
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances in the role of interfacial liquids in electrochemical reactions
Rani
Baidoun
 a,
Gexu
Liu
b and
Dohyung
Kim
*a
a,
Gexu
Liu
b and
Dohyung
Kim
*a
aDepartment of Chemical and Biomolecular Engineering, University of Pennsylvania, Philadelphia, PA 19104, USA. E-mail: dohyungk@seas.upenn.edu
bDepartment of Materials Science and Engineering, University of Pennsylvania, Philadelphia, PA 19104, USA
First published on 28th February 2024
Abstract
The interfacial liquid, situated in proximity to an electrode or catalyst, plays a vital role in determining the activity and selectivity of crucial electrochemical reactions, including hydrogen evolution, oxygen evolution/reduction, and carbon dioxide reduction. Thus, there has been a growing interest in better understanding the behavior and the catalytic effect of its constituents. This minireview examines the impact of interfacial liquids on electrocatalysis, specifically the effects of water molecules and ionic species present at the interface. How the structure of interfacial water, distinct from the bulk, can affect charge transfer kinetics and transport of species is presented. Furthermore, how cations and anions (de)stabilize intermediates and transition states, compete for adsorption with reaction species, and act as local environment modifiers including pH and the surrounding solvent structure are described in detail. These effects can promote or inhibit reactions in various ways. This comprehensive exploration provides valuable insights for tailoring interfacial liquids to optimize electrochemical reactions.
 Rani Baidoun | Rani Baidoun obtained his BS degree in Chemical and Biomolecular Engineering from Lehigh university. Currently, he is a second year Ph.D. candidate in the Chemical and Biomolecular Engineering Department of the University of Pennsylvania. His research focuses on interfacial microenvironments at the electrode/electrolyte interface as well as electrochemical bond cleavage. |
 Gexu Liu | Gexu Liu obtained her BS degree in Material and Nano Science from University of Waterloo and BEng degree in Material Science and Technology from Beijing Jiaotong University. Currently, she is a master's student at University of Pennsylvania, focusing on interfacial liquids and their impact on electrochemical reactions. |
 Dohyung Kim | Dohyung Kim is an Assistant Professor in the Department of Chemical and Biomolecular Engineering at the University of Pennsylvania. He received his B.S. in Materials Science and Engineering from Seoul National University in 2012 and a Ph.D. in Materials Science and Engineering from the University of California, Berkeley in 2018. Before joining Penn in 2022, he was a postdoctoral scholar at the Department of Chemical Engineering, Stanford University. His research focuses on gaining fundamental insights into electrochemical processes occurring at solid–liquid interfaces and applying such knowledge to various areas of energy, chemicals, materials, and the environment. |
1. Introduction
The field of electrocatalysis has so far greatly advanced with a focus on the design and engineering of materials that would act as efficient catalysts for various important applications.1–8 While further efforts are needed to improve the activity and stability of catalyst materials and find more earth-abundant and cost-effective alternatives, there also has been growing recognition of the importance of the interfacial liquid found at the catalyst-electrolyte interface for electrochemical applications.6,9 The interfacial liquid, which includes species such as cations, anions, water, and other compounds found in the electric double layer (EDL) and near the surface, is considered an integral part of (photo)electrochemical systems.9–12It has been demonstrated that these species within the liquid interface have an undeniable impact on important electrocatalytic reactions such as the hydrogen evolution/oxidation reactions (HER/HOR), oxygen evolution/reduction reactions (OER/ORR), and carbon dioxide reduction reaction (CO2RR).13–16 These reactions are key to promoting sustainable energy conversion and storage as well as reducing and reversing greenhouse gas emissions by generating value-added products with renewables.6,17,18 In electrochemical systems, these reactions take place as follows:
| HER/HOR:2H+ + 2e− ⇄ H2 |
| OER/ORR: 2H2O ⇄ O2 + 4H+ + 4e− |
| CO2RR: CO2 + 2H+ + 2e− → CO + H2O |
| \quad\quad\quad\quad\quad\quad 2CO2 + 12H+ + 12e− → C2H4 + 4H2O |
They often involve a large number of elementary steps. Furthermore, for the ORR and CO2RR, there exist multiple reaction pathways, such as the 2e− pathway for ORR to produce H2O2 and a multielectron reduction pathway for CO2 to produce hydrocarbons and oxygenates. For a better understanding of all the details regarding the mechanisms of those reactions as well as the catalyst design considerations, readers are encouraged to go over other excellent perspectives and reviews published previously.13–16,19–21
This minireview focuses on how the interfacial liquid affects the rates and selectivity of these reactions. Understanding the behavior of water molecules at the solid–liquid interface, distinct from the bulk, is crucial for water-splitting electrochemistry. Ionic species are critical components of the EDL that form against a charged solid and their presence near the active sites can significantly facilitate or inhibit reactions in numerous ways that include intermediate (de)stabilization,22,23 local environment modification (e.g., pH, solvent structure),24,25 and active site modification (e.g., electronic structure, accessibility).26 While the recognition of such catalytic effects at electrochemical interfaces is not new,9,27 recent advances in experimental and theoretical approaches have significantly enhanced our understanding of the impacts of interfacial liquids on electrochemical reactions. This minireview highlights the latest findings in this evolving field. Advancements in both catalyst synthesis and the manipulation of interfacial liquid effects are poised to elevate the efficiencies of electrochemical reactions to unprecedented levels.
In this minireview, we delve into the latest advances and current insights in three distinct sections, offering a detailed overview of the electrocatalytic impacts of structured interfacial water, solvated and desolvated cations, and specifically and non-specifically adsorbing anions, as summarized in Table 1. Due to the growing importance of interfacial liquids in electrocatalytic reactions, there have been recent reviews particularly focused on the cation effects in CO2 electroreduction.28–31 Given the ubiquity of interfacial liquids and their far-reaching implications in various essential reactions, our objective is to offer a more thorough examination of all species within the interfacial liquid. We describe their general effects and diverse influences on activity and selectivity across different reactions. Regarding the mechanisms, we present varying perspectives, many of which are actively under investigation. This minireview is meticulously curated to provide a comprehensive overview of our latest understanding in this area and propose avenues for future research to advance our knowledge.
| Reactions | Interfacial liquid components | Effects |
|---|---|---|
| HER | Water molecules | • More rigid water structures lead to higher kinetic barriers due to higher reorganization energy requirements |
| • Structured water facilitates H/OH transport leading to higher HER activities | ||
| Cations | • Smaller alkali metal cations lead to higher HER activity on Pt-group metals and lower HER activity on coinage metals | |
| • Cations stabilizing HER transition states either directly or indirectly (e.g., via adsorbed OH) | ||
| • Cations can alter the water reorganization energy | ||
| OER/ORR | Cations | • Larger alkali metal cations lead to increase in OER |
| • Cations can stabilize or destabilize key OER and ORR intermediates affecting reaction rates | ||
| • Larger alkali metal cations disrupt water networks which facilitates the rearrangement of water molecules during with charge transfer | ||
| • Cations also influence the local pH with higher pH values leading to higher OER activities | ||
| • Cations influence the formation of catalytically active phases under operation for OER | ||
| Anions | • Anions exhibiting poisoning effects and decreasing ORR rates | |
| CO2RR/CORR | Water molecules | • Hydrophobic environments and breakage of water networks hinder HER and favor CO2RR |
| • Stabilize key reaction intermediates via H-bond interactions | ||
| Cations | • Larger alkali metal cations increase CO2RR/CORR rates through local electric field modulation and direct intermediate stabilization | |
| • Alkali metal cations buffer the local pH, therefore controlling the reactant concentration (i.e., CO2) near the surface | ||
| Anions | • Halides donate partial negative charge to the catalyst surface enhancing the interaction with reaction intermediates | |
| • Supporting electrolyte anions, such as bicarbonate, also function as an additional proton and CO2 source |
2. Structured water at the interface
In most electrochemical systems containing aqueous electrolytes, water is the predominant species. The arrangement and orientation of water molecules within the interfacial liquid have been shown to have a significant impact on various electrochemical reactions, such as the HER and CO2RR.32 Therefore, gaining insights into the dynamic behavior of interfacial water as well as its overall structure by the hydrogen bonding network is vital in order to better control these electrochemical reactions. In this section, we elucidate the relationship between the structure of the interfacial water and the activity and selectivity of HER and CO2RR, while also delving into the underlying molecular mechanisms.2.1. Hydrogen evolution reaction
Efforts to understand the impact of water molecules on HER have primarily focused on alkaline media systems, where the rates are sluggish.32,33 It has been suggested that the interfacial microenvironment, specifically the structure of water molecules, serves as an important descriptor of HER kinetics in alkaline media.34 In recent discourse, two primary viewpoints have emerged concerning the mechanism by which the structure of interfacial water molecules influences the rates of HER. The first view posits that the hydrogen-bonded interfacial water may introduce structural rigidity difficult to accommodate HER intermediates via reorganization and slow the kinetics. On the other hand, there have been works emphasizing the connectivity of the hydrogen-bonded interfacial water network that facilitates the transfer of proton/hydroxides and therefore increases HER activity.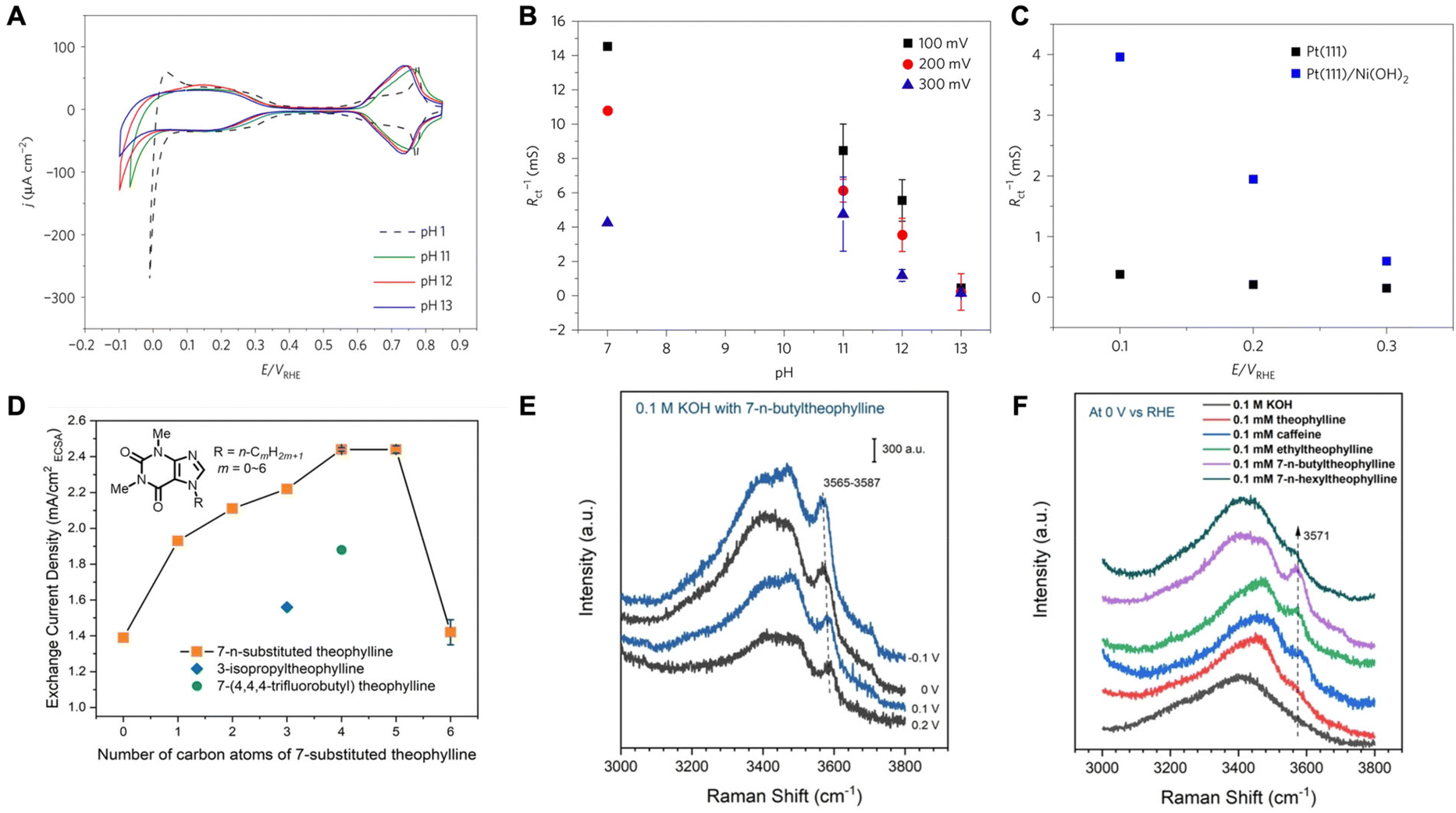 | ||
| Fig. 1 (A) Comparison of cyclic voltammograms for Pt(111) at different pH values. (B) Inverse of charge transfer resistance for hydrogen adsorption as a function of solution pH. (C) The inverse of charge transfer resistance for hydrogen adsorption as a function of potential for Pt(111) and Ni(OH)2 decorated Pt(111). Reproduced from ref. 33 with permission from Springer Nature, copyright 2017. (D) HER/HOR activities on polycrystalline (pc) Pt in H2-saturated 0.1 M KOH with 0.1 mM theophylline derivatives. (E) In situ SHINERS of interfacial water on a Pt(pc) electrode in 0.1 M KOH with 0.1 mM 7-n-butyltheophylline at −0.1 to 0.2 V. (F) In situ SHINERS of interfacial water on a Pt(pc) electrode in 0.1 M KOH with 0.1 mM various theophylline derivatives at 0 V. Reproduced from ref. 35 with permission from John Wiley and Sons, copyright 2022. | ||
They went on to further show that by manipulating the pzfc and the resulting interfacial water rigidity, HER kinetics in alkaline media can be improved. Through laser-induced temperature-jump measurements, it was found the electric field strength decreases due to a negative shift in the pzfc when Ni(OH)2 is adsorbed on Pt. This brings the H-UPD and HER potential region closer to the pzfc and makes the interfacial water less rigid resulting in the improved kinetics for Pt/Ni(OH)2 (Fig. 1C). These results highlight the significance of interfacial water structure on the kinetics of HER.
In another work, Intikhab et al. studied the effects of double-layer dopants such as caffeine on the HER/HOR reactions in alkaline media.36 They demonstrated that both HER and HOR rates increased by 5-fold on a Pt(111) surface in alkaline media in the presence of caffeine as the double layer dopant. They also demonstrated an increase in HER/HOR rates on other Pt single crystals as well as polycrystalline surfaces and nanoscale surfaces such as Pt/C. For instance, on Pt(pc), the exchange current densities increased from 0.30 mA cm−2 to 0.86 mA cm−2, whereas on Pt(110), the exchange current densities increased from 1.03 mA cm−2 to 2.21 mA cm−2. Furthermore, the authors demonstrate that caffeine adsorbs on the surface of the catalyst as evidenced by H-UPD region suppression and remains stable up to potentials of 1.0 V vs. RHE. The authors then studied the effects of caffeine in acidic environments (pH = 1) where they demonstrated that HER/HOR kinetics are hindered rather than enhanced. They explain that because the electric field in the H-UPD region is stronger in alkaline media than in acidic media, the caffeine molecule disrupts this electric field and therefore lowers the barrier for water reorganization. In acidic media however, since the electric field is weak in the H-UPD region (close to the pzc), water reorganization does not play a role in impacting the rates of these reactions, and therefore caffeine only acts as a hindrance through site blocking.
Based on a similar school of thought, Zhao and coworkers explored the enhancement of HER/HOR by introducing caffeine-derivative organic compounds that act as interfacial water structure disruptors.35 They determined that 7-n-butyltheophylline increased the HER activity the most by inducing surface hydrophobicity on Pt (Fig. 1D). Through in situ surface enhanced infrared absorption spectroscopy (SEIRAS), they determined that 7-n-butyltheophylline is strongly bound to the Pt surface with an adsorption energy comparable to CO adsorption on Pt. Due to its interfacial specificity, shell-isolated nanoparticle enhanced Raman spectroscopy (SHINERS) was employed to understand the interfacial water structure in different coordination environments. It is observed that a sharp band appears upon adsorption of 7-n-butyltheophylline. This band corresponds to weakly H-bonded water molecules. The band red shifts as well as increases in intensity with decreasing potential suggesting the presence of these water molecules in the immediate proximity of the electrode surface, especially in the presence of 7-n-butyltheophylline than other derivatives (Fig. 1E and F). The authors propose that alkyl groups on adsorbed 7-substituted theophylline act as structure breakers of interfacial water by disrupting their H-bond network, therefore decreasing the activation energy of the HOR/HER. The weakening of H-bonds within interfacial water molecules makes it less energetically costly to change their configuration to stabilize the intermediates of the HER/HOR reaction. This H-bonding network disruption by alkyl chain adsorbates is local and only reactions occurring on unoccupied Pt sites close to the adsorbates would be impacted.
Rebollar et al. further studied caffeinated systems and the consequences of the HER/HOR reactions on Pt surfaces.37 The authors demonstrated once more that the presence of caffeine hinders HER/HOR in acidic media while boosting the reaction in alkaline media. The authors attributed this change in activity to the pzfc shift and its effects. The pzfc shifted to negative values in the presence of caffeine indicating that in alkaline media caffeine disrupts the electric field allowing for an enhancement in HER/HOR. Furthermore, while the pzfc was very close to the RHE in a caffeine-containing base, it actually shifted further away from the RHE in a caffeine-containing acid, also explaining the slower kinetics in acid in the presence of caffeine. The authors also demonstrated that the presence of caffeine enhanced alkaline HER/HOR regardless of the pH and inhibited HER/HOR in acidic media. Overall, they were able to confirm the pH effects on HER/HOR that may be associated with how close the pzfc is to the RHE potential that reduces the kinetic barriers associated with solvent dynamics suggested by Ledezma-Yanez et al.33 However, they also suggest that there might be other mechanistic reasons to the widely varying degree of HER/HOR kinetics across different pHs and the enhancement in alkaline media caused by adsorbed molecules, such as the caffeine, as kinetic isotope experiments show that solvent dynamics are fast in acid and slow in base regardless of the pzfc shift by the adsorbed caffeine.
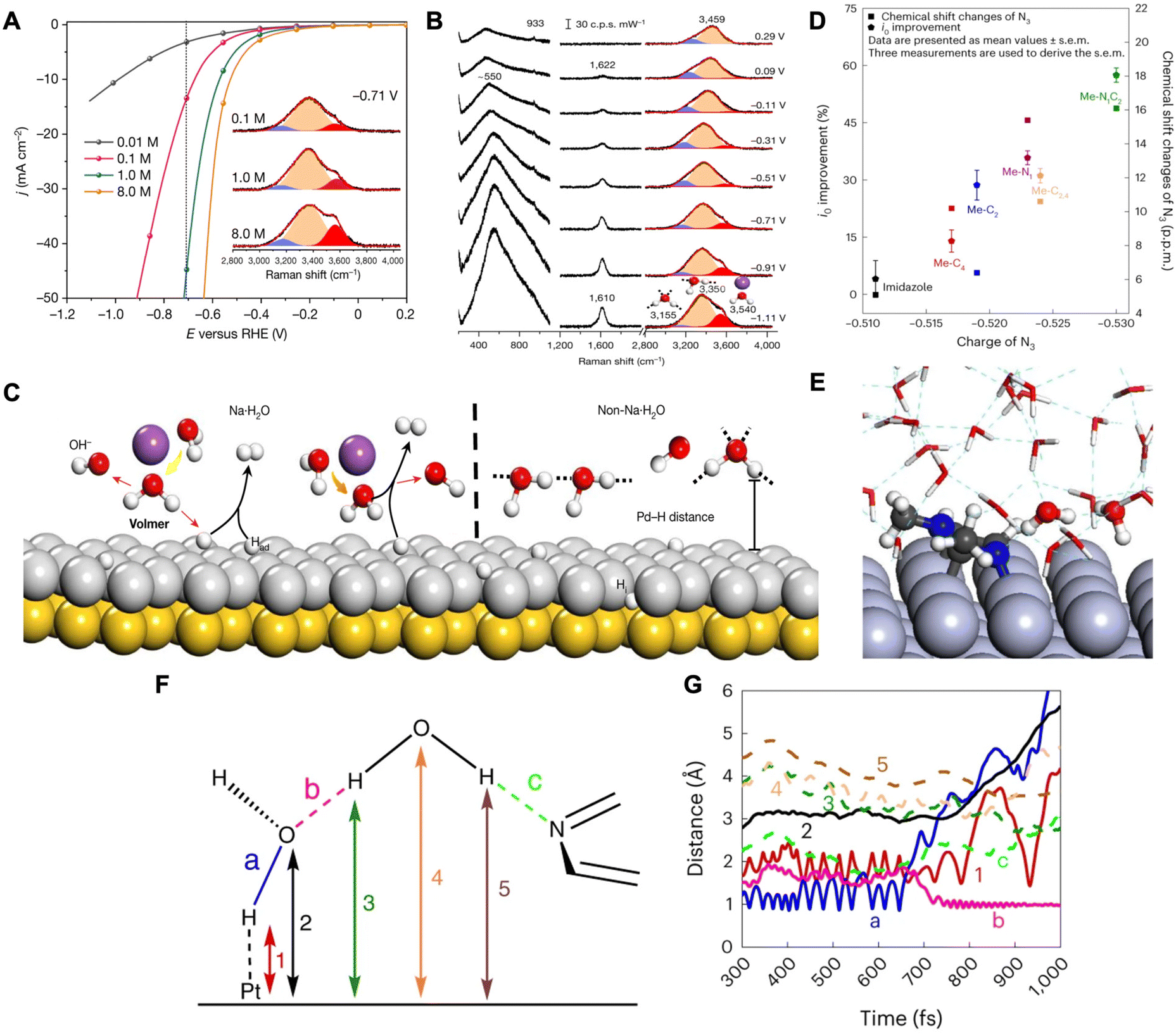 | ||
| Fig. 2 (A) HER profiles for solutions with different Na+ cations and Raman spectra for O–H stretching modes (inset). (B) In situ Raman spectra of interfacial water on a Pd(111) electrode in a 0.1 M NaClO4 electrolyte of pH 11. Gaussian fitting of three O–H stretching modes for each spectrum is shown in blue, orange, and red. (C) Schematic for the interfacial water dissociation on an Au(111) coated with Pd(111) surface. Reproduced from ref. 38 with permission from Springer Nature, copyright 2021. (D) Improvements in the i0 (left y-axis) and 14N3/15N3 chemical shift changes (right y-axis) of N-methylimidazoles between two different media as a function of N3 charge. Note: N3 charge was calculated using DFT. (E) Atomic configuration of an equilibrated H2O–Pt(100) system including Me–N1C2 in alkaline medium. (F) Schematic of an interfacial water dimer composed of H2O(H-down)1st and an N3 of Me–N1C2 bonded with an H2O(H-down)2nd. (G) Time evolution of bond distances during the Volmer step at the H2O–Pt(100) interface with Me–N1C2 in alkaline medium. Reproduced from ref. 39 with permission from Springer Nature, copyright 2023. | ||
In another work by Sun and coworkers, it was demonstrated that the addition of N-methylimidazoles at the platinum–water interface promotes HER rates by facilitating the diffusion of hydroxides through an ordered interfacial water structure.39 They propose that HER/HOR kinetics of Pt in acid and base media are dependent on the diffusion of protons and hydroxides, respectively, through the H-bonding network of interfacial water by the Grotthuss mechanism. The Grotthuss mechanism involves proton migration within the H-bonding network of water, where a proton jumps from one stationary oxygen atom to a neighboring oxygen atom through the cleavage and formation of bonds.40,41 The authors performed rotating disk electrode (RDE), density functional theory (DFT), and 14N and 15N Nuclear Magnetic Resonance (NMR) studies in order to understand the effects of N-methylimidazoles on HER. From RDE measurements, a trend was found that the greater the number of methyl groups, the higher the exchange current density leading to higher HER/HOR rates. With DFT determining the partial charges of N-methylimidazole combined with RDE and NMR results, they show that Me–N1C2 (1,2-dimethylimidazole) exhibits the most negative N3 (pyridinic nitrogen of N-methylimidazole) and the strongest N3–H2O bond leading to the highest exchange current density (i0) (Fig. 2D). Furthermore, the authors conducted a combination of in situ attenuated total reflectance surface-enhanced infrared reflection absorption spectroscopy (ATR-SEIRAS) and AIMD to understand N-methylimidazole's interaction with interfacial water. In situ ATR-SEIRAS spectra for Me–N1C2-free electrolyte demonstrate water reorganization from the H-up, proton donor position to the H-down, proton acceptor position due to the negatively charged Pt surface. Furthermore, when adding Me–N1C2, the spectra do not exhibit changes indicating that the strong H-bonded interfacial water near the Pt remains intact.
AIMD simulations were conducted on the Pt(100)–water interface with and without Me–N1C2. Without the Me–N1C2, the water molecules closest to the Pt (first layer) were organized in the H-down orientation (denoted as: H2O(H-down)1st) due to the negatively charged surface and the O–H dipole. These H2O(H-down)1st (proton acceptors) form H-bonds with H2O(H-down)2nd (proton donors) and therefore construct the interfacial H-bond network. When Me–N1C2 was introduced, the energetically favorable binding configuration was the parallel binding of the imidazole ring. In this configuration, the H2O(H-down)2nd is an H-bond donor to both the H2O(H-down)1st and the N3 (Fig. 2E). Metadynamic simulations show that, as potential is applied in the absence of Me–N1C2, the H2O(H-down)1st adsorbs to the surface and dissociates and the generated hydroxide is chemisorbed onto the surface. When Me–N1C2 is introduced, the hydroxide that is generated is hydrogenated by the H2O(H-down)2nd that is bound to Me–N1C2. This replenishes the H2O(H-down)1st as indicated by a decrease in the H-bond length between H2O(H-down)1st and H2O(H-down)2nd (Fig. 2F and ‘b’ in 2G). Thereafter, the hydroxide continues diffusing through the Grotthuss mechanism. The catalytic enhancement observed with Me–N1C2 was attributed to its ability to bring the second layer of water molecules closer to the surface through its interactions, thus facilitating hydroxide diffusion within the water network.
There have been other views on the effect of cations on the H-bonding network at the electrode–electrolyte interface. Li et al. performed AIMD simulations to illustrate the differences between the structure of the EDL in acidic and alkaline media.34 They found that the closest ion plane (CIP) in alkaline media is closer to the electrode surface and has a higher cation concentration than in acidic media. The crowded cations in alkaline media lose their solvation shell and cause a water gap above the CIP. This gap, in turn, disrupts the H-bond network and therefore disrupts the proton “highway” decreasing HER activity. Theoretical vibrational density of states (VDOS) calculations and in situ SEIRAS further reveal the scarcity of H-bond networks in the gap region. The authors conclude that the discontinuity in the H-bonding network in alkaline media due to cation presence plays a large role in the sluggishness of HER. Furthermore, the addition of a more oxophillic Ru to the Pt surface promotes OH adsorption which decreases the water gap and facilitates H-bonding connectivity thereby improving HER activity.
A follow-up work by Su et al. explored the pH effect of HER/HOR on various metal catalysts.42 Instead of a decrease in HER/HOR rates with increasing pH, they observed an inflection point where the rates of HER/HOR begin to increase once more after a specific pH. Using a triple-path microkinetic model, it was revealed that the formation of OHad promotes the HOR/HER kinetics by improving the H-bond network in the EDL instead of decreasing intermediate reaction energy barriers. The authors claim that since the electrode is more negatively charged in HER/HOR potential regions at higher pH (i.e., higher pzfc at higher pH), the surface becomes crowded with cations which in turn weakens the H-bonding between the water molecules close to the surface and the ones further away in the EDL. The presence of OHad, however, reduces the strength of cation–water interactions due to OHad–cation interactions and regenerates some of the H-bonded water networks. This effect of OHad would be more pronounced on catalysts with stronger hydroxide binding energies and such was experimentally observed for more oxophillic catalysts.
It is evident that the structure of interfacial water critically affects the HER in diverse media and on various catalyst materials. Despite all research efforts, the precise mechanism by which interfacial water affects HER (or HOR) remains a topic of ongoing debate. More research is needed to develop a comprehensive understanding of how interfacial water molecules and their collective structure influence HER and to be able to control this environment for specific applications.
2.2. Carbon dioxide reduction
Numerous variables have been reported to affect the electrocatalytic reduction of CO2.43 Those include the bulk and local pH and the electrolyte composition and concentration. In this section, however, we focus on the effects of interfacial water on the CO2RR. Typically, research efforts have focused on introducing ionic or organic additives to the electrolyte which create a hydrophobic interfacial environment. This decreases water diffusion to the catalyst and breaks down the water network hindering HER and boosting CO2RR.44,45 While these are the main effects of interfacial water discussed in the literature, it is also important to note that water molecules play other roles regarding the CO2RR, such as enhancing the stability of key intermediates.46Mohandas et al. performed in situ SEIRAS experiments together with linear sweep voltammetry to reveal how organic additives such as DMF break down the water structure and the H-bonding network impeding HER, a competing reaction of CO2RR.44 Through a negative shift in onset potentials, the authors demonstrated the universality of DMF's role in the suppression of HER for various catalyst materials in argon-saturated electrolytes. However, in CO2-saturated electrolytes, DMF positively shifted the onset potential for the CO2RR and produced a CO faradaic efficiency (FE) of 94% on a polycrystalline Au electrode when 15 mol% DMF was introduced into the electrolyte. Through in situ SEIRAS and the deconvolution of O–H stretching bands, it was revealed that DMF accumulates at the interface which in turn modifies interfacial water structure by creating H-bonding interactions between the O and N of DMF molecules and interfacial water. The authors concluded that DMF-induced disruption in interfacial water structure as well as water repulsion from the interface are the main reason for HER inhibition and an increase in CO selectivity.
Ge and coworkers studied potential-driven dynamics of the interfacial microenvironment with quaternary ammonium cationic surfactant additives for the co-reduction of CO2 and H2O.47 It was revealed through EIS phase angle measurements that the structure of these surfactants on the surface of electrodes dynamically changes with an increase in applied potential from a random distribution to a nearly ordered assembly. Furthermore, cetyltrimethylammonium bromide (CTAB) showed the highest double layer capacitance (Cdl) indicating that longer alkyl-chain surfactants are easier to adsorb. With CTAB as an electrolyte additive, various types of silver catalysts were tested for CO2 reduction and all were found to gain in CO selectivity over H2. In situ SEIRAS and in situ surface enhanced Raman spectroscopy (SERS) were utilized to probe the interfacial water structure in the system with and without CTAB. In the system with CTAB, the formation of a hydrophobic microenvironment was found. Furthermore, the presence of CTAB did not allow water molecules to reorient into the two H-down, proton acceptor, ordered structure. Finally, through molecular dynamics (MD) simulations, it was found that there are fewer water H-bonds in a system with CTAB than in a system without CTAB. The theoretical and experimental results demonstrate that these surfactants adsorbed to the surface create a highly hydrophobic–aerophilic environment which increases the selectivity of CO2 to CO while simultaneously decreasing the reactivity of water dissociation and therefore HER activity.
Since the CO2RR occurs in competition with the HER, most literature discusses the effect of additives from the perspective of creating a hydrophobic interfacial environment repelling water molecules and hindering HER while simultaneously bolstering CO2RR. However, there has been limited research on how water molecules themselves could in fact promote CO2RR. Meng et al. explored the effect of interfacial water molecules on the CO2RR activity and selectivity using DFT and AIMD simulations with boron-doped bismuth catalysts (B@Bi and B2@Bi).46 From partial density of states (PDOS) calculations, the peaks of C 2p, O 2p, B 2p, and Bi 5d orbitals were found close to the Fermi level in aqueous electrolytes. These results indicate that water molecules exhibit promoter effects on CO2 activation through H-bonding as well as enhance the CO2 adsorption energy. Furthermore, it was found that the presence of water molecules affects the thermodynamic potential determining step (PDS). Boron-doped bismuth demonstrated lower overpotential requirements for the production of methane in a system that includes 30 H2O than the gas-phase system. Similar results were also found for the production of ethylene. The authors attribute this to the H-bond interactions of water molecules with key reaction intermediates.
3. Thermodynamics and kinetics of reaction pathways influenced by cations
Alkali metal cations (AMCs) have been shown to exist in a coordinated hydrated environment within the electrolyte.9 Many characteristics of the AMCs, such as the charge density (i.e. cation size) and the hydration degree and energy, have a significant effect on various electrochemical reactions such as the HER/HOR, OER/ORR, and the CO2RR.9 The exact mechanism by which AMCs affect these different reactions varies from reaction to reaction. Thermodynamically, AMCs alter intermediate stabilization via direct non-covalent and indirect electrostatic interactions.22,23 Kinetically, AMCs alter solvent reorganization energies to facilitate fast charge transfers.48 In this section, we describe the effects of AMCs on the reaction rates and selectivity of HER/HOR, OER/ORR, and CO2RR, as well as their underlying mechanisms.3.1. Hydrogen evolution reaction
A study by Goyal et al. suggested that cations play a crucial role in stabilizing the transition state of the rate-determining Volmer step (H2O + e− + * → *H + OH−) through interactions with the dissociating water molecules (*H–OHδ–M+).53 They found that HER activity is enhanced on Au electrodes as the cation concentration increased at moderately alkaline conditions (Fig. 3A). Also, chronoamperometry studies reveal that as pH increases, the current at each overpotential also increases and Tafel slopes decrease indicating activity enhancement. They attribute this behavior to the indirect effect of increased pH leading to an increase in the near surface cation concentration as a result of the change in the local field strength. However, there is a volcano-type relationship where HER activity diminishes at very high pH and cation concentrations due to blockage of the catalyst surface (Fig. 3B). Thus, it seems that there is an optimal concentration of cations for promoting HER through the stabilization of transition state during water dissociation.
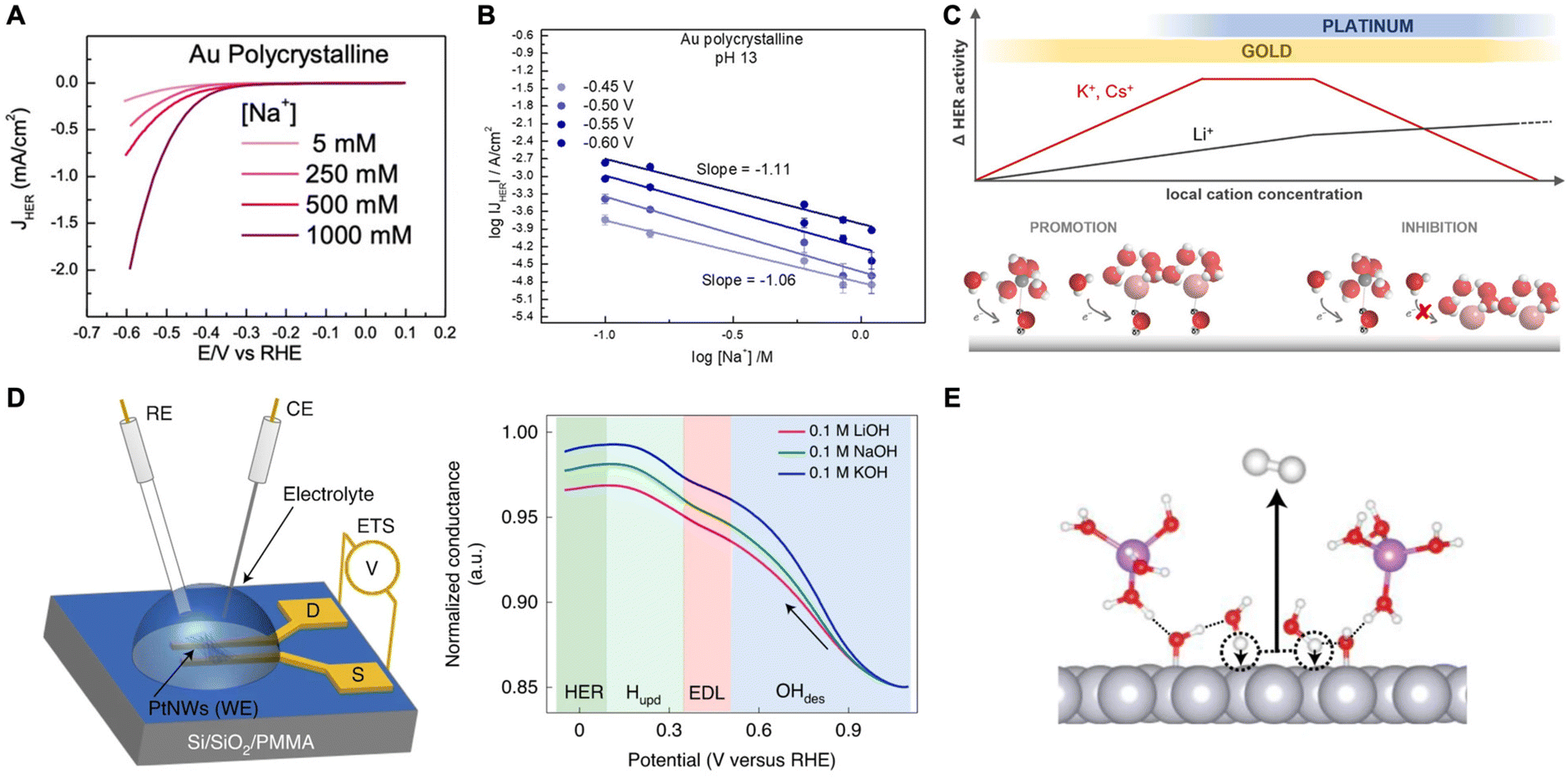 | ||
| Fig. 3 (A) Cyclic voltammograms obtained for HER on a polycrystalline Au electrode in an alkaline environment (pH 11) at different Na+ concentrations. (B) Reaction order plot of HER rates as a function of Na+ concentration at pH 13 showing an inhibition of HER. Reproduced from ref. 53 with permission from John Wiley and Sons, copyright 2021. (C) Schematic representation illustrating the effects of local pH and AMC concentration on HER activity on Au and Pt electrodes. Reproduced from ref. 54 with permission from the American Chemical Society, copyright 2021. (D) Schematic of Pt nanowires for ETS measurements, where RE, CE, WE are the reference, counter, and working electrodes, respectively, and S and D are the source and drain terminals, respectively (left). Normalized ETS conductance plot as a function of potential showcasing the behavior of ions at the different regimes (right). (E) Schematic demonstrating the promoting effects of OHads on the alkaline Volmer step. Reproduced from ref. 55 with permission from Springer Nature, copyright 2022. | ||
A follow-up study was conducted on Au and Pt electrodes to further elucidate the cation effect on HER.54 Using Tafel analysis coupled with cyclic voltammetry (CV) on gold, they found that weakly hydrated cations such as K+ increase HER rates on Au catalysts, but only at low overpotentials (i.e., low local alkalinity, low pH). At higher overpotentials (i.e., high near-surface AMC concentration), the AMCs become detrimental due to overcrowding at the outer Helmholtz plane (OHP) further confirming the previous trend.53 However, with Pt electrodes, HER inhibition by weakly hydrated cations was observed already at low pH conditions suggesting strong Pt–AMC interactions. The authors proposed a model to explain the aforementioned inverted trend between Pt and Au (Fig. 3C). At low local pH and AMC concentration (i.e. a promotion regime), weakly hydrated cations better stabilize the transition state of water dissociation. However, these cations tend to interact with the surface more which then turns their promoting effects into hindering ones at higher local pH and AMC concentrations. In the inhibition regime, a negative reaction order is observed with increased cation concentration. Such is the case for Au. However, for Pt which interacts more strongly with cations, HER occurs mostly in the inhibition regime where strongly hydrated Li+ is favored over weakly hydrated K+ or Cs+.
In another study, Shah et al. utilized a combination of EIS and electrical transport spectroscopy (ETS) to study the effect of near-surface AMCs on HER rates on Pt catalysts in alkaline media.55 As stated previously, the authors show that the smaller cations better stabilize the adsorbed hydroxide species in the H-UPD region favoring high OHad coverage on the Pt surface. Moreover, due to the high polarity of OHad, water dissociation is easier to occur, therefore boosting the Volmer-step kinetics and HER activity. From ETS analysis, they concluded that the lowest conductance was at the OHad regime due to the strongly bonded OH on the Pt surface reducing the conductance of Pt nanowires (Fig. 3D). As the potential was scanned negative, the conductance increased monotonically, first due to the replacement of OH on the surface with water molecules (EDL regime) and thereafter the replacement of water molecules and residual OH molecules with adsorbed hydrogen (H-UPD regime). Eventually, conductance levels off due to the saturation of the electrode surface with Hads in the HER region. This trend was observed for all three different electrolytes with different AMCs (Li+, Na+, and K+). However, the one with Li+ showed the least increase in conductance indicating that solvated Li+ stabilizes the OH adsorbates more than other cations. The authors further utilized DFT and AIMD simulations to confirm this trend, together with capacitance measurements that exhibit values following Li+ > Na+ > K+ at the HUPD and HER regime related to the remaining adsorbed OH. Furthermore, it was shown that adsorbed hydroxides act as H-bond acceptors/donors and therefore stabilize the near-surface water molecules decreasing their barrier for dissociation (Fig. 3E). These results indicate that HER activity is dependent on the dissociation of water and the trend of Li+ > Na+ > K+ is due to AMC's influence on the amount of adsorbed hydroxides on Pt.
3.2. Oxygen evolution and reduction reactions
AMCs have been shown to also affect OER rates for RuO2 (110) electrodes in alkaline solutions.23 The trend was similar where larger cations (e.g., K+) resulted in higher OER activity compared to smaller cations (e.g., Li+). Rao et al. explained that structure-breaking larger cations with less compact hydration shells lead to more isolated water molecules in the surroundings. This leads to a more reactive interfacial OH− not stabilized by icelike water structures. Furthermore, the water molecules within the hydration shell of a larger cation are less acidic (i.e., large pKa) with reduced interactions for stabilizing adsorbed oxygen intermediates. These effects combined improve the kinetics of the rate-limiting step of Oads + OH− → OOHads + e− (Fig. 4A). Thus, in this case, the improved OER activity by larger AMCs is caused by the destabilization of a key OER intermediate.
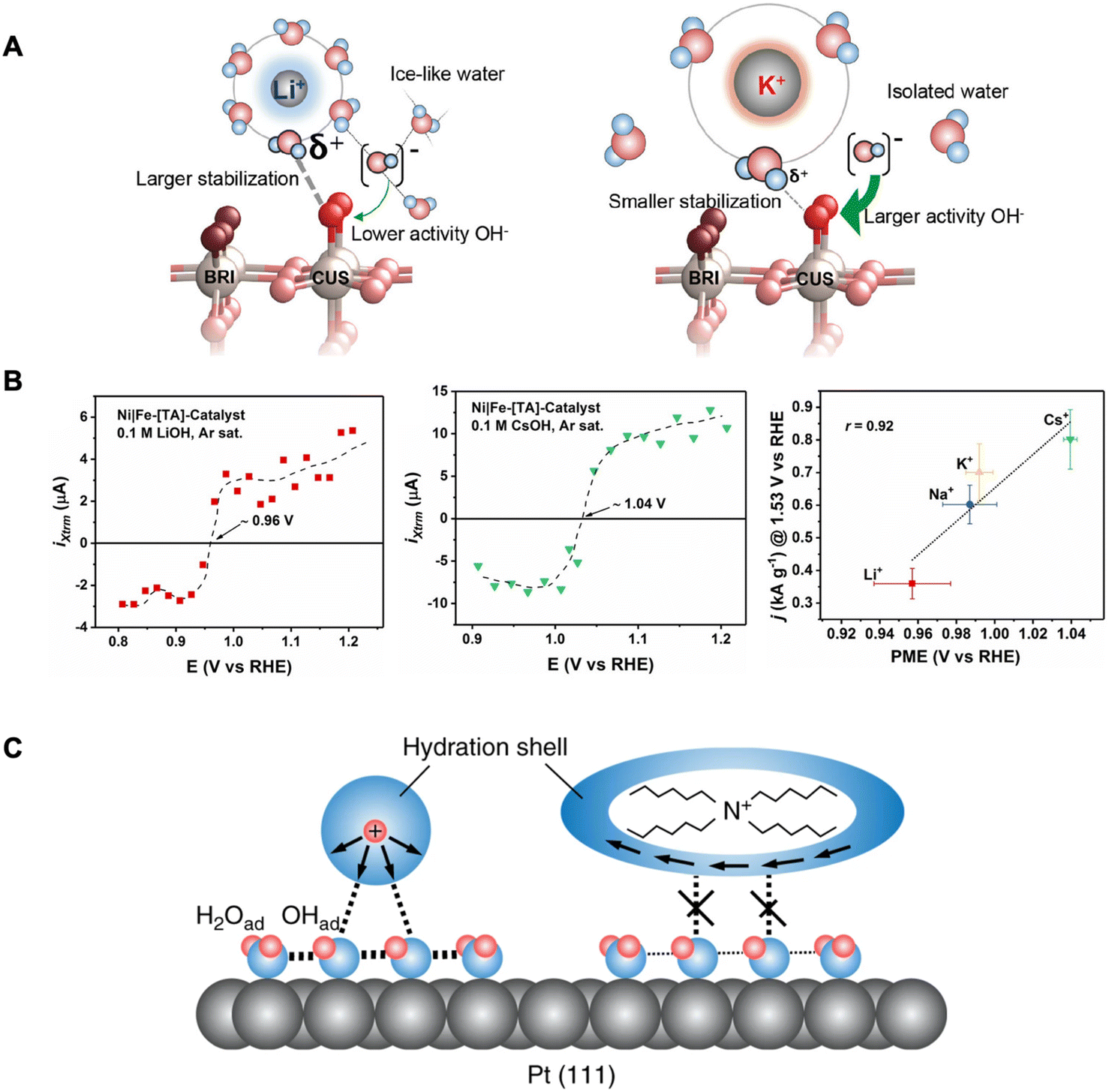 | ||
| Fig. 4 (A) Schematic of the RuO2(110) electrified interface in Li+-containing electrolyte (left) and K+-containing electrolyte (right). Reproduced from ref. 23 with permission from the American Chemical Society, copyright 2021. (B) LICT data demonstrating the current transients as a function of applied potential for different AMCs (left and middle) and electrocatalytic activity dependence on the PME in the presence of different AMCs. Reproduced from ref. 25 with permission from John Wiley and Sons, copyright 2022. (C) Schematic of the interfacial environment in the presence of hydrophilic and hydrophobic cations. Reproduced from ref. 60 with permission from Springer Nature, copyright 2018. | ||
With the same goal of elucidating how cations can affect ORR, Kumeda et al. studied the ORR activity on single-crystal Pt surfaces in acidic electrolytes, however with the presence of organic hydrophobic tetraalkylammonium (TAA) cations with varying alkyl chain lengths.60 Their immediate observations were that the longer alkyl chain (n) TAA cations (i.e., higher hydrophobicity), the higher the ORR activity following the trend: THA+ (n = 6) ≫ TBA+ (n = 4) > TEA+ (n = 2) > TMA+ (n = 1) ≈ HClO4. Furthermore, in situ IR and X-ray CTR measurements demonstrate that in the presence of THA+, less OHads coverage is found on Pt(111). They reason that hydrophobic cations such as THA+ destabilize the OHads which is known to hinder ORR.64 This occurs by THA+ strengthening the hydrogen bonding in the primary hydration shell and restricting their coordination to species outside this shell (Fig. 4C). This weakens the interaction between the hydration shell around the THA+ and OHads layer (with co-adsorbed H2O) resulting in a more efficient interface for ORR.
3.3. Carbon dioxide and carbon monoxide reduction
 | ||
| Fig. 5 (A) Schematic of hydrated cation electrostatic interactions with intermediates having large dipole moments (left), the effect of cation size on various product formations (right). (B) The energy required to bring different solvated cations from the bulk to the OHP at the Cu(111) facet. Reproduced from ref. 67 with permission from the American Chemical Society, copyright 2017. (C) Schematic of the electrode–electrolyte interface showing the accumulation of hydrated K+ at the OHP. Reproduced from ref. 68 with permission from Springer Nature, copyright 2022. | ||
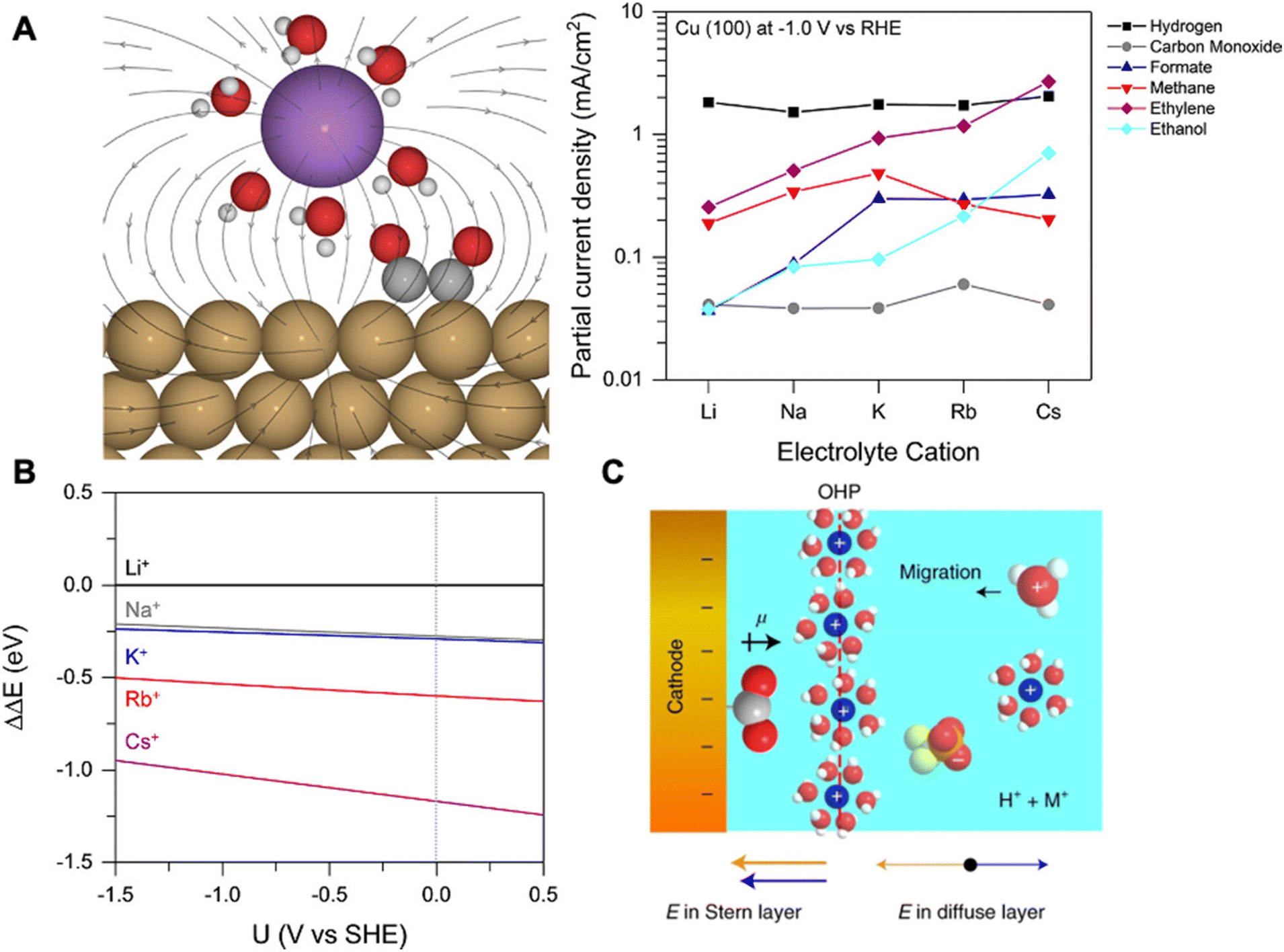
Ringe et al. applied a multi-scale comprehensive model combining ab initio and continuum electrolyte models to understand the cation effects of CO2RR.69 Employing a modified Poisson Boltzmann method within the continuum framework, the authors observed significant alterations in the surface charge density and associated electric fields due to repulsive interactions among hydrated cations in the Helmholtz layer. Specifically, they found that weakly hydrated cations (e.g., Cs+) with smaller hydrated cation radii exhibit weaker repulsion and accumulate at the OHP at a higher concentration. This induces a higher surface charge density and a stronger electric field, leading to enhanced CO2RR. Their model closely matched the experimental trends seen for Ag and Cu electrodes in CO2RR.
In another work, Gu et al. elucidated the cation effects on CO2RR in highly acidic environments (pH = 1.0) with SnO2/C, Au/C, and Cu/C nanoparticle catalysts.68 The authors demonstrate that hydrated alkali cations hinder hydronium ion migration decreasing HER while simultaneously promoting CO2RR through reaction intermediate stabilization. In the presence of K+ in acidic electrolytes, HER was surprisingly well suppressed with formic acid (max FE 88%) and CO (max FE 91%) as major products for SnO2/C, and Au/C, respectively. For Cu/C, a plethora of products were detected, including formic acid, CO, methane, ethylene, and ethanol, with ethylene FE as high as 25%. Simulations based on the Poisson–Nernst–Planck (PNP) model showed that hydrated K+ and hydronium ions compete for adsorption creating a chemically inert hydrated K+ layer formed at the OHP (Fig. 5C). This layer shields the electric field from the cathode, therefore suppressing hydronium ion migration and HER. Moreover, the key intermediates for CO2RR (e.g., *CO2, *OCCO) are stabilized by dipole-field interactions between the intermediate dipole (pointing away from the catalyst surface) and electric field created by cations within the Stern layer (pointing towards the catalyst surface) enhancing CO2RR (Fig. 5C). Furthermore, the authors reached a similar conclusion where the increase in intrinsic cation size (e.g., Cs+) results in a higher concentration of cations at the OHP and the stronger stabilization of those intermediates.
 | ||
| Fig. 6 (A) Cathodic and anodic scans recorded in argon and CO2 atmospheres with different concentrations of Cs+. (B) Schematic demonstrating the interaction of the cation with the reaction intermediate (*CO2−) as well as a reaction mechanism for the CO2RR. Reproduced from ref. 65 with permission from Springer Nature, copyright 2021. (C) Schematic demonstrating the replacement of methyl4Nads by specifically adsorbed K+. (D) Coverage of specifically adsorbed AMCs at −0.45 V vs. RHE measured by integrating the area of the 1482 cm−1 band as a function of bulk AMC concentration. Reproduced from ref. 66 with permission from Springer Nature, copyright 2022. | ||
Qin et al. reached a similar conclusion demonstrating cation-coordinated inner-sphere CO2 reduction by studying the effects of AMCs on CO2RR on Au surfaces using atomic-level simulations.70 Through slow-growth (SG) AIMD methods, the authors generated a comprehensive free energy diagram for the CO2RR for the first time. It was observed that, in the presence of K+ at the Au–water interface, CO2 reduction is favorable with the CO2 activation step only exhibiting a 0.66 eV barrier by cation coordination. Simultaneously, the competing HER is significantly suppressed, displaying a 1.27 eV barrier in the rate-limiting Volmer step (H2O + e− + * → *H + OH−). Furthermore, the simulations confirmed that *COatop (linearly bonded CO) is the active intermediate in CO2 electroreduction while *CObridge (bridge bonded CO) is a kinetically inert spectator.
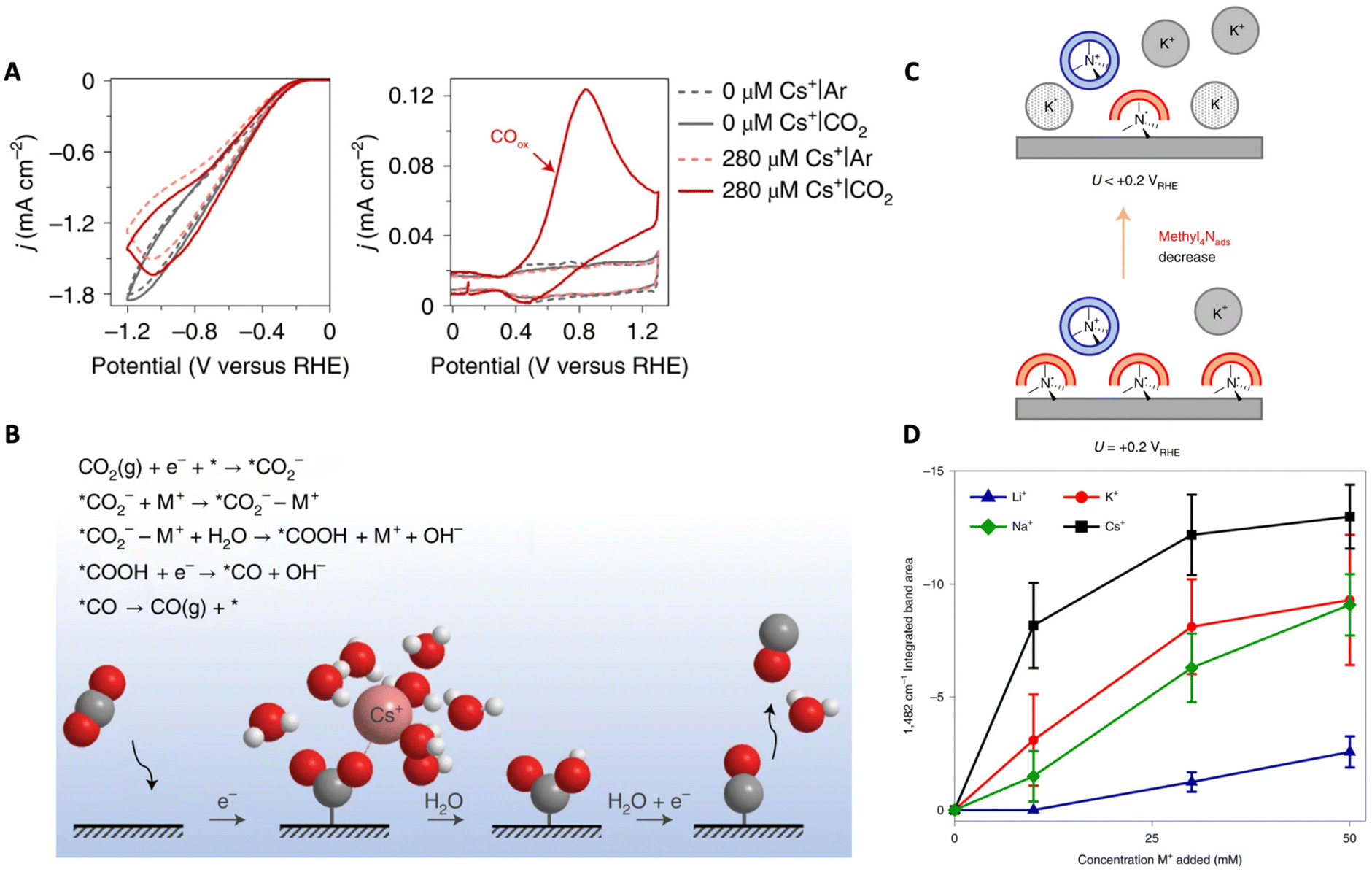
Partial dehydration and specific adsorption of AMCs at the interface have been described in other works as well. Ovalle et al. studied the effect of AMCs on the CO2 reduction to CO on polycrystalline Au by using tetramethylammonium (methyl4N+) as a vibrational probe.66 Using ATR-SEIRAS and probing the asymmetric deformation band of CH3 of methyl4N+ at the Au–electrolyte interface, the authors showed that, with an increase in K+ concentration, a negative band attributed to the non-hydrated specifically adsorbed methyl4N+ (1482 cm−1) grows due to their displacement by K+ (Fig. 6C). Using this band as a spectroscopic measure for the coverage of specifically adsorbed cations, the Li+ < Na+ < K+ < Cs+ trend was found (Fig. 6D). Furthermore, they were able to associate the degree to which these cations displace methyl4Nads (i.e. degree of cation coverage) with their free energies of hydration. This observation demonstrates that metals with a soft hydration shell have a higher tendency to specifically adsorb on the electrode surface. Further, the partial current density for CO was shown to increase with increasing cation coverage. Thus, the authors suggest that direct cation–intermediate interactions may be possible with these specifically adsorbed cations.
Shin et al. employed density functional theory in a classical explicit solvent (DFT-CES) to study the interactions of AMCs with key intermediates of CO2RR.71 From these calculations they identified 6 crucial intermediates (*CO2, *COOH, *CHO, *OCCO, *OCCOH, and *HOCCOH) that can be coordinated to a cation during the production of C1 and C2 products such as CO, CH4, and C2H4. They also performed electrochemical measurements which revealed that the activity trend for CO and C2H4 formation on electrodes like Ag and Cu follows the order: Cs+ > Rb+ > K+ > Na+ > Li+, while for CH4 formation the trend is the opposite. These results coupled with results by Chan and coworkers demonstrating the dependence of CO2RR products on pH,72 the authors suggest that the rate-determining step (RDS) for CO and C2H4 formation involves a cation-coupled electron transfer (CCET), while the RDS for CH4 formation involves a proton-coupled electron transfer (PCET). The variation in cation-dependent activity is attributed to the differing stabilization capabilities of various AMCs as mentioned in previous studies. Furthermore, the authors demonstrate first-order kinetics on the surface charge density for CO and C2H4 formation which is in line with what is expected assuming the cation-coupled electron transfer RDS.
As part of this effort, Kim et al. demonstrated a novel approach, so-called the nanoparticle/ordered-ligand interlayer (NOLI), which almost exclusively reduces CO2 to CO due to the cation effect.73 NOLI contains Ag nanoparticles which have tetradecylphosphonic acid (TDPA) ligands “floating” in the vicinity under negative bias. At the interlayer between the Ag NP surface and ligand, CO2RR to CO occurs with approximately 97% CO selectivity as well as 2 orders of magnitude increase in intrinsic activity when compared to polycrystalline Ag foil. Utilizing X-ray absorption near edge structure (XANES) and AIMD simulations, the authors showed the presence of dehydrated cations (e.g., K+) within the interlayer where the phosphonate group (from TDPA ligands) anchors the dehydrated cation. Through the first-principles free energy calculations, this structure was found to facilitate the activation of CO2 by the dehydrated cation. Furthermore, even with strongly solvated cations such as Li+ which is normally not favored for CO2RR, having the NOLI structure allowed achieving a 70% CO selectivity (in contrast with the Ag foil only at 3%). Similar improvements were demonstrated with other metals, such as Au and Pd. While it is evident that AMCs affect the rates and selectivity of CO2RR, their impacts could be strengthened by partially removing their hydration shells and allowing direct interaction with intermediates.
Building upon this previous study, Ayemoba et al. experimentally studied the buffering effect by probing the pH at the Au–electrolyte interface using ATR-SEIRAS.75 They managed to probe the pH through the inversely proportional relationship between H+ concentration and the ratio of CO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HCO3− peak integrated intensity. The authors observe an increase in pH during CO2RR regardless of the AMC. However, with larger AMCs, the pH increased minimally supporting the earlier work by Singh et al.74
:HCO3− peak integrated intensity. The authors observe an increase in pH during CO2RR regardless of the AMC. However, with larger AMCs, the pH increased minimally supporting the earlier work by Singh et al.74
Zhang et al. used a rotating ring-disc electrode (RRDE) with an Au disk and Pt ring to detect local pH changes during the CO2RR.76 They observed a −86 mV pH−1 shift in the CO (which is a product of the Au disk) oxidation peak by the Pt-ring which they used to directly probe the pH changes near the electrode surface. Utilizing this technique, they found that local basicity decreased with the cation identity following the trend: Li+ > Na+ > K+ > Cs+ in agreement with previous studies.74,75 They also concluded that larger cations having larger buffering capability result in a lower local pH and therefore enhance CO2RR.
Even though cationic pH buffering arguments might seem reasonable, latest studies suggest that it might not be the case. For example, Monteiro et al. argue that AMC pH buffering does not play a major role in the enhancement of the CO2RR.65 They suggest that, if local pH effects are key, the CO2RR should still take place, even in the absence of cations, in a pH = 3 solution. However, they find that the absence of AMCs in fact eliminates the CO2RR from taking place at all, which contradicts the pH buffering theory and supports other theories for the cation effects.
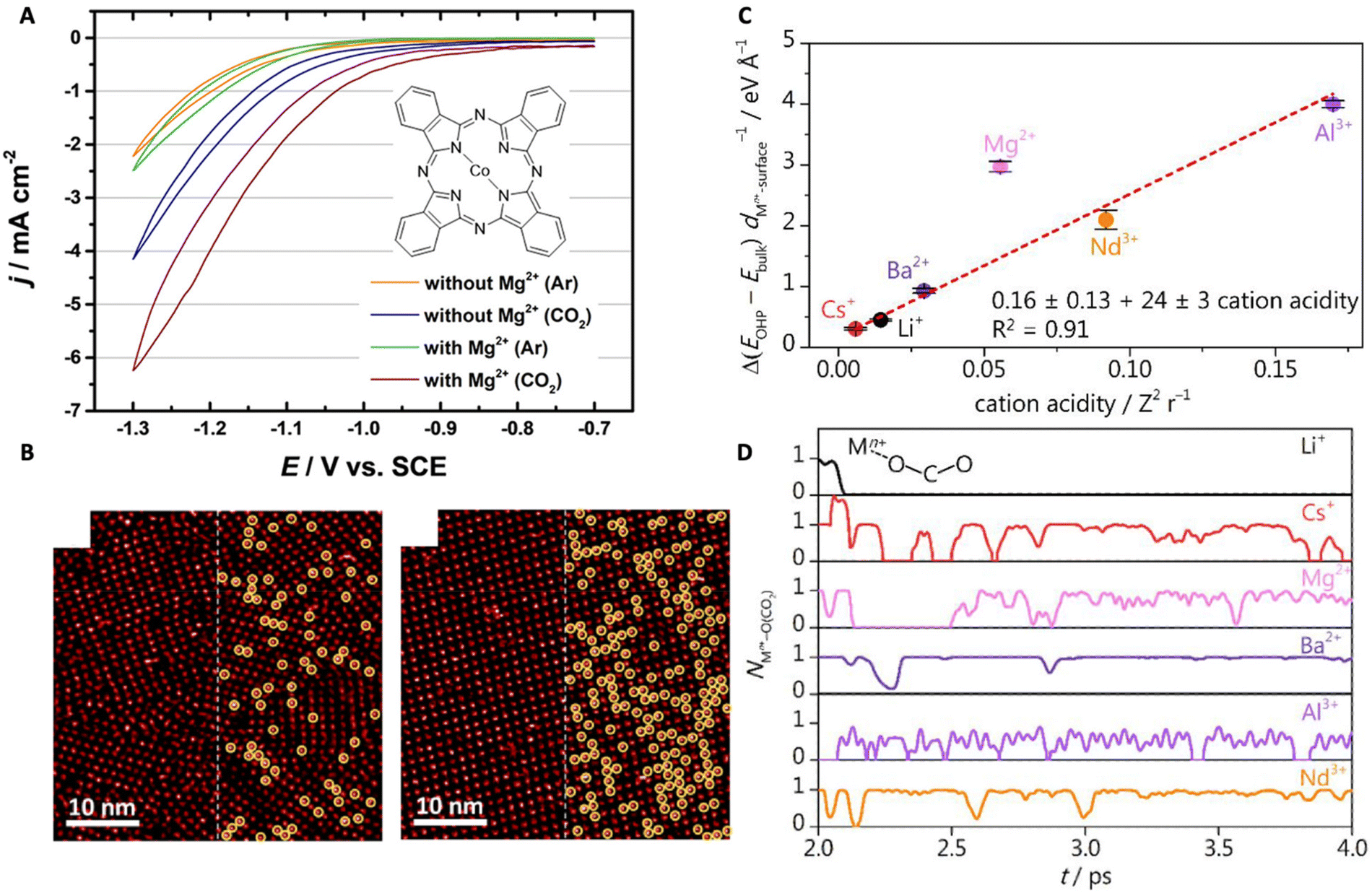 | ||
| Fig. 7 (A) Cyclic voltammograms of the CoPc-modified Au(111) electrode showing the effects of Mg2+ on CO2RR (the inset shows the chemical structure of the CoPc molecule). (B) STM images of the self-assembled monolayer of CoPc on the gold surface in the CO2 environment at different concentrations of Mg2+: 0.005 M Mg2+ (left), 0.03 M Mg2+ (right). Reproduced from ref. 77 with permission from the American Chemical Society, copyright 2022. (C) The relationship between the thermodynamic driving force for cation accumulation and cation acidity. (D) AIMD simulation demonstrating the coordination between the cation and CO2. Reproduced from ref. 24 with permission from the American Chemical Society, copyright 2022. | ||
Monteiro et al. studied the effects of various cations, that are AMCs and multivalent cations, on the CO2RR in a mildly acidic (bulk pH 3) condition.24 The authors found that strongly acidic cations (e.g., Ce3+, Nd3+) only favor CO2RR at low overpotentials till they start to undergo hydrolysis (i.e., at high overpotentials where the local pH shifts basic) favoring HER. At high overpotentials, a higher CO activity was observed for electrolytes containing less acidic cations, such as Cs+, Li+, Ba2+, and Ca2+. A quantitative comparison among them led to a CO activity trend that is Ca2+ < Li+ < Ba2+ < Cs+. This complex behavior is due to the various factors that are involved in cation effects. AIMD simulations show that softly hydrated cations accumulate at the OHP at a higher concentration, and the lower the acidity, the larger their accumulation for intermediate stabilization (Fig. 7C). This is the reason why Cs+ and Ba2+ are better for CO2RR. However, for the short-range coordinative interaction between the cation and *CO2− that facilitates CO2 activation, trivalent cations (e.g., Nd3+) are more effective intrinsically (Fig. 7D). However, they also promote water dissociation at basic pHs leading to faster kinetics for HER at high overpotentials and thus, were only effective for CO2RR at low overpotentials.
Pérez-Gallent and coworkers explored the effects of AMCs on the CORR on Cu single crystals where they showed that the surface structure of Cu as well as the applied potential influence the cation effects.78 From online electrochemical mass spectrometry (OLEMS) and high-performance liquid chromatography (HPLC), they showed that cation effects are potential-dependent. Larger cations (e.g., Cs+) increase the selectivity towards ethylene production at low overpotentials while methane is favored at more negative potentials, a trend found in all Cu structures. The authors also showed that the cation effects are structure sensitive with the onset potential for ethylene formation most positive on Cu(100). The RDS for CO to methane or ethylene formation on Cu(100) is CO hydrogenation, and this process is significantly more efficient when alkaline cations are present for a CO dimer (to *OCCOH) as opposed to a CO monomer (to *CHO) based on DFT. Furthermore, FTIR studies showed the presence of a hydrogenated dimer intermediate (*OCCOH) at low overpotentials. The formation of this intermediate depends on cation size as it is detected in the presence of Li+, Na+, and K+, but not in the presence of Rb+ and Cs+. DFT calculations explained the FTIR observations suggesting that the potential necessary to form *OCCOH from *CO in the presence of Cs+ is more negative compared to smaller cations such as Li+. Moreover, the activation energy required for any intermediate formation (*CO, 2*CO, *OCCO, *CHO, and *OCCOH) is decreased in the presence of cations highlighting the intermediate stabilization effects of AMCs. Overall, these results demonstrate the clear effect of AMCs on the CORR with larger cations tending to promote the pathways with intermediates more effectively than smaller cations.
Similarly, Malkani et al. studied the effects of AMCs on the CORR on polycrystalline Cu in alkaline media.79 The authors demonstrated that larger AMCs (not considering the hydration shell size) promote CO reduction on Cu(poly) in alkaline media. They observed a trend where the rates of both CORR and HER increase with cation size. Moreover, the FE for CORR products increased with increasing AMC size. In situ SEIRAS investigations showed that the CO adsorption distribution varies with different cations: the ratio of CO adsorbed on step sites to CO adsorbed on terraces increased from Li+ to K+ then leveled off. This indicates the existence of an interaction between AMCs and the CO adsorbed. They also measured Stark tuning rates (indicative of EDL electric field strength) which exhibited the following trend: Cs+·xH2O ∼ Rb+·xH2O ∼ K+·xH2O > Na+·xH2O > Li+·xH2O. This suggests that the thickness of the double layer is thinner for cations like K+ compared to Li+. Thus, the interfacial electric field seems mostly responsible for the enhanced CORR activity from Li+ to K+. However, such an effect plateaus beyond K+ while the CORR continuously improves suggesting a non-electric field (NEF) component within the cation effect on CORR. Further investigations with cations such as the K+ chelated crown ethers were conducted to show how the electric and NEF components of the cation effect could be affected by the chemical structure and charge distribution of cations.
4. Anion effects on electrocatalytic reactions
4.1. Water electrochemistry (HER/HOR/OER/ORR)
It is also of great importance to understand how anions affect the ORR activity as future technologies such as proton exchange membrane fuel cells (PEMFCs) utilize Nafion membranes which are comprised of sulfonate groups. Nafion membranes are essential in these systems as they provide a proton conductive path as well as act as a binder in the catalyst layer.84 Therefore, understanding how anions influence ORR is essential to be able to design efficient devices, such as fuel cells and metal–air batteries, for future sustainability applications.
Jusys and Behm conducted an in-depth study on the effects of anions on the ORR on annealed gold films in different electrolytes such as sulfuric and perchloric acids as well as sodium hydroxide.86 The authors observed ORR activity inhibition due to the adsorption of the anionic species onto the surface of the electrode and limiting the number of active sites. In general, ORR activity followed the trend: NaOH > HClO4 > H2SO4 as evidenced by a more positive onset potential in alkaline media as well as in perchloric acid solutions compared to sulfuric acid electrolytes. Further results from potentiodynamic studies revealed that in acidic media the ORR proceeds towards the formation of superoxides while in alkaline media the reaction favors the 4e− pathway at high overpotentials. The enhanced activity and selectivity observed in the 4e− pathway under alkaline electrolyte conditions is due to a densely packed OHads layer. This adlayer may serve as a scaffold for the ORR, potentially through an outer-sphere ORR process. In contrast, at lower potentials and, thus, diminished OHads coverages, the reaction seems to progress through the interaction between O2,ads and the Au site.
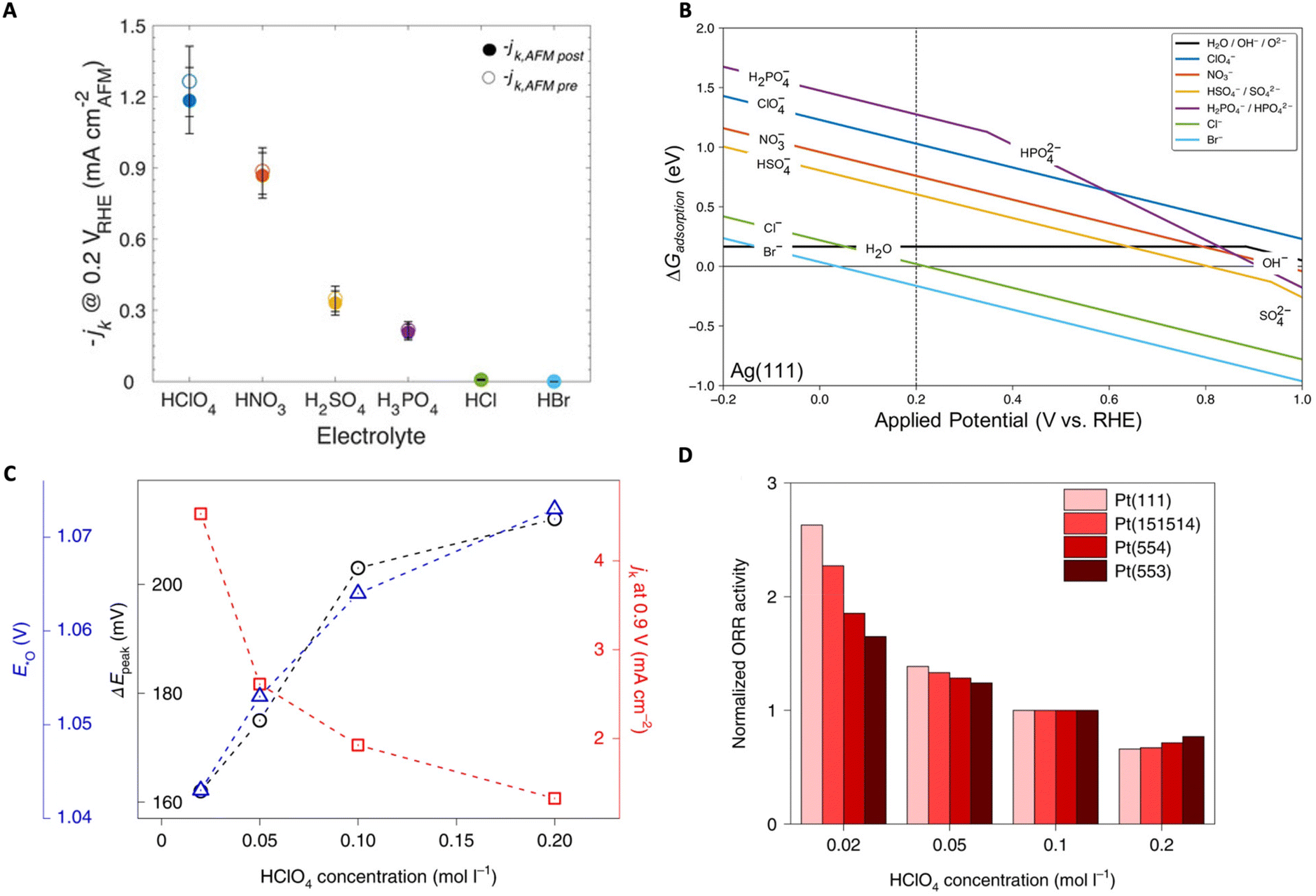
In another study, Zeledón et al. combined electrochemical and physical characterization, revealing the different effects of acidic electrolyte anions on ORR activity and selectivity on Ag and Pd catalyst thin films.83 Through CV and Tafel slope analysis, they revealed that on Ag (weak oxygen binding) the ORR activity trend follows: HClO4 > HNO3 > H2SO4 > H3PO4 > HCl ≫ HBr, and on Pd (strong oxygen binding) the trend follows: HClO4 > H2SO4 > HNO3 > H3PO4 > HCl ≫ HBr (Fig. 8A). The authors claim that the observed trends are related to competitive anion adsorption as well as non-covalent interactions. To understand these effects, the authors conducted DFT calculations of the adsorption free energy of each anion. From these calculations they found that Cl− and Br− were chemisorbed to the surface while the anions NO3−, HSO4−, SO42−, H2PO4−, and HPO42− may physisorb onto the surface (Fig. 8B). Their results indicate that, in the presence of the two halides (Cl− and Br−), surface active sites are blocked and therefore decrease the total number of active sites available for molecular oxygen to interact with the surface, thus hindering the ORR.
 | ||
| Fig. 8 (A) Average ORR specific activity normalized by exposed surface area before (empty circles) and after (filled circles) AFM measurement for Ag. (B) Adsorption free energy of anions as a function of applied potential (vs. RHE) on an Ag(111) surface. Reproduced from ref. 83 with permission from John Wiley and Sons, copyright 2021. (C) Plot showing the peak-to-peak potential difference between the anodic and cathodic peak of the *O ↔ *OH transition (black), the specific activities (red), and peak potentials of *O from CV (blue) as a function of HClO4 concentration. (D) Normalized ORR activities on different Pt surfaces as a function of HClO4 concentration. Reproduced from ref. 87 with permission from Springer Nature, copyright 2022. | ||
Kamat et al. further studied the effect of various acid anions on the ORR/OER on Pt(poly) catalyst.82 The ORR activity trend was observed to follow HClO4 > HNO3 > H2SO4, with each acid's onset potential becoming less positive. Similarly, the OER activity trend was noted as HClO4 > HNO3 ∼ H2SO4. To further understand these trends, the adsorption free energies of the anions were calculated using DFT where it was shown that the adsorption energies followed the trend: ClO4− > NO3− ∼ SO42− demonstrating that certain anions strongly adsorb to the catalyst surface and hinder ORR through active site blocking. Furthermore, in HClO4, there is less anion adsorption leading to the increase in OER rates as well due to higher availability of active sites. These results demonstrate that the anionic effects are predominantly due to competitive adsorption rather than non-adsorbed anion–intermediate interactions.
Working with non-precious-metal catalysts, specifically a Fe/N/C catalyst, Holst-Olesen and coworkers studied the anionic effects on the ORR in various acidic electrolytes.26 They found that the anions had less poisoning effects on the Fe/N/C compared to the Pt. On Pt(poly), ORR activity decreased in the order HClO4 > H2SO4 > H3PO4 > HCl as previously mentioned, while for the Fe/N/C catalyst, the activity followed the trend H3PO4 > HClO4 ∼ H2SO4 > HCl. Using DFT calculations, it was shown that for both Pt(111) and Fe/N/C catalysts, phosphoric acid interacts strongly with the catalyst compared to the expected ORR intermediates and, therefore likely to decrease ORR activity. However, the observed enhancement of ORR on Fe/N/C in the presence of phosphoric acid can be attributed to the 2D structure of Fe/N/C where, despite anion adsorption, the opposite side can function as the ORR active site. Further DFT calculations demonstrated that the stronger binding acid (H3PO4) altered the FeN4 site and decreased the thermodynamic barrier for the RDS for ORR.
Luo and Koper focused on the effect of non-specifically adsorbed (NSA) anions (e.g., methanesulfonic acid (MSA), perfluorosulfonic acid (PFSA) ionomers) on the ORR activity of single crystal Pt electrodes and found a general inhibitory effect.87 They utilized the electrochemical transition between *O ↔ *OH, where both are key ORR intermediates, as a kinetic descriptor to shed light on the effect of NSA anions on the ORR rates. It was observed that, with higher concentration of NSA anions, the ORR activity on Pt (111) declined as well as the rate of *O ↔ *OH transition measured by the peak-to-peak separation (ΔEpeak) in CV (Fig. 8C). Further studies utilizing Nafion-covered Pt(111) catalysts demonstrated that Nafion also suppresses ORR together with the declined reversibility of the *O ↔ *OH transition. Furthermore, they conducted similar studies on stepped Pt surfaces where not only the kinetic descriptor predictions were verified (i.e., ORR activity decreased with HClO4 concentration) but also Pt (111) was shown to exhibit the highest sensitivity to the anion inhibitory effects (i.e., (111) terraces are most sensitive) (Fig. 8D). The authors also demonstrate that this kinetic descriptor applies well to cases where cations are present. It is speculated that a strong interaction between the anions (or cations) and the *OH layer exists causing slowed kinetics for the *O ↔ *OH transition, but as stated by the authors, further in-depth investigations are necessary.
4.2. Carbon dioxide reduction
In another work, Huang et al. studied the effects of halide anions on the CO2RR on single crystal Cu catalysts such as Cu(100) and Cu(111) in unbuffered 0.1 M KClO4 as well as KA (where A = halide) electrolytes.94 The authors focused on how the halides affect CO2RR selectivity, specifically for the C2+ products. The FE of C2+ products increased following a similar aforementioned anionic trend: ClO4− < Cl− < Br− < I− (Fig. 9A) and this was due to the partial current densities increasing for ethylene and ethanol. In the unbuffered KI electrolyte, the FEs for ethylene and ethanol were the highest at 50% and 16%, respectively, with the overall FE for C2 and C3 products reaching 74%. The authors also found that in the presence of I−, more COads led to increasing C2+ production. In situ Raman spectroscopy showed a red shift in the C–O stretching mode from 2087 cm−1 to 2060 cm−1 when varying the electrolyte from KClO4 to KI demonstrating the effect of anions on the environment of CO (Fig. 9B). The reason for the enhancement in C2+ formation was explained as a combined effect of a higher *CO population and the modified electronic structure of local Cu sites due to the adsorbed I−.
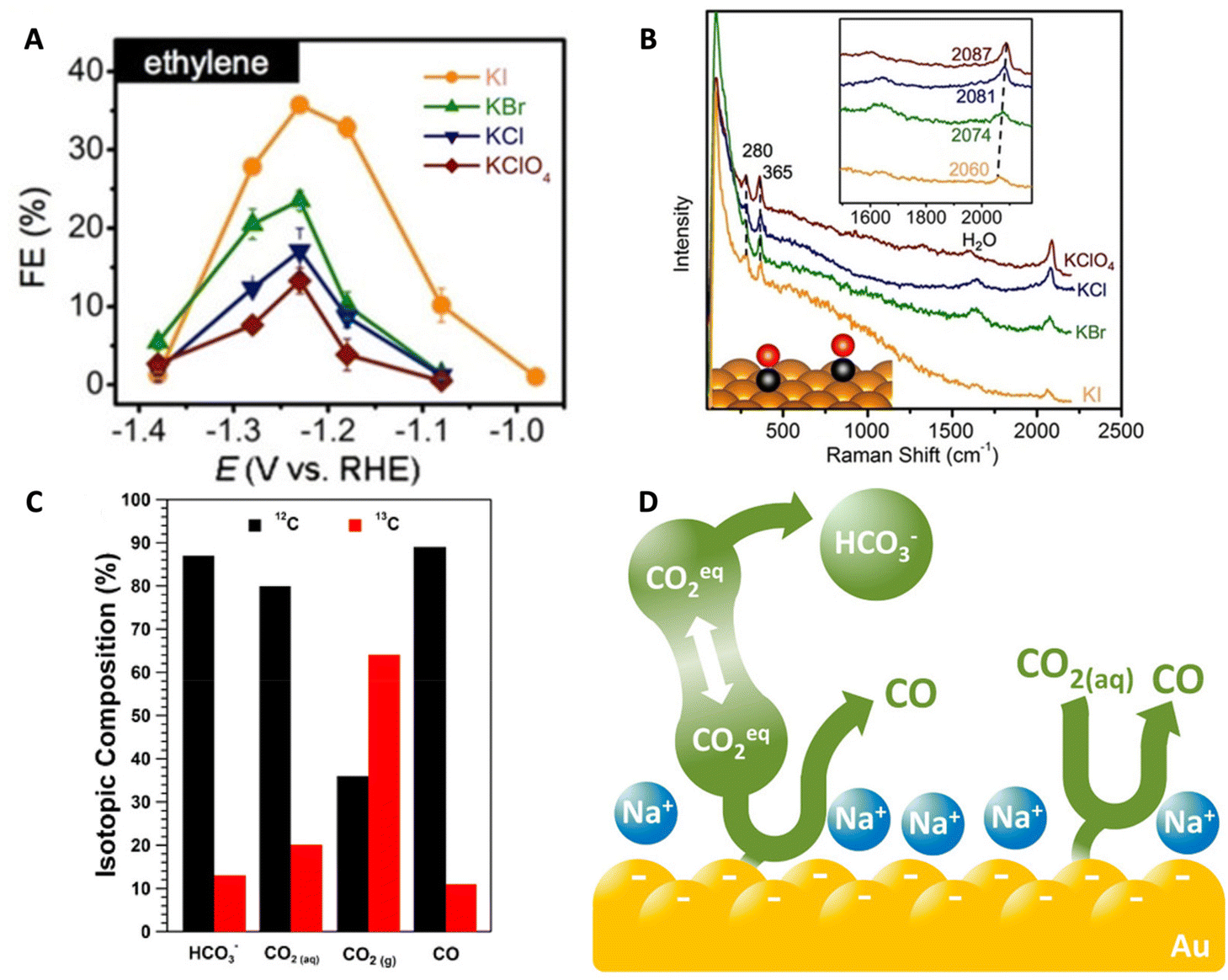 | ||
| Fig. 9 (A) FE for ethylene produced on a Cu(111) catalyst in 0.1 M HClO4, KCl, KBr, and KI electrolytes. (B) In situ Raman spectra of a Cu(pc) catalyst during CO2RR in the same electrolytes. Top inset shows the red shift of the CO stretching band and the bottom inset is a schematic of the CO adsorbed onto the Cu(pc) surface. Reproduced from ref. 94 with permission from John Wiley and Sons, copyright 2018. (C) Isotopic compositions of HCO3−, CO2(aq), CO2(g), and CO produced during the CO2RR. (D) Schematic demonstrating that the main source of CO2 is the HCO3− through which there is a rapid equilibrium with CO2 within the electrolyte. Reproduced from ref. 12 with permission from the American Chemical Society, copyright 2017. | ||
Studying the effects of halide anions on the CO2RR has been a challenge since halides have been shown to affect the morphology of the catalyst surface during CO2 reduction.88,94 The convolution of morphological changes and halide anion presence renders it difficult to determine the origin of CO2RR activity enhancement. To deconvolute these variables, Yuan et al. pretreated CuO-nanosheets (Cu–ONS) electrodes in order to reach a morphologically stable surface state prior to CO2 reduction investigations.90 In addition, they added a supporting K2SO4 electrolyte to minimize any further morphological changes that may occur during the CO2 reduction. It was found that the FE of C2 products, specifically ethylene and ethanol, increases with increasing halide concentration with a maximum FE of C2+ products ∼84.5% in I−-containing media. This was due to the partial current densities increasing for ethylene, ethanol, and propanol with increasing I− concentration. The authors utilized in situ ATR-SEIRAS and found that, in a halide-containing electrolyte, the COads band red-shifted to lower wavenumbers with increasing halide concentration. Similar to the earlier works, they explain that when halides specifically adsorb onto the surface, negative charges from the anions transfer to the COads which enhances CO adsorption strength and C–C coupling.
Zhang et al. studied the effects of halides on nanoporous Ag catalysts.89 In the presence of F−, Cl−, and Br−, the CO FE is enhanced with the maximum FE found in the presence of Br−. This was due to an enhancement in CO partial current density while that of H2 showed the opposite trend. Interestingly, in the presence of I−, the Ag catalyst demonstrated the poorest catalytic activity until −0.69 V vs. RHE. However, at potentials more negative than −0.89 V vs. RHE, the CO FE and partial current density was the highest surpassing the effects of Br− and reaching a maximum FE of 90.3%. To further explore how halides affect the CO2RR, the authors studied the Tafel kinetics. In theory, the RDS for the CO2 to CO reaction is a single electron transfer step resulting in *CO2− with a Tafel slope of 118 mV dec−1. All Tafel slopes calculated for the systems containing halides were smaller than this theoretical value indicating a change in the RDS from a single electron transfer step to the proton transfer chemical step. Moreover, the Tafel slopes decreased with increasing anion size indicating accelerated kinetics of the CO2RR. With DFT calculations, they found that the binding affinity of the key intermediate COOH increased with increasing halide size with a maximum value of −191.45 meV in the presence of I−. This suggests that halide anions, especially I−, promote increased adsorption of COOH thereby decreasing the required overpotential for the CO2RR.
Then, the authors utilized isotopic labeling coupled with the potential square-wave approach to gain insight into the mechanism of CO2RR. The purge consisted of 13CO2 while the electrolyte was comprised of 0.5 M NaH12CO3. They found that the unlabeled 12COads was observed initially, indicating that the carbon source is from the H12CO3− within the electrolyte and not the headspace 13CO2 purge. In fact, utilizing a combination of MS and ATR-FTIR, they were able to determine that the composition of the dissolved CO2 was 80% 12CO2(aq) matching closely with the isotopic composition of bicarbonate indicating a rapid equilibrium (faster than CO2(g) diffusion from the headspace) between CO2(aq) and bicarbonate (Fig. 9C). Further MS studies demonstrated that about 89% of the CO produced was 12CO further demonstrating that the CO2(aq) equilibrated with bicarbonate is the major carbon source for the CO2RR. Lastly, the authors conducted a kinetic analysis in which a bicarbonate first-order dependence was observed. The work describes the role of bicarbonate as a buffer as well as a CO2(aq) source leading to an increase in local CO2(aq) concentration and therefore enhancing the CO2RR (Fig. 9D).
It is important to mention that, while water is an important source, the supporting electrolyte also contributes to the proton donation process in the CO2RR. Resasco et al. found that the formation rates of H2 and CH4 strongly depend on the type and concentration of anions used.96 The authors propose that this is due to the proton donating capability of certain anions whose pKa is lower than water. Furthermore, a study conducted by Wuttig et al. found that the proton sources for CO2RR on Au were bicarbonate, hydronium, and/or carbonic acid where the first electron transfer to form adsorbed H is rate limiting for H2 evolution.97
Focused on a broader range of anions, Hong et al. studied the effects of anions such as Cl−, HPO42−, SO42−, and HCO3− on the CO2RR on Au sputtered gas diffusion electrode (Au/GDL) at a neutral pH.95 The electrolyte containing Cl− exhibited the highest CO FE out of all the other electrolytes reaching 82%, while the lowest CO FE was observed in electrolyte containing HPO42− with 11%. The H2 FE followed the opposite trend and the total current densities were similar, except for HPO42−, whose H2 FE was the highest with total current density nearly doubling indicating a substantial enhancement for HER. Structural characterizations ensured that the changes in CO2RR activities in different electrolytes were due to the intrinsic properties of each anion. The authors propose that anion adsorption suppresses HER and promotes CO2RR. Thus, higher CO2RR activities are expected for anions with high binding strength to the surface. This idea explains the trend of increasing CO2RR following: Cl− > SO42− > HPO42−. However, a discrepancy in this theory was observed by the fact that HPO42− binds more strongly than SO42− and yet does not promote CO2RR as much as SO42−. The authors attribute this discrepancy to the intrinsic characteristics of HPO42− and its ability to deprotonate further into PO43− lowering the local pH and providing protons for the enhancement of the HER rather than the CO2RR. Therefore, the authors claim that the identity of the anion is not the only factor that affects the CO2RR but also its ability to deprotonate.
5. Summary and outlook
It is clear that electrochemical interfaces involve numerous components extending beyond the solid surface and its active sites in reactions like CO2RR, HER/HOR, and OER/ORR. The growing interest in the role of interfacial liquids has led researchers to delve into their influence. These endeavors have provided insights into how these reactions truly unfold and can be enhanced at molecular levels. By deepening our understanding of these systems, we can explore strategies to manipulate interfacial liquids, thereby advancing existing technologies and fostering the development of new ones for a more sustainable future.However, despite notable advancements in recent years, there remains much to unravel. Our current experimental methodologies face limitations in probing the constituents of the interfacial liquid and the electric double layer at high precision in terms of concentration, distribution, coordination, and orientation. Consequently, many mechanisms have primarily been explored through theoretical approaches. Yet, even in theory, the computational demands of considering the motions and interactions of all constituents simultaneously have led to simplified models that may lack comprehensiveness. Moreover, fully understanding the dynamic structure and properties of interfacial liquids proves challenging due to a lack of tools offering sufficient spatial and temporal resolution. These challenges have led to the formulation of various hypotheses to explain commonly observed effects or phenomena. Future research should prioritize the development of advanced experimental and theoretical methods that can thoroughly interrogate the intricacies of electrochemical interfaces, with a specific emphasis on the liquid phase. Additionally, the insights gleaned regarding interfacial liquid effects should be integral considerations in the design and application of catalyst materials for various electrochemical applications.
Author contributions
R. B. and D. K. conceptualized the manuscript. R. B., G. L., and D. K. wrote the original draft. R. B., G. L., and D. K. contributed to the revision of the manuscript. D. K. supervised the writing and revision of the manuscript, and provided the resources.Conflicts of interest
The authors declare no competing interests.Acknowledgements
The authors acknowledge the start-up support from the University of Pennsylvania as well as the generous support from Hanwha for this work.References
- P. C. K. Vesborg, B. Seger and I. Chorkendorff, J. Phys. Chem. Lett., 2015, 6, 951–957 CrossRef CAS PubMed.
- S. Bai, M. Yang, J. Jiang, X. He, J. Zou, Z. Xiong, G. Liao and S. Liu, npj 2D Mater. Appl., 2021, 5, 78 CrossRef CAS.
- J. Zhu, L. Hu, P. Zhao, L. Y. S. Lee and K.-Y. Wong, Chem. Rev., 2020, 120, 851–918 CrossRef CAS PubMed.
- Y. Lei, Z. Wang, A. Bao, X. Tang, X. Huang, H. Yi, S. Zhao, T. Sun, J. Wang and F. Gao, Chem. Eng. J., 2023, 453, 139663 CrossRef CAS.
- M. Shao, Q. Chang, J.-P. Dodelet and R. Chenitz, Chem. Rev., 2016, 116, 3594–3657 CrossRef CAS PubMed.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- D. Gao, R. M. Arán-Ais, H. S. Jeon and B. Roldan Cuenya, Nat. Catal., 2019, 2, 198–210 CrossRef CAS.
- B. M. Hunter, H. B. Gray and A. M. Müller, Chem. Rev., 2016, 116, 14120–14136 CrossRef CAS PubMed.
- M. M. Waegele, C. M. Gunathunge, J. Li and X. Li, J. Chem. Phys., 2019, 151, 160902 CrossRef PubMed.
- L. Su, J. L. Weaver, M. Groenenboom, N. Nakamura, E. Rus, P. Anand, S. K. Jha, J. S. Okasinski, J. A. Dura and B. Reeja-Jayan, ACS Appl. Mater. Interfaces, 2021, 13, 9919–9931 CrossRef CAS PubMed.
- X. Ding, D. Scieszka, S. Watzele, S. Xue, B. Garlyyev, R. W. Haid and A. S. Bandarenka, ChemElectroChem, 2022, 9, e202101088 CrossRef CAS.
- M. Dunwell, Q. Lu, J. M. Heyes, J. Rosen, J. G. Chen, Y. Yan, F. Jiao and B. Xu, J. Am. Chem. Soc., 2017, 139, 3774–3783 CrossRef CAS PubMed.
- Y. Zheng, A. Vasileff, X. Zhou, Y. Jiao, M. Jaroniec and S.-Z. Qiao, J. Am. Chem. Soc., 2019, 141, 7646–7659 CrossRef CAS PubMed.
- R. Kortlever, J. Shen, K. J. P. Schouten, F. Calle-Vallejo and M. T. M. Koper, J. Phys. Chem. Lett., 2015, 6, 4073–4082 CrossRef CAS PubMed.
- L. R. L. Ting and B. S. Yeo, Curr. Opin. Electrochem., 2018, 8, 126–134 CrossRef CAS.
- N. Wang, S. Ma, P. Zuo, J. Duan and B. Hou, Adv. Sci., 2021, 8, 2100076 CrossRef CAS PubMed.
- L. Ge, H. Rabiee, M. Li, S. Subramanian, Y. Zheng, J. H. Lee, T. Burdyny and H. Wang, Chem, 2022, 8, 663–692 CAS.
- X. Tian, X. F. Lu, B. Y. Xia and X. W. Lou, Joule, 2020, 4, 45–68 CrossRef CAS.
- A. Kulkarni, S. Siahrostami, A. Patel and J. K. Nørskov, Chem. Rev., 2018, 118, 2302–2312 CrossRef CAS PubMed.
- J. Song, C. Wei, Z.-F. Huang, C. Liu, L. Zeng, X. Wang and Z. J. Xu, Chem. Soc. Rev., 2020, 49, 2196–2214 RSC.
- A. Raveendran, M. Chandran and R. Dhanusuraman, RSC Adv., 2023, 13, 3843–3876 RSC.
- A. C. Garcia, T. Touzalin, C. Nieuwland, N. Perini and M. T. M. Koper, Angew. Chem., Int. Ed., 2019, 58, 12999–13003 CrossRef CAS PubMed.
- R. R. Rao, B. Huang, Y. Katayama, J. Hwang, T. Kawaguchi, J. R. Lunger, J. Peng, Y. Zhang, A. Morinaga, H. Zhou, H. You and Y. Shao-Horn, J. Phys. Chem. C, 2021, 125, 8195–8207 CrossRef CAS.
- M. C. O. Monteiro, F. Dattila, N. López and M. T. M. Koper, J. Am. Chem. Soc., 2022, 144, 1589–1602 CrossRef CAS PubMed.
- S. Hou, L. Xu, X. Ding, R. M. Kluge, T. K. Sarpey, R. W. Haid, B. Garlyyev, S. Mukherjee, J. Warnan, M. Koch, S. Zhang, W. Li, A. S. Bandarenka and R. A. Fischer, Angew. Chem., Int. Ed., 2022, 61, e202201610 CrossRef CAS PubMed.
- K. Holst-Olesen, M. Reda, H. A. Hansen, T. Vegge and M. Arenz, ACS Catal., 2018, 8, 7104–7112 CrossRef CAS.
- V. R. Stamenkovic, D. Strmcnik, P. P. Lopes and N. M. Markovic, Nat. Mater., 2017, 16, 57–69 CrossRef CAS PubMed.
- H. Khani, A. R. Puente Santiago and T. He, Angew. Chem., Int. Ed., 2023, e202306103 CAS.
- B. Deng, M. Huang, X. Zhao, S. Mou and F. Dong, ACS Catal., 2022, 12, 331–362 CrossRef CAS.
- Y. J. Sa, C. W. Lee, S. Y. Lee, J. Na, U. Lee and Y. J. Hwang, Chem. Soc. Rev., 2020, 49, 6632–6665 RSC.
- X. Lu, W. Tu, Y. Zhou and Z. Zou, Adv. Energy Mater., 2023, 13, 2300628 CrossRef CAS.
- C. Cheng, M. Deng, L. Li and Z. Wei, Sci. China: Chem., 2022, 65, 1854–1866 CrossRef CAS.
- I. Ledezma-Yanez, W. D. Z. Wallace, P. Sebastián-Pascual, V. Climent, J. M. Feliu and M. T. M. Koper, Nat. Energy, 2017, 2, 17031 CrossRef CAS.
- P. Li, Y. Jiang, Y. Hu, Y. Men, Y. Liu, W. Cai and S. Chen, Nat. Catal., 2022, 5, 900–911 CrossRef CAS.
- K. Zhao, X. Chang, H.-S. Su, Y. Nie, Q. Lu and B. Xu, Angew. Chem., Int. Ed., 2022, 61, e202207197 CrossRef CAS PubMed.
- S. Intikhab, L. Rebollar, Y. Li, R. Pai, V. Kalra, M. H. Tang and J. D. Snyder, ACS Catal., 2020, 10, 6798–6802 CrossRef CAS.
- L. Rebollar, S. Intikhab, S. Zhang, H. Deng, Z. Zeng, J. D. Snyder and M. H. Tang, J. Catal., 2021, 398, 161–170 CrossRef CAS.
- Y.-H. Wang, S. Zheng, W.-M. Yang, R.-Y. Zhou, Q.-F. He, P. Radjenovic, J.-C. Dong, S. Li, J. Zheng, Z.-L. Yang, G. Attard, F. Pan, Z.-Q. Tian and J.-F. Li, Nature, 2021, 600, 81–85 CrossRef CAS PubMed.
- Q. Sun, N. J. Oliveira, S. Kwon, S. Tyukhtenko, J. J. Guo, N. Myrthil, S. A. Lopez, I. Kendrick, S. Mukerjee, L. Ma, S. N. Ehrlich, J. Li, W. A. Goddard, Y. Yan and Q. Jia, Nat. Energy, 2023, 8, 859–869 CrossRef CAS.
- K. Lourenssen, J. Williams, F. Ahmadpour, R. Clemmer and S. Tasnim, J. Energy Storage, 2019, 25, 100844 CrossRef.
- S. N. Suarez, J. R. Jayakody, S. G. Greenbaum, T. Zawodzinski Jr. and J. J. Fontanella, J. Phys. Chem. B, 2010, 114, 8941–8947 CrossRef CAS PubMed.
- L. Su, J. Chen, F. Yang, P. Li, Y. Jin, W. Luo and S. Chen, J. Am. Chem. Soc., 2023, 145, 12051–12058 CrossRef CAS PubMed.
- S. Sharifi Golru and E. J. Biddinger, Chem. Eng. J., 2022, 428, 131303 CrossRef CAS.
- N. Mohandas, T. N. Narayanan and A. Cuesta, ACS Catal., 2023, 13, 8384–8393 CrossRef CAS.
- B. Yang, H. Lang, Z. Liu, S. Wang, Z. Men and C. Sun, J. Mol. Liq., 2021, 324, 114996 CrossRef CAS.
- Y. Meng, Z. Xu, Z. Shen, Q. Xia, Y. Cao, Y. Wang and X. Li, J. Mater. Chem. A, 2022, 10, 6508–6522 RSC.
- W. Ge, Y. Chen, Y. Fan, Y. Zhu, H. Liu, L. Song, Z. Liu, C. Lian, H. Jiang and C. Li, J. Am. Chem. Soc., 2022, 144, 6613–6622 CrossRef CAS PubMed.
- B. Huang, R. R. Rao, S. You, K. Hpone Myint, Y. Song, Y. Wang, W. Ding, L. Giordano, Y. Zhang, T. Wang, S. Muy, Y. Katayama, J. C. Grossman, A. P. Willard, K. Xu, Y. Jiang and Y. Shao-Horn, JACS Au, 2021, 1, 1674–1687 CrossRef CAS PubMed.
- S. Xue, B. Garlyyev, S. Watzele, Y. Liang, J. Fichtner, M. D. Pohl and A. S. Bandarenka, ChemElectroChem, 2018, 5, 2326–2329 CrossRef CAS.
- Y. Taji, A. Zagalskaya, I. Evazzade, S. Watzele, K.-T. Song, S. Xue, C. Schott, B. Garlyyev, V. Alexandrov, E. Gubanova and A. S. Bandarenka, Nano Mater. Sci., 2022 DOI:10.1016/j.nanoms.2022.09.003.
- J. T. Bender, A. S. Petersen, F. C. Østergaard, M. A. Wood, S. M. J. Heffernan, D. J. Milliron, J. Rossmeisl and J. Resasco, ACS Energy Lett., 2023, 8, 657–665 CrossRef CAS.
- L. Jiao, E. Liu, S. Mukerjee and Q. Jia, ACS Catal., 2020, 10, 11099–11109 CrossRef CAS.
- A. Goyal and M. T. M. Koper, Angew. Chem., Int. Ed., 2021, 60, 13452–13462 CrossRef CAS PubMed.
- M. C. O. Monteiro, A. Goyal, P. Moerland and M. T. M. Koper, ACS Catal., 2021, 11, 14328–14335 CrossRef CAS PubMed.
- A. H. Shah, Z. Zhang, Z. Huang, S. Wang, G. Zhong, C. Wan, A. N. Alexandrova, Y. Huang and X. Duan, Nat. Catal., 2022, 5, 923–933 CrossRef CAS.
- X. Chen, I. T. McCrum, K. A. Schwarz, M. J. Janik and M. T. M. Koper, Angew. Chem., Int. Ed., 2017, 56, 15025–15029 CrossRef CAS PubMed.
- N. Perini and E. A. Ticianelli, J. Catal., 2019, 378, 277–282 CrossRef CAS.
- A. M. Raventos and R. Kortlever, Electrochim. Acta, 2022, 415, 140255 CrossRef.
- R. A. Márquez, K. Kawashima, Y. J. Son, G. Castelino, N. Miller, L. A. Smith, C. E. Chukwuneke and C. B. Mullins, ACS Energy Lett., 2023, 8, 1141–1146 CrossRef.
- T. Kumeda, H. Tajiri, O. Sakata, N. Hoshi and M. Nakamura, Nat. Commun., 2018, 9, 4378 CrossRef PubMed.
- M. Görlin, J. Halldin Stenlid, S. Koroidov, H.-Y. Wang, M. Börner, M. Shipilin, A. Kalinko, V. Murzin, O. V. Safonova, M. Nachtegaal, A. Uheida, J. Dutta, M. Bauer, A. Nilsson and O. Diaz-Morales, Nat. Commun., 2020, 11, 6181 CrossRef PubMed.
- J. D. Michael, E. L. Demeter, S. M. Illes, Q. Fan, J. R. Boes and J. R. Kitchin, J. Phys. Chem. C, 2015, 119, 11475–11481 CrossRef CAS.
- B. Garlyyev, S. Xue, M. D. Pohl, D. Reinisch and A. S. Bandarenka, ACS Omega, 2018, 3, 15325–15331 CrossRef CAS PubMed.
- D. Strmcnik, K. Kodama, D. van der Vliet, J. Greeley, V. R. Stamenkovic and N. M. Marković, Nat. Chem., 2009, 1, 466–472 CrossRef CAS PubMed.
- M. C. O. Monteiro, F. Dattila, B. Hagedoorn, R. García-Muelas, N. López and M. T. M. Koper, Nat. Catal., 2021, 4, 654–662 CrossRef CAS.
- V. J. Ovalle, Y.-S. Hsu, N. Agrawal, M. J. Janik and M. M. Waegele, Nat. Catal., 2022, 5, 624–632 CrossRef CAS.
- J. Resasco, L. D. Chen, E. Clark, C. Tsai, C. Hahn, T. F. Jaramillo, K. Chan and A. T. Bell, J. Am. Chem. Soc., 2017, 139, 11277–11287 CrossRef CAS PubMed.
- J. Gu, S. Liu, W. Ni, W. Ren, S. Haussener and X. Hu, Nat. Catal., 2022, 5, 268–276 CrossRef CAS.
- S. Ringe, E. L. Clark, J. Resasco, A. Walton, B. Seger, A. T. Bell and K. Chan, Energy Environ. Sci., 2019, 12, 3001–3014 RSC.
- X. Qin, T. Vegge and H. A. Hansen, J. Am. Chem. Soc., 2023, 145, 1897–1905 CrossRef CAS PubMed.
- S.-J. Shin, H. Choi, S. Ringe, D. H. Won, H.-S. Oh, D. H. Kim, T. Lee, D.-H. Nam, H. Kim and C. H. Choi, Nat. Commun., 2022, 13, 5482 CrossRef CAS PubMed.
- G. Kastlunger, L. Wang, N. Govindarajan, H. H. Heenen, S. Ringe, T. Jaramillo, C. Hahn and K. Chan, ACS Catal., 2022, 12, 4344–4357 CrossRef CAS.
- D. Kim, S. Yu, F. Zheng, I. Roh, Y. Li, S. Louisia, Z. Qi, G. A. Somorjai, H. Frei, L.-W. Wang and P. Yang, Nat. Energy, 2020, 5, 1032–1042 CrossRef CAS.
- M. R. Singh, Y. Kwon, Y. Lum, J. W. Ager III and A. T. Bell, J. Am. Chem. Soc., 2016, 138, 13006–13012 CrossRef CAS PubMed.
- O. Ayemoba and A. Cuesta, ACS Appl. Mater. Interfaces, 2017, 9, 27377–27382 CrossRef CAS PubMed.
- F. Zhang and A. C. Co, Angew. Chem., Int. Ed., 2020, 59, 1674–1681 CrossRef CAS PubMed.
- Y.-Q. Wang, X.-H. Dan, X. Wang, Z.-Y. Yi, J. Fu, Y.-C. Feng, J.-S. Hu, D. Wang and L.-J. Wan, J. Am. Chem. Soc., 2022, 144, 20126–20133 CrossRef CAS PubMed.
- E. Pérez-Gallent, G. Marcandalli, M. C. Figueiredo, F. Calle-Vallejo and M. T. M. Koper, J. Am. Chem. Soc., 2017, 139, 16412–16419 CrossRef PubMed.
- A. S. Malkani, J. Li, N. J. Oliveira, M. He, X. Chang, B. Xu and Q. Lu, Sci. Adv., 2020, 6, eabd2569 CrossRef CAS PubMed.
- D. V. Tripkovic, D. Strmcnik, D. van der Vliet, V. Stamenkovic and N. M. Markovic, Faraday Discuss., 2009, 140, 25–40 RSC.
- E. Lamy-Pitara, S. El Mouahid and J. Barbier, Electrochim. Acta, 2000, 45, 4299–4308 CrossRef CAS.
- G. A. Kamat, J. A. Zamora Zeledón, G. T. K. K. Gunasooriya, S. M. Dull, J. T. Perryman, J. K. Nørskov, M. B. Stevens and T. F. Jaramillo, Commun. Chem., 2022, 5, 20 CrossRef CAS PubMed.
- J. A. Z. Zeledón, G. A. Kamat, G. T. K. K. Gunasooriya, J. K. Nørskov, M. B. Stevens and T. F. Jaramillo, ChemElectroChem, 2021, 8, 2467–2478 CrossRef.
- F. Zhou, Y. Yan, S. Guan, W. Guo, M. Sun and M. Pan, Int. J. Energy Res., 2020, 44, 10155–10167 CrossRef CAS.
- G. You, W. Zhu and Z. Zhuang, Int. J. Hydrogen Energy, 2019, 44, 13373–13382 CrossRef CAS.
- Z. Jusys and R. J. Behm, ChemPhysChem, 2019, 20, 3276–3288 CrossRef CAS PubMed.
- M. Luo and M. T. M. Koper, Nat. Catal., 2022, 5, 615–623 CrossRef CAS.
- A. S. Varela, W. Ju, T. Reier and P. Strasser, ACS Catal., 2016, 6, 2136–2144 CrossRef CAS.
- Y. Zhang, L. Liu, L. Shi, T. Yang, D. Niu, S. Hu and X. Zhang, Electrochim. Acta, 2019, 313, 561–569 CrossRef CAS.
- T. Yuan, T. Wang, G. Zhang, W. Deng, D. Cheng, H. Gao, J. Zhao, J. Yu, P. Zhang and J. Gong, Chem. Sci., 2022, 13, 8117–8123 RSC.
- H. Wang, E. Matios, C. Wang, J. Luo, X. Lu, X. Hu and W. Li, Nano Lett., 2019, 19, 3925–3932 CrossRef CAS PubMed.
- M. G. Kibria, C.-T. Dinh, A. Seifitokaldani, P. De Luna, T. Burdyny, R. Quintero-Bermudez, M. B. Ross, O. S. Bushuyev, F. P. García de Arquer, P. Yang, D. Sinton and E. H. Sargent, Adv. Mater., 2018, 30, 1804867 CrossRef PubMed.
- I. T. McCrum, S. A. Akhade and M. J. Janik, Electrochim. Acta, 2015, 173, 302–309 CrossRef CAS.
- Y. Huang, C. W. Ong and B. S. Yeo, ChemSusChem, 2018, 11, 3299–3306 CrossRef CAS PubMed.
- S. Hong, S. Lee, S. Kim, J. K. Lee and J. Lee, Catal. Today, 2017, 295, 82–88 CrossRef CAS.
- J. Resasco, Y. Lum, E. Clark, J. Z. Zeledon and A. T. Bell, ChemElectroChem, 2018, 5, 1064–1072 CrossRef CAS.
- A. Wuttig, M. Yaguchi, K. Motobayashi, M. Osawa and Y. Surendranath, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E4585–E4593 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |