
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The existence of a strongly bonded layer in associating liquids within silica pores – a spectral and molecular dynamics study†
Natalia
Soszka
 a,
Magdalena
Tarnacka
*b,
Barbara
Hachuła
a,
Patryk
Włodarczyk
c,
Roman
Wrzalik
b,
Marek
Hreczka
cd,
Marian
Paluch
b and
Kamil
Kamiński
b
a,
Magdalena
Tarnacka
*b,
Barbara
Hachuła
a,
Patryk
Włodarczyk
c,
Roman
Wrzalik
b,
Marek
Hreczka
cd,
Marian
Paluch
b and
Kamil
Kamiński
b
aInstitute of Chemistry, University of Silesia in Katowice, Szkolna 9, 40-006 Katowice, Poland
bAugust Chełkowski Institute of Physics, University of Silesia in Katowice, 75 Pułku Piechoty 1a, 41-500 Chorzów, Poland. E-mail: magdalena.tarnacka@us.edu.pl
cLukasiewicz Research Network – Institute of Non-Ferrous Metals, Sowinskiego 5, 44-100 Gliwice, Poland
dDepartment of Mechatronics, Silesian University of Technology, Akademicka 10A, 44-100 Gliwice, Poland
First published on 19th February 2024
Abstract
The properties of confined materials are assumed to be governed by the phenomena occurring at the interface, especially the formation of an irreversible adsorption layer (IAL), which has been widely discussed and detected in the case of thin polymer films and silica nanoparticles. In this paper, we present a novel experimental approach allowing us to reveal the formation of an IAL in two phenyl alcohols infiltrated into various mesoporous silica templates. The proposed methodology (based on evaporation) allowed us to detect the alterations in the OH and aromatic CH stretching vibration bands in infrared spectra, which were considered as evidence of the existence of IAL in constrained systems. Such interpretation was also confirmed by complementary molecular dynamics (MD) simulations that indicated the creation of much stronger hydrogen bonds between alcohols and silanol units than between alcohols themselves. Moreover, computation allowed us to identify additional enormously strong π-stacking interactions between phenyl rings stabilizing the interfacial layer. MD simulations also shed new light on the clustering process of both alcohols under confinement. Simulation and experimental data presented in this paper allowed a much deeper understanding of the processes occurring at the interface-formation of IAL and the association phenomenon at the nanoscale level.
I. Introduction
Soft materials under nanometric spatial constraint have attracted over recent years a growing interest from both scientific and industrial perspectives, which arises due to their (often) significantly different behavior from that of their bulk counterparts, just to mention differences in phase transition,1,2 dynamics,3–5 or the tendency to crystallization.6,7 This makes them promising agents in developing a novel generation of drug carriers,8–10 sensors,11 coatings,12 fuel cells,13etc. Changes in basic parameters of confined materials are considered to be an effect of mainly two factors: finite size and surface effects. However, despite continuing research efforts, the detailed understanding of the confinement effect is yet to be fully addressed (i.e., the contribution of different factors allowing the prediction of the behavior of partially restricted systems).One can recall that a majority of recent studies suggested the dominant influence of the latter issue (surface effects), which consists of, e.g., interactions14,15 (and surface polarity16,17) as well as roughness,15,18 curvature19 and wettability, might be the dominant factor. Nevertheless, it seems that one of the most interesting surface effects affecting/governing the behavior of confined materials is the formation of a fraction of molecules irreversibly adsorbed at the interface of the applied constrained medium (assigned as an irreversibly adsorbed layer, IAL). The occurrence of an IAL was first shown in the case of annealing experiments of capped thin polystyrene films performed by Napolitano and Wübbenhorst.20 The investigation revealed the shift of the segmental relaxation process with time due to the density perturbation leading to the thickening of ‘the layer of chains irreversibly adsorbed onto the substrate’ (Guiselin brushes) upon annealing, resulting in a change in the glass transition temperature, Tg (even recovery of the bulk-like behavior).20–23 It was assumed that the surface coverage increases as the new coming molecules more loosely interact with the surface and are adsorbed by fewer segments forming loops, tails, and trains,20,24 depending on the polymer rigidity and, also sometimes, on the interfacial dynamics.25 This was attributed to the formation of a defined IAL of molecules due to a decreasing free volume over time. Those observations also agreed with theoretical studies taking into account various free volume-based models, i.e., free volume diffusion model26,27 or a cooperative free volume model,28–30 which clearly show that the detected changes in the segmental dynamics and Tg might result from either the diffusion of free volume holes or free volume-dependent segmental relaxation behavior of confined systems, respectively. Herein, it is also worth stressing that the formation of the IAL was also reported in the case of materials cast on silica nanoparticles.31–34 Surprisingly, many of the above-mentioned effects observed for various thin polymer films (considered as one-dimensional confinement) are also recognized in higher-dimensional spatially constrained systems such as porous materials (two-dimensional confinement). One can mention the changes in dynamics upon annealing observed for various low-35,36 and high-molecular-weight glass37,38 formers infiltrated within either silica or anodized aluminum oxide (AAO) porous template characterized by different pore size, d = 10–160 nm. It should be also highlighted that in some cases, even the recovery of the bulk-like properties after prolonged annealing were reported.39,40 However, despite those similarities between the behavior of thin polymer films and porous materials, we still do not know whether these effects have the same molecular origin. Up to now, the existence of IAL has been experimentally confirmed only in the case of the former systems; as for the latter, this issue has yet to be addressed. This raises the question of whether it is possible to obtain/extract this layer in membranes, and if formed, whether it is governed by the same physics as in the case of thin films and, thus, is a fundamental feature of spatially confined systems.
Herein, we address some of those questions as we investigated two monohydroxy phenyl-substituted alcohols (PhAs), 2-phenyl-1-ethanol (2Ph1E) and 3-phenyl-1-propanol (3Ph1P, see their chemical structures in Fig. 1), infiltrated into silica mesopores of d = 4–8 nm by means of Fourier transform infrared (FTIR) spectroscopy. However, to verify the existence of IAL in porous systems, we propose a slightly different approach in contrast to the Guiselin brushes experiments.20,41–43 As porous materials are characterized by highly curved interface when compared to thin films (see Fig. S1 in ESI†), instead of removing the liquid inside the pores by ‘washing out’, the substances were evaporated from the silica templates. This approach would allow us to avoid the presence of any solvent, which might remain in the system after the procedure. The choice of materials for this study was not accidental. We chose two associating liquids (characterized by a different length of alkyl chain; see Fig. 1), which relatively easily evaporate at ambient pressure. Moreover, in this study, we applied two types of silica membranes, native (of untreated hydrophilic interface) and silanized (of treated hydrophobic interface), to distinguish the role of specific interactions (in this case, hydrogen bonding, H-bonding) in the formation of IAL. The nanoporous silica templates were prepared by electrochemical etching of silicon wafers and subsequent thermal oxidation, according to the procedure reported in ref. 44 and 45.
 | ||
| Fig. 1 Infrared spectra of bulk 2Ph1E (a) and 3Ph1P (b) together with those obtained for PhAs infiltrated into native silica templates of d = 4 nm measured at T = 297 K in the spectral region 3800–2600 cm−1. The spectra were normalized to the OH stretching vibration peak. Additionally, in each case, a spectrum of empty native silica template is included. | ||
II. Experimental
Materials
2-Phenyl-1-ethanol (2Ph1E) of purity ≥ 98% and 3-phenyl-1-propanol (3Ph1P) of purity 98% were purchased from Sigma Aldrich. Before any experiments, the samples were dried under a stream of liquid nitrogen. The chemical structures of compounds are presented in Fig. 1.Silica (SiO2) membranes
Mesoporous silica templates characterized by pore diameter d = 4–8 nm were prepared by electrochemical etching of silicon wafers and subsequent thermal oxidation. For more information, see ref. 45.Sample preparation
Before filling, SiO2 membranes were dried in an oven at T = 373 K under a vacuum in order to remove any volatile impurities from the nanochannels. After cooling, the templates were placed in a small glass flask containing alcohol. The whole system was maintained for t = 1.5 h at T = 323 K in a vacuum (10−2 bar) to allow infiltration of alcohols into the mesopores. After filling, the excess sample located at the surface of the membrane was removed.Fourier transform infrared (FTIR) spectroscopy
Temperature- and time-dependent measurements were performed using a Nicolet iS50 spectrometer (Thermo Scientific). Every spectrum was an average of 16 scans collected with a resolution of 2 cm−1. During low-temperature measurements (T = 297–153 K), the bulk of each monohydroxy alcohol (MA) was placed between two CaF2 windows with 3.5 μm spacers made of polyethylene terephthalate in order to obtain a uniform thickness of samples. The spectra were recorded every 4 K, with a cooling rate of 4 K min−1. Throughout the measurements, liquid nitrogen was passed through the spectrometer to avoid atmospheric H2O and CO2 in the spectra. A Linkam heating/cooling stage (Linkam Scientific Instruments Ltd, Surrey, UK) was used to collect the spectra. The annealing spectra of bulk substances were recorded using a GladiATR (Pike Technologies) with a single reflection monolithic diamond. To study the evaporation process, 2Ph1E was measured at T = 353 K and 3Ph1P at T = 363 K in a time scale of 1 hour. For infiltrated systems, the temperature-dependent experiments were performed in the temperature range T = 297–153 K with the use of a heating/cooling stage, collected every 4 K at a rate of 4 K min−1. As backgrounds, the spectra of empty silica membranes were used in order to obtain data without bands originating from empty pores. The annealing measurements were performed at T = 353 K and T = 363 K for 2Ph1E and 3Ph1P, respectively. For each system, the sample was heated up from room temperature to the desired annealing temperature, where spectra were collected every 1 minute for 1 hour.Raman spectroscopy
The Raman experiment for bulk and infiltrated 2Ph1E was performed using a WITec confocal Raman microscope (CRM alpha 300R) featuring an air-cooled solid-state laser (λ = 532 nm, P = 10 mW). An Olympus LMPlanFI 50× objective was chosen. The collected Raman spectra were accumulated by 30 scans with an integration time of 2 s and a resolution of 2 cm−1.Density functional theory (DFT) calculations
DFT calculations were performed with the ORCA 5.0.4 package.46–50 All the geometry optimizations were done with the B97M-D4 functional and 6-31G(2df,2dp) basis set. Energy evaluations were done using hybrid B97 functional with additional D4 corrections and 6-311G(2df,2pd) basis set. The interaction energies were calculated as a difference between the energy of the optimized dimer and the sum of the energy of isolated molecules, according to the following equation: | (1) |
Molecular dynamics calculations
Molecular dynamics study was performed with the Gromacs 202351–54 program package. The initial pore structures were modeled in PoreMS software.55 Pores with two different sizes were simulated. The length of the pores was equal to 8 nm (12 nm for larger ones), while the inner diameter was equal to 4 nm (8 nm for larger ones). The pore was placed in a box with a length of 12 nm (16 nm for larger ones). On both sides of the pore, there was empty space (2 nm length on each side) prepared for the molecular reservoir. The pore and additional empty space were further filled with alcohol molecules (2Ph1E or 3Ph1P). The initial simulation was done with the isothermal-isobaric ensemble (NPT), where the pressure was set to 1 bar in the z direction and temperature to 293 K (c-rescale mode as barostat and v-rescale mode as thermostat were used). The positions of silica net atoms were frozen in the x and y directions while they were free in the z direction during simulation (which was the direction of applied pressure). The initial simulation time was equal to 5 ns, which was sufficient for density equilibration. In the next step, the pore structure filled with alcohol, without additional space outside the pore (reservoir), was cut and set as the starting configuration for the next production runs. The production runs were done with frozen silica net atom positions and canonical ensemble (NVT) with the v-rescale thermostat type and temperature set to 293 K. The NVT simulation time was equal to 20 ns, which was sufficient for producing a well-equilibrated system. In all simulations, AMBER force field was used, which is accurate for the silica–organic materials systems.56 The bulk systems of 2Ph1E and 3Ph1P were simulated with the NPT ensemble in a cubic box (500 molecules each). The initial structures for bulk simulations were prepared by the Packmol program.57 The maps of distribution functions were done with the use of the TRAVIS program58–60 for trajectory analysis. Cluster analysis was performed in Gromacs software. The clusters were identified based on the criterion of the distance between the acceptor and donor atoms, which must be below 0.35 nm.III. Results and discussion
To monitor all changes within the examined materials, we performed FTIR measurements to probe their intermolecular dynamics, where special attention was mainly paid to the bands assigned to the stretching vibrations of (i) the hydroxy (OH) groups that are not involved in intermolecular hydrogen bonds (HBs) (named free OH) (νOH free ∼ 3550 cm−1), (ii) H-bonded OH units (νOH bonded ∼ 3330 cm−1), as well as (iii) the aromatic CH moieties (νCH aromatic ∼ 3100–3000 cm−1), see Fig. 1. The scope of this article is as follows. At first, we studied the freshly prepared confined materials in comparison to the bulk; then, the evaporation experiments were performed; and at the end, experimental results were compared with the outcome of the molecular dynamics simulations. To our best knowledge, this study is the first one in the literature that reveals/confirms the formation of a layer strongly bonded to the curved surface with properties similar to IAL, which complements our knowledge about this phenomenon, which was mostly studied in case of thin films61–65 or nanoparticles.66–69Fig. 1 shows the FTIR spectra collected for both bulk compounds and MAs infiltrated into the native silica mesopores at room temperature; for comparison, the spectrum of the ‘empty’ native silica membrane is also included. Both MAs, 2Ph1E and 3Ph1P, are characterized by a similar position of the bands assigned to the stretching vibrations of H-bonded OH units (ca. 3330 cm−1), while their widths (the full width at half maximum, FWHM) are slightly different (FWHM2Ph1E > FWHM3Ph1P; see Fig. S2†). This indicates that 2Ph1E exhibits a slightly higher heterogeneity in the distribution of HB strength than 3Ph1P.70 Interestingly, only a few differences occurred in the investigated systems after the incorporation of the examined PhAs into the silica mesopores (see Fig. S3†). We draw the reader's attention to four of them.
Firstly, a sharp peak at ∼3750 cm−1 originating from the stretching vibration of ‘free’ (non-associated) OH (Si-OH) groups in the spectrum of an ‘empty’ native silica membrane is not detected in the spectra of confined MAs (grey line in Fig. 1). This is a clear indication of the effective infiltration of the studied alcohols.
