L2@SERS-NS are equal, but in one case the peptide is linked to the gold surface through the N-terminus and in the other case through the C-terminus (see Table 1). This makes part of the peptide more or less available for the interaction with PD-L1. However, the poor targeting behaviors of both L1@SERS-NS and L2@SERS-NS in Fig. 7 are similar. The very good targeting result observed for L3@SERS-NS, in which the peptide is the same, shows that the presence of the PEG chain and the short charged amino acid sequence strongly enhances the targeting activity of the nanostructure, reaching almost 90% of cell targeting.
The importance of the amino acid sequence of the peptide can be seen by comparing the behaviors of L3@SERS-NS and L4@SERS-NS. These two nanostructures have the same design ((SERS-NS)–PEG–KKKGG–peptide), but differ for the sequence of amino acid of the peptide, which is randomized (scrambled) in L4@SERS-NS. The low activity of L4@SERS-NS shows the importance of an appropriate amino acid sequence of the targeting peptide of L3@SERS-NS.
These results confirm the conclusions of MD calculations, which predicted that the amino acid sequence of CLP002, and not that of sCLP002, was appropriate for the targeting of PD-L1.
The above results can be compared also with those of other nanostructures used for targeting other antigens, like EGFR13,14,22 and integrins.12 In these examples, the presence of the PEG chain and the charged sequence is always beneficial for the targeting activity, showing that the sequence (SERS-NS)–PEG–KKKGG–peptide is a general structure for obtaining a good targeting activity using peptides as targeting units.
3. Conclusions
Immune check points are important protein structures for immune therapies. Among them, PD-L1, expressed by many tumor cells, is a check point with important properties because it deactivates the killer activity of CD8+ T-lymphocyte cells. Thus, PD-L1-targeting molecules allow its activity to be blocked, preserving the T-cells’ killer function. The SERS nanostructures functionalized with an engineered peptide are shown to be good candidates to block PD-L1 activity. Molecular dynamic calculations helped in understanding the activity of the studied peptide in targeting the site of the interaction of PD-L1 with PD-1 and also showed the poor activity of a scrambled version of the same peptide. Incubation with MDA-MB-231 breast adenocarcinoma cancer cells, which express a high level of PD-L1, allowed quantification of the targeting activity of the functionalized SERS nanostructures exploiting the SERS signals of the nanostructures. It is found that the peptide modified with a short sequence of charged amino acids and a long PEG chain linked to the SERS nanostructures is a very good structure for blocking PD-L1.The results also allow us to conclude, from the present and previous studies,12–14,22 that structures obtained by sequentially binding to SERS nanostructures a PEG chain, a charged short peptide sequence and the active targeting peptide ((SERS-NS)-PEG-KKKGG-peptide) are good for exploiting the peptide's activity against tumor antigens.
4. Experimental section
4.1. Materials and Instrumentation
Unless differently specified, all chemicals were commercial products and used without further purification. 9-Fluorenylmethoxycarbonyl (Fmoc) amino acids (Sigma-Aldrich), Rink Amide AM LL resin (loading 0.35 mmol g−1, Novabiochem) and chemicals for the solid phase peptide synthesis were provided by Merck KGaA. Rink Amide MBHA resin (loading 0.52 mmol g−1) and Fmoc-8-amino-3,6-dioxaoctanoic acid (Fmoc–O2oc–OH) were purchased from Iris Biotech GmbH. S-Trityl-thioPEG N-hydroxysuccinimide ester (TrtS–PEG–NHS, MW of around 3000 Da) was provided by Rapp Polymer. Sulforhodamine 101-bis-cysteamide (TR-SH) was used as a SERS reporter.23 Bovine serum albumin (BSA) was purchased from Sigma-Aldrich.Unless otherwise indicated, peptides and conjugates were analysed using an Agilent 6100 Series single quadrupole LC/MS system coupled on-line via a UV-vis detector (detection at 220 and 280 nm) to the electrospray ionization (ESI) source and operating in positive mode. Samples were separated using a Kinetex column (100 × 4.6 mm, 3.5 μm XB-C18) combining mobile phases A (aqueous 0.1% trifluoroacetic acid (TFA)) and B (0.1% TFA in acetonitrile) to form binary gradients. Semi-preparative HPLC was performed using a Shimadzu series LC-6A chromatographer equipped with two independent pump units, a UV-vis detector and a Jupiter column (250 × 10 mm, 10 μm). Elutions were carried out with the same mobile phases described above using binary gradients. Mass spectral analysis of L3 and L4 was performed using a Xevo-G2S Q-TOF instrument (Waters, Milford, MA, USA) operating in positive mode with the ESI technique.
