
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparations of spherical nanoparticles of chiral Cinchona alkaloid-based bridged silsesquioxanes and their use in heterogeneous catalysis of enantioselective reactions†
David
Tetour
 a,
Marika
Novotná
a,
Jan
Tatýrek
a,
Veronika
Máková
b,
Martin
Stuchlík
b,
Christopher
Hobbs
b,
Michal
Řezanka
b,
Monika
Müllerová
c,
Vladimír
Setnička
d,
Kristýna
Dobšíková
d and
Jana
Hodačová
*a
a,
Marika
Novotná
a,
Jan
Tatýrek
a,
Veronika
Máková
b,
Martin
Stuchlík
b,
Christopher
Hobbs
b,
Michal
Řezanka
b,
Monika
Müllerová
c,
Vladimír
Setnička
d,
Kristýna
Dobšíková
d and
Jana
Hodačová
*a
aDepartment of Organic Chemistry, Faculty of Chemical Technology, University of Chemistry and Technology, Prague, Technická 5, 166 28 Prague 6, Czech Republic. E-mail: Jana.Hodacova@vscht.cz
bDepartment of Nanochemistry, Institute for Nanomaterials, Advanced Technologies and Innovation, Technical University of Liberec, Studentská 1402/2, 461 17 Liberec 1, Czech Republic
cInstitute of Chemical Process Fundamentals, The Czech Academy of Sciences, Rozvojová 135, 165 02 Prague 6, Czech Republic
dDepartment of Analytical Chemistry, Faculty of Chemical Engineering, University of Chemistry and Technology, Prague, Technická 5, 166 28 Prague 6, Czech Republic
First published on 7th March 2024
Abstract
Two spherical nanoparticulate materials were prepared by base-catalyzed sol–gel hydrolysis/self-condensation of the bis-Cinchona alkaloid-phthalazine-based bridged bis(triethoxysilanes). For the purpose of comparing the catalytic properties, two compact materials were also prepared from the same precursors using a fluoride-catalyzed sol–gel process. All materials were characterized by SEM, TEM, solid-state 29Si NMR and 13C NMR, TGA, and FTIR. The prepared silsesquioxane-based materials were studied as potential heterogeneous catalysts for selected enantioselective reactions. The spherical material with regularly incorporated bis-quinine-phthalazine chiral units exhibited good to excellent enantioselectivities in osmium-catalyzed dihydroxylations of alkenes. Enantioselectivities observed in dihydroxylations of aromatic trans-alkenes were as excellent as those observed with the homogeneous catalyst (DHQ)2–PHAL. One compact and one nanoparticulate material was successfully recycled and reused five times without loss of enantioselectivity. Furthermore, both quinine-based and cinchonine-based materials were tested as heterogeneous organocatalysts for chlorolactonization of 4-arylpent-4-enoic acids. The materials showed only moderate enantioselectivities; however, these are the first heterogeneous catalysts for enantioselective chlorolactonization published so far.
Introduction
Chirality is an important aspect of biologically active compounds. Their activity is based on interaction with enzymes, receptors, or other binding molecules that exist in living organisms in an enantiomerically pure form. Binding affinity typically differs for individual enantiomers or diastereomers of a chiral molecule, resulting in different biological properties of these stereoisomers. The ability to synthesize the desired stereoisomer of a chiral compound is a key aspect in the development and production of pharmaceuticals, diagnostics, vitamins, fragrances, and agrochemicals. In particular, absolute configuration of a molecule is of vital importance in drug design and development. While one enantiomer of a drug exhibits the desired effect, the other enantiomer can have reduced, no, or even deleterious effects.To synthesize a single enantiomer of a chiral compound, several synthetic approaches can be chosen: i. a target compound can be prepared as a racemate and then resolved into individual enantiomers; ii. a chiral auxiliary, which controls the stereochemical course of the reaction, can be introduced into a starting compound and then removed from the molecule after the stereoselective reaction; iii. an enantiomerically pure stoichiometric reagent can be used to transform an achiral starting compound to an enantiomerically pure product; iv. the stereochemical course of the reaction can be controlled with an enantiomerically pure catalyst.1–5 The latest approach is highly preferred because the enantiomerically pure compound is usually highly limited and expensive, whilst only used in small amounts in the catalytic setup.
Development of catalysts for enantioselective reactions has been a subject of extensive research since the middle of the last century. Since then, a large number of successful catalytic systems have been reported, including metal complexes with enantiomerically pure ligands, organocatalysts and natural enzymes.5,6 The vast majority of enantioselective catalysts function as homogeneous catalysts, although the use of heterogeneous catalysts is more advantageous. Solid catalysts can be easily separated from the reaction mixture and eventually recycled or used in continuous processes. It is therefore not surprising that attention has focused on the development of solid heterogeneous catalysts for enantioselective reactions in recent years. A large variety of heterogeneous chiral catalysts have been introduced, including chiral polymers, metal–organic frameworks, and hybrid organosilica materials.7 By far the most common method of preparing a solid heterogeneous catalyst is immobilization of a chiral catalyst onto an insoluble solid support, which can be either an inorganic solid or an organic polymer.8–11 Silicas and silica-based mesoporous materials (e.g. MCM-41, SBA-15) have been widely used as the support materials due to their strength, stability, and inertness.12,13 The disadvantage of these materials is low loading and irregular distribution of chiral organic molecules on the surface of the material. In addition, pores of a mesoporous material can be blocked by the attached molecules. Another approach to prepare organosilica materials is co-condensation of organic trialkoxysilane with a source of inorganic SiO2 (e.g. TEOS).14 When this one-pot procedure is done in the presence of surfactant, periodically ordered mesoporous organosilica (PMO) can be obtained.15–17 However, the disadvantage of this method is that the mesoporous material contains only a minor amount of the chiral organic component.14 A few successful catalysts of enantioselective reactions were prepared by co-condensation of two organic bis(trialkoxysilanes), where the chiral bridged bis(trialkoxysilane) was condensed with an excess of a smaller achiral bridged bis(trialkoxysilane) in the presence of a surfactant to form the functionalized PMO.18–21