Furthermore, surprisingly, the νOH bonded bands of confined MAs are only slightly blue-shifted (shifted towards higher wavenumber) relative to the bulk samples, which suggests a shortening of the OH bond lengths correlated with the weakening of the existing HBs. This fact can result from increased steric repulsion between alcohol molecules due to their confinement. One can also mention that the detected blue-shift was widely noticed in the case of other confined MAs, i.e., 2-ethyl-1-hexanol, 2-ethyl-1-butanol, and 5-methyl-3-heptanol,71 as well as confined water and its isotopic derivatives.72–79 In the latter case, the observed spectral effect was explained by the formation of weak H-bonding interactions between OH groups (interfacial water molecules) with the silica oxygen atoms, which exhibit weaker H-bond accepting ability compared to water molecules.79
Thirdly, the constrained systems exhibit a larger FWHM of their νOH bonded bands compared to bulk ones, suggesting a greater heterogeneity of the environments experienced by the confined MA molecules. Interestingly, the broadening of these bands in the analyzed spectra is more pronounced in the lower wavenumber region. Therefore, at first glance, it may be assumed that an increase in the νOH bonded bandwidth arises from the presence of OH groups engaged in stronger HBs under confinement.
Lastly, the confined samples show a higher intensity of the νOH free bands compared to bulk samples, which indicates that the confinement affects the association process of MAs and reduces the formation of H-bonded aggregates. It should be highlighted that the same observations were found in the case of MAs incorporated within silanized silica mesopores (see Fig. S4†).
Taking into account the above-mentioned findings, one can assume that, surprisingly, the differences in the strength and the population of HBs occurring in bulk and confined MAs are relatively small despite the application of silica templates characterized by small pore size of d = 4 nm. Note that the same behavior was also observed in the case of PhAs within silica templates of d = 8 nm. We assume that this interesting behavior might be a result of two scenarios: (i) the mean size of the nanoaggregates formed by the examined MAs is not affected by the spatial restriction80–82 or (ii) the recorded signal is dominated by the ‘core’ molecules, which are characterized by bulk-like properties.83–85 In this context, one can recall that previous studies on a series of PhAs diluted in carbon tetrachloride (CCl4) showed that they form nanoaggregates consisting of 2–3 molecules (independent of their molecular weight).70 It should be noted that these results agree well with molecular dynamics computations, supported by X-ray diffraction, which revealed that PhAs organize into dimeric and trimeric H-bonded clusters, maintained by additional weak interactions.86 Thus, it might indicate that the former scenario is valid. However, as the latter cannot be completely ignored, we decided to remove the ‘core’ molecules from the examined confined systems. For this purpose, the studied porous materials were heated to the chosen temperature (T = 353–363 K) to evaporate the molecules that are not strongly interacting with the interface of membranes. Note that in the case of bulk compounds, the complete evaporation at those temperatures was accomplished in less than one hour (t = 40 min; see Fig. S5†); thus, the same procedure was applied in the case of the examined incorporated MAs.
Fig. 2(a) and Fig. S7† show the FTIR spectra collected before (t = 0 min) and after (t = 60 min, longer than required to remove the bulk sample) the evaporation process of 2Ph1E infiltrated into native silica mesopores. Data for confined 3Ph1P are shown in Fig. S8.† As one can see, a systematic reduction of the intensity of all bands in the recorded spectra was detected upon evaporation. However, despite this fact, FTIR spectra collected after the evaporation revealed the presence of all three characteristic regions previously observed for both bulk and confined MAs (before the evaporation); however, with some differences. This finding might be shown in a better way in Fig. 3, where we compare all available FTIR spectra together. Note that data collected after the evaporation did not exhibit the presence of a sharp peak at ∼3750 cm−1, characteristic for an ‘empty’ native silica membrane, which clearly shows that molecules of examined alcohols are still present within the porous materials after the evaporation (Fig. 3). The same behavior was also observed in the case of PhAs within silica templates of d = 8 nm, please see Fig. S6.† Therefore, we assume that all changes observed within the examined porous materials after evaporation are solely related to the formation of the interfacial layer strongly bonded to the pore walls.
 | ||
| Fig. 2 (a and c) FTIR spectra before and after the evaporation process of 2Ph1E infiltrated into native silica mesopores (d = 4 nm) at t = 0 min and t = 60 min in the (a) 3730–2600 cm−1 and (c) 3130–2990 frequency regions. (b and d) Raman spectra of 2Ph1E within native silica nanopores at room temperature after evaporation at T = 353 K in the (b) 3300–2700 cm−1 and (d) 1700–1500 cm−1 spectral regions. | ||
 | ||
| Fig. 3 Comparison of FTIR spectra recorded in the spectral region 3800–2600 cm−1 at T = 297 K of bulk 2Ph1E (a) and 3Ph1P (b), MAs infiltrated into native silica mesopores of d = 4 nm before and after evaporation, and bulk MAs within 0.1 M solution of CCl4. For comparison, in each case, the FTIR spectrum of an ‘empty’ native silica membrane is also included. Data for the bulk samples measured within 0.1 M solution of CCl4 were taken from ref. 70. Data collected for PhAs within silica templates of d = 8 nm are shown in Fig. S6.† | ||
The most significant changes are observed in both νOH bonded and νOH free band ranges (Fig. 2(a) and Fig. S7†). In the case of the νOH bonded band, one can see a very broad structure of relatively small amplitude, whereas for the νOH free band, aside from broadening, a pronounced shift towards higher wavenumber can be observed as well. Interestingly, the same changes occurring within the ‘residual’ νOH band were previously also observed for MAs diluted in CCl4 solution70 (those data are included in Fig. 3). As can be observed, the νOH free bands in both spectra are located in a similar spectral range, which might imply that there are some non-H-bonded alcohol molecules present in the confined samples. This observation agrees with the results presented for water confined in periodic mesoporous (organo)silicas, which showed that in the adsorbed layer, non-bonded OH groups and van der Waals water–water interactions may still exist.87 In this context, one can also assume that the observed very broad and blurred νOH bonded band of confined 2Ph1E after evaporation also somehow resembles the one recorded for dissolved PhAs, which was previously assigned to the dominant presence of small H-bonded alcohol aggregates.70 Such complex character of the signal of evaporated samples provides direct evidence of the existence of different specific interactions occurring at the silica/alcohol interface, i.e., O–H⋯O or O–H⋯π HBs.
In the next step, we focused on the νCH aromatic bands (located at 3100–3000 cm−1), which were well visible in the spectra after the annealing process and, interestingly, they shifted towards higher wavenumbers after evaporation for both confined MAs (Fig. 2(c) and S7(d)†). This also clearly proves two issues: (1) there are some molecules of alcohols present within porous materials after the performed evaporation (of duration longer than required for the bulk sample) and (2) these molecules have a different conformation from those observed for the freshly prepared samples due to the adsorption processes (of physical and/or chemical nature). Thus, this spectral region might be the fingerprint of the remaining IAL within the examined mesoporous matrices.
To identify the mechanism of the adsorption processes occurring at the silica/alcohol interface, i.e., physisorption or chemisorption, we also performed Raman measurements on infiltrated 2Ph1E at room temperature (T = 297 K) before and after the annealing process. As shown in Fig. 2(b and d), the positions of the νCH aromatic peaks (the band at 3058 cm−1) moved to a higher wavenumber (3063 cm−1) after evaporation. Moreover, the change in the band's subtle structure located at ∼1610 cm−1 (the stretching vibrations of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C groups) was also visible (Fig. 2(d)). On the other hand, no new/additional bands associated with creating a new type of bond, being the manifestation of the chemisorption process, were observed in the Raman spectra of infiltrated 2Ph1E after evaporation (Fig. S9†). These findings clearly indicate structural/conformational changes of MAs occurring in confinement after annealing. What is more, they are an effect of the adsorption processes of a physical nature (physical adsorption). In this context, it can be added that gravimetric studies conducted for aliphatic alcohols incorporated into MCM-41 mesoporous templates also indicated that the physisorption process occurs in confinement.88
C groups) was also visible (Fig. 2(d)). On the other hand, no new/additional bands associated with creating a new type of bond, being the manifestation of the chemisorption process, were observed in the Raman spectra of infiltrated 2Ph1E after evaporation (Fig. S9†). These findings clearly indicate structural/conformational changes of MAs occurring in confinement after annealing. What is more, they are an effect of the adsorption processes of a physical nature (physical adsorption). In this context, it can be added that gravimetric studies conducted for aliphatic alcohols incorporated into MCM-41 mesoporous templates also indicated that the physisorption process occurs in confinement.88
It should be highlighted that identical spectral effects were observed during the evaporation of both alcohols from silanized silica templates (Fig. S7(c, d) and S8(c, d)†). Surprisingly, in the case of silanized systems, the remaining signal in FTIR spectra originating from MA molecules was still detected after t = 60 min, just like for native templates (Fig. S6(c, d) and S7(c, d)†). Moreover, the νCH aromatic bands were also shifted for both MAs incorporated within silanized silica templates after evaporation. However, the intensity of the analyzed signals was smaller when compared to that coming from the untreated templates. For better clarity, a comparison of FTIR spectra for 2Ph1E within silanized membranes of d = 4 nm is presented in Fig. S10.†
In order to gain deeper insight into the behavior of studied alcohols in pores, with special emphasis on the molecules directly interacting with the pore walls, MD simulations were carried out. In Fig. 4(c), the simulated model is shown. The initial model is composed of an 8 nm-length silica pore with a diameter of d = 4 nm and additional reservoirs on both ends of the pore filled with the studied alcohol. The initial simulation was performed in the NPT at temperature T = 293 K and pressure p = 1 bar, which was applied in the z direction (anisotropic) to obtain proper alcohol density inside the pore. As one can see in Fig. 4(c), after the equilibration, the 2Ph1E alcohol density in the pore is approximately 30% lower than in the reservoir outside the pore (0.67 g cm−3versus 1 g cm−3), where there is a typical bulk density. Importantly, this result agrees very well with the data reported in ref. 89, where it was shown that the free volume inside pores is higher with respect to the bulk material. After the initial NPT simulation, the box was limited to the pore only (without additional molecular reservoirs), and NVT production runs with constant volume were performed. In the last step, the evaporation of alcohol from the pore was simulated by performing NVT simulations with reduced alcohol density inside the pore with 75%, 50%, and 25% initial density. This procedure mimics the evaporation experiment described above. In Fig. 4(a) and (b), one can observe the xy plane cut through the center of the pore with the mapped alcohol density. It is clearly seen how the reduction of the amount of alcohol molecules from 100% to 25% is associated with the formation of an alcohol layer on the inner surface of the silica pore. Moreover, there are slight differences between 2Ph1E and 3Ph1P. In the case of 2Ph1E, the formation of the inner layer is more pronounced than in the case of 3Ph1P (its formation becomes visible at 75% of initial density). This is caused by the differences in the H-bond interaction energies between alcohol molecules as well as between alcohol–silica molecules.
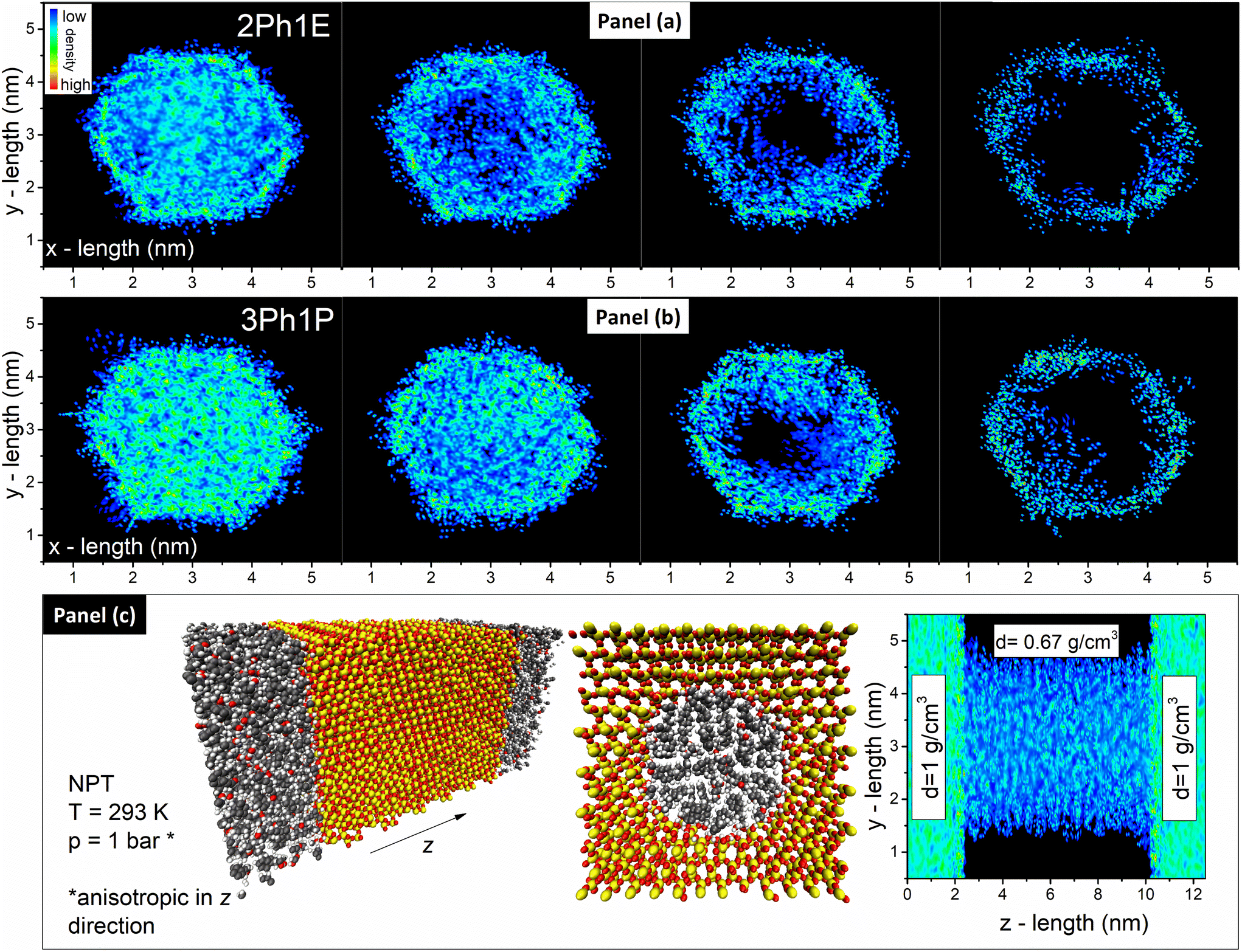 | ||
| Fig. 4 (a) Density maps of 2Ph1E within pores of d = 4 nm in the xy plane. From left to right, maps for 100%, 75%, 50%, and 25% of the initial density are presented. (b) Density maps of 3Ph1P within pores of d = 4 nm in the xy plane. From left to right, maps for 100%, 75%, 50%, and 25% of the initial density are shown. (c) Silica pore filled with alcohol: model and density map for an equilibrated model in the xz plane with marked average density in the different zones. | ||
Moreover, the impact of the pore size on the formation of the alcohol layer at the silica pore interface was checked by performing additional MD simulations. The simulation was performed on the silica pore characterized by d = 8 nm filled with 2Ph1E molecules (see Fig. 5(a)). It was found that the elimination of alcohol molecules from the pore interior led to the formation of the same alcohol layer as in the case of the pore with a diameter d = 4 nm. Although the process of layer formation occurs in both systems, a significant difference in alcohol density inside different pores was found. In the case of d = 4 nm, the average alcohol density is equal to 0.67 g cm−3, while it is equal to 0.86 g cm−3 in the case of d = 8 nm (versus 1 g cm−3 in the bulk system). The formation of the alcohol layer, as well as a comparison of alcohol density in the xy-plane for 2Ph1E for both systems filled with molecules at 25% of initial density, is shown in Fig. S11.†
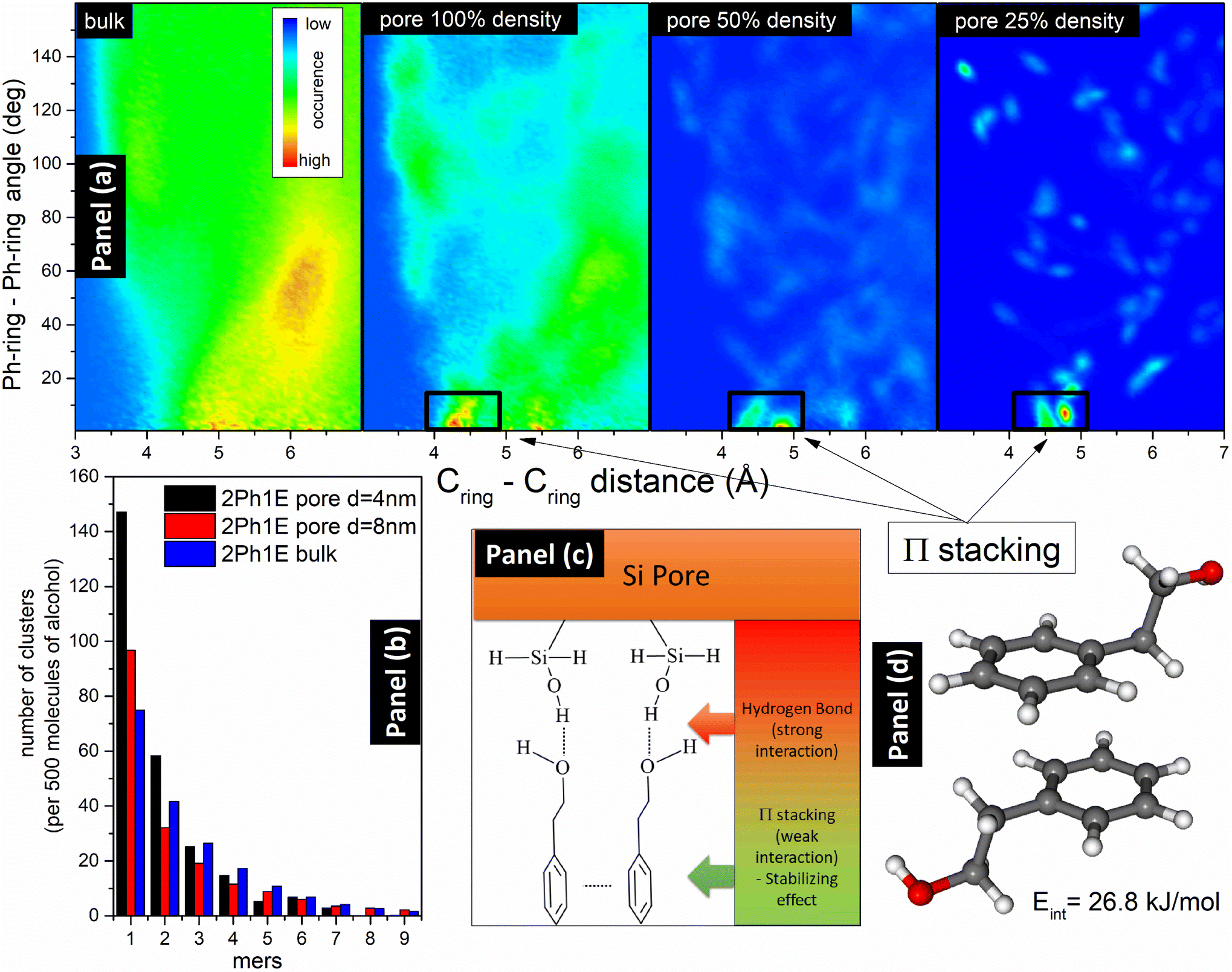 | ||
| Fig. 5 (a) Maps of 2Ph1E presenting phenyl ring distance versus dihedral angle between phenyl rings in the pore system. The marked boxes are the areas of sandwich-type π-stacking interactions. (b) Histogram comparing the formation of clusters in the 2Ph1E alcohol in bulk and in pores with diameters d = 4 nm and d = 8 nm. (c) Model representing interactions at the pore–alcohol interface and the role of HBs and π–π interactions. (d) Energy of π-stacking interaction between two 2Ph1E molecules calculated in the DFT model. | ||
MD simulations have also been used to obtain HB cluster distributions for 2Ph1E and 3Ph1P alcohols. In Fig. 5(b), a comparison of histograms for 2Ph1E in the bulk and pores with d = 4 nm and d = 8 nm is presented. As one can see, the alcohol molecules form the strongest clusters in the bulk system and the weakest in the small pore of d = 4 nm. The clustering ability of alcohol molecules in large pores of d = 8 nm diameter is between that in small pores of d = 4 nm and the bulk system. More distributions of clusters are presented in Fig. S12.† Interestingly, the distribution of self-assemblies formed by alcohol–alcohol interactions in the pores is similar to the distribution of clusters in a bulk system. However, the average cluster size is slightly lower in the pores for both alcohols. This result is quite consistent with the FTIR data that indicated a very small change in the position of the νOH bonded band in the pores with respect to the bulk sample. In the case of 2Ph1E, we have an average cluster size, a, equal to 3.7 molecules in the bulk system and 3.2 molecules within the pores, which can be related to the 30% lower density. For 3Ph1P, the situation is similar, where a = 4.5 molecules and a = 3.3 molecules within bulk and pores, respectively. In bulk 3Ph1P, 20.6% of molecules are non-H-bonded (monomers), while in 2Ph1E, there are 14.9% monomers, which means a higher association degree of 2Ph1E than of 3Ph1P. The next comparison is related to all HBs in silica pores (i.e., silica–silica, alcohol–alcohol, and alcohol–silica) with initial and reduced alcohol density (100% and 50%). 2Ph1E in both density scenarios (100% and 50%) has more clusters than 3Ph1P. For an initial 100% density, there are only 6.9% free OH groups in the entire 2Ph1E pore system, while this value is 22.2% for 3Ph1P. However, when we compare HBs existing only in the core region between alcohol molecules, the percentage of free OH groups is higher in the pores than in the bulk for both systems (30% free OH groups in 2Ph1E and 33% in 3Ph1P). This result also agrees with the FTIR data, showing an increase in the νOH free band intensity for confined samples compared to bulk samples, thus the existence of a larger amount/number of monomers under confinement. When the density is reduced to 50% of the initial value, and we take into account all HBs (silica–silica, alcohol–alcohol, and silica–alcohol), there are 14% free OH groups in 2Ph1E, while 30% in 3Ph1P. During the reduction of alcohol density in the pores, the differences between 2Ph1E and 3Ph1P systems become smaller, which can indicate that the strength of the silica–alcohol bond and tendency to form layer are similar for both systems, while alcohol–alcohol HBs are much weaker in 3Ph1P. The same behavior was observed for 2Ph1E in the pore with diameter d = 8 nm. There are 10.6% free OH groups in the entire pore system, while 19.3% free OH groups in the core area (alcohol–alcohol). The data related to the average densities and amounts of free OH groups in all studied systems are presented in Table S1.†
The formation of the layer caused by the physisorption process is related to the H-bond interactions between hydroxyl groups from alcohol and silica. Therefore, in the next step, we performed an analysis of H-bond cluster formation in bulk alcohols and in pores fully filled with alcohol and after partial evaporation (50% of density). Moreover, we have used DFT calculations to obtain interaction energies between alcohol molecules and between alcohol and silica. It was found that the H-bond energy between silica and alcohol is higher than between two alcohol molecules, but only when the silica group is a donor and alcohol is an acceptor of the H-bond. The interaction energy, Eint, between two 2Ph1E molecules is equal to Eint = 42.6 kJ mol−1. When SiOH or SiO2H2 groups are donors and alcohol is the acceptor, the interaction energies are equal to Eint,SiOH = 45.3 kJ mol−1 and Eint,SiO2H2 = 50.2 kJ mol−1, respectively. In the opposite situation, when the alcohol is a proton donor, and SiOH or SiO2H2 are acceptors, the interaction energies are equal to Eint,SiOH = 30.5 kJ mol−1 and Eint,SiO2H2 = 32.1 kJ mol−1, respectively. This is a clear indication that from the energetic point of view, the most stable are connections of alcohol with the pore SiOH and SiO2H2 groups when the alcohol is an acceptor of hydrogen. In the aromatic alcohols, there are also present π-stacking interactions;90,91 thus, we have calculated the energies of two possible π-stacking formations. The first one is a sandwich type, which is slightly displaced, and the second one is a T-shape π-stacking. The energy of the sandwich-type interaction is equal to Eint,sandwich = 26.8 kJ mol−1, while the energy of the T-shape reaches Eint,T-shape = 16.4 kJ mol−1. The sandwich-type interaction is shown in Fig. 5(d). The energy of sandwich π-stacking is two times smaller than the energy of the H-bond. However, when alcohols are connected by HBs to the inner layer of silica pores, these interactions can