
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biomolecular interactions on densely coated nanoparticles: a single-molecule perspective†
Swayandipta
Dey‡
 *ab,
Rodrigo
Rivas-Barbosa‡
c,
Francesco
Sciortino
c,
Emanuela
Zaccarelli
cd and
Peter
Zijlstra
*ab
*ab,
Rodrigo
Rivas-Barbosa‡
c,
Francesco
Sciortino
c,
Emanuela
Zaccarelli
cd and
Peter
Zijlstra
*ab
aEindhoven University of Technology, Department of Applied Physics and Science Education, Postbus 513, 5600 MB, Eindhoven, The Netherlands. E-mail: s.dey@tue.nl; p.zijlstra@tue.nl
bInstitute for Complex Molecular Systems and Eindhoven Hendrik Casimir Institute, Eindhoven University of Technology, The Netherlands
cDipartmento di Fisica, Universita’ di Roma “La Sapienza”, Piazzale Moro 5, Roma I-00185, Italy
dCNR Institute of Complex Systems, Uos Sapienza, Piazzale Aldo Moro 2, 00185 Roma, Italy
First published on 1st February 2024
Abstract
DNA-modified gold nanoparticles (AuNPs) play a pivotal role in bio-nanotechnology, driving advancements in bio-sensing, bio-imaging, and drug delivery. Synthetic protocols have focused on maximizing the receptor density on particles by fine-tuning chemical conditions, particularly for DNA. Despite their significance, the understanding of hybridization kinetics on functionalized AuNPs is lacking, particularly how this kinetics depends on DNA density and to what extent it varies from particle-to-particle. This study explores the molecular mechanisms of DNA hybridization on densely coated AuNPs by employing a combination of single-molecule microscopy and coarse-grained molecular dynamics simulations providing a quantification of the molecular rate constants for single particles. Our findings demonstrate that DNA receptor density and the presence of spacer strands profoundly impact association kinetics, with short spacers enhancing association rates by up to ∼15-fold. In contrast, dissociation kinetics are largely unaffected by receptor density within the studied range. Single-particle analysis directly reveals variability in hybridization kinetics, which is analyzed in terms of intra- and inter-particle heterogeneity. A coarse-grained DNA model that quantifies hybridization kinetics on densely coated surfaces further corroborates our experimental results, additionally shedding light on how transient base pairing within the DNA coating influences kinetics. This integrated approach underscores the value of single-molecule studies and simulations for understanding DNA dynamics on densely coated nanoparticle surfaces, offering guidance for designing DNA-functionalized nanoparticles in sensor applications.
1. Introduction
DNA-modified gold nanoparticles (AuNPs) have enabled new directions in the field of bio-nanotechnology ever since the surface modification strategies of Mirkin1 and Alivisatos2 were introduced. This has sparked tremendous research efforts to functionalize gold particles with the desired density of ligands via different routes such as thiol-mediated chemistry, or gold binding sequences.3–8 Such solution-based bio-functionalization protocols generally yield high grafting densities of functional ligands, which appear beneficial for colloidal stability and drive the multivalent interactions of the particles with each other and with functional surfaces. Experimental and theoretical investigations have further identified the role of ionic strength, pH, sequence length, molecular crowding, etc. in the grafting process.9–11 Surface tethered DNA strands in these constructs have shown several unique properties such as extremely sharp and tuneable melting temperatures compared to free DNA in solution.12 In addition, numerous applications of multivalent DNA–AuNPs have been explored in the areas of bio-sensing, bio-imaging, self-assembly, medical diagnostics, and drug delivery.13–18 With such diverse interests in these DNA-modified nanoconjugates, comprehending the molecular mechanisms of DNA hybridization on AuNPs is pivotal for the rational design of these hybrids. Carefully adjusting the affinity and length of the DNA strands on the nanoparticles is known to affect the response toward target molecules or surfaces. Until now, several reports have been published on the thermodynamics and kinetics of DNA hybridization on planar gold films,19–22 and on DNA-functionalized colloidal AuNPs.23 Using ensemble-based characterization techniques and by extracting binding enthalpy and free energy values, these studies further explored the effect of various DNA linker molecules on hybridization efficiency.20,24 Studies performed individually by Chen et al. and Takashima et al. have indicated the effect of DNA surface density and the presence of macromolecular crowders on the interfacial hybridization kinetics on DNA-coated AuNPs.21,23These reports, however focused on ensemble-averaged hybridization in suspension and therefore lacked information on inter- and intra-particle variability and the sources of such heterogeneity. Implementing single-particle and single-molecule detection methodologies to study DNA hybridization was recently pioneered for both dielectric and gold particles.25,26 These studies demonstrated that statistical analyses can reveal sources of heterogeneity, thus offering a window into the polydispersity of a system. As might be expected, using quantitative data derived from single-molecule experiments, currently existing numerical models can be validated and further refined, thereby enhancing the accuracy of our overall understanding of complex interfacial biomolecular interactions.
Such numerical models in the form of all-atomistic simulations27 have enabled the study of the conformational changes of single-stranded DNA both near and tethered to surfaces,28,29 as well as the influence of surfaces on the hybridization process.30 Unfortunately, due to its demanding computational cost, all-atom simulations are limited to small systems of one complementary pair of short DNA strands. A particularly recent exception is the work by Cholko and Chang,31 who modelled systems with up to 5 tethered receptor strands to study surface-mediated hybridization for polar and hydrophobic surfaces. Simple coarse-grained models encapsulating many nucleotides in a single bead have been employed to corroborate the optimal receptor length32 and were pushed to simulate higher coating densities.33 More detailed models, where nucleotides are represented by several beads, have shown that the pathways for DNA hybridization of surface-bounded strands depend on the repetitiveness of the sequence.34
In this work, we investigated the effect of DNA grafting density on the hybridization kinetics at the single-particle and single-molecule levels. Our previous work26 has introduced the use of DNA–PAINT to study molecular interactions at the single-particle and single-molecule level. Here, we expand this methodology and we (1) quantify the molecular rate constants for single particles by correlating ensemble quantification with single-molecule data, (2) introduce molecular dynamics simulations to facilitate the interpretation of the data, and (3) quantify particle-to-particle heterogeneity. Using single-molecule microscopy35 we find that the association rate varies by more than one order of magnitude depending on the design of the DNA coating, whereas the dissociation process is not perturbed. The experimental findings align well with the results obtained from our numerical simulations, where the model provides detailed information on transient base-pairing in the coating layer that is not accessible experimentally. This study showcases the added value of single-molecule studies combined with numerical simulations and provides guiding principles for the design of novel sensors utilizing densely packed DNA-functionalized nanoparticles.
2. Results & discussion
Fig. 1a represents the schematic outline of the single-molecule microscopy based on qPAINT (quantitative Points Accumulation for Imaging Nanoscale Topography),35 which we use to monitor transient DNA hybridization events occurring on the surface of individual gold nanorods (Au NRs). This allows us to monitor stochastic single-molecule binding behaviour and hybridization kinetics between dye-labelled DNA probes in solution and ssDNA receptors on the surface of Au NRs (Fig. 1b). | ||
Fig. 1 Outline of qPAINT based microscopy. (a) Schematic illustration of the optical microscopy setup based on total internal reflection fluorescence microscopy. A typical field of view where each diffraction limited spot corresponds to one-photon photoluminescence from individual Au NRs is shown below. (b) The zoomed inset depicts the close-up of a single Au NR functionalized with receptors. The probes bind transiently to the receptors on the Au NR surface resulting in fluorescence intensity bursts (c) SEM image of citrate-capped Au nanorods dried on a substrate. (d) A representative fluorescence intensity time-trace of a single Au where individual bursts depict binding events. Bursts above a threshold level (dotted blue line) are extracted, taking into account intermittent dark frames due to dye blinking. (e) Histograms of tbright and tdark, fitted with a single exponential function y(t) = A(−t/![[small tau, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0d4.gif) ), where A is a constant and is the fitted average bright and dark time. ), where A is a constant and is the fitted average bright and dark time. | ||
The conjugation chemistry of DNA functionalization on Au nanorods is based on the covalent thiol–Au linkage between thiol-terminated DNA sequences and the Au surface. This surface modification strategy allows nanoparticles to be functionalized with thiolated oligonucleotides, creating unique DNA-directed nano-assemblies for innovative biosensing applications. In this work, we start the sample preparation by first modifying the surface of the glass substrate with a silanizing agent (see details under Methods, ESI†). We used citrate-capped Au NRs (80 nm × 40 nm, SEM image shown in Fig. 1c) that were spin-coated at a low density on glass substrates. Following this, the Au NRs were functionalized with a monolayer of commercially available 30 nt thiolated sequences (hereinafter referred to as “receptor”), mixed with variable dilution ratios of either short 10 nt or long 30 nt thiolated sequences (hereinafter referred to as “spacer”), keeping the total DNA concentration constant throughout different sample dilutions (see details of all the oligo sequences used under Materials & methods, ESI†). This mixed monolayer with spacer sequences not only provides an anti-fouling coating in suppressing non-specific interactions on the particle surface, but also enables us to vary the number of receptors per particle. The Atto655-labelled DNA (hereinafter referred to as “probe”) is a 10 nt long sequence from which 9 bases from the 5′-end are complementary to the receptor. Using these DNA mixed-monolayers we explored the molecular mechanisms of interfacial hybridization kinetics quantitatively at the single-particle and single-molecule level.
The intensity-time trajectories resulting from transient hybridization events (as observed by the fluorescence bursts in Fig. 1d) on individual Au NRs are recorded. The temporal analysis of these individual bursts of events yields tdark (dark-time or time between two consecutive events) and tbright (bright times or bound-state lifetime). By employing a threshold level analysis on fluorescence intensity-time trajectories for individual particles, the dark and bright times are extracted automatically (zoomed inset of time-trace, Fig. 1d). Finally, the distribution of these dark and bright times is fitted with a single-exponential function (Fig. 1e), thereby yielding the mean dark (dark) and bright (bright) times for each single particle.
To perform a quantitative analysis of association and dissociation rates, we first determine the average loading of receptor strands on the Au NRs using protocols described before.7,37–39 Briefly, gold nanoparticles are coated with thiolated receptors at a low pH (∼3.0, citrate buffer). To this end Atto532-labeled 30 nt receptors are mixed with either 10 nt or 30 nt spacers, and then mixed with the gold particles. After 30 minutes of incubation excess DNA is removed by centrifugation at a speed of 4500 RPM for 5 minutes. This was followed by the addition of excess 2-mercaptoethanol to remove the fluorescently labelled receptors from the particle surface. Note that there was a net loss of ∼10% DNA, probably due to sticking of DNA to the walls of the Eppendorf tubes. To calculate the average number of receptors per particle we use N = cDNA/cAuNR, wherein cDNA is determined from the fluorescence intensity of Atto532 (measured at 550 nm). The concentration of Au NRs is obtained using Beer–Lambert's law by using the extinction peak at the longitudinal surface plasmon resonance (at 650 nm) and the molar extinction coefficient provided by the supplier Nanopartz. Considering the Au NRs as spherically-capped cylinders, the receptor density (nm−2) is further calculated using the average surface area of the Au NRs.
Fig. 2a shows the experimentally determined average number of receptors (N) per particle and the corresponding receptor density (nm−2) as a function of the receptor fractions used in the solution. It is found that the total number of receptors increases nearly linearly with the percentage of receptors in solution, irrespective whether the receptors are mixed with 10 nt or 30 nt spacer strands. For a coating without any spacer strands we find a receptor density of ∼0.05 nm−2, equivalent to a DNA footprint of 20 nm2. Under these buffer conditions, we assume ssDNA will adopt a “mushroom-like” conformation for which a similar molecular footprint of ∼16 nm2 was reported before.26
 | ||
| Fig. 2 (a) Ensemble quantification of total number of average receptors (N, left y-axis) and receptor density (nm−2, right y-axis) as a function of % receptor DNA fraction. (b) Experimentally calculated event frequency (Hz) values as a function of receptor density for DNA mixed monolayers (10 nt spacer, blue and 30 nt spacer, red). The ratiometric estimate indicates the expected trend if the association rate were independent of receptor density, see the main text for details. The green dotted line indicates the event frequency for a non-complementary probe. The right most data points in panel (a) and (b) corresponding to 100% receptor fraction and 0.05 nm−2 represent the receptor only sample. | ||
We then perform qPAINT analysis on the AuNR samples with varying receptor densities and different spacer lengths. The observed frequency of binding events is shown in Fig. 2b as a function of receptor density. The experimental values of event frequencies are calculated from the dark times as obtained from the single-particle intensity-time trajectories. The ratiometric estimate (black solid line) of event frequency is obtained from the measured N in Fig. 2a.35,36 Although we would expect a nearly linear scaling of event frequency as a function of receptor density, the experimental results clearly reveal otherwise a surprising trend, particularly observed for 10 nt spacer. For the 30 nt spacer, the event frequency scales nearly as expected, with an event frequency that decreases as the receptor density reduces. However, the trend is very different for the 10 nt spacer. This difference is directly attributed to the different degrees of steric hindrance (or, surface accessibility) observed for different DNA mixed monolayers (10 nt spacer vs. 30 nt spacer). This essentially means that the event frequency depends strongly on the chain length of the spacers where short spacers provide higher accessibility for the probes to interact and bind with the surface-immobilized receptors as compared to longer spacers. Furthermore, the specificity of DNA hybridization events was verified by performing control experiments with a non-complementary probe sequence (see Fig. 2b and Fig. S1†).
Fig. 3 shows the same data but converted to single-particle association (kon) and dissociation (koff) rates in different mixed monolayers. To obtain the values of kon, we used that kon = (dark × N × cprobe)−1. The value of N was obtained from the solution-phase experiments shown in Fig. 2a, where the value was corrected for the presence of the glass substrate that shields an estimated 30% of receptors. The probe concentration (cprobe) was kept constant at 200 pM for all sample conditions. It is interesting to observe here that the change in kon follows a strong dependence with the receptor density for the 10 nt spacer, which directly reveals the effect of receptor density on the surface accessibility (Fig. 3a). The effect of steric hindrance gradually diminishes when receptors are mixed with spacers, which is observed as an increase of kon as the receptor density decreases. This increment is significantly visible for 10 nt short spacers (red dotted line), while the effect is minimal for 30 nt spacers (blue dotted line). For the lowest receptor density, in combination with 10 nt spacer strand, we find that the mean absolute kon increases ∼15× compared to the highest receptor density (i.e. 100% receptor fraction, no spacers). The value of kon for a low receptor density with 10 nt spacer is of similar magnitude as the values obtained earlier using single-molecule imaging on DNA origami.36
 | ||
| Fig. 3 (a) Effect of receptor density on the (a) association (kon) and (b) dissociation (koff) processes of transient DNA hybridization events in mixed monolayers, as extracted from single particle qPAINT measurements. The association rates (kon) show strong dependence with the receptor density as well as the different degree in surface accessibility arising from the different spacer lengths, with nearly exponential increment (red dotted line) for 10 nt spacer and linear (blue dotted line) for 30 nt spacer. The right most data point in both the panels corresponding to a receptor density of 0.05 nm−2 represent receptor only sample. | ||
We now turn our attention to the dissociation process, displayed in Fig. 3b. Within the experimental error the change in dissociation rate (koff = τbright−1) does not exhibit any strong correlation with the receptor density or with the chain length of spacer sequences. The nearly constant koff values obtained for both 10 nt and 30 nt spacers indicate that the dissociation process is neither strictly governed by the surface packing density nor by the different degrees of surface accessibility.
Our single-particle and single-molecule approach provides direct access to the heterogeneity of association and dissociation processes, enabling us to disentangle contributions from intra- and inter-particle effects. Studies into heterogeneity are scarce, but a prior ensemble-averaged study observed a strongly multi-exponential behavior in the dissociation kinetics.40 This was attributed to multiple complex dissociation pathways arising due to interfacial heterogeneities. Ensemble-averaged studies however do not reveal whether the heterogeneity originates from intra- or inter-particle differences. We use the single-molecule approach to first analyze the intra-particle heterogeneity, followed by the inter-particle heterogeneity.
Intra-particle heterogeneity
We analyzed the cumulative distribution functions (CDFs) obtained for the bright times on individual particles for different sample conditions (see Fig. S2†). Irrespective of the receptor density we find that the CDFs exhibit dissociation pathways corresponding single, double, and multi-exponential behavior. This indicates the existence of intra-particle heterogeneity in the dissociation kinetics, where individual receptors on the particle exhibit a different koff resulting in a heterogeneous dissociation pathway. Interestingly, the intra-particle heterogeneity is not strongly correlated to the receptor density, indicating that variability in the surface facets and/or surface charge density between the particles may be the dominant contributor.To check whether the intra-particle heterogeneity is modulated by the buffer conditions (particularly the presence of mono- or bivalent salts that form bridges between DNA strands) we performed qPAINT under two different buffer conditions (10 mM Tris and PBS + 0.5 M NaCl, see Fig. S3†). We did not observe any significant changes, so we conclude that the effects we observe are dominated by the particle surface chemistry (modulated by surface faceting) rather than electrostatic effects.
Inter-particle heterogeneity
Despite the presence of intra-particle heterogeneity the dominant contributor to heterogeneity is however inter-particle heterogeneity of both kon and koff. To quantify this, we correlate both parameters for two dilution factors (9% and 50% receptor fraction) with 10 nt and 30 nt spacers, see Fig. 4. The distribution of rate constants is accumulated across 3 different sample batches with data collected on an average of over 300 individual particles per condition. The most obvious source of particle-to-particle differences are uncertainties due to a limited number of events. We collect 100–300 events per particle, indicating a counting error of no more than 10% so this is not a dominant factor.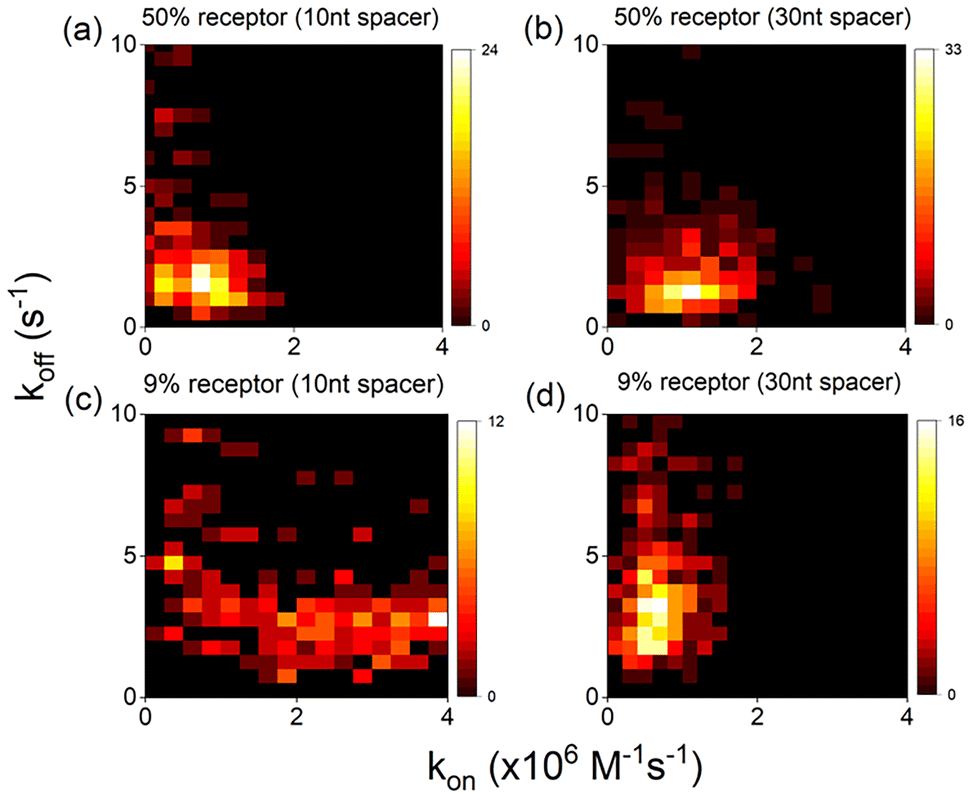 | ||
| Fig. 4 Heatmaps (a–d) showing correlation between experimental kon and koff values for different coating conditions, averaged over >300 particles and >3 sample preparations per condition. | ||
The next obvious source of heterogeneity is related to the surface chemistry of the AuNPs which could be linked to the particle-size distribution, aspect ratio, shape, surface charge and crystal facets. From particle size distribution analysis, we find that the particle's surface area's vary by ∼15% (coefficient of variation, CV), which cannot explain the much larger variability in rate constants of ∼70% (see Fig. S3†). This is rather dominated by the heterogeneity in the molecular coating which could be caused by particle-to-particle variability in surface charge. But what is interesting to note here is the different degree of heterogeneity observed under different sample conditions. We observe that the inter-particle heterogeneity increases for reduced receptor densities, and this is significantly more visible for samples mixed with short 10 nt spacer. In Fig. 4c this is clearly visible as an increased spread of kon for the mixed monolayer with 10 nt spacers, the same degree of spread was observed for even lower receptor densities. This difference in heterogeneity for the two spacer lengths is surprising but could be attributed to particle-to-particle variations in patchiness. Patchy particles might contain islands of densely packed receptor strands that exhibit a reduced kon as explained above, whereas homogeneously coated particles would exhibit receptor strands that are more accessible thereby displaying an increased kon. Despite this increased heterogeneity the trend of increasing average kon with decreasing receptor strand density is clear from Fig. 3a.
To gain a deeper insight into the observed trends we performed molecular dynamics simulations of DNA using the coarse grained oxDNA2 model.41 The model describes DNA strands using three interaction sites per nucleotide as shown in Fig. 5(a). In short, the interactions between nucleotides include stacking, excluded volume, electrostatics and of course sequence dependent hydrogen bonding, which altogether reproduce structural and thermodynamic properties of single and double stranded DNA. Our systems consisted of freely diffusing probes, and surface bound receptors and spacers placed in a square grid, modelling what would be only a small volume element (31.5 nm × 31.5 nm × 31.5 nm) on a flat surface. We simulated two kinds of systems: (i) surfaces coated only with receptors at different surface densities (0.1 nm−2, 0.2 nm−2, 0.28 nm−2) and (ii) surfaces with an overall coating density of 0.1 nm−2 diluted with 10 or 30 nucleotides long spacers at two receptor fractions (9%, 50%). Information regarding the bonding of the receptors and spacers to the surface and their arrangement is given in the ESI Simulation methods (Fig. S5†). We chose larger surface densities with respect to experiments to study the interfacial binding behavior under enhanced crowding effects. To increase the overall number of binding and unbinding events, we set the temperature in the simulation to T = 50 °C and use a probe concentration cprobe = 5.3 mM. The high temperature decreases the lifetime of the binding events, whereas the high probe concentration increases the chances of hybridization. Due to the differences in temperature and coating surface density, we cannot make a direct quantitative comparison between experimental and simulated association and dissociation rates, nonetheless the scaling of the rates as a function of receptor density can be compared. An outline of the model, the simulated systems and the tracking of binding events is shown in Fig. 5.
 | ||
| Fig. 5 Outline of the simulations. (a) Illustration of the three interaction sites of the oxDNA model: the backbone sites are represented with large spherical beads, the stacking and base sites lay inside the cyan ellipsoidal beads; the stacking (base) site is coloured pink (orange) in the probes first base. (b) Simulated system with a 0.1 nm−2 surface density: 100 receptors (blue) bound to a flat surface with an area of ∼31.5 × 31.5 nm2, and 100 freely diffusing probes (yellow). (c) Tracking of the number of paired bases of an individual probe in time; four binding events of different duration are detected; the binding events shown occurred between a single tracked probe that bound to different receptors. The red dashed line at ∼7.3 paired bases indicates the average number of bases paired during binding events. | ||
We monitored in time the number of bases paired for each probe-receptor combination, and from there reconstructed the binding events. For every binding event we know its starting and ending times. The koff is calculated from fitting the histogram of the duration of the binding events to an exponential distribution with mean bright time as the rate parameter (t; ) = ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−t/). The kon is calculated from the times in between the starting of two consecutive binding events. For every start of a binding event, a kon is calculated in the following way: kon = (cprobe(nb) × (Nr − nb) × Δt)−1, with nb is the number of already existing probe-receptor bonds, Nr is the total number of receptors, and Δt is the time in between the start of two consecutive binding events. The kon reported is the average over all binding events. Additional information about the definition of a binding event using our model is given in the Simulation methods section (Fig. S6–S8†).
exp(−t/). The kon is calculated from the times in between the starting of two consecutive binding events. For every start of a binding event, a kon is calculated in the following way: kon = (cprobe(nb) × (Nr − nb) × Δt)−1, with nb is the number of already existing probe-receptor bonds, Nr is the total number of receptors, and Δt is the time in between the start of two consecutive binding events. The kon reported is the average over all binding events. Additional information about the definition of a binding event using our model is given in the Simulation methods section (Fig. S6–S8†).
Keeping in mind that the surface density is ∼1.5× larger than the experimental setup, we first discuss the koff and kon for a 0.1 nm−2 surface density diluted with spacers, shown in Fig. 6(a). In good agreement with the experimental measurements (Fig. 3b), the simulations in Fig. 6(a) in the bottom panel show similar values of koff regardless of the dilution and spacer strands used.
 | ||
| Fig. 6 Simulated kon (top) and koff (bottom) (A) as function of the fraction of receptors for a 0.1 nm−2 surface density diluted with 10 nt (blue) or 30 nt (red) long spacers, and (B) as function of the of the surface density (100% receptors). | ||
As discussed in an earlier section based on the experimental data, koff is found to be nearly independent of surface receptor density. In contrast, the top panel shows an increase in kon as the receptor density is reduced by incorporating spacer strands of 10 nt or 30 nt. In particular, the increase in kon is considerably larger when using the 10 nt spacers, again in good agreement with the trend observed in the experiments (Fig. 3a). As mentioned earlier, the larger increase of kon for the short spacers is due to the length difference between receptors and spacers: the binding site of the 30 nt receptors is more accessible to probes due to the length difference with the 10 nt spacer strands.