To prepare the materials with the maximum amount of organic molecules incorporated, the bridged silsesquioxanes must be prepared by hydrolysis/self-condensation of the bridged organic bis(trialkoxysilanes) of general formula (R′O)3Si-R-Si(OR′)3, where R is chiral organic unit. Unlike in the materials prepared by the co-condensation method, the chiral organic unit is repeated regularly in the structures of those materials.22 Despite high expectations for this type of materials, only a small number of such chiral materials have been reported so far and only a few of them have been used to catalyze enantioselective reactions. The first catalysts based on the bridged silsesquioxanes were reported by Moreau et al.23–26 The enantiomerically pure (1R,2R)-cyclohexane-1,2-diamine organic unit was incorporated into the materials and their rhodium complexes were used to catalyze hydrogen transfer reduction of acetophenone with enantioselectivities in the range of 14–58% ee. Hybrid material that was prepared by self-condensation of rhodium complex of (2R,3R)-1,4-bis(diphenylphosphaneyl)butane-2,3-diyl bis{[3-(triethoxysilyl)propyl]carbamate} in the absence of surfactant by Moreau et al.27 catalyzed hydrogenation of (Z)-2-acetamido-3-phenylacrylic acid with 34% ee. The first bridged silsesquioxane-based PMO was reported by Polarz et al.,28 who prepared PMO by hydrolysis/self-condensation of (S)-1-[3,5-bis(tri-iso-propoxysilyl)phenyl]ethan-1-ol in the presence of Pluronic P-123 and then modified it with AlIII. The obtained material successfully catalyzed enantioselective carbonyl–ene reaction with enantioselectivities up to 74% ee. Recently, we prepared the bridged silsesquioxanes possessing the bis-cinchonine–phthalazine chiral unit, whose osmium complexes catalyzed Sharpless dihydroxylation of alkenes with enantioselectivities up to 98% ee.29
Various Cinchona alkaloid derivatives are used as ligands or organocatalysts in homogeneous catalysis.30,31 Discovery of the (DHQ)2–PHAL and (DHQD)2–PHAL ligands (Fig. 1) by Sharpless et al.32,33 proved to be a key improvement in homogeneous osmium-catalyzed enantioselective dihydroxylation of alkenes, which became applicable to a wide range of substrates. Although other similar ligands containing two Cinchona alkaloids attached to an aromatic linker have been developed, these two ligands are the most universal to date. In addition to the osmium-catalyzed dihydroxylations of alkenes, (DHQ)2–PHAL or (DHQD)2–PHAL was also used as an organocatalyst of enantioselective conjugate addition of alkynones to β-diketones,34 dichlorination of allylic alcohols35,36 and amides,37 chlorolactonization of 4-substituted 4-pentenoic acids,38,39 and decarboxylative trichloromethylation of Morita–Baylis–Hillman adducts.40,41
 | ||
| Fig. 1 Structures of (DHQ)2–PHAL and (DHQD)2–PHAL. | ||
Soon after the pioneering work of the Sharpless’ team, research began to prepare analogous solid catalysts that could be easily isolated from the reaction mixture and recycled. Since then, many heterogeneous catalysts have been prepared in which Cinchona alkaloid derivatives are bound to insoluble organic polymers,42–48 silicones49–51 or grafted onto prefabricated silica materials.52–56 However, these materials often contain, as linkers between the alkaloid derivative and the support, functional groups that are hydrolytically labile under conditions of Sharpless dihydroxylation, causing leaching of the ligand from the solid material. Salvadori et al.57 clearly demonstrated that even low solution concentration of the alkaloid derivative, which is a product of the hydrolysis of the material, could be responsible for high ee values in Sharpless dihydroxylation rather than the insoluble material. In addition, it has been shown that in some organic polymers the alkaloid derivative is only physically trapped and can be easily washed out into the solution.58,59 Proof of hydrolytic stability of the heterogenized ligands for Sharpless dihydroxylation is a key experiment to confirm the heterogeneous nature of catalysis.57 However, most published articles do not report this experiment.
Herein, we present the preparation of novel spherical nanoparticles that are chiral bridged silsesquioxanes in nature and contain bis-quinine–phthalazine or bis-cinchonine–phthalazine derivatives as chiral units. Further, their application as heterogenized chiral ligands in osmium-catalyzed enantioselective alkene dihydroxylation and as solid organocatalysts in enantioselective chlorolactonization of 4-arylpent-4-enoic acids are demonstrated.
Experimental
General
Melting points were determined on an Electrothermal IA 9100 melting point apparatus. Optical rotation was measured on a Bellingham & Stanley ADP450 Polarimeter at 25 °C; [α]D is reported in 10−1 deg cm2 g−1 and concentration in g per 100 mL. FTIR spectra of samples soluble in CHCl3 and CH2Cl2 were measured as a film on KBr glass using a Nicolet 6700 instrument. Liquid NMR spectra were recorded on a Varian Mercury Plus spectrometer (1H NMR at 299.97 MHz), an Agilent-MR DDR2 spectrometer (1H NMR at 399.94 MHz; 13C NMR at 100.58 MHz; 19F at 376.29 MHz), and a JEOL JNM-ECZR 500 MHz (1H NMR at 500.16 MHz; 29Si NMR at 99.37 MHz). All NMR measurements were carried out at rt. Chemical shifts δ are reported in ppm, coupling constants J are given in Hz. 1H NMR and 13C NMR spectra were referenced to the signals of residual non-deuterated solvents (CDCl3δH 7.26, δC 77.16; DMSO δH 2.50, δC 39.52), 29Si NMR spectra to tetramethylsilane and 19F NMR spectra to trichlorofluoromethane as internal standards. Mass spectra were measured on a Thermo Scientific LTQ Orbitrap Velos spectrometer with electrospray ionization. TLC analyses were carried out on aluminium foils coated with Silica gel 60 F254 (Merck) and compounds were detected under UV light (λ = 254 nm) or visualized by a treatment with 3% aqueous KMnO4 and heating. Silica gel (Fluka, 63–200 μm) was used for a preparative column chromatography. Silica gel (Fluka 40–60 μm) was used for flash column chromatography, which was performed on a Büchi Pure C-810 Flash instrument. HPLC analyses were done on an Ecom instrument (pump LCP4100, UV detector LCD2083, software CSW32) using the YMC Chiral Cellulose-SB (250 × 4.6 mm; 5 μm) and YMC Chiral ART Amylose-SA (250 × 4.6 mm; 5 μm) columns.Solid-state NMR (SS-NMR) spectra were measured at 9.39 T using a JEOL JNK-ECZ400R/M1 NMR spectrometer (400 MHz, JEOL, Japan). For all measured samples, the dry material was packed into 3.2 mm ZrO2 sample tube rotors and spectra recorded at rt. Solid-state 13C (100.53 MHz) NMR spectra were referenced externally to γ-glycine (174.1 ppm), with spectra for samples obtained utilizing CP/MAS (with high-powered 1H decoupling) at 20 kHz using optimized relaxation delays of 2 s and a contact time of 2.0 ms. Solid-state 29Si (79.43 MHz) NMR spectra were referenced externally to tetrakis(trimethylsilyl)silane (−9.8 ppm), with spectra for samples obtained utilizing CP/MAS (with high-powered 1H decoupling) at 6 kHz using optimized relaxation delays of 2 s and a contact time of 5.0 ms. Spectra were subsequently analyzed using MestReNova software (V. 14.3.1 Mestrelab Research).
The content of individual structural types (T1, T2, T3) was obtained by peak deconvolution and integration of the 29Si CP/MAS NMR spectra. Degree of condensation c was calculated using the equation:

The mass of Si in the material was determined from the weighted average of Si mass fraction present in Tn subunits, whereby Tn ratios were obtained from 29Si CP/MAS NMR. Similarly, the average molecular weight of the material was determined from the weighted average of Tn subunit molecular weights, whereby Tn ratios were obtained from 29Si CP/MAS NMR.
Thermogravimetric analyses were performed using a Q500 analyser (TA Instruments, New Castle, USA). A sample was placed on a platinum pan and heated at a rate of 10 °C min−1 in a flow (60 mL min−1) of nitrogen![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :oxygen (molar ratio 80:20) mixture up to 800 °C. Residual weight (mr) was used (after correction to absorbed water) to calculation of content of Si in the materials (wSi), assuming the incombustible residue was pure SiO2:
:oxygen (molar ratio 80:20) mixture up to 800 °C. Residual weight (mr) was used (after correction to absorbed water) to calculation of content of Si in the materials (wSi), assuming the incombustible residue was pure SiO2:
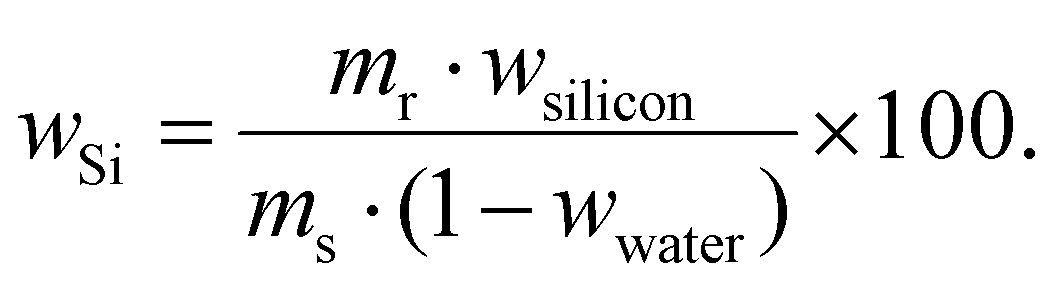
Scanning electron microscopy was performed on a ZEISS Ultra Plus instrument (Sigma Family, Jena, Germany) supplemented by an EDS detector X-Max 20 Oxford Instruments. Samples were applied to the metal targets containing carbon tape either in the form of a powder or as a suspension in distilled water. Thanks to the implemented nitrogen probe, it was not necessary to gild the samples. All samples were viewed as secondary electron images with the use of the following parameters: EHT = 1 kV, low-current mod 7–20 μm aperture with decrease of extraction voltage up to 3 kV. To protect the samples against disruption the working distance was determined 1.5–4 mm. Histograms were obtained from 100 measured diameters in the ImageJ program.
A transmission electron microscope JEOL JEM 2200FS equipped with an energy dispersive spectrometer Oxford Instruments was used for detailed structural characterization. A powder sample in the form of a suspension in propan-2-ol was dropped on the copper grid with supportive carbon and formvar layers and freely dried. Observation was performed in transmission mode using accelerating voltage of 200 kV.
An Nicolet iZ10 FTIR spectrometer (Thermo Scientific, USA) with the ATR sampling technique was used to obtain FTIR spectra of solid samples. The sample was placed on a diamond crystal and was measured in the spectral range 4000–400 cm−1 with a resolution of 4 cm−1. Averaging 16 background scans and 8 sample scans were used to obtain the spectrum. Atmospheric and baseline corrections were done using the Omnic software.
The ECD spectra of solid-state samples were recorded on a J-815 (JASCO Corporation, Tokyo, Japan) in the spectral range of 220–500 nm at a scanning speed of 50 nm min−1, a response time of 2 s, and a resolution of 0.1 mm. The spectrometer was purged during the measurements by gaseous nitrogen (purity >99.99%, Siad, Czech Republic). Due to the small amount of sample M2 (6.8 mg), it was diluted with KCl (0.1360 g), the spectrum of matrix was recorded under identical conditions and subtracted. The obtained spectra represent five spectral accumulations. The corresponding absorbance values were computed using the Spectra analysis module of Spectra Manager (JASCO Corporation, Tokyo, Japan) based on the photomultiplier HT voltage values.
Organic solvent nanofiltration (OSN)60 was carried out using solvent-resistant stirred cell Millipore (for 47 mm membranes) equipped with 100 kDa regenerated cellulose ultrafiltration discs Millipore (Ultracel®) and PTFE encapsulated O-rings Teflex (FEP/Viton) with nitrogen as a driving gas. The pressure was adjusted to 1–5 mbar so that the eluent exits the cell dropwise.
Syntheses of compounds
10,11-Didehydroquinine (3),61 10,11-didehydrocinchonine (4),61 1,4-dichlorophthalazine,62 (3-azidopropyl)triethoxysilane,63 Cu/C catalyst,64 precursor 2,29 4-phenylpent-4-enoic acid,65 4-(4-methoxyphenyl)pent-4-enoic acid,65 4-[4-(trifluoromethyl)phenyl]pent-4-enoic acid66 and analytical standards of racemates for catalytic experiments were synthesized according to literature procedures. Synthetic procedures and analytical data for all synthesized compounds can be found in the ESI.† Other chemicals and solvents were used as received from commercial sources (Sigma-Aldrich, Fluorochem, Penta). Anhydrous tetrahydrofuran and absolute ethanol were obtained from a PureSOLV PS-MD-7 (VWE International) drying columns. Dioxane was dried over sodium and freshly distilled prior to use.Materials preparations
000 rpm for 15 min, the solid was separated and treated with 1 M solution of NH4NO3 in ultrasonic bath at 45 °C for 30 min. The solid was separated by centrifugation at 21000 rpm for 15 min and the washing cycle with NH4NO3 was repeated. The solid was 10 times washed with water (centrifugation at 21000 rpm for 15 minutes between the washing cycles) and lyophilized at −53 °C and 14 Pa for 4 h. Material M1 was obtained as a white solid in 62% yield (121.2 mg; based on an average molecular weight of 1052.50 g mol−1 calculated from T1, T2 and T3 units, see above).
000 rpm for 10 min, the solid was separated and washed three times with acetone (10 mL) with centrifugation at 11000 rpm for 15 min between the washing cycles. The solid was separated, stirred at 500 rpm in acetone (20 mL) and refluxed for 2 hours. After cooling to rt, the solid was separated by centrifugation at 11000 rpm for 15 min and washed three times with acetone (10 mL) – centrifugation at 11000 rpm for 15 min between the washing cycles. The solid was lyophilized at −53 °C and 14 Pa for 6 h. Material M2 was obtained as a white solid in 52% yield (68.2 mg; based on an average molecular weight of 987.76 g mol−1 calculated from T1, T2 and T3 units, see above).
Catalytic studies
Leaching experiment
A mixture of material M3 (13.5 mg, 0.0133 mmol), CH3SO2NH2 (25.7 mg, 0.266 mmol), K2CO3 (112.0 mg, 0.80 mmol), K3Fe(CN)6 (267.0 mg, 0.80 mmol), distilled water (0.4 mL) and t-BuOH (0.4 mL) was vigorously stirred at rt for 16 h, then diluted with t-BuOH (0.6 mL) and water (0.6 mL) and filtered through a PTFE syringe filter (0.1 μm). A solution of potassium osmate (0.48 mg, 0.00133 mmol) in water (135 μL) was added to the filtrate. After 10 min of vigorous stirring at rt, 1-phenylcyclohexene (44 μL, 42 mg, 0.266 mmol) was added and the mixture was stirred at rt for 16 h. The mixture was diluted with water (3 mL) and extracted with ethyl acetate (3 × 3 mL). The combined organic layers were washed with brine (2 mL), dried over anhydrous Na2SO4, filtered and evaporated in vacuo. The crude product was purified by flash column chromatography in hexane/ethyl acetate (gradient from 0 to 38%) giving 1-phenylcyclohexane-1,2-diol in 96% yield (49 mg) with er 56:44.
Materials recycling
A mixture of material M (0.06 mmol), CH3SO2NH2 (113.0 mg, 1.18 mmol), K2CO3 (489.0 mg, 3.54 mmol), K3Fe(CN)6 (1.16 g, 3.54 mmol), potassium osmate (2.17 mg, 0.006 mmol), distilled water (2.4 mL) and t-BuOH (2.8 mL) was vigorously stirred at rt for 10 min. (E)-Methyl 3-phenylprop-2-enoate (119.3 mg, 1.18 mmol) was added and the mixture was stirred at rt for 48 h. The ratio of starting alkene to product was determined by GLC analysis. The reaction mixture was diluted with water (15 mL) and ethyl acetate (10 mL). Compact material M3 was isolated by filtration using a PTFE membrane (0.2 μm), thoroughly washed with water (10 mL) and ethyl acetate (15 mL), and dried in vacuo. Nanoparticulate material M1 was isolated by OSN:60 The reaction mixture was diluted with ethyl acetate (20 mL) and water (20 mL) and was subjected to OSN until the residual retentate volume of cca 1–2 mL was reached. The retentate was suspended in ethyl acetate/water (1:1) mixture (50 mL) and again subjected to OSN until the residual retentate volume of cca 1–2 mL was reached. The washing-filtration cycle was repeated three times. The final portion of the retentate was withdrawn and evaporated to dryness in vacuo.
The filtrate phases were separated and the aqueous one was extracted with ethyl acetate (2 × 10 mL). The combined organic phases were washed with brine (10 mL), dried over anhydrous Na2SO4, filtered and evaporated in vacuo. The crude product was purified by flash column chromatography in hexane/ethyl acetate (gradient from 10 to 50%) giving methyl (2R,3S)-2,3-dihydroxy-3-phenylpropanoate as a white solid (analytical data are available in the ESI†).
The recovered material M was used in the next cycle. Weights of chemicals were recalculated according to the weight of the recovered material M to maintain the same molar ratios of the substances as in the first cycle and the whole procedure was repeated. The results are summarized in Table 3.
Results and discussion
Based on the successful use of bis-Cinchona alkaloid-phthalazine in homogeneous enantioselective catalysis, we decided to incorporate this structural unit into the bridged silsesquioxane to prepare a solid catalyst that could be easily removed from the reaction mixture by filtration and possibly recycled. The sol–gel hydrolysis/self-condensation of the bridged organic bis(trialkoxysilanes) was selected as the material preparation method with the aim of introducing the maximum number of chiral units into the material. Furthermore, a hydrolytically stable triazole ring was chosen as a linker between the alkaloid derivative and the silsesquioxane network to prevent leaching of the ligand from the solid material.To prepare the materials, we used derivatives of two Cinchona alkaloids, quinine and cinchonine. In the first stage, we succeeded in the preparation of two bridged silsesquioxanes containing the cinchonine derivative and used them as heterogenized ligands in enantioselective dihydroxylation of alkenes.29 The reported cinchonine-based materials were obtained either as a compact bulk material or as an irregularly nanostructured material. In this article, we describe follow-up research leading to quinine-based materials and optimization of the sol–gel procedure, which enables us to prepare nanosphere-shaped materials.
Synthesis of bis(triethoxysilane) precursors
Two bis(triethoxysilane) precursors, bis-quinine–phthalazine derivative 1 and bis-cinchonine–phthalazine derivative 2 (Fig. 2), were synthesized and then subjected to the sol–gel self-condensation to give hybrid organosilica materials. | ||
| Fig. 2 Structures of precursors. | ||
The same synthetic strategy as in our previous paper29 was used to synthesize bridged bis(trialkoxysilanes) 1 and 2. Organic triethoxysilanes were chosen since ethanol by-product is formed during the sol–gel process and the lower reactivity of the ethoxy groups allows for a greater synthetic control to tailor the required nanostructures.22 Synthesis of precursors 1 and 2 started from Cinchona alkaloids. The alkene double bond presented in the alkaloid structure was transformed to a triple bond by a two-step procedure consisting of bromine addition and dehydrobromination.9,16 10,11-Didehydro Cinchona alkaloid 3 or 4 then reacted with 1,4-dichlorophthalazine in the presence of base giving diyne 5 or 6, respectively (Scheme 1). The triethoxysilyl groups were introduced into the precursor structure via copper-catalyzed alkyne–azide cycloaddition (CuAAC)67,68 of bis-Cinchona alkaloid-phthalazine diyne 5 or 6 with (3-azidopropyl)triethoxysilane.
 | ||
| Scheme 1 Synthesis of precursors. | ||
The previously described experimental procedure29 was used to prepare cinchonine-based precursor 2. In the preparation of 10,11-didehydroquinine 3, the original procedure61 was modified to avoid electrophilic bromination of the aromatic ring and to increase yield of the bromination step. The reaction was done at higher temperature (0 °C instead of −20 °C) using only one molar equivalent of bromine (contrary to original two equivalents) giving 10,11-didehydroquinine 3 in 65% overall yield. The next step was substitution of the chlorine atoms in 1,4-dichlorophthalazine for two 10,11-didehydroquinine molecules leading to diyne 5. While synthesis of cinchonine-based diyne 6 proceeded smoothly using the procedure reported for preparation of (DHQ)2–PHAL,32 the preparation of quinine-based diyne 5 required optimization of the reaction conditions. When the reaction was carried out according to above mentioned procedure in toluene using a mixture of potassium carbonate and potassium hydroxide as base, it gave low and non-reproducible yields, probably due to the heterogeneous nature of the reaction mixture. The use of sodium hydride in tetrahydrofuran led to a significant improvement in yield and reproducibility of the reaction. The last step in the precursor 1 synthesis was the CuAAC reaction of diyne 5 with (3-azidopropyl)triethoxysilane. To avoid undesired hydrolysis of the ethoxysilyl groups, the CuAAC reaction must be done under anhydrous conditions with exclusion of acids, bases or strong nucleophiles. The best results were obtained using the copper-in-charcoal (Cu/C) catalyst64 in anhydrous dioxane.
Preparation of materials
Both bridged bis(triethoxysilanes) 1 and 2 were subjected to sol–gel hydrolysis/condensation under various experimental conditions. Two nanoparticulate materials (M1 and M2) were prepared by base-catalyzed sol–gel process and two compact materials (M3 and M4) were prepared by fluoride-catalyzed sol–gel process, whereby M1 and M3 are based on bridged bis(triethoxysilane) 1 and M2 and M4 are based on bridged bis(triethoxysilane) 2.The reaction conditions were optimized to obtain nanomaterials M1 and M2 from bis(triethoxysilane) precursors 1 and 2, respectively. Specifically, reaction time, temperature, surfactant type, ratio of hybrid precursor:surfactant, solvent volume, and catalyst used were varied. Several different types of suitable surfactants were chosen to test their ability to form nanospheres: Triton X-100, Pluronic P123, and CTAB, which are well known to promote the formation of nanospheres from various types of inorganic or organic–inorganic precursors.69,70 Using CTAB as a surfactant and NaOH as a catalyst was found to be the best conditions for the formation of M1 nanoparticles from bis(triethoxysilane) precursor 1.
Nanoparticles with an average size of 108 nm were formed under these conditions (Fig. 3), which is similar to the previously reported synthesis of chiral bridged organosilane nanoparticles.70
 | ||
| Fig. 3 Characterization of the material M1: (a) SEM image (scale bar 200 nm) (b) histogram of particles diameters, (c) 29Si NMR spectrum and (d) composition of siloxane fraction defined as the total amount of individual Tn structure units in molar % obtained from signal intensities of 29Si NMR spectra and calculated degree of condensation c (in %). | ||
On the other hand, Pluronic 123 as a surfactant with 25% aqueous ammonia as a catalyst was shown to be the best conditions for the preparation of M2 nanoparticles from bis(triethoxysilane) precursor 2. Despite the similar published synthesis,70 we experienced no fused nanoparticle formation and well resolved nanospheres with an average size of 132 nm formed (see Fig. 4a).
 | ||
| Fig. 4 Characterization of the material M2: (a) SEM image (scale bar 1 μm) (b) histogram of particles diameters, (c) 29Si NMR spectrum and (d) composition of siloxane fraction defined as the total amount of individual Tn structure units in molar % obtained from signal intensities of 29Si NMR spectra and calculated degree of condensation c (in %). | ||
The compact material M3 was prepared from bis(triethoxysilane) precursor 1 similar to our previously published procedure29 to compare its efficiency to catalyze enantioselective reactions. The previously described29 material M4 (compact material based on bis(triethoxysilane) precursor 2) was used for the comparison to nanoparticulate systems as well.
Materials characterization
All the prepared materials M1–M3 were thoroughly characterized using SEM, 29Si SS-NMR (Fig. 3–5), TEM, 13C NMR, FTIR, TGA, and ECD (see ESI†). Characterization of material M4 was published in our previous paper.29 The calculated degree of condensation (Fig. 3d, 4d and 5c) ranges between 83 and 96% and is similar to that reported previously for organosilane nanoparticles.29,71,72 | ||
| Fig. 5 Characterization of the material M3: (a) SEM image (scale bar 200 nm) (b) 29Si NMR spectrum and (c) composition of siloxane fraction defined as the total amount of individual Tn structure units in molar % obtained from signal intensities of 29Si NMR spectra and calculated degree of condensation c (in %). | ||
SEM images from M1 and M2 display spherical nanoparticles with varying diameters (Fig. 3a and 4a), with SEM used to measure the nanoparticle diameter distribution (Fig. 3b and 4b), resulting in an average particle size of 108 ± 43 and 132 ± 53 nm for M1 and M2, respectively (see ESI†). In contrast, SEM images for M3 displayed a compact solid material (Fig. 5a). 29Si SS-NMR spectra show three predominant peaks originating from the polycondensed silica species T1, T2, and T3 units for M1 (Fig. 3c), with only T2 and T3 units observed for M2 and M3 (Fig. 4c and 5b), used to calculate the degree of polycondensation.
Calculations based on the ratio of Tn units from 29Si CP/MAS SS-NMR (Fig. 3d, 4d and 5c) showed that the percentage of silicon is 5.33, 5.69 and 5.37 for materials M1, M2 and M3, respectively. The calculations were not corrected to the presence of remaining ethoxy groups, as the determination of their amount from 13C SS-NMR is very imprecise. On the other hand, TGA (see ESI†) was used as an alternative method to ascertain the Si content in the materials. TGA revealed that the materials contain only a small amount of water and that all of them are stable up to ca. 200 °C before their decomposition begins. None of the materials contain any indication of residual surfactant being present. The calculated percentage of silicon is 5.48, 6.48 and 6.25 for materials M1, M2 and M3, respectively. The higher silicon content according to TGA is caused by the encapsulation of unburnt carbonaceous material in the SiO2 matrix, which was proved by Raman spectroscopy of residual material (data not shown).
13C CP/MAS SS-NMR together with FTIR (see ESI†) confirmed the presence of the organic component of the materials M1–M3. Positions and intensities of 13C CP/MAS SS-NMR peaks in the materials are similar to those measured for the corresponding precursors in liquid state. Moreover, FTIR shows typical peaks for aromatics (C–H ∼3130 and ∼3070, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C ∼1580 and ∼1510 cm−1), aliphatics (CH2 ∼2930 and ∼2870, C–H ∼1460 cm−1) and the silica part as well (Si–O–C ∼1090, ∼1030 and ∼940, Si–O–Si ∼1050 cm−1), which is in good agreement with previously published results.29
C ∼1580 and ∼1510 cm−1), aliphatics (CH2 ∼2930 and ∼2870, C–H ∼1460 cm−1) and the silica part as well (Si–O–C ∼1090, ∼1030 and ∼940, Si–O–Si ∼1050 cm−1), which is in good agreement with previously published results.29
Catalytic studies
The prepared materials were tested in the heterogeneous catalysis of enantioselective reactions. In order to assess the influence of the nanosphere shape of the materials on enantioselectivity of the reactions, catalytic properties of the spherical materials M1 and M2 with analogous compact materials M3 and M4 were compared.One of the reactions studied, and chosen was Sharpless dihydroxylation of alkenes. Since it is well known from homogeneous catalysis that ligands based on derivatives of quinine or quinidine provide better enantioselectivities than ligands based on derivatives of cinchonine or cinchonidine,73 this reaction was only studied with materials M1 and M3, which contain quinine derivative. The starting alkenes (see Table 1) were chosen to cover different structural types of alkenes (mono-, di-, trisubstituted; cis-, trans-; aromatic or aliphatic). As can be seen from the results presented in Table 1, the spherical nanomaterial M1 provided higher enantioselectivities for all studied alkenes and, in most cases, also higher yields of dihydroxylation reactions than the compact material M3. The highest increase in enantioselectivity when using spherical nanoparticles M1 compared to the compact material M3 was achieved in the dihydroxylation of aliphatic alkenes trans-hex-2-ene (trans-disubstituted double bond) and 2-methyl-hept-2-ene (trisubstituted double bond).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Alkene | Product | Material M1 | Material M3 | ||
| Yielda [%] | er | Yielda [%] | er | |||
| a Isolated yield after column chromatography. b Obtained from HPLC on a column with chiral stationary phase. | ||||||
| 1 | Methyl cinnamate | Methyl 2,3-dihydroxy-3-phenylpropanoate | 80 | 100:0 |
82 | 99:1 |
| 2 | trans-Stilbene | 1,2-Diphenylethane-1,2-diol | 67 | 100:0 |
72 | 98:2 |
| 3 | trans-Hex-2-ene | Hexane-2,3-diol | 65 | 85:15 |
54 | 75:25 |
| 4 | 2-Methylhept-2-ene | 2-Methylheptane-2,3-diol | 61 | 92:8 |
49 | 78:22 |
| 5 | 1-Phenylcyclohexene | 1-Phenylcyclohexane-1,2-diol | 95 | 98:2 |
88 | 93:7 |
| 6 | 2-Phenylpropene | 2-Phenylpropane-1,2-diol | 95 | 90:10 |
79 | 85:15 |
| 7 | Styrene | 1-Phenylethane-1,2-diol | 89 | 90:10 |
76 | 83:17 |
| 8 | Dodec-1-ene | Dodecane-1,2-diol | 72 | 80:20 |
54 | 75:15 |
| 9 | 1H-Indene | 2,3-Dihydro-1H-indene-1,2-diol | 90 | 61:39 |
54 | 59:41 |
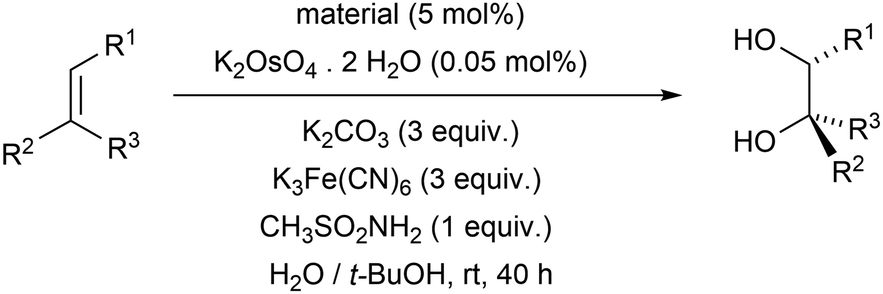
In agreement with the results of homogeneous Sharpless dihydroxylations using the (DHQ)2–PHAL ligand,32 the best enantioselectivities were observed for aromatic trans-alkenes, while cis-alkene provided the worst enantioselectivity The comparison of enantioselectivities obtained in heterogeneous catalysis in the presence of material M1 with those achieved in homogeneous catalysis in the presence of the (DHQ)2–PHAL ligand32 is presented in Table 2.
| Entry | Starting alkene | M1 | (DHQ)2–PHAL32 |
|---|---|---|---|
| 5 mol% | 1 mol% | ||
| er | er | ||
| a Ref. 75. b Ref. 76. | |||
| 1 | Methyl cinnamate | 100:0 |
99.5:0.5a |
| 2 | trans-Stilbene | 100:0 |
99.8:0.2 |
| 3 | trans-Hex-2-ene | 85:15 |
n.a. |
| 4 | trans-But-2-ene | n.a. | 86:14 |
| 5 | 2-Methylhept-2-ene | 92:8 |
97.5:2.5 |
| 6 | 1-Phenylcyclohexene | 98:2 |
98.5:1.5 |
| 7 | 2-Phenylpropene | 90:10 |
96.5:3.5 |
| 8 | Styrene | 90:10 |
98.5:1.5 |
| 9 | Dodec-1-ene | 80:20 |
90:10 |
| 10 | 1H-Indene | 61:39 |
68.5:31.5b |
Since we did not find the literature data for dihydroxylation of trans-hex-2-ene under homogeneous conditions (Table 2, line 3), we present for comparison the result of dihydroxylation of trans-but-2-ene (Table 2, line 4), which is structurally similar and contains the same type of double bond (trans-disubstituted). The data presented in Table 2 show that heterogenized ligand M1 exhibits the same excellent enantioselectivities as (DHQ)2–PHAL for trans-disubstituted alkenes, and only slightly lower enantioselectivities for tri- and monosubstituted as well as terminal alkenes. However, it should be noted that the homogeneous catalysis study utilized 1 mol% of (DHQ)2–PHAL ligand,32 whereas this work employed an amount of material (M1 or M3) that corresponds to 5 mol% of the incorporated ligand. A larger amount of solid catalyst was needed to achieve reasonable reaction times. Since the same enantiomers of the products predominate in our experiments as in analogous homogeneous experiments and the same trend of dependence of enantioselectivity on the character of the alkene double bond was observed, we can conclude that the same mechanism of chirality transfer from the ligand to the substrate applies as in homogeneous catalysis. It is generally accepted that Sharpless dihydroxylation proceeds via a [3 + 2] cycloaddition mechanism within the U-shaped ligand cavity.74
It was confirmed that the observed enantioselectivities are not due to leaching of the ligand from the solid material. Using the protocol published by Salvadori et al.,57 a leakage test was performed with material M3 for the dihydroxylation reaction of 1-phenylcyclohexene. The obtained er 56:44 proved that the high enantioselectivities are not the result of homogeneous catalysis caused by the washed-out ligand.
An important aspect of heterogeneous catalysts is the possibility of recycling them. Recyclability of materials M1 and M3 was studied for dihydroxylation of methyl cinnamate. After the dihydroxylation reaction, the material was isolated, thoroughly washed, dried, and the recovered material was used in the next cycle. While compact material M3 was isolated from the reaction mixture by filtration using a PTFE membrane filter (0.2 μm), it was impossible to isolate nanoparticulate material M1 by simple filtration or centrifugation. Finally, organic solvent nanofiltration (OSN)60 was successfully used to isolate material M1. The results of the recycling experiments are summarized in Table 3 and show that enantioselectivity of the methyl cinnamate dihydroxylation reaction remained consistent over five cycles.
It is difficult to compare the results obtained by us with previously published results of dihydroxylation of alkenes using heterogeneous catalysts in which the bis-Cinchona alkaloid-phthalazine ligand was grafted onto a silica gel52–56 or polymer support.48 Although excellent enantioselectivities were reported in all cases, Sherrington59 and Salvadori57 later demonstrated that leaching of the ligand from these solids occurs under Sharpless dihydroxylation conditions and that low concentrations of the eluted ligand are responsible for high enantioselectivities of the dihydroxylation reactions.57 To the best of our knowledge, a structurally similar heterogenized ligand with confirmed hydrolytic stability has not yet been reported by other authors. Two hydrolytically stable non-spherical bis-cinchonine–phthalazine-based bridged silsesquioxanes that we published recently29 showed lower enantioselectivities than the materials presented in this paper.
The second reaction studied was chlorolactonization of 4-arylpent-4-enoic acids. In 2010, Borhan et al.38 developed a methodology for enantioselective chlorolactonization that utilizes (DHQD)2–PHAL as an organocatalyst. They used 1,3-dichloro-5,5-dimethylhydantoin (DCDMH) or 1,3-dichloro-5,5-diphenylhydantoin (DCDPH) as a chlorine source, one equivalent of benzoic acid as an additive and 0.1 equivalent of organocatalyst (DHQD)2–PHAL to achieve good enantioselectivities at −40 °C. The same reaction conditions and DCDMH as a chlorine source were utilized to evaluate the catalytic properties of materials M1–M4 in chlorolactonizations of three 4-arylpent-4-enoic acids. However, the reactions progressed too slowly at −40 °C when the reported amount of solid organocatalyst (10 mol%) was employed. At higher temperatures, the chlorolactonization reactions proceeded with low enantioselectivities. As a result, the catalyst amount was increased to 30 mol%.
The obtained results (Table 4) show that chlorolactonizations of 4-phenylpent-4-enoic and 4-[4-(trifluoromethyl)phenyl]pent-4-enoic acids proceeded with moderate enantioselectivity. Spherical materials M1 and M2 provided slightly higher enantioselectivity than compact materials M3 and M4. The difference between the quinine-based material M1 and the cinchonine-based material M2 was minimal, with material M2 exhibiting higher enantioselectivity in chlorolactonization of 4-[4-(trifluoromethyl)phenyl]pent-4-enoic acid. Chlorolactonization of 4-(4-methoxyphenyl)pent-4-enoic acid provided in all cases no or only negligible enantioselectivity. This result is in agreement with the outcome of the chlorolactonization reaction catalyzed by the homogeneous catalyst (DHQD)2–PHAL38 (see Table 5, entry 2).
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Aryl | Material M1 | Material M2 | Material M3 | Material M4 | ||||
| Yielda [%] | er | Yielda [%] | er | Yielda [%] | er | Yielda [%] | er | ||
| a Isolated yield after column chromatography. b Obtained from HPLC on a column with chiral stationary phase. | |||||||||
| 1 | Phenyl | 83 | 74:26 |
67 | 74:26 |
75 | 72:28 |
58 | 64:36 |
| 2 | 4-Methoxyphenyl | 64 | 50:50 |
66 | 52:48 |
64 | 50:50 |
59 | 51:49 |
| 3 | 4-(Trifluoromethyl)phenyl | 64 | 69:31 |
52 | 74:26 |
45 | 63:37 |
45 | 63:37 |

Based on the comparison of the results of chlorolactonization reactions catalyzed by material M2 and (DHQD)2–PHAL (Table 5), it can be concluded that the studied materials catalyzed chlorolactonization with significantly lower enantioselectivity than the homogeneous catalyst (DHQD)2–PHAL.38 Similar to the dihydroxylation of alkenes, also in chlorolactonization of 4-arylpent-4-enoic acids, the same enantiomer of the product predominates in our experiments as in homogeneous catalysis. This observation may lead to the conclusion that the same mechanism of chirality transfer from the organocatalyst to the reaction product as proposed by Borhan et al.39 for homogeneous catalysis is applied
Conclusion
A synthesis method was developed for the bridged bis(triethoxysilane) 1, which contains the bis-quinine–phthalazine structural unit. This precursor, along with the previously published analogous cinchonine-based bis(triethoxysilane) 229 were employed in the preparations of solid bridged silsesquioxanes using a sol–gel technique. The reaction conditions were optimized to obtain spherical nanoparticulate materials M1 and M2. The quinine-based material M1 was successfully used as a heterogenized chiral ligand in Sharpless dihydroxylations of alkenes, providing higher enantioselectivities than structurally similar compact material M3. Excellent enantioselectivities, comparable to those obtained with homogeneous ligand (DHQ)2–PHAL,32 were observed for aromatic trans-alkenes. Furthermore, we confirmed that the material can be recycled in at least five cycles. We thus prepared the first heterogeneous analogue of (DHQ)2–PHAL ligand for enantioselective dihydroxylation of alkenes with a regular distribution of chiral organic units within the material structure, which is hydrolytically stable under Sharpless dihydroxylation conditions.
Both quinine-based material M1 and cinchonine-based material M2 were tested as solid organocatalysts in chlorolactonizations of 4-arylpent-4-enoic acids. Chlorolactonizations of 4-phenylpent-4-enoic and 4-[4-(trifluoromethyl)phenyl]pent-4-enoic acids proceeded with only moderate enantioselectivities that were lower than those obtained under homogeneous organocatalysis using (DHQD)2–PHAL.38 Notwithstanding, to the best of our knowledge, materials M1–M4 are the first heterogeneous catalysts reported for enantioselective chlorolactonization reaction. Once again, the analogous compact materials M3 and M4 gave lower enantioselectivities than the spherical materials M1 and M2, indicating that the morphology of the materials affects their catalytic activity. To further improve the catalytic properties of the materials, it would be necessary to prepare these bridged silsesquioxanes in the form of PMOs, which is, however, a very challenging task.
Author contributions
D. T. – investigation, formal analysis, writing; M. N. – investigation; J. T. – investigation; V. M. – investigation, formal analysis, writing; M. S. – investigation; C. H. – investigation; M. Ř. – funding acquisition, methodology, supervision, formal analysis, writing; M. M. – investigation; V. S. – supervision, formal analysis; K. D. – investigation; J. H. – conceptualization, funding acquisition, methodology, project administration, supervision, writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Czech Science Foundation, grant number 18-09824S, by the Ministry of Education, Youth and Sports of the Czech Republic and the European Union – European Structural and Investment Funds in the frames of Operational Programme Research, Development and Education – project Hybrid Materials for Hierarchical Structures (HyHi, Reg. No. CZ.02.1.01/0.0/0.0/16_019/0000843) and by the Research Infrastructure NanoEnviCz, supported by the Ministry of Education, Youth and Sports of the Czech Republic under Project No. LM2023066.References
- R. S. Atkison, Stereoselective Synthesis, Wiley, 1995 Search PubMed.
- Stereoselective Synthesis, ed. J. G. de Vries, G. A. Molander and P. A. Evans, Thieme, New York, Stuttgart, and Delhi, 2011 Search PubMed.
- R. E. Gawley and J. Aubé, Principles of Asymmetric Synthesis, Elsevier, 2nd edn, 2012 Search PubMed.
- Stereoselective Synthesis of Drugs and Natural Products, ed. V. Andrushko and N. Andrushko, Wiley, Hoboken, 2013 Search PubMed.
- E. J. Corey and L. Kürti, Enantioselective Chemical Synthesis: Methods, Logic, and Practice, Direct Book Publishing, Dallas, 2010 Search PubMed.
- Catalytic Asymmetric Synthesis, ed. T. Akiyama and I. Ojima, Wiley-VCH, Weinheim, 2022 Search PubMed.
- K. Ding and Y. Uozumi, Handbook of Asymmetric Heterogeneous Catalysis, Wiley-VCH, 2008 Search PubMed.
- C. E. Song and S.-G. Lee, Chem. Rev., 2002, 102, 3495–3524 CrossRef CAS PubMed.
- Q.-H. Fan, Y.-M. Li and A. S. C. Chan, Chem. Rev., 2002, 102, 3385–3466 CrossRef CAS PubMed.
- C. Li, H. Zhang, D. Jiang and Q. Yang, Chem. Commun., 2007, 547–558 RSC.
- A. Zamboulis, N. Moitra, J. J. E. Moreau, X. Cattoën and M. Wong Chi Man, J. Mater. Chem., 2010, 20, 9322–9338 RSC.
- Q. Yang, J. Liu, L. Zhang and C. Li, J. Mater. Chem., 2009, 19, 1945–1955 RSC.
- M. Ferré, R. Pleixats, M. Wong Chi Man and X. Cattoën, Green Chem., 2016, 18, 881–922 RSC.
- F. Hoffmann, M. Cornelius, J. Morell and M. Fröba, Angew. Chem., Int. Ed., 2006, 45, 3216–3251 CrossRef CAS PubMed.
- N. Mizoshita, T. Tani and S. Inagaki, Chem. Soc. Rev., 2011, 40, 789–800 RSC.
- S. S. Park, M. S. Moorthy and C.-S. Ha, NPG Asia Mater., 2014, 6, e96 CrossRef CAS.
- M. W. A. MacLean, L. M. Reid, X. Wu and C. M. Crudden, Chem. – Asian J., 2015, 10, 70–82 CrossRef CAS PubMed.
- X. Liu, P. Wang, Y. Yang, P. Wang and Q. Yang, Chem. – Asian J., 2010, 5, 1232–1239 CrossRef CAS PubMed.
- K. Liu, R. Jin, T. Cheng, X. Xu, F. Gao, G. Liu and H. Li, Chem. – Eur. J., 2012, 18, 15546–15553 CrossRef CAS PubMed.
- T. Seki, K. McEleney and C. M. Crudden, Chem. Commun., 2012, 48, 6369–6371 RSC.
- R. Jin, K. Liu, D. Xia, Q. Qian, G. Liu and H. Li, Adv. Synth. Catal., 2012, 354, 3265–3274 CrossRef CAS.
- J. G. Croissant, X. Cattoën, J.-O. Durand, M. Wong Chi Man and N. M. Khashab, Nanoscale, 2016, 8, 19945–19972 RSC.
- A. Adima, J. J. E. Moreau and M. Wong Chi Man, J. Mater. Chem., 1997, 7, 2331–2333 RSC.
- A. Adima, J. J. E. Moreau and M. Wong Chi Man, Chirality, 2000, 12, 411–420 CrossRef CAS PubMed.
- C. Bied, D. Gauthier, J. J. E. Moreau and M. Wong Chi Man, J. Sol-Gel Sci. Technol., 2001, 20, 313–320 CrossRef CAS.
- A. Brethon, J. J. E. Moreau and M. Wong Chi Man, Tetrahedron: Asymmetry, 2004, 15, 495–502 CrossRef CAS.
- A. Brethon, C. Bied, J. J. E. Moreau and M. Wong Chi Man, J. Sol-Gel Sci. Technol., 2009, 50, 141–151 CrossRef CAS.
- A. Kuschel and S. Polarz, J. Am. Chem. Soc., 2010, 132, 6558–6565 CrossRef CAS PubMed.
- D. Tetour, T. Paška, V. Máková, B. Nikendey Holubová, J. Karpíšková, M. Řezanka, J. Brus and J. Hodačová, J. Catal., 2021, 404, 493–500 CrossRef CAS.
- K. Kacprzak and J. Gawroński, Synthesis, 2001, 961–998 CAS.
- L. Bernardi and M. Fochi, in Sustainable catalysis without metals or other endangered elements - Part 2, The Royal Society of Chemistry, Cambridge, 2016, pp. 1–43 Search PubMed.
- K. B. Sharpless, W. Amberg, Y. L. Bennani, G. A. Crispino, J. Hartung, K. S. Jeong, H. L. Kwong, K. Morikawa and Z. M. Wang, J. Org. Chem., 1992, 57, 2768–2771 CrossRef CAS.
- H. C. Kolb, M. S. VanNieuwenhze and K. B. Sharpless, Chem. Rev., 1994, 94, 2483–2547 CrossRef CAS.
- M. Bella and K. A. Jørgensen, J. Am. Chem. Soc., 2004, 126, 5672–5673 CrossRef CAS PubMed.
- K. C. Nicolaou, N. L. Simmons, Y. Ying, P. M. Heretsch and J. S. Chen, J. Am. Chem. Soc., 2011, 133, 8134–8137 CrossRef CAS PubMed.
- M. R. Monaco and M. Bella, Angew. Chem., Int. Ed., 2011, 50, 11044–11046 CrossRef CAS PubMed.
- B. Soltanzadeh, A. Jaganathan, Y. Yi, H. Yi, R. J. Staples and B. Borhan, J. Am. Chem. Soc., 2017, 139, 2132–2135 CrossRef CAS PubMed.
- D. C. Whitehead, R. Yousefi, A. Jaganathan and B. Borhan, J. Am. Chem. Soc., 2010, 132, 3298–3300 CrossRef CAS PubMed.
- R. Yousefi, A. Sarkar, K. D. Ashtekar, D. C. Whitehead, T. Kakeshpour, D. Holmes, P. Reed, J. E. Jackson and B. Borhan, J. Am. Chem. Soc., 2020, 142, 7179–7189 CrossRef CAS PubMed.
- T. Nishimine, H. Taira, E. Tokunaga, M. Shiro and N. Shibata, Angew. Chem., Int. Ed., 2016, 55, 359–363 CrossRef CAS PubMed.
- M. V. Enevoldsen, J. Overgaard, M. S. Pedersen and A. T. Lindhardt, Chem. – Eur. J., 2018, 24, 1204–1208 CrossRef CAS PubMed.
- B. M. Kim and K. B. Sharpless, Tetrahedron Lett., 1990, 31, 3003–3006 CrossRef CAS.
- D. Pini, A. Petri, A. Nardi, C. Rosini and P. Salvadori, Tetrahedron Lett., 1991, 32, 5175–5178 CrossRef CAS.
- B. B. Lohray, A. Thomas, P. Chittari, J. R. Ahuja and P. K. Dhal, Tetrahedron Lett., 1992, 33, 5453–5456 CrossRef CAS.
- D. Pini, A. Petri and P. Salvadori, Tetrahedron: Asymmetry, 1993, 4, 2351–2354 CrossRef CAS.
- B. B. Lohray, E. Nandanan and V. Bhushan, Tetrahedron Lett., 1994, 35, 6559–6562 CrossRef CAS.
- C. E. Song, E. J. Roh, S.-G. Lee and I. O. Kim, Tetrahedron: Asymmetry, 1995, 6, 2687–2694 CrossRef CAS.
- C. E. Song, J. W. Yang, H. J. Ha and S.-G. Lee, Tetrahedron: Asymmetry, 1996, 7, 645–648 CrossRef CAS.
- M. S. DeClue and J. S. Siegel, Org. Biomol. Chem., 2004, 2, 2287–2298 RSC.
- D. Cancogni, A. Mandoli, R. P. Jumde and D. Pini, Eur. J. Org. Chem., 2012, 1336–1345 CrossRef CAS.
- B. T. Kumpuga and S. Itsuno, Asian J. Org. Chem., 2019, 8, 251–256 CrossRef CAS.
- B. B. Lohray, E. Nandanan and V. Bhushan, Tetrahedron: Asymmetry, 1996, 7, 2805–2808 CrossRef CAS.
- C. E. Song, J. W. Yang and H.-J. Ha, Tetrahedron: Asymmetry, 1997, 8, 841–844 CrossRef CAS.
- H. M. Lee, S.-W. Kim, T. Hyeon and B. M. Kim, Tetrahedron: Asymmetry, 2001, 12, 1537–1541 CrossRef CAS.
- I. Motorina and C. M. Crudden, Org. Lett., 2001, 3, 2325–2328 CrossRef CAS PubMed.
- S. M. Sarkar, M. E. Ali, M. L. Rahman and M. M. Yusoff, J. Nanomater., 2014, 123680 Search PubMed.
- A. Mandoli, D. Pini, M. Fiori and P. Salvadori, Eur. J. Org. Chem., 2005, 1271–1282 CrossRef CAS.
- A. Petri, D. Pini and P. Salvadori, Tetrahedron Lett., 1995, 36, 1549–1552 CrossRef CAS.
- L. Canali, C. E. Song and D. C. Sherrington, Tetrahedron: Asymmetry, 1998, 9, 1029–1034 CrossRef CAS.
- P. Marchetti, M. F. Jimenez Solomon, G. Szekely and A. G. Livingston, Chem. Rev., 2014, 114, 10735–10806 CrossRef CAS PubMed.
- K. M. Kacprzak, W. Lindner and N. M. Maier, Chirality, 2008, 20, 441–445 CrossRef CAS PubMed.
- T. M. H. Vuong, J. Weimmerskirch-Aubatin, J.-F. Lohier, N. Bar, S. Boudin, C. Labbé, F. Gourbilleau, H. Nguyen, T. T. Dang and D. Villemin, New J. Chem., 2016, 40, 6070–6076 RSC.
- G. Singh, S. S. Mangat, J. Singh, A. Arora and R. K. Sharma, Tetrahedron Lett., 2014, 55, 903–909 CrossRef CAS.
- B. H. Lipshutz and B. R. Taft, Angew. Chem., Int. Ed., 2006, 45, 8235–8238 CrossRef CAS PubMed.
- K. D. Ashtekar, M. Vetticatt, R. Yousefi, J. E. Jackson and B. Borhan, J. Am. Chem. Soc., 2016, 138, 8114–8119 CrossRef CAS PubMed.
- L. Zhou, C. K. Tan, X. Jiang, F. Chen and Y.-Y. Yeung, J. Am. Chem. Soc., 2010, 132, 15474–15476 CrossRef CAS PubMed.
- N. Moitra, J. J. E. Moreau, X. Cattoën and M. Wong Chi Man, Chem. Commun., 2010, 46, 8416–8418 RSC.
- K. Bürglová, N. Moitra, J. Hodačová, X. Cattoën and M. Wong Chi Man, J. Org. Chem., 2011, 76, 7326–7333 CrossRef PubMed.
- K. Zheng and A. R. Boccaccini, Adv. Colloid Interface Sci., 2017, 249, 363–373 CrossRef CAS PubMed.
- S. Omar and R. Abu-Reziq, Appl. Sci., 2020, 10, 5960 CrossRef CAS.
- L.-C. Hu, Y. Yonamine, S.-H. Lee, W. E. van der Veer and K. J. Shea, J. Am. Chem. Soc., 2012, 134, 11072–11075 CrossRef CAS PubMed.
- Y. Fatieiev, J. G. Croissant, K. Julfakyan, L. Deng, D. H. Anjum, A. Gurinov and N. M. Khashab, Nanoscale, 2015, 7, 15046–15050 RSC.
- H. C. Kolb, P. G. Andersson and K. B. Sharpless, J. Am. Chem. Soc., 1994, 116, 1278–1291 CrossRef CAS.
- E. J. Corey and M. C. Noe, J. Am. Chem. Soc., 1996, 118, 319–329 CrossRef CAS.
- C. Bengtsson, H. Nelander and F. Almqvist, Chem. Eur. J., 2013, 19, 9916–9922 CrossRef CAS PubMed.
- M. H. Junttila and O. O. E. Hormi, J. Org. Chem., 2009, 74, 3038–3047 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr06234a |
| This journal is © The Royal Society of Chemistry 2024 |