stabilize energetically the layer. This effect was found during molecular dynamics study, where in simulations mimicking evaporation, sandwich π-stacking interactions are abundant (see Fig. 5(a)). Therefore, the stability of the inner layer is greatly enhanced by the strong HBs with silica and by the additional π-stacking interactions of alcohols connected to the silica by HBs. The visualization of such stable connection of alcohol with silica surface is presented in Fig. 5(c). It should be noted that this finding correlates well with the results of FTIR measurements, i.e., these π-stacking interactions occurring between the studied MAs can be responsible for the shift of the νCH aromatic bands towards higher wavenumbers after evaporation of confined MAs. Hence, as observed from FTIR data and molecular dynamics simulations, the formation of the interfacial layer induces and strengthens π-stacking interactions between alcohols.
IV. Conclusions
In summary, we conducted FTIR measurements for two MAs infiltrated into native and silanized silica mesopores (d = 4–8 nm) to investigate any differences in the nature of interactions under these conditions (the impact of confinement on intermolecular interactions). Initially, the spectra of both systems (bulk vs. confinement) showed only little discrepancies, indicating that the ‘core’ layer of the molecule dominates in infiltrated MAs. Interestingly, this finding was well supported by the MD simulations that revealed the small impact of the nanoscale confinement on the association phenomenon in both alcohols. To remove the bulk-like fraction (core layer), we proposed a solvent-free approach depending on the evaporation process. In this way, the layer at the interface was revealed for the first time in 2D-constrained systems. Importantly, the obtained ‘residual’ band was characterized by a new subtle structure compared to that in the bulk and confined MAs. The changes especially observed in the high-frequency region of IR spectra suggested the differences in the arrangement of both free and associated OH groups. The source of the observed differences was the formation of a layer adsorbed on the interface (IAL). It should be noted that these changes were obtained regardless of the kind/type of silica templates used (non-treated/treated). What is more, the Raman spectra of 2Ph1E in native silica mesopores before and after annealing clearly indicate the formation of IAL due to the physisorption mechanism, not chemisorption. Importantly, these suppositions were further confirmed by MD simulations, which indicated the formation of the interfacial layer (considered as IAL due to its resistance to the evaporation process) attached to the silica pores that is stabilized by the effective H-bonds and enormously strong π-stacking interactions. In conclusion, we present a pioneering work with a novel approach to examining strongly bonded interfacial layers formed in pores of similar characteristics to IAL investigated in thin films or silica nanoparticles.Data and materials availability
The ESI† file is available free of charge at https://pubs.acs.org. It contains additional figures and tables, including the schematic structure of silica mesopore templates, IR spectra of bulk and infiltrated MAs at room temperature, IR spectra of bulk MAs before and after evaporation, IR spectra of MAs infiltrated into native silica pores (d = 4 nm and 8 nm) before and after annealing, IR spectra of empty silanized silica template, Raman spectra of bulk and 2Ph1E incorporated into nanopores before and after annealing at 297 K, distribution of 2Ph1E molecules in xy plane within d = 8 nm nanopore at different levels of initial density, the density of 2Ph1E molecules in the pore for the equilibrating NPT, schematic existence of strongly bonded alcohol layer at the silica interface for silica mesopores (d = 4 and 8 nm) obtained from molecular dynamics calculations, histograms comparing the formation of clusters in MAs in mesopores with d = 8 nm with 100% and 50% alcohol densities, and a table including average densities of alcohols, and the amounts of free OH groups in the studied systems.Author contributions
N. S. – writing – original draft, investigation, formal analysis, visualization. M. T. – writing – original draft, conceptualization, funding acquisition. B. H. – formal analysis, methodology. P. W. – investigation, formal analysis. R. W. – investigation. M. H. – investigation. M. P. – methodology. K. K. – supervision, funding acquisition, writing – review & editing.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
The experimental part of this research was funded by National Science Centre, Poland [Dec. no 2019/33/B/ST3/00500]. MD simulations of this work were funded by the National Science Centre, Poland [Dec. no 2022/47/B/ST4/00236]. For the purpose of Open Access, the author has applied a CC-BY public copyright licence to any Author Accepted Manuscript (AAM) version arising from this submission.References
- D. T. Limmer and D. Chandler, J. Chem. Phys., 2012, 137, 1841–1844 CrossRef PubMed.
- P. Huber, J. Phys.: Condens. Matter, 2015, 27, 103102 CrossRef PubMed.
- L. Petychakis, G. Floudas and G. Fleischer, Europhys. Lett., 1997, 40, 685–690 CrossRef CAS.
- H. Duran, A. Gitsas, G. Floudas, M. Mondeshki, M. Steinhart and W. Knoll, Macromolecules, 2009, 42, 2881–2885 CrossRef CAS.
- M. Krutyeva, A. Wischnewski, M. Monkenbusch, L. Willner, J. Maiz, C. Mijangos, A. Arbe, J. Colmenero, A. Radulescu, O. Holderer, M. Ohl and D. Richter, Phys. Rev. Lett., 2013, 110, 1–5 Search PubMed.
- C. L. Jackson and G. B. McKenna, Chem. Mater., 1996, 8, 2128–2137 CrossRef CAS.
- C. Alba-Simionesco, G. Dosseh, E. Dumont, B. Frick, B. Geil, D. Morineau, V. Teboul and Y. Xia, Eur. Phys. J. E: Soft Matter Biol. Phys., 2003, 12, 19–28 CrossRef CAS PubMed.
- W. Yan, V. K. S. Hsiao, Y. B. Zheng, Y. M. Shariff, T. Gao and T. J. Huang, Thin Solid Films, 2009, 517, 1794–1798 CrossRef CAS.
- G. Jeon, S. Y. Yang and J. K. Kim, J. Mater. Chem., 2012, 22, 14814–14834 RSC.
- A. Hashemi, M. Ezati, J. Mohammadnejad, B. Houshmand and S. Faghihi, Int. J. Nanomed., 2020, 15, 4471–4481 CrossRef CAS PubMed.
- S. Sotiropoulou, V. Vamvakaki and N. A. Chaniotakis, Biosens. Bioelectron., 2005, 20, 1674–1679 CrossRef CAS PubMed.
- S. P. Adiga, C. Jin, L. A. Curtiss, N. A. Monteiro-Riviere and R. J. Narayan, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2009, 1, 568–581 CAS.
- M. Tarnacka, A. Chrobok, K. Matuszek, S. Golba, P. Maksym, K. Kaminski and M. Paluch, ACS Appl. Mater. Interfaces, 2016, 8, 29779–29790 CrossRef CAS PubMed.
- A. Schönhals, F. Rittig and J. Kärger, J. Chem. Phys., 2010, 133, 094903 CrossRef PubMed.
- M. Tarnacka, M. Wojtyniak, A. Brzózka, A. Talik, B. Hachuła, E. Kamińska, G. D. Sulka, K. Kaminski and M. Paluch, Nano Lett., 2020, 20, 5714–5719 CrossRef CAS PubMed.
- W. K. Kipnusu, M. M. Elmahdy, M. Elsayed, R. Krause-Rehberg and F. Kremer, Macromolecules, 2019, 52, 1864–1873 CrossRef CAS.
- R. Renou, A. Szymczyk and A. Ghoufi, J. Chem. Phys., 2014, 140, 044704 CrossRef PubMed.
- A. Panagopoulou, C. Rodríguez-Tinoco, R. P. White, J. E. G. Lipson and S. Napolitano, Phys. Rev. Lett., 2020, 124, 27802 CrossRef CAS PubMed.
- A. Talik, M. Tarnacka, M. Wojtyniak, E. Kaminska, K. Kaminski and M. Paluch, J. Mol. Liq., 2020, 298, 111973 CrossRef CAS.
- S. Napolitano and M. Wübbenhorst, Nat. Commun., 2011, 2, 260 CrossRef.
- S. Napolitano, V. Lupaşcu and M. Wübbenhorst, Macromolecules, 2008, 41, 1061–1063 CrossRef CAS.
- S. Napolitano, C. Rotella and M. Wübbenhorst, ACS Macro Lett., 2012, 1, 1189–1193 CrossRef CAS PubMed.
- C. Rotella, S. Napolitano, S. Vandendriessche, V. K. Valev, T. Verbiest, M. Larkowska, S. Kucharski and M. Wübbenhorst, Langmuir, 2011, 27, 13533–13538 CrossRef CAS PubMed.
- P. Gin, N. Jiang, C. Liang, T. Taniguchi, B. Akgun, S. K. Satija, M. K. Endoh and T. Koga, Phys. Rev. Lett., 2012, 109, 1–5 CrossRef PubMed.
- J. M. Y. Carrillo, S. Cheng, R. Kumar, M. Goswami, A. P. Sokolov and B. G. Sumpter, Macromolecules, 2015, 48, 4207–4219 CrossRef CAS.
- S. Napolitano and D. Cangialosi, Macromolecules, 2013, 46, 8051–8053 CrossRef CAS.
- D. Cangialosi, V. M. Boucher, A. Alegría and J. Colmenero, J. Chem. Phys., 2011, 135, 014901 CrossRef PubMed.
- R. P. White and J. E. G. Lipson, Macromolecules, 2016, 49, 3987–4007 CrossRef CAS.
- R. P. White and J. E. G. Lipson, ACS Macro Lett., 2017, 6, 529–534 CrossRef CAS PubMed.
- A. Debot, R. P. White, J. E. G. Lipson and S. Napolitano, ACS Macro Lett., 2019, 8, 41–45 CrossRef CAS PubMed.
- N. Bizmark, M. A. Ioannidis and D. E. Henneke, Langmuir, 2014, 30, 710–717 CrossRef CAS PubMed.
- S. Milani, F. B. Bombelli, A. S. Pitek, K. A. Dawson and J. Rädler, ACS Nano, 2012, 6, 2532–2541 CrossRef CAS PubMed.
- X. Li, O. Niitsoo and A. Couzis, J. Colloid Interface Sci., 2014, 420, 50–56 CrossRef CAS PubMed.
- Z. U. A. Arain, S. Al-Anssari, M. Ali, S. Memon, M. A. Bhatti, C. Lagat and M. Sarmadivaleh, Petroleum, 2020, 6, 277–285 CrossRef.
- M. Tarnacka, E. Kamińska, M. Paluch and K. Kamiński, J. Phys. Chem. Lett., 2022, 13, 10464–10470 CrossRef CAS PubMed.
- M. Tarnacka, E. Kaminska, K. Kaminski, C. M. Roland and M. Paluch, J. Phys. Chem. C, 2016, 120, 7373–7380 CrossRef CAS.
- M. Tarnacka, O. Madejczyk, K. Kaminski and M. Paluch, Macromolecules, 2017, 50, 5188–5193 CrossRef CAS.
- S. Zimny, M. Tarnacka, M. Geppert-Rybczyńska and K. Kamiński, J. Phys. Chem. C, 2023, 127, 23034–23043 CrossRef CAS.
- K. Adrjanowicz and M. Paluch, Phys. Rev. Lett., 2019, 122, 176101 CrossRef CAS PubMed.
- M. Tarnacka, M. Dulski, M. Geppert-Rybczyńska, A. Talik, E. Kamińska, K. Kamiński and M. Paluch, J. Phys. Chem. C, 2018, 122, 28033–28044 CrossRef CAS.
- M. F. Thees, J. A. McGuire and C. B. Roth, Soft Matter, 2020, 16, 5366–5387 RSC.
- N. Jiang, M. Sen, W. Zeng, Z. Chen, J. M. Cheung, Y. Morimitsu, M. K. Endoh, T. Koga, M. Fukuto, G. Yuan, S. K. Satija, J. M. Y. Carrillo and B. G. Sumpter, Soft Matter, 2018, 14, 1108–1119 RSC.
- C. Housmans, M. Sferrazza and S. Napolitano, Macromolecules, 2014, 47, 3390–3393 CrossRef CAS.
- C. Iacob, J. R. Sangoro, P. Papadopoulos, T. Schubert, S. Naumov, R. Valiullin, J. Kärger and F. Kremer, Phys. Chem. Chem. Phys., 2010, 12, 13798–13803 RSC.
- W. K. Kipnusu, W. Kossack, C. Iacob, M. Jasiurkowska, J. R. Sangoro and F. Kremer, Z. Phys. Chem., 2012, 226, 797–805 CrossRef CAS.
- S. Grimme, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 211–228 CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, 1–15 Search PubMed.
- C. Bannwarth, E. Caldeweyher, S. Ehlert, A. Hansen, P. Pracht, J. Seibert, S. Spicher and S. Grimme, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2021, 11, 1–49 Search PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, 1327 Search PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- H. Bekker, H. J. C. Berendsen, E. J. Dijkstra, S. Achterop, R. van Drunen, D. van der Spoel, A. Sijbers and H. Keegstra, et al., in Physics computing 92, ed. R. A. de Groot and J. Nadrchal, World Scientific, Singapore, 1993, pp. 252–256 Search PubMed.
- E. Lindahl, B. Hess and D. van der Spoel, J. Mol. Model, 2001, 7, 306–317 CrossRef CAS.
- D. Van Der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. C. Berendsen, J. Comput. Chem., 2005, 26, 1701–1718 CrossRef CAS PubMed.
- B. Hess, C. Kutzner, D. van der Spoel and E. Lindahl, J. Chem. Theory Comput., 2008, 4, 435–447 CrossRef CAS PubMed.
- H. Kraus, J. Rybka, A. Höltzel, N. Trebel, U. Tallarek and N. Hansen, Mol. Simul., 2021, 47, 306–316 CrossRef CAS.
- K. Kanhaiya, M. Nathanson, P. J. in ‘t Veld, C. Zhu, I. Nikiforov, E. B. Tadmor, Y. K. Choi, W. Im, R. K. Mishra and H. Heinz, J. Chem. Theory Comput., 2023, 19, 8293–8322 CrossRef CAS PubMed.
- L. Martinez, R. Andrade, E. G. Birgin and J. M. Martinez, J. Comput. Chem., 2009, 30, 2157–2164 CrossRef CAS PubMed.
- M. Brehm and B. Kirchner, J. Chem. Inf. Model., 2011, 51, 2007–2023 CrossRef CAS PubMed.
- O. Hollõczki, M. Macchiagodena, H. Weber, M. Thomas, M. Brehm, A. Stark, O. Russina, A. Triolo and B. Kirchner, ChemPhysChem, 2015, 16, 3325–3333 CrossRef PubMed.
- M. Brehm, M. Thomas, S. Gehrke and B. Kirchner, J. Chem. Phys., 2020, 152, 164105 CrossRef CAS PubMed.
- W. Ren, Y. Li, Y. Tang, J. Xu, C. Zhang, O. K. C. Tsui and X. Wang, ACS Macro Lett., 2023, 12, 854–859 CrossRef CAS PubMed.
- C. Rodríguez-Tinoco, D. N. Simavilla, R. D. Priestley, M. Wübbenhorst and S. Napolitano, ACS Macro Lett., 2020, 9, 318–322 CrossRef PubMed.
- S. Napolitano, Soft Matter, 2020, 16, 5348–5365 RSC.
- A. B. Unni, R. Mroczka, J. Kubacki and K. Adrjanowicz, J. Mol. Liq., 2022, 368, 120675 CrossRef.
- A. B. Unni, R. Winkler, D. M. Duarte, K. Chat and K. Adrjanowicz, J. Phys. Chem. B, 2022, 126, 8072–8079 CrossRef PubMed.
- K. Randazzo, M. Bartkiewicz, B. Graczykowski, D. Cangialosi, G. Fytas, B. Zuo and R. D. Priestley, Macromolecules, 2021, 54, 10224–10234 CrossRef CAS.
- Y. Qiang, K. T. Turner and D. Lee, Macromolecules, 2023, 56, 122–135 CrossRef CAS.
- M. A. Khazaei, D. Bastani, A. Mohammadi and A. Kordzadeh, Langmuir, 2022, 38, 12421–12431 CrossRef CAS PubMed.
- H. Li, X. Chen, D. Shen, F. Wu, R. Pleixats and J. Pan, Nanoscale, 2021, 13, 15998–16016 RSC.
- N. Soszka, B. Hachuła, M. Tarnacka, E. Kamińska, J. Grelska, K. Jurkiewicz, M. Geppert-Rybczyńska, R. Wrzalik, K. Grzybowska, S. Pawlus, M. Paluch and K. Kamiński, Phys. Chem. Chem. Phys., 2021, 23, 23796–23807 RSC.
- A. Talik, M. Tarnacka, M. Geppert-Rybczyńska, B. Hachuła, R. Bernat, A. Chrzanowska, K. Kaminski and M. Paluch, J. Colloid Interface Sci., 2020, 576, 217–229 CrossRef CAS PubMed.
- J. R. Bailey and M. M. McGuire, Langmuir, 2007, 23, 10995–10999 CrossRef CAS PubMed.
- M. Baum, F. Rieutord, F. Juranyi, C. Rey and D. Rébiscoul, Langmuir, 2019, 35, 10780–10794 CrossRef CAS PubMed.
- S. A. Yamada, J. Y. Shin, W. H. Thompson and M. D. Fayer, J. Phys. Chem. C, 2019, 123, 5790–5803 CrossRef CAS.
- S. Le Caër, S. Pin, S. Esnouf, Q. Raffy, J. P. Renault, J. B. Brubach, G. Creff and P. Roy, Phys. Chem. Chem. Phys., 2011, 13, 17658–17666 RSC.
- X. F. Huang, Q. Wang, X. X. Liu, S. H. Yang, C. X. Li, G. Sun, L. Q. Pan and K. Q. Lu, J. Phys. Chem. C, 2009, 113, 18768–18771 CrossRef CAS.
- R. Musat, J. P. Renault, M. Candelaresi, D. J. Palmer, S. Le Caër, R. Righini and S. Pommeret, Angew. Chem., Int. Ed., 2008, 47, 8033–8035 CrossRef CAS PubMed.
- S. A. Yamada, S. T. Hung, W. H. Thompson and M. D. Fayer, J. Chem. Phys., 2020, 152, 154704 CrossRef CAS PubMed.
- H. S. Senanayake, J. A. Greathouse, A. G. Ilgen and W. H. Thompson, J. Chem. Phys., 2021, 154, 104503 CrossRef CAS PubMed.
- A. Ghoufi, J. Phys. Chem. B, 2020, 124, 11501–11509 CrossRef CAS PubMed.
- A. Ghoufi, I. Hureau, R. Lefort and D. Morineau, J. Phys. Chem. C, 2011, 115, 17761–17767 CrossRef CAS.
- A. Ghoufi, I. Hureau, D. Morineau, R. Renou and A. Szymczyk, J. Phys. Chem. C, 2013, 117, 15203–15212 CrossRef CAS.
- C. L. Jackson and G. B. McKenna, J. Non-Cryst. Solids, 1991, 131–133, 221–224 CrossRef CAS.
- J. Y. Park and G. B. McKenna, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 6667–6676 CrossRef CAS.
- K. Adrjanowicz, K. Kolodziejczyk, W. K. Kipnusu, M. Tarnacka, E. U. Mapesa, E. Kaminska, S. Pawlus, K. Kaminski and M. Paluch, J. Phys. Chem. C, 2015, 119, 14366–14374 CrossRef CAS.
- J. Grelska, K. Jurkiewicz, A. Nowok and S. Pawlus, Phys. Rev. E, 2023, 108, 024603 CrossRef CAS PubMed.
- B. Malfait, A. Moréac, A. Jani, R. Lefort, P. Huber, M. Fröba and D. Morineau, J. Phys. Chem. C, 2022, 126, 3520–3531 CrossRef CAS.
- P. J. Branton, P. G. Hall and K. S. W. Sing, Adsorption, 1995, 1, 77–82 CrossRef CAS.
- W. K. Kipnusu, M. Elsayed, W. Kossack, S. Pawlus, K. Adrjanowicz, M. Tress, E. U. Mapesa, R. Krause-Rehberg, K. Kaminski and F. Kremer, J. Phys. Chem. Lett., 2015, 6, 3708–3712 CrossRef CAS PubMed.
- T. Chen, M. Li and J. Liu, Cryst. Growth Des., 2018, 18, 2765–2783 CrossRef CAS.
- J. H. Deng, J. Luo, Y. L. Mao, S. Lai, Y. N. Gong, D. C. Zhong and T. B. Lu, Sci. Adv., 2020, 6, 1–9 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr06187f |
| This journal is © The Royal Society of Chemistry 2024 |