UV-vis spectra were recorded with a Cary5000 spectrometer (Agilent) in 0.2 or 1 cm quartz cells. DLS and zeta-potential measurements were performed with a Malvern Nano-ZS instrument equipped with a 633 nm He–Ne Laser. SERS spectra were recorded with a Renishaw inVia micro-Raman spectrometer under excitation at 633 nm (0.8 mW) with a 20× objective for 1 s. SERS spectra of the colloidal solutions were recorded with a 10× objective for 10 s.
Fourier-transform infrared (FTIR) absorption spectra of the peptide powders were recorded with a Thermo Fisher FTIR is50 spectrometer (Thermo Fisher Scientific Co., Madison, WI, USA) in attenuated total reflection (ATR) mode using a diamond single reflection cell. Each spectrum was obtained by mediating over 64 scans with a resolution of 2 cm−1. Circular dichroism experiments were performed using a Jasco-1500 CD spectrometer (Jasco, Tokyo Japan) using quartz cuvettes of 1 mm optical length in the far UV region (spectral range: 200–260 nm). Each spectrum was obtained by mediating over 4 scans. For both peptides, 100 μM water solution was used.
4.2. Synthesis of peptides and conjugates
Peptides were synthesized on an Advanced Chemtech 348 Ω peptide synthesizer starting from Rink amide MBHA resin (0.05 mmol) or Rink amide AM resin (0.03 mmol). The side chains of Fmoc-amino acids were protected by the tert-butyl group (threonine and tyrosine), the tert-butyloxycarbonyl group (tryptophan), the trityl group (asparagine, histidine and cysteine) and the 2,2,4,6,7-pentamethyl-dihydroxybenzofuran-5-sulfonyl group (arginine). Couplings were carried out in DMF for 45–60 min using an excess of the Fmoc-amino acid (4 eq.) and in the presence of N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl)uroniumhexafluorophosphate, N-hydroxybenzotriazole and N,N,N-ethyldiisopropylamine (4:4:12 eq.). Fmoc deprotection was achieved with 20% piperidine in DMF (5 + 15 min). PEG conjugates (L3 and L4) were prepared by reacting TrtS–PEG–NHS (0.012 mmol in 0.5 mL of DMF) with the amino terminal group of the peptide (0.003 mmol) still attached to the solid support. The mixture was stirred for 24 h at RT; after that the solution containing the unreacted PEG was filtered off and the resin was repeatedly washed with DMF and DCM. Cleavage and deprotection were carried out by treating the dry peptide resin with a TFA:triisopropylsilane:H2O:1,2-ethanedithiol (94:1:2.5:2.5 v/v/v/v) mixture for 90 min at room temperature. The resin was filtered off, the filtrate was reduced to a small volume and the crude peptide was precipitated by the addition of cold diethyl ether. After purification by semi-preparative HPLC, the peptides and the PEG conjugates were characterized as shown in Table S1 and Fig. S1 and S2 of the ESI.†
4.3. Synthesis of the functionalized SERS nanostructures
The synthesis of the gold nanoparticles was performed by laser ablation of a pure gold target (99.9%) in a diluted solution of NaCl in water (10 μM) with 6 ns pulses and a 10 Hz repetition rate using a Nd:YAG laser at 1064 nm with a fluence of 2 J cm−2. The solution was stirred during the ablation. TEM images show (in the inset in Fig. 1) that the nanoparticles have a diameter of about 20 nm, as previously reported.23 The nanoparticles are negatively charged due to the oxidation of a small portion of the gold surface that occurred in the plasma environment of ablation.19 A negative ζ-potential of −30 mV is usually measured and creates a very good stability of the colloidal solution.23Fitting the localized surface plasmon resonance at 522 nm allows the nanoparticle concentration in the colloidal solution to be obtained,8 which is of the order of 2–5 nM.
The SERS nanostructures were obtained by mixing 5 μL of a 35 μM solution of TR-SH in methanol with 2 mL of the colloidal solution of gold nanoparticles. One observes a change of the color of the solution from red to dark violet caused by the aggregation of the nanoparticles. Centrifugation at 3000 rcf allowed the excess of the SERS reporter to be discarded.
The functionalization with the engineered peptides was performed by mixing the solution of SERS-NS with a mM solution of the peptides and stirring the solution for 1 hour at room temperature. The presence of a thiol group on the engineered peptide allowed it to be linked to the gold surface of the nanostructures. Concentrations of the nanostructure solutions are given with the nanoparticles present in the solutions, obtained by comparing the extinction spectra in the interband spectral transitions region at about 400 nm of the final solution used for the incubations with the initial one whose concentration was obtained by fitting the spectrum with BEM calculations.8 Centrifugation at 3000 rcf was used to discard the excess peptide. The absorption difference of the peptide solution before and after the functionalization of the nanostructures was used to determine the number of peptides per nanoparticle. Dynamic light scattering allowed the evaluation of the hydrodynamic nanostructure dimensions to be of the order of 160 nm and ζ-potentials of about −20 mV.
