
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA thiolated copper-hydride nanocluster with chloride bridging as a catalyst for carbonylative C–N coupling of aryl amines under mild conditions: a combined experimental and theoretical study†
Anish Kumar
Das‡
 a,
Sourav
Biswas‡
a,
Amit
Pal
a,
Surya Sekhar
Manna
b,
Avirup
Sardar
a,
Pradip Kumar
Mondal
c,
Basudev
Sahoo
*a,
Biswarup
Pathak
*b and
Sukhendu
Mandal
*a
a,
Sourav
Biswas‡
a,
Amit
Pal
a,
Surya Sekhar
Manna
b,
Avirup
Sardar
a,
Pradip Kumar
Mondal
c,
Basudev
Sahoo
*a,
Biswarup
Pathak
*b and
Sukhendu
Mandal
*a
aSchool of Chemistry, Indian Institute of Science Education and Research Thiruvananthapuram, Kerala 695551, India. E-mail: sukhendu@iisertvm.ac.in; basudev@iisertvm.ac.in
bDepartment of Chemistry, Indian Institute of Technology Indore, Madhya Pradesh 453552, India. E-mail: biswarup@iiti.ac.in
cElettra Sincrotrone Trieste, Area Science Park, 341494 Basovizza, Italy
First published on 9th January 2024
Abstract
Atomically precise copper nanoclusters (Cu NCs), an emerging class of nanomaterials, have garnered significant attention owing to their versatile core–shell architecture and their potential applications in catalytic reactions. In this study, we present a straightforward synthesis strategy for [Cu29(StBu)12(PPh3)4Cl6H10][BF4] (Cu29) NCs and explore their catalytic activity in the carbonylative C–N coupling reaction involving aromatic amines and N-heteroarenes with dialkyl azodicarboxylates. Through a combination of experimental investigations and density functional theory studies, we elucidate the radical mechanisms at play. The crucial step in the catalytic process is identified as the decomposition of diisopropyl azodicarboxylates on the surface of Cu29 NCs, leading to the generation of oxyacyl radicals and the liberation of nitrogen gas. Subsequently, an oxyacyl radical abstracts a hydrogen atom from aniline, initiating the formation of an aminyl radical. Finally, the aminyl radical reacts with another oxyacyl radical, culminating in the synthesis of the desired carbamate product. This detailed analysis provides insights into the intricate catalytic pathways of Cu29 NCs, shedding light on their potential for catalyzing carbonylative C–N coupling reactions.
Introduction
Since the discovery of nanoclusters (NCs), gold (Au) and silver (Ag) NCs have emerged due to their relatively superior stability compared to the lighter congeners of transition elements.1–3 However, the progressing research interest has now developed strategies to synthesize copper (Cu) NCs, the most abundant and lightest congener of group 11.4–11 Discrete molecular properties and quantum confinement make these NCs more promising for applications than their nanoparticle counterparts.2,12 In addition to this, their precise structural architecture, high surface-to-volume ratio, low half-cell reduction potential, and high chemical reactivity appear advantageous for opening up their unique opportunities in catalytic activity.13–17 Generally, metal NCs consist of a core architecture of metal atoms surrounded by metal-ligand motifs. The interconnectivity between metals and ligands creates a different core configuration that preferentially governs the geometric architecture of the overall NCs and creates active catalytic sites with fewer coordination bonds, which are inaccessible in any other contemporary materials.18–24The history of CuH complexes dates back to the early 1800s when they were first discovered.25,26 Initially, their application was primarily focused on catalytic reactions involving the reduction of intermediates.27 However, it took several more decades for these complexes to find their applications as catalysts in organic media. