
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ultralight aerogels via supramolecular polymerization of a new chiral perfluoropyridine-based sulfonimidamide organogelator†
Giampiero
Proietti
a,
Anton
Axelsson
a,
Antonio J.
Capezza
 b,
Yogesh
Todarwal
a,
Julius
Kuzmin
a,
Mathieu
Linares
c,
Patrick
Norman
a,
Zoltán
Szabó
a,
Christofer
Lendel
a,
Richard T.
Olsson
b and
Peter
Dinér
*a
b,
Yogesh
Todarwal
a,
Julius
Kuzmin
a,
Mathieu
Linares
c,
Patrick
Norman
a,
Zoltán
Szabó
a,
Christofer
Lendel
a,
Richard T.
Olsson
b and
Peter
Dinér
*a
aDepartment of Chemistry, KTH Royal Institute of Technology, 10044 Stockholm, Sweden. E-mail: diner@kth.se
bDepartment of Fiber and Polymer Technology, KTH Royal Institute of Technology, 10044 Stockholm, Sweden
cPDC Center for High Performance Computing, KTH Royal Institute of Technology, 10044, Stockholm, Sweden
First published on 7th March 2024
Abstract
Chiral and enantiopure perfluorinated sulfonimidamides act as low-molecular weight gelators at low critical gelation concentration (<1 mg mL−1) via supramolecular polymerization in nonpolar organic solvents and more heterogenic mixtures, such as biodiesel and oil. Freeze-drying of the organogel leads to ultralight aerogel with extremely low density (1 mg mL−1). The gelation is driven by hydrogen bonding resulting in a helical molecular ordering and unique fibre assemblies as confirmed by scanning electron microscopy, CD spectroscopy, and computational modeling of the supramolecular structure.
Introduction
Small organic molecules can form supramolecular assemblies that grow into long, anisotropic, three-dimensional networks.1–7 The formation of these supramolecular polymers is driven by non-covalent secondary intermolecular interactions, such as hydrogen bonding, π–π interactions, and van der Waals forces. At a sufficiently high concentration, the organic molecules can form fibres that entangle into networks that immobilize the solvent (Fig. 1A).8–10 The spontaneous formation of these so-called organogels allows reversible assembly/disintegration of the network via energy input such as heating and light.11–16 Classical motifs for organogelators include derivatives of e.g., fatty acids,17,18 amino acids,19 cholesterols,20 sugars,21–23 and anthryl derivatives (Fig. 1B),24 but the development of new classes of organogelators remains an important task since functionalized materials of organogels have found use in many applications25–28 such as photoelectronics,29,30 drug delivery,31 and as scaffolds for cell cultures in tissue engineering. Sulfonimidamides (SIAs) are the aza-analogues of sulfonamides and were first described by Levchenko in 1960.32–35 In the last two decades, an increased interest in the use of sulfonimidamides as isosteres in pharmacological applications36–40 has led to the development of new methods for the synthesis of sulfonimidamides and their derivatives.41–53 Recently, we developed a synthesis of enantiopure sulfonimidamides via a light-mediated reaction between perfluoroaryl azides (PPFA) and sulfinamides (SA).54 | ||
| Fig. 1 (A) Simplified cartoon highlighting the process of supramolecular gelation. (B) Classic motifs for organogelators. (C) Synthesis of perfluorinated SIA-based gelators, highlighting critical functional groups for gelation (this work). | ||
Serendipitously, we discovered that one of the synthesized compounds, (S)-SIA, exhibited gel-forming properties as revealed by the gelation of the reaction mixture. Due to its ability to gelate nonpolar solvents, we set out to investigate the self-assembly properties of low-molecular weight gelators (LMWG, MW = 285 g mol−1) that are based on this novel chiral sulfonimidamide scaffold (S)-SIA.
In this study, both enantiomers, (S)-SIA and (R)-SIA, display reversibility in the assembly and disassembly of the supramolecular structure upon heating and cooling of the system. Scanning electron microscopy revealed the fibrous structure of the network and its different structural features depended on the solvents and drying technique used. Molecular dynamics simulation of the supramolecular assembly revealed the helical supramolecular structure, which was verified via a comparison between calculated and experimental circular dichroism (CD) spectra.
Results and discussion
In our quest to develop photo-mediated reactions between perfluorinated aromatic azides and chiral sulfinamides via nitrene intermediates, we discovered that one of the chiral sulfonimidamides, (S)-SIA, had the ability to gel the reaction medium (α,α,α-trifluorotoluene, CF3Ph) (Fig. 1C). Previously, many organogelators based on small, chiral organic molecules have been studied,55–62 but to the best of our knowledge, chiral sulfonimidamides have not previously been investigated as a scaffold for organogelators. Therefore, we set out to investigate the gelating properties, supramolecular structure, and functions of these sulfonimidamide compounds. The enantiomeric pairs of sulfonimidamide, (S)-SIA and (R)-SIA, were both able to form organogels in aromatic and aliphatic nonpolar organic solvents such as toluene, o-dichlorobenzene (ODCB), heptane, dodecane, cyclohexane, chloroform etc. (Fig. 2A and Fig. S2†). The critical gel concentration (CGC) was below 1 mg mL−1 for highly nonpolar aliphatic solvents, such as heptane, cyclohexane and dodecane (0.6–0.9 mg mL−1), while the CGC was one order of magnitude higher for aromatic solvents (7–14 mg mL−1) and even higher for the more polar chloroform (28 mg mL−1) (Fig. 2B). Gelation was induced either via thermal heating or by sonication63 and irrespective of the gelation technique, all gels formed were clear and transparent except for cyclohexane, which formed an opaque gel at higher concentrations (Fig. S2†). The SIA-organogelator also formed gels of mixed nonpolar liquids such as kerosene, biodiesel (NExBTL), gasoline (95) and motor oil, indicating its potential use as a potential phase-selective gelator of oil-spill remediations (Fig. S2†).64–67 Solvents containing hydrogen-bond acceptors, such as ethyl acetate, tetrahydrofuran, and alcohols (ethanol, isopropyl alcohol, 1-octanol, oleyl alcohol), led to dissolution and no gel formation was observed even at higher concentrations (0.1 M). The gelation correlates with the hydrogen bonding term in the Hansen solubility parameters (HSP) for the investigated solvents, while no clear correlation was found for the dielectric constant (cf. PhCF3 and EtOAc) suggesting that intermolecular hydrogen bonds play a pivotal role in the formation of the gels. Several features in the sulfonimidamide are critical for its gelating properties. Subtle modifications of its structure, such as methylation of the primary amino group, replacement of the t-butyl group with a tolyl group, and removal of the pyridine nitrogen from the perfluorinated ring led to complete loss of the gelation abilities (see Fig. S3†). The rheological properties of the gel in heptane were investigated using dynamic shear oscillation. The storage modulus (G′) and loss modulus (G′′) of the gel were monitored in the frequency range from 0.001 to 100 Hz (Fig. 2B). The results show that the sample of (S)-SIA had a stable storage modulus (G′) within the frequency range tested and presented an increasing trend above 10 Hz (high frequencies). The loss tangent (tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ) at 1.0 Hz was 0.18 and was always below 0.2 over the entire frequency range tested (0.001–100 Hz). Furthermore, the G′ is about one order of magnitude larger than the G′′, and no cross-section was found in the frequency sweep, indicating that the relaxation rate of the sample is slow and conforms with the typical behavior of a gel with a high elastic response.68–70 Overall, the results demonstrate the formation of a stable solid-like network gel. Fig. 2B shows that the gel has a storage modulus of ca. 90 Pa (at 1.0 Hz), which is in the range of that for previously reported strong protein nanofibril hydrogels.71,72
δ) at 1.0 Hz was 0.18 and was always below 0.2 over the entire frequency range tested (0.001–100 Hz). Furthermore, the G′ is about one order of magnitude larger than the G′′, and no cross-section was found in the frequency sweep, indicating that the relaxation rate of the sample is slow and conforms with the typical behavior of a gel with a high elastic response.68–70 Overall, the results demonstrate the formation of a stable solid-like network gel. Fig. 2B shows that the gel has a storage modulus of ca. 90 Pa (at 1.0 Hz), which is in the range of that for previously reported strong protein nanofibril hydrogels.71,72
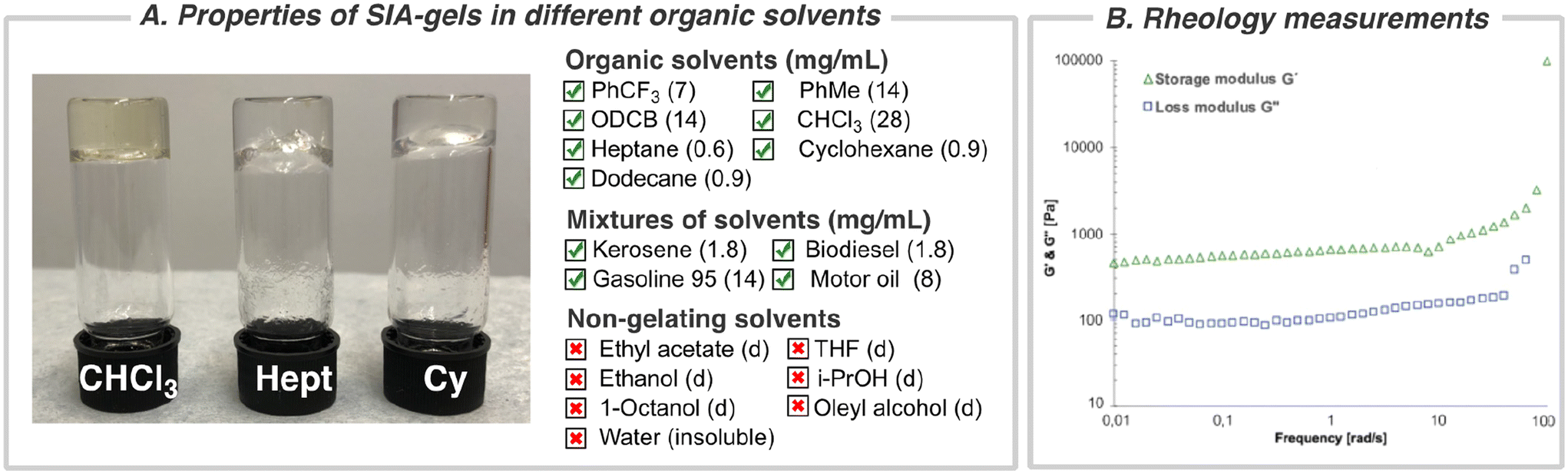 | ||
| Fig. 2 (A) Solvent scope for gelation and critical gel concentration (mg mL−1); abbreviations: (d) = dissolved, gelation in chloroform (CHCl3), heptane (Hept), and cyclohexane (Cy). (B) Rheology measurements of (S)-SIA. Storage (green triangles) and loss (blue squares) moduli of (S)-SIA (0.9 mg mL−1) in heptane as a function of frequency. The frequency sweep was performed at a strain of 3 × 10−6 MPa (accordingly within the linear region from the amplitude sweep test) and temperature (T = 293 K). | ||
To gain insight about the structure of the organogel, the samples were air-dried and the morphologies of the formed xerogels in different solvents were examined by SEM.
In heptane and PhCF3, the fibres were fused together into a film and no individual fibres of the network were distinguishable (Fig. S4†). However, in the xerogels formed from chloroform the SEM analysis revealed a more detailed picture of the fibrous network. The morphology contained three distinct features: a network built from about 100 nm wide fibres (Fig. 3A–C), twisted bundles of threads, and rolled-up piles of fibres on the surface of the fibrous network. No helical topology could be distinguished for the individual fibres at this level of resolution, but for the bundled fibres there is a clear handedness of the bundles, suggesting a P-type macroscopic chirality of the helix built from the (S)-enantiomer. The opposite handedness of the bundled fibres was observed from the opposite enantiomer, suggesting an opposite macroscopic helical conformation (M-type) originating already on the level of the individual fibres (Fig. S5†). At 30k× magnification, the diameter of the bundled fibres was determined to be 4.1 ± 0.3 μm and the finer individual fibres within these bundled fibres have diameters of 80 ± 9 nm (Fig. 3B, see arrows).
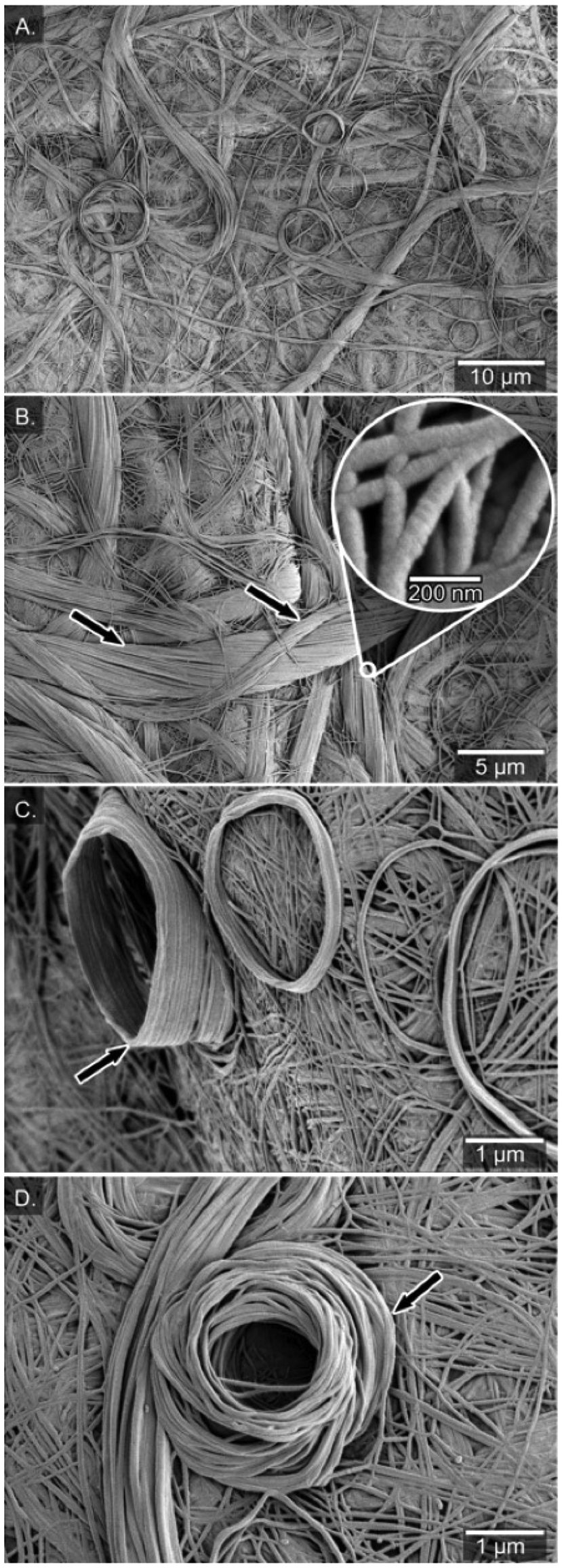 | ||
| Fig. 3 (A) Air-dried xerogel from (S)-SIA in chloroform (14 mg mL−1). (B) Bundles of fibres with clear handedness of the bundles with a P-type macroscopic chirality from the (S)-enantiomer. (C) Side-view of rolled-up fibres deposited on the surface of the fibrous network. (D) Top-view of rolled-up fibres. | ||
A peculiar feature in the fibrous network of the xerogel was the rolled-up fibres deposited on the surface of the fibrous network (Fig. 3C & D), mimicking a rolled-up rope on a dock. Similar lantern-shaped structures have been observed in xerogels of gold(I) complexes of functionalized N-heterocyclic carbenes.73 The number of turns in the rolled piles differs, from a few turns up to many turns (15), leading to a cone-like structure (up to 1500 nm high, see Fig. 3C). The diameter of the rolled-up piles ranges from roughly 3 to 10 μm (Fig. 3C & D) and the rolls were likely formed during the drying process of the gel into the xerogel. Previously, Mears et al. have shown the influence of the drying process in sample preparation of hydrogels and that the SEM analysis does not always show the primary network of fibers but rather depicts the results of aggregation of the fibers.74 Along these lines, the diversity of morphology in xerogels from chloroform (carpet-like network, bundled twisted fibres and rolled-up piles) probably originates from air-drying processes in which the volume decreases drastically upon solvent evaporation and forms a more dense material.
To have a more realistic view of the network structure in the organogel, the formation of aerogels via freeze-drying was attempted. Neither chloroform nor heptane are suitable solvents for freeze-dying, but fortunately organogels from cyclohexane produced a brittle aerogel upon lyophilization without any significant loss of volume (about 10%), which is in stark contrast to the loss of volume observed in the xerogel from chloroform (Fig. 4A and Fig. S6, 7†). Therefore, we assume that the formed aerogel structure from cyclohexane better reflects the three-dimensional network organization of the organogel in solution. A more detailed analysis of the morphology from (R)-SIA aerogel using scanning electron microscopy (SEM) shows a highly porous material (Fig. 4A) that consists of individual fibres or fibres that are fused together either into bundled fibres or fishnet-type structures (Fig. 4B). The individual fibres have a diameter of 60 ± 10 nm, which is slightly thinner than that in the xerogel from chloroform. The light -weight of the aerogel is demonstrated by resting the material (1 mg) on the seed head of a dandelion (Fig. 4A, inset). The estimated density of the aerogel is roughly 1 mg cm−3, which is one order of magnitude higher than that of aerographene,75–77 which is one of the most light weight materials in the world. Both the (R)- and the (S)-enantiomer showed a similar structure of the formed material (Fig. S6 and 7†), but no direct indication of helical structure in the individual fibres was observed using SEM (except for the macroscopic twisting in the bundled fibres in chloroform). Freeze-drying non-gelated solutions in concentrations below the critical gel concentration (0.1 mg mL−1) led to larger loss of volume (compared to the solution sample) and a less homogenic fibre structure (see Fig. S8†).
 | ||
| Fig. 4 (A) Freeze-dried aerogel of (R)-SIA from cyclohexane. Inset: freeze-dried (R)-SIA (1 mg mL−1) resting on the seed head of a dandelion. (B) View of porous aerogel where the scale bar in the inset in B represents 500 nm. | ||
CD spectroscopy measures the differential absorption of circularly polarized light and is a powerful tool to determine molecular conformations and configurations of chiral supramolecular systems.78 Therefore, we aimed to use CD spectroscopy to establish if a chiral helical arrangement of the molecules is responsible for the supramolecular polymerization of the organogels. The CD spectra were measured at different concentrations of the organogelator ((S)-SIA) in heptane, both below and slightly above the critical gel concentration. At concentrations ten times below the CGC (0.1 mg mL−1), the CD spectra of the two enantiomers show a strong antipodal maximum/minimum at 240 nm corresponding to the CD signal of the non-assembled monomer of the respective enantiomers (Fig. 5A). At higher concentrations (0.5–1.0 mg mL−1), the spectra change with the appearance of strong antipodal CD peaks at 216 nm, 235 nm, 250 nm and 270 nm (Fig. 5A). The emergence of these strong CD signals in the range of the critical gel concentration indicates the formation of a supramolecular structure with a helical arrangement. The two enantiomers of the SIA show the same spectra, but with opposite signs of all peaks, which suggests opposite chirality in the supramolecular structure. In a similar manner, the maximum of the UV-vis absorbance spectra shows a small shift (5 nm) to longer wavelengths at the higher concentrations (0.5 and 1.0 mg mL−1) compared to that at the lower concentration (0.1 mg mL−1) (Fig. 5B). The rate of the of organogelation was investigated using CD spectroscopy by allowing a heated solution of the monomer (65 °C) to cool down to 25 °C while recording the CD spectrum (Fig. 5C & D). During the first three scans (corresponding to the time required for the instrument to adjust to the lower temperature, 140 s), no significant changes were observed, and the spectra corresponded to monomers in solution. After 210 seconds, the signals corresponding to a supramolecular structure from the gel rapidly builds up and after 500 seconds no significant changes were observed. This shows rapid formation of the supramolecular structure (in minutes) that also leads to the gel formation at concentrations above the critical gel concentration. The UV-vis spectra show the same small redshift (6 nm) during the supramolecular polymerization that was observed at concentrations below and above the critical gel concentration (Fig. S9†). In addition, the temperature dependence was measured by variable-temperature CD spectroscopy by stepwise increasing the temperature from 15 to 65 °C in 5 °C increments (Fig. 5E). At the lower temperatures (15–25 °C), the supramolecular structure is present as indicated by the strong signals (at 216 nm, 235 nm, 250 nm and 270 nm) that gradually decrease upon an incremental temperature increase. The UV-vis spectra at high (65 °C) and low temperature (15 °C) once again show the same redshift (6 nm, see Fig. S10†) as seen for previous monomer-to-supramolecular transitions. The magnitude of the CD signal was quantified by calculating the absorptive dissymmetry factor (gabs), which is the ratio of molar CD to molar extinction coefficient for unpolarized light. The plot shows a large difference in |gabs| at high and low temperatures (15 and 65 °C, respectively), which suggests that there is a large increase in chiral anisotropy upon going from monomers (65 °C) to the aggregated supramolecular polymer (Fig. S11†).
 | ||
| Fig. 5 (A) CD spectra of (R)-SIA and (S)-SIA at different concentrations (0.1, 0.5 and 1.0 mg mL−1) in heptane. (B) UV-vis spectra of (R)-SIA and (S)-SIA at different concentrations (0.1, 0.5 and 1.0 mg mL−1) in heptane. (C) & (D) Time-dependence of the formation of the macrocyclic structure of (S)-SIA upon cooling from 65 °C to 25 °C (140 s) as measured by CD and absorbance spectroscopy. (E) Temperature-dependence of the CD spectra upon incremental heating of (S)-SIA from 15 °C to 65 °C. (F) The melting temperature, Tm, was determined by taking the middle point of the min–max signal from CD spectroscopy (250 nm) and 19F NMR spectroscopy. | ||
The melting temperature Tm of the supramolecular polymer is reached at 38 °C (measured halfway from the minimum to maximum of the CD signal at 250 nm). At about 45–50 °C, the strong CD signals disappear and reach a plateau and the CD spectrum overlaps with the signals observed at lower concentrations (Fig. 5F). This suggests a disassembly of the ordered supramolecular structure into monomers at a critical upper temperature of 45 °C.
In addition to studying the formation of the supramolecular structure using CD spectroscopy, 19F-NMR spectroscopy was used to study the gel formation by monitoring the 19F NMR spectra of (S)-SIA (0.5 mg mL−1) in the temperature range of 65–15 °C in heptane (Fig. S13†). At 65 °C, the two 19F signals (−92.55 and −152.22 ppm) correspond to the concentration of the monomer species as previously measured by CD spectroscopy at the same temperature (no bisignate signal, see Fig. 5E). The stacked plot of the spectra clearly shows that the signals of the monomer are decreasing at lower temperatures (from 65–15 °C) without the appearance of any new signals or signal peak shifts. This suggests immediate formation of larger aggregates that due to their large molecular weight and slow tumbling rate have a short NMR spin–spin relaxation time (T2), which results in large broadening and disappearance of their NMR signals. The plot of the transition from monomer to the supramolecular structure has the same appearance as that measured by CD spectroscopy, except the expected shift to a lower melting point temperature (35 °C) due to the lower concentration of (S)-SIA (0.5 mg mL−1) (Fig. 5F). In addition, the concentration-dependence of the monomer was measured using 19F NMR spectroscopy upon dilution of the total concentration at a fixed temperature (293 K, Fig. S15–17†). Both the temperature- and concentration-dependent curves show the shape of sharp transitions from monomers to an aggregated structure that is the hallmark of a cooperative polymerization mechanism.79–81
To correlate the observed CD spectrum to the supramolecular structure leading to gel formation, we designed and built a model structure based on a two-layer unit comprised of eight (R)-SIA molecules and calculated the CD spectra with the use of VeloxChem program.82 To create a hydrogen-bonded network, four molecules of (R)-SIA were assembled into a one-layer unit (see Fig. S21 in ESI†) and 16 such monomer units were stacked vertically to form an M-type helical fibre. In the formation of this fibre, the hydrogen-bonded network was completed with the formation of inter-layer hydrogen bonds (see Fig. S20b† for a dimer illustration).
After a molecular mechanics (MM) structure optimization of the solvated fibre in heptane, the central dimer unit was extracted and its CD spectrum was determined using time-dependent density functional theory (TDDFT). Details about the MM and TDDFT calculations can be found in the ESI.† In Fig. 6A, the resulting theoretical CD spectrum of the dimer is shown. The numerous transitions underlying the spectral bands are of π–π* character. A detailed analysis of the CD spectrum intensities (shown in Table S2†) and nature of the transitions (shown in Fig. S31†) reveals that several strongly contributing transitions are of mixed intra- and intermolecular character, and, furthermore, the intermolecular transitions take place not only within a layer but also in between layers. Following a comparison with the corresponding experimental spectrum, it is noted that the agreement between theory and experiment is striking. In both cases, we observe four signature bands with the lowest band showing a negative Cotton effect positioned at 278 nm in the theoretical spectrum (compared to 270 nm in the experiment). The second and third bands are positive and negative and positioned around 250 and 235 nm, respectively, both in the theoretical and experimental spectra. Whilst the overall excellent agreement between the theoretical and experimental CD spectra might be fortuitous and associated with error cancellation, given the molecular dimer model and the computational approximations made in the calculations, the overlap of the qualitative features of the fingerprinting bands provides a strong indication that the theoretical model correctly represents the underlying microscopic supramolecular organization.
 | ||
| Fig. 6 (A) Theoretical and experimental CD spectra of (R)-SIA. (B) Intra- and inter-layer hydrogen bond distances as well as fibril diameters. (C) Solvated fibril. (D) Two fibrils oppositely directed to cancel the dipole moment. (E & F) Fiber thickness, observed from different view angles. All illustrations and MD trajectory analyses were performed with the VIAMD software.83 | ||
Based on the discussed fibrillar structure, nanofibers were built of progressive thickness ranging up to clusters of 12 helical fibrils. Each fibril in turn comprised 32 layers of SIAs and the separate fibrils were organized in alternating directions as to zero the macroscopic dipole moment of the bundled fibre (see Fig. 6D–F). Molecular dynamics (MD) simulations of the bundled fibres in solution were performed with the employment of the same MM force field that was used in the structure optimization discussed above (see the ESI† for further details). These MD simulations allowed us to assess the strength of the hydrogen-bonded network and ultimately the thermal stability of the fibres. We observed significant fibre disintegrations already at a temperature of 150 K, which is far lower than the melting temperature observed in the experiment. Given that our proposed fibrillar structure saturates the hydrogen bonding in a 3D network and gives rise to a correct CD response signal, we are inclined to believe that the observed discrepancy in thermal stability is due to inexact force-field parameters for non-bonded interactions and/or a too simplistic fibre bundle model. From the MD trajectories, the parameters of fibrillar molecular diameters (d1–d3 and d4) were estimated (see Fig. 6B & F, respectively). Based on the kernel density estimation (KDE) plot, we attribute a value of 2.16 Å for the intra-layer hydrogen bond distance d1 and the inter-layer hydrogen bond distance, d2, equals 1.92 Å (Fig. S24 and 25†). The fibril diameter, d3, is measured to 17.4 Å and the fibre thickness, d4, is estimated to 80.1 Å (Fig. S26 and 27†). The theoretical diameters of the modelled fibres are similar to those of the experimentally determined diameters from SEM (60–80 nm).
Conclusion
We have developed and characterized a novel organogelator scaffold that forms a gel and solidifies at very low critical concentrations (1 mg mL−1) in nonpolar organic solvents and can thus be foreseen to open up a diverse array of applications. The organogelator is based on a chiral, perfluoropyridine-containing sulfonimidamide, which forms a supramolecular structure where the chirality of the organogelator translates into a macroscopic chiral arrangement of thin and very uniform ca. 60–80 nm thick polymer fibers encapsulating the solvent and the freeze-dried material forms ultralight aerogels. The supramolecular arrangement of the layered monomers was confirmed by comparison of experimental and calculated CD spectra and a plausible supramolecular structure consisting of twelve monomeric bundles was modelled by means of molecular mechanics.Author contributions
G. P., A. A. and P. D. formulated the research problem, developed the synthesis and characterization of the gelating properties. P. D. conceived and directed the project. A. C. and R. O. performed the SEM characterization of the xerogels and aerogels. C. L. performed the CD spectroscopy studies and Y. T., M. L. and P. N. performed the computational studies. Z. S. performed the VT-NMR studies. All authors discussed the results and approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
P. D. acknowledges financial support from the Carl Trygger Foundation (CTS:19-80) for a postdoctoral fellowship for A. A. Our work was supported by the Swedish Research Council (grant no. 2023-04482, 2023-5171) and the Swedish e-Science Research Centre (SeRC). Computational resources were provided by the National Academic Infrastructure for Supercomputing in Sweden (NAISS) A. C. acknowledges financial support from The Bo Rydins Foundation (grant no. F 30/19).References
- E. R. Draper and D. J. Adams, Chem, 2017, 3, 390–410 CAS.
- X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165–13307 CrossRef CAS PubMed.
- L. A. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104, 1201–1218 CrossRef CAS PubMed.
- N. M. Sangeetha and U. Maitra, Chem. Soc. Rev., 2005, 34, 821–836 RSC.
- P. Terech and R. G. Weiss, Chem. Rev., 1997, 97, 3133–3160 CrossRef CAS PubMed.
- R. G. Weiss, J. Am. Chem. Soc., 2014, 136, 7519–7530 CrossRef CAS PubMed.
- C. D. Jones, H. T. D. Simmons, K. E. Horner, K. Liu, R. L. Thompson and J. W. Steed, Nat. Chem., 2019, 11, 375–381 CrossRef CAS PubMed.
- E. Yashima, N. Ousaka, D. Taura, K. Shimomura, T. Ikai and K. Maeda, Chem. Rev., 2016, 116, 13752–13990 CrossRef CAS PubMed.
- S. Yang, L. Zhao, C. Yu, X. Zhou, J. Tang, P. Yuan, D. Chen and D. Zhao, J. Am. Chem. Soc., 2006, 128, 10460–10466 CrossRef CAS PubMed.
- A. Aggeli, I. A. Nyrkova, M. Bell, R. Harding, L. Carrick, T. C. B. McLeish, A. N. Semenov and N. Boden, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 11857–11862 CrossRef CAS PubMed.
- C. D. Jones and J. W. Steed, Chem. Soc. Rev., 2016, 45, 6546–6596 RSC.
- S. Ziane, S. Schlaubitz, S. Miraux, A. Patwa, C. Lalande, I. Bilem, S. Lepreux, B. Rousseau, J. F. Le Meins, L. Latxague, P. Barthélémy and O. Chassande, Eur. Cells Mater., 2012, 23, 147–160 CrossRef CAS PubMed ; discussion 160.
- H. Wang and Z. Yang, Nanoscale, 2012, 4, 5259–5267 RSC.
- T.-Y. Cheng, M.-H. Chen, W.-H. Chang, M.-Y. Huang and T.-W. Wang, Biomaterials, 2013, 34, 2005–2016 CrossRef CAS PubMed.
- L. Latxague, M. A. Ramin, A. Appavoo, P. Berto, M. Maisani, C. Ehret, O. Chassande and P. Barthélémy, Angew. Chem., Int. Ed., 2015, 54, 4517–4521 CrossRef CAS PubMed.
- E. V. Alakpa, V. Jayawarna, A. Lampel, K. V. Burgess, C. C. West, S. C. J. Bakker, S. Roy, N. Javid, S. Fleming, D. A. Lamprou, J. Yang, A. Miller, A. J. Urquhart, P. W. J. M. Frederix, N. T. Hunt, B. Péault, R. V. Ulijn and M. J. Dalby, Chem, 2016, 1, 298–319 CAS.
- N. Pilpel, Chem. Rev., 1963, 63, 221–234 CrossRef CAS.
- F. H. Gaskins, J. G. Brodnyan and W. Philippoff, Trans. Soc. Rheol., 1969, 13, 17–38 CAS.
- K. Hanabusa, J. Tange, Y. Taguchi, T. Koyama and H. Shirai, J. Chem. Soc., Chem. Commun., 1993, 390–392 RSC.
- N. Ramanathan, A. L. Currie and J. R. Colvin, Nature, 1961, 190, 779–781 CrossRef CAS.
- J. Morris, J. Bietsch, K. Bashaw and G. Wang, Gels, 2021, 7, 24 CrossRef CAS PubMed.
- S. Yamasaki and H. Tsutsumi, Bull. Chem. Soc. Jpn., 1994, 67, 906–911 CrossRef CAS.
- S. Yamasaki and H. Tsutsumi, Bull. Chem. Soc. Jpn., 1994, 67, 2053–2056 CrossRef CAS.
- T. Brotin, R. Utermöhlen, F. Fages, H. Bouas-Laurent and J.-P. Desvergne, J. Chem. Soc., Chem. Commun., 1991, 416–418 RSC.
- B. O. Okesola, V. M. P. Vieira, D. J. Cornwell, N. K. Whitelaw and D. K. Smith, Soft Matter, 2015, 11, 4768–4787 RSC.
- A. Dawn, T. Shiraki, S. Haraguchi, S.-i. Tamaru and S. Shinkai, Chem. – Asian J., 2011, 6, 266–282 CrossRef CAS PubMed.
- A. R. Hirst, B. Escuder, J. F. Miravet and D. K. Smith, Angew. Chem., Int. Ed., 2008, 47, 8002–8018 CrossRef CAS PubMed.
- B. O. Okesola and D. K. Smith, Chem. Soc. Rev., 2016, 45, 4226–4251 RSC.
- S. S. Babu, V. K. Praveen and A. Ajayaghosh, Chem. Rev., 2014, 114, 1973–2129 CrossRef CAS PubMed.
- S. Ghosh, V. K. Praveen and A. Ajayaghosh, Annu. Rev. Mater. Res., 2016, 46, 235–262 CrossRef CAS.
- F. Rodríguez-Llansola, J. F. Miravet and B. Escuder, Chem. Commun., 2011, 47, 4706–4708 RSC.
- E. S. Levchenko, I. E. Sheinkman and A. V. Kirsanov, Zh. Obshch. Khim., 1960, 30, 1941–1946 CAS.
- E. S. Levchenko, N. Y. Derkach and A. V. Kirsanov, Zh. Obshch. Khim., 1962, 32, 1208–1212 CAS.
- E. S. Levchenko, E. S. Kozlov and A. V. Kirsanov, Zh. Obshch. Khim., 1962, 32, 882–886 CAS.
- E. S. Levchenko and A. V. Kirsanov, Zh. Obshch. Khim., 1960, 30, 1553–1561 CAS.
- G. C. Nandi and P. I. Arvidsson, Adv. Synth. Catal., 2018, 360, 2976–3001 CrossRef CAS.
- P. K. Chinthakindi, T. Naicker, N. Thota, T. Govender, H. G. Kruger and P. I. Arvidsson, Angew. Chem., Int. Ed., 2017, 56, 4100–4109 CrossRef CAS PubMed.
- F. Sehgelmeble, J. Janson, C. Ray, S. Rosqvist, S. Gustavsson, L. I. Nilsson, A. Minidis, J. Holenz, D. Rotticci, J. Lundkvist and P. I. Arvidsson, ChemMedChem, 2012, 7, 396–399 CrossRef CAS PubMed.
- F. Izzo, M. Schäfer, P. Lienau, U. Ganzer, R. Stockman and U. Lücking, Chem. – Eur. J., 2018, 24, 9295–9304 CrossRef CAS PubMed.
- U. Lücking, Org. Chem. Front., 2019, 6, 1319–1324 RSC.
- E. Wojaczyńska and J. Wojaczyński, Chem. Rev., 2020, 120, 4578–4611 CrossRef PubMed.
- T. Hisashi, W. Itaru and M. Teruaki, Bull. Chem. Soc. Jpn., 1965, 38, 1989–1993 CrossRef.
- C. R. Johnson, E. U. Jonsson and A. Wambsgans, J. Org. Chem., 1979, 44, 2061–2065 CrossRef CAS.
- Y. Chen and J. Gibson, RSC Adv., 2015, 5, 4171–4174 RSC.
- T. Q. Davies, A. Hall and M. C. Willis, Angew. Chem., Int. Ed., 2017, 56, 14937–14941 CrossRef CAS PubMed.
- F. Liu, H. Wang, S. Li, G. A. L. Bare, X. Chen, C. Wang, J. E. Moses, P. Wu and K. B. Sharpless, Angew. Chem., Int. Ed., 2019, 58, 8029–8033 CrossRef CAS PubMed.
- S. Greed, E. L. Briggs, F. I. M. Idiris, A. J. P. White, U. Lücking and J. A. Bull, Chem. – Eur. J., 2020, 26, 12533–12538 CrossRef CAS PubMed.
- H. Yu, Z. Li and C. Bolm, Angew. Chem., Int. Ed., 2018, 57, 15602–15605 CrossRef CAS PubMed.
- J. Wen, H. Cheng, S. Dong and C. Bolm, Chem. – Eur. J., 2016, 22, 5547–5550 CrossRef CAS PubMed.
- F. Izzo, M. Schäfer, R. Stockman and U. Lücking, Chem. – Eur. J., 2017, 23, 15189–15193 CrossRef CAS PubMed.
- C. Worch, I. Atodiresei, G. Raabe and C. Bolm, Chem. – Eur. J., 2010, 16, 677–683 CrossRef CAS PubMed.
- O. García Mancheño and C. Bolm, Beilstein J. Org. Chem., 2007, 3, 25 Search PubMed.
- T. Q. Davies, M. J. Tilby, J. Ren, N. A. Parker, D. Skolc, A. Hall, F. Duarte and M. C. Willis, J. Am. Chem. Soc., 2020, 142, 15445–15453 CrossRef CAS PubMed.
- G. Proietti, J. Kuzmin, A. Z. Temerdashev and P. Dinér, J. Org. Chem., 2021, 86, 17119–17128 CrossRef CAS PubMed.
- C. Kulkarni, J. A. Berrocal, M. Lutz, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2019, 141, 6302–6309 CrossRef CAS PubMed.
- A. K. Patterson, L. H. El-Qarra and D. K. Smith, Chem. Commun., 2022, 58, 3941–3944 RSC.
- C.-W. Liu, M. Su, X.-L. Li, T. Xue, N. Liu, J. Yin, Y.-Y. Zhu and Z.-Q. Wu, Soft Matter, 2015, 11, 5727–5737 RSC.
- P. Jana and S. Kanvah, Langmuir, 2020, 36, 2720–2728 CrossRef CAS PubMed.
- B. Yue, L. Yin, W. Zhao, X. Jia, M. Zhu, B. Wu, S. Wu and L. Zhu, ACS Nano, 2019, 13, 12438–12444 CrossRef CAS PubMed.
- S. Cicchi, G. Ghini, L. Lascialfari, A. Brandi, F. Betti, D. Berti, P. Baglioni, L. Di Bari, G. Pescitelli, M. Mannini and A. Caneschi, Soft Matter, 2010, 6, 1655–1661 RSC.
- E. L. Bonifazi, A. S. Mac Cormack, V. M. Busch, M. L. Japas, L. Di Bari and P. H. Di Chenna, J. Sol-Gel Sci. Technol., 2022, 102, 30–40 CrossRef CAS.
- R. K. Das, R. Kandanelli, J. Linnanto, K. Bose and U. Maitra, Langmuir, 2010, 26, 16141–16149 CrossRef CAS PubMed.
- D. Bardelang, Soft Matter, 2009, 5, 1969–1971 RSC.
- Y. Cui, M.-C. Li, Q. Wu, J. A. Pojman and D. G. Kuroda, ACS Appl. Mater. Interfaces, 2017, 9, 33549–33553 CrossRef CAS PubMed.
- M. Xue, D. Gao, K. Liu, J. Peng and Y. Fang, Tetrahedron, 2009, 65, 3369–3377 CrossRef CAS.
- S. R. Jadhav, P. K. Vemula, R. Kumar, S. R. Raghavan and G. John, Angew. Chem., Int. Ed., 2010, 49, 7695–7698 CrossRef CAS PubMed.
- A. M. Vibhute, V. Muvvala and K. M. Sureshan, Angew. Chem., Int. Ed., 2016, 55, 7782–7785 CrossRef CAS PubMed.
- M. Jiménez-Rosado, J. F. Rubio-Valle, V. Perez-Puyana, A. Guerrero and A. Romero, J. Cleaner Prod., 2021, 286, 124948 CrossRef.
- M. Jiménez-Rosado, V. Perez-Puyana, A. Guerrero and A. Romero, Ind. Crops Prod., 2022, 185, 115128 CrossRef.
- E. Álvarez-Castillo, S. Oliveira, C. Bengoechea, I. Sousa, A. Raymundo and A. Guerrero, J. Food Eng., 2021, 288, 110255 CrossRef.
- X. Yang, E. Bakaic, T. Hoare and E. D. Cranston, Biomacromolecules, 2013, 14, 4447–4455 CrossRef CAS PubMed.
- X. Ye, A. J. Capezza, X. Xiao, C. Lendel, M. S. Hedenqvist, V. G. Kessler and R. T. Olsson, ACS Nano, 2021, 15, 5341–5354 CrossRef CAS PubMed.
- T. H. T. Hsu, J. J. Naidu, B.-J. Yang, M.-Y. Jang and I. J. B. Lin, Inorg. Chem., 2012, 51, 98–108 CrossRef CAS PubMed.
- L. L. E. Mears, E. R. Draper, A. M. Castilla, H. Su, Z. Zhuola, B. Dietrich, M. C. Nolan, G. N. Smith, J. Doutch, S. Rogers, R. Akhtar, H. Cui and D. J. Adams, Biomacromolecules, 2017, 18, 3531–3540 CrossRef CAS PubMed.
- J. Ma, W. Li, Y. Fan, J. Yang, Q. Yang, J. Wang, W. Luo, W. Zhou, N. Nomura, L. Wang and W. Jiang, ACS Appl. Mater. Interfaces, 2019, 11, 46386–46396 CrossRef CAS PubMed.
- H. Sun, Z. Xu and C. Gao, Adv. Mater., 2013, 25, 2554–2560 CrossRef CAS PubMed.
- H. Hu, Z. Zhao, W. Wan, Y. Gogotsi and J. Qiu, Adv. Mater., 2013, 25, 2219–2223 CrossRef CAS PubMed.
- T. Taniguchi and T. Usuki, in Supramolecular Chemistry, John Wiley & Sons, Ltd., 2012 Search PubMed.
- H. M. M. ten Eikelder, A. J. Markvoort, T. F. A. de Greef and P. A. J. Hilbers, J. Phys. Chem. B, 2012, 116, 5291–5301 CrossRef CAS PubMed.
- H. M. M. ten Eikelder and A. J. Markvoort, Acc. Chem. Res., 2019, 52, 3465–3474 CrossRef CAS PubMed.
- F. Oosawa and M. Kasai, J. Mol. Biol., 1962, 4, 10–21 CrossRef CAS PubMed.
- Z. Rinkevicius, X. Li, O. Vahtras, K. Ahmadzadeh, M. Brand, M. Ringholm, N. H. List, M Scheurer, M. Scott, A. Dreuw and P. Norman, WIREs Comput. Mol. Sci., 2020, 10(5), e1457 CrossRef CAS.
- R. Skånberg and I. Hotz, J. Chem. Inf. Model., 2023, 63(23), 7382–7391 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr06460c |
| This journal is © The Royal Society of Chemistry 2024 |