However, if this was to be the single reason, we would then expect, for the systems diluted with 30 nt spacers, to have the same kon irrespective of the introduction of the spacers since both strands are equally long. Nevertheless, we do observe a small but non-negligible increase in kon even for the 30 nt spacer strands. We therefore hypothesize that inter-strand interactions (receptor–receptor, receptor–spacer and spacer–spacer) do occur, and that similarly to the 10 nt spacer case, these interactions modulate the accessibility of the receptors. To study this in more detail we ran additional simulations of the receptor and 30 nt spacer systems but in the absence of probe strands.
From the simulations in absence of probe strands, we tracked the center of mass z-coordinate of the last 9 bases of the receptors as shown in Fig. S9.† We observe no difference between the extension of the receptors and spacers species at any dilution: on average the binding site distribution is at the same distance from the particle surface irrespective of the conditions.
We therefore turned our attention to possible base pairing between strands. We monitored the number of times base pairing occurred between individual bases, taking notice on the average energy contribution of each pair. Fig. S10† shows the energy-weighted base pairing occurrence maps between the surface strands for different dilution conditions. The most energetic contribution in surface strand interactions comes at about halfway the backbone, between bases 17–18 and 18–19 (3′–5′), which corresponds to C–G base pairings. Being consecutive bases, the pairing cannot be of a single receptor with itself but must be with another receptor; these receptor–receptor pairings decrease with dilution due to the increasing distance between them (the layout of the receptor/spacer plays an important role). Interactions between receptors and spacers are in general different, but the maps do not give evidence that surface strand base pairing leads to an increase in the kon with dilution.
We are hinted to believe that the mechanism is not due (at least not exclusively) to surface inter-strand interactions, but instead attributed to the probe-receptor interaction volume that changes with dilution. Let us first consider the 30 nt spacer system with 9% receptor fraction, the distance between receptors is such that they barely interact (as can also be seen from the base interaction maps Fig. S9†), we can say that the receptors interaction volumes, where probes can come and bind, do not overlap; any probe that approaches the interaction volume of an individual receptor will bind to it and it alone. Now, as we move to the 50% receptor system, the receptors interaction volumes begin to overlap; when a probe gets inside the shared interaction volume of two nearby receptors, both receptors compete to bind with it; in other words, the decrease in kon with receptors fraction comes not (only) from inaccessibility of the probes to reach the complementary sites of the receptors, but from a reduction in the effective interaction volume of the receptors by its nearest neighbours. For the 100% receptor system, the effective interaction volume is further reduced, and hence the decrease in kon. The trends in both rates with dilution are qualitatively equivalent to the experimental measurements, we believe the quantitative differences come jointly from the larger surface density (∼1.5) and temperature used in simulation versus experiments. Note that the estimated simulated value of koff is nearly five orders of magnitude higher than the measured value (see Fig. S13†), which can be largely attributed to the difference in temperature (T = 20 °C for the experiments, and T = 50 °C for the simulations, see ESI† for a quantification). Therefore, overall we observe a difference in absolute values of kon and koff between our experiments and the model, but the scaling of these rate constants exhibit a similar trend as a function of receptor density.
Now let us move our attention to what happens to the rates when increasing the total density of surface strands (receptor + spacer), shown in Fig. 6(b). From the bottom panel, we see that koff remains the same when doubling the initial coating (receptor) density 0.1 nm−2 to 0.2 nm−2, again supporting the idea that the dissociation process is independent of the coating conditions; however, when we further increase the density to 0.28 nm−2, more than four times the experimental value, we do detect a rise in koff: binding events are on average shorter. To investigate the possible reason behind the koff increase only at the densely packed system, we looked at the average number of bases paired during binding events: Fig. S11† shows that the average number of bases paired is ∼7.3 for all cases again but the 0.28 nm−2 system with a lower number ∼7.15 of bases paired. We inquired at the individual base level this discrepancy, counting the fraction of times each base pairs during the binding events, as shown in Fig. S12.† We see that the six bases furthest away from the surface pair a similar fraction of times independently of the surface density; instead, for the three bases closer to the surface, there is no difference between the 0.1 and 0.2 nm−2 systems but there is with the denser 0.28 nm−2: these last three bases pair less frequently, in fact, the closer to the surface the bases are, the larger the difference becomes. We conclude again that crowding effects come into play at these very high densities: neighbour receptors decrease the accessibility of the probes 3′ three bases, inhibiting their base pairing, and effectively decreasing the melting temperature, hence the higher koff for the densest system. Another feature that can be seen from the base-level contribution is the effect of stacking: the occurrence of the 9th position (3′ to 5′) A base is higher than that of the last A 3′ base due to the extra stacking provided by the receptor's 10th base.
As a limiting case, we simulated a system with only one single receptor (surface density ∼0.001 nm−2). The average number of bases paired during binding events is ∼7.29 (see Fig. S11†), a base pairing occurrence fraction similar to all the other systems but the 0.28 nm−2.
Moving to the association rate, it can be seen from Fig. 6(b) that kon decreases with density by similar motives, the increase in shared interaction volume plus the excessively high crowding in the surface inhibits probes from hybridizing with the complementary sites.
3. Conclusions
In this study, densely coated DNA-modified AuNPs were used as a model system to investigate the effect of receptor density and mixed monolayers on the DNA hybridization kinetics. We employed a synergistic approach combining single-molecule microscopy with molecular dynamics simulations based on a coarse-grained oxDNA2 model. Both experiments and simulations reveal that DNA receptor density and the presence of spacer strands profoundly impact association kinetics, with short spacers enhancing association rates by up to ∼15-fold. This was attributed purely due to differences in the degree of surface accessibility arising not just as a function of receptor density, but also from the length of spacers used. In contrast, dissociation kinetics are largely unaffected by receptor density within the studied range.Our single-particle and single-molecule approach uniquely provides insight into intra- and inter-particle heterogeneity. Intra-particle heterogeneity was observed in the multi-exponential distribution of bright times on single nanoparticles. We observed nanoparticles that exhibit mono-, bi-, and multi-exponential distributions of the bright time, which we attributed to local variations in underlying surface chemistry and surface faceting that affects the accessibility of binding sites on the particle. Inter-particle heterogeneity was however more pronounced, particularly for mixed monolayers of DNA with a short spacer strand. This was attributed to a larger variation in the shared interaction volume between receptor and probe sequences, when particles are coated with shorter spacers, particularly at lower receptor densities. Additionally, the variation in surface charge on the particles could result in patchy particles, and the inter-particle variation in the degree of patchiness could result in such heterogeneity. Methods like nanoparticle tracking analysis42 and tunable resistive pulse sensing43 could in the future be used to quantify the heterogeneity in surface charge of bio-functionalized colloidal nanoparticles. However, correlating dispersion in zeta-potential with receptor density (N) in individual NPs, require advanced correlation methods.
Overall, we demonstrated that a synergistic approach between single-molecule microscopy and molecular dynamics simulations provide novel insights into DNA hybridization pathways across the bio-nano interfaces. It is worth mentioning that the integrated approach of ensemble-averaged and single-molecule quantification, combined with molecular dynamics simulations can be extended to other model systems such as silver or glyconanoparticles.
Prospective research could explore ways to further optimize design parameters like receptor density and spacers to achieve tailored interactions of DNA-coated nanoparticles in applications ranging from bio-sensing to drug delivery. By understanding the intricate relationship between receptor density, spacer molecules, and association kinetics, one can rationally design nanoparticles with precisely tuned properties for their intended applications.
Author contributions
S. D. and P. Z. conceived the research project and designed the experiments. S. D. performed the experiments and conducted the data analysis. R. R.-B. conducted the simulations. F. S., E. Z. and P. Z. supervised the research. All authors discussed the results and contributed to writing the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement No 864772). R. R.-B., F. S., E. Z., and P. Z. acknowledge funding from the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska-Curie program (ITN SuperCol, Grant Agreement 860914).References
- C. A. Mirkin, R. L. Letsinger, R. C. Mucic and J. J. Storhoff, Nature, 1996, 382, 607–609 CrossRef CAS PubMed.
- C. J. Loweth, W. B. Caldwell, X. Peng, A. P. Alivisatos and P. G. Schultz, Angew. Chem., Int. Ed., 1999, 38, 1808–1812 CrossRef CAS PubMed.
- D. Y. Choi, S. Kim, J.-W. Oh and J.-M. Nam, Bull. Korean Chem. Soc., 2022, 43, 1298–1306 CrossRef CAS.
- B. Malile, J. Brkic, A. Bouzekri, D. J. Wilson, O. Ornatsky, C. Peng and J. I. L. Chen, ACS Appl. Bio Mater., 2019, 2, 4316–4323 CrossRef CAS PubMed.
- C. J. Ackerson, M. T. Sykes and R. D. Kornberg, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 13383–13385 CrossRef PubMed.
- B. Liu and J. Liu, J. Am. Chem. Soc., 2017, 139, 9471–9474 CrossRef CAS PubMed.
- X. Zhang, M. R. Servos and J. Liu, J. Am. Chem. Soc., 2012, 134, 7266–7269 CrossRef CAS PubMed.
- H. Pei, F. Li, Y. Wan, M. Wei, H. Liu, Y. Su, N. Chen, Q. Huang and C. Fan, J. Am. Chem. Soc., 2012, 134, 11876–11879 CrossRef CAS PubMed.
- R. Jin, G. Wu, Z. Li, C. A. Mirkin and G. C. Schatz, J. Am. Chem. Soc., 2003, 125, 1643–1654 CrossRef CAS PubMed.
- S. E. Seo, T. Li, A. J. Senesi, C. A. Mirkin and B. Lee, J. Am. Chem. Soc., 2017, 139, 16528–16535 CrossRef CAS PubMed.
- Q. Lei, C. Ren, X. Su and Y. Ma, Sci. Rep., 2015, 5, 9217 CrossRef CAS PubMed.
- A. K. R. Lytton-Jean and C. A. Mirkin, J. Am. Chem. Soc., 2005, 127, 12754–12755 CrossRef CAS PubMed.
- A. A. Saeed, J. L. A. Sánchez, C. K. O'Sullivan and M. N. Abbas, Bioelectrochemistry, 2017, 118, 91–99 CrossRef CAS PubMed.
- E. Ferrari, Biosensors, 2023, 13(3), 411 CrossRef CAS PubMed.
- A. Jayagopal, K. C. Halfpenny, J. W. Perez and D. W. Wright, J. Am. Chem. Soc., 2010, 132, 9789–9796 CrossRef CAS PubMed.
- M.-E. Kyriazi, D. Giust, A. H. El-Sagheer, P. M. Lackie, O. L. Muskens, T. Brown and A. G. Kanaras, ACS Nano, 2018, 12, 3333–3340 CrossRef CAS PubMed.
- H. Chai, M. Wang, L. Tang and P. Miao, Anal. Chim. Acta, 2021, 1165, 338543 CrossRef CAS PubMed.
- S. Dey, M. Dolci and P. Zijlstra, ACS Phys. Chem. Au, 2023, 3, 143–156 CrossRef CAS PubMed.
- L. Zhang, Z. Li, X. Zhou, G. Yang, J. Yang, H. Wang, M. Wang, C. Liang, Y. Wen and Y. Lu, J. Electroanal. Chem., 2015, 757, 203–209 CrossRef CAS.
- B. Lang, J. Chem. Thermodyn., 2010, 42, 1435–1440 CrossRef CAS.
- C. Chen, W. Wang, J. Ge and X. S. Zhao, Nucleic Acids Res., 2009, 37, 3756–3765 CrossRef CAS PubMed.
- A. W. Peterson, R. J. Heaton and R. M. Georgiadis, Nucleic Acids Res., 2001, 29, 5163–5168 CrossRef CAS PubMed.
- A. Takashima and M. Oishi, RSC Adv., 2015, 5, 76014–76018 RSC.
- D. Zhu, P. Song, J. Shen, S. Su, J. Chao, A. Aldalbahi, Z. Zhou, S. Song, C. Fan, X. Zuo, Y. Tian, L. Wang and H. Pei, Anal. Chem., 2016, 88, 4949–4954 CrossRef CAS PubMed.
- T. Andrian, S. Pujals and L. Albertazzi, Nanoscale Adv., 2021, 3, 6876–6881 RSC.
- M. Horáček, D. J. Engels and P. Zijlstra, Nanoscale, 2020, 12, 4128–4136 RSC.
- W. D. Cornell, P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz, D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell and P. A. Kollman, J. Am. Chem. Soc., 1995, 117, 5179–5197 CrossRef CAS.
- R. M. Elder and A. Jayaraman, J. Chem. Phys., 2014, 140, 155103 CrossRef.
- V. Barone, I. Cacelli, A. Ferretti, S. Monti and G. Prampolini, Phys. Chem. Chem. Phys., 2009, 11, 10644–10656 RSC.
- S. Monti, I. Cacelli, A. Ferretti, G. Prampolini and V. Barone, J. Phys. Chem. B, 2010, 114, 8341–8349 CrossRef CAS PubMed.
- T. Cholko and C. A. Chang, J. Phys. Chem. B, 2021, 125, 1746–1754 CrossRef CAS PubMed.
- A. Jayaraman, C. K. Hall and J. Genzer, Biophys. J., 2006, 91, 2227–2236 CrossRef CAS PubMed.
- A. Jayaraman, C. K. Hall and J. Genzer, J. Chem. Phys., 2007, 127, 144912 CrossRef PubMed.
- M. J. Hoefert, E. J. Sambriski and J. José de Pablo, Soft Matter, 2011, 7, 560–566 RSC.
- R. Jungmann, M. S. Avendaño, M. Dai, J. B. Woehrstein, S. S. Agasti, Z. Feiger, A. Rodal and P. Yin, Nat. Methods, 2016, 13, 439–442 CrossRef CAS PubMed.
- R. Jungmann, C. Steinhauer, M. Scheible, A. Kuzyk, P. Tinnefeld and F. C. Simmel, Nano Lett., 2010, 10, 4756–4761 CrossRef CAS PubMed.
- B. L. Baldock and J. E. Hutchison, Anal. Chem., 2016, 88, 12072–12080 CrossRef CAS PubMed.
- L. M. Demers, C. A. Mirkin, R. C. Mucic, R. A. Reynolds, R. L. Letsinger, R. Elghanian and G. Viswanadham, Anal. Chem., 2000, 72, 5535–5541 CrossRef CAS PubMed.
- X. Zhang, B. Liu, N. Dave, M. R. Servos and J. Liu, Langmuir, 2012, 28, 17053–17060 CrossRef CAS PubMed.
- L. Vanjur, T. Carzaniga, L. Casiraghi, M. Chiari, G. Zanchetta and M. Buscaglia, Biophys. J., 2020, 119, 989–1001 CrossRef CAS PubMed.
- B. E. K. Snodin, F. Randisi, M. Mosayebi, P. Šulc, J. S. Schreck, F. Romano, T. E. Ouldridge, R. Tsukanov, E. Nir, A. A. Louis and J. P. K. Doye, J. Chem. Phys., 2015, 142, 234901 CrossRef PubMed.
- G. Midekessa, K. Godakumara, K. Dissanayake, M. M. Hasan, Q. U. Reshi, T. Rinken and A. Fazeli, Membranes, 2021, 11, 779 CrossRef CAS PubMed.
- R. Vogel, A. K. Pal, S. Jambhrunkar, P. Patel, S. S. Thakur, E. Reátegui, H. S. Parekh, P. Saá, A. Stassinopoulos and M. F. Broom, Sci. Rep., 2017, 7, 17479 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr06140j |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2024 |