4.4. Molecular dynamics simulations
Molecular dynamics (MD) simulations were carried out using the GROMACS v5.0.7 software package24 and the gromos53A6 force field25 parameters were used for the peptide, as already successfully used for similar systems.12–14,22 A constant pressure and temperature ensemble (NPT) was used for all simulations and periodic boundary conditions were applied. Following our protocol, there was a two-step energy minimization. In the first step, only the solvent was energy minimized, whereas in the second step the solute (peptide and protein) was considered.After equilibration for 150 ps at 300 K (time step of 0.5 fs), production runs of 100 ns were performed with a time step of 2 fs. The particle mesh Ewald algorithm (PME)26,27 was used for electrostatic interactions (cutoff = 1.4 nm).
Explicit simple point charge (SPC)28 water molecules were used to solvate the simulation box and a chloride ion (in the presence of the peptide only) or two sodium ions (for the PD-L1/peptide system) were added to ensure electroneutrality. A cutoff distance was used for the van der Waals interactions (1.4 nm). In all the simulations, the velocity rescale scheme29 was used to keep the temperature constant (σT = 0.6 ps) and the Berendsen algorithm30 was used under isotropic conditions for pressure coupling (σP = 1 ps).
For the peptide MD simulations, a cubic box of 6.3 × 6.3 × 6.3 nm3 containing 8065 water molecules was used. For studying the PD-L1/peptide complex, a cubic box of 7.3 × 7.3 × 7.3 nm3 containing 12054 water molecules was used.
The conformational clusters were calculated using the Gromacs ‘cluster’ tool by applying a cutoff of 0.13 nm for the root mean square deviation, while the solvent accessible surface (SAS) was obtained using the Gromacs ‘sasa’ tool.
The Visual Molecular Dynamics (VMD) program31 was used for structure visualization.
4.5. In vitro targeting
MDA-MB-231 breast cancer cells expressing a high level of PD-L1 were grown in T75 flasks until about 80% confluency. The cells were detached with trypsin, washed with PBS and about 10,000 cells were plated in 8 well-chamber slides. After 24 h, the cells were incubated with Ln@SERS-NS solutions, in which the concentrations of nanoparticles were 0.05, 0.1, 0.25, 0.5 and 1 nM for 2 h at 37 °C. After 2 h, the cells were washed twice with PBS, then immediately fixed with 2% paraformaldehyde for 1 h and dried.SERS measurements were performed with a Renishaw inVia micro-Raman spectrometer using a 20× objective and under excitation at 633 nm with a He–Ne laser. Each spectrum was collected for 10 s in a static configuration recording the spectral region at around 1500 cm−1 where the two main peaks of the SERS reporter are present. A Matlab code was used for the determination of the Pearson correlation coefficient of each spectrum after subtraction of the spectral background.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The project P-DiSC#06BIRD2020-UNIPD is acknowledged. The authors thank Filippo Pannocchia and Claudia Bravo for their help in different steps of the work. Fabrizio Mancin is acknowledged for making available the Malvern Nano-ZS instrument.References
- R. Mahjub, S. Jatana, S. E. Lee, Z. Qin, G. Pauli, M. Soleimani, S. Madadi and S. D. Li, J. Controlled Release, 2018, 288, 239–263 CrossRef PubMed.
- Y. Jiang, Y. Li and B. Zhu, Cell Death Dis., 2015, 6, e1792 CrossRef CAS PubMed.
- W. P. Zou and L. P. Chen, Nat. Rev. Immunol., 2008, 8, 467–477 CrossRef CAS PubMed.
- C. Boutros, A. Tarhini, E. Routier, O. Lambotte, F. L. Ladurie, F. Carbonnel, H. Izzeddine, A. Marabelle, S. Champiat, A. Berdelou, E. Lanoy, M. Texier, C. Libenciuc, A. M. M. Eggermont, J. C. Soria, C. Mateus and C. Robert, Nat. Rev. Clin. Oncol., 2016, 13, 473–486 CrossRef CAS PubMed.
- N. Q. Gong, N. C. Sheppard, M. M. Billingsley, C. H. June and M. J. Mitchell, Nat. Nanotechnol., 2021, 16, 25–36 CrossRef CAS PubMed.
- X. Yu, C. Fang, K. Zhang and C. X. Su, Pharmaceutics, 2022, 14, 1581 CrossRef CAS PubMed.
- Y. Moon, M. K. Shim, J. Choi, S. Yang, J. Kim, W. S. Yun, H. Cho, J. Y. Park, Y. Kim, J. K. Seong and K. Kim, Theranostics, 2022, 12, 1999–2014 CrossRef CAS PubMed.
- L. Litti and M. Meneghetti, Phys. Chem. Chem. Phys., 2019, 21, 15515–15522 RSC.
- L. Litti, A. Colusso, M. Pinto, E. Ruli, A. Scarsi, L. Ventura, G. Toffoli, M. Colombatti, G. Fracasso and M. Meneghetti, Sci. Rep., 2020, 10, 15805 CrossRef CAS PubMed.
- F. Bertorelle, M. Pinto, R. Zappon, R. Pilot, L. Litti, S. Fiameni, G. Conti, M. Gobbo, G. Toffoli, M. Colombatti, G. Fracasso and M. Meneghetti, Nanoscale, 2018, 10, 976–984 RSC.
- V. Amendola and M. Meneghetti, J. Mater. Chem., 2007, 17, 4705–4710 RSC.
- F. Biscaglia, G. Ripani, S. Rajendran, C. Benna, S. Mocellin, G. Bocchinfuso, M. Meneghetti, A. Palleschi and M. Gobbo, ACS Appl. Nano Mater., 2019, 2, 6436–6444 CrossRef CAS.
- F. Biscaglia, S. Rajendran, P. Conflitti, C. Benna, R. Sommaggio, L. Litti, S. Mocellin, G. Bocchinfuso, A. Rosato, A. Palleschi, D. Nitti, M. Gobbo and M. Meneghetti, Adv. Healthcare Mater., 2017, 6, 1700596 CrossRef PubMed.
- F. Biscaglia, I. Caligiuri, F. Rizzolio, G. Ripani, A. Palleschi, M. Meneghetti and M. Gobbo, Nanoscale, 2021, 13, 10544–10554 RSC.
- J. Yang and L. Q. Hu, Med. Res. Rev., 2019, 39, 265–301 CrossRef CAS PubMed.
- X. Lin, X. Lu, G. S. Luo and H. Xiang, Eur. J. Med. Chem., 2020, 186, 111876 CrossRef CAS PubMed.
- H. Liu, Z. Zhao, L. Zhang, Y. K. Li, A. Jain, A. Barve, W. Jin, Y. L. Liu, J. Fetse and K. Cheng, J. Immunother. Cancer, 2019, 7, 270 CrossRef PubMed.
- J. S. Suk, Q. G. Xu, N. Kim, J. Hanes and L. M. Ensign, Adv. Drug Delivery Rev., 2016, 99, 28–51 CrossRef CAS PubMed.
- H. Muto, K. Yamada, K. Miyajima and F. Mafuné, J. Phys. Chem. C, 2007, 111, 17221–17226 CrossRef CAS.
- E. C. Le Ru and P. G. Etchegoin, in Principles of Surface-Enhanced Raman Spectroscopy, Elsevier, Amsterdam, 2009 Search PubMed.
- K. M. Zak, R. Kitel, S. Przetocka, P. Golik, K. Guzik, B. Musielak, A. Domling, G. Dubin and T. A. Holak, Structure, 2015, 23, 2341–2348 CrossRef CAS PubMed.
- C. Mazzuca, B. Di Napoli, F. Biscaglia, G. Ripani, S. Rajendran, A. Braga, C. Benna, S. Mocellin, M. Gobbo, M. Meneghetti and A. Palleschi, Nanoscale Adv., 2019, 1, 1970–1979 RSC.
- M. Meneghetti, A. Scarsi, L. Litti, G. Marcolongo, V. Amendola, M. Gobbo, M. Di Chio, A. Boscaini, G. Fracasso and M. Colombatti, Small, 2012, 8, 3733–3738 CrossRef CAS PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindah, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- C. Oostenbrink, A. Villa, A. E. Mark and W. F. Van Gunsteren, J. Comput. Chem., 2004, 25, 1656–1676 CrossRef CAS PubMed.
- D. M. York, T. A. Darden and L. G. Pedersen, J. Chem. Phys., 1993, 99, 8345–8348 CrossRef CAS.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS.
- H. J. Berendsen, J. P. Postma, W. F. Van Gunsteren and J. Hermans, Intermolecular Forces, Reidel Publ. Co., Dordrecht, 1981 Search PubMed.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- H. J. C. Berendsen, J. P. M. Postma, W. F. Van Gunsteren, A. Dinola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics Modell., 1996, 14, 33–38 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization of peptides, mass spectra, UV CD and IR spectra of peptides, and MD simulations. See DOI: https://doi.org/10.1039/d4nr00172a |
| This journal is © The Royal Society of Chemistry 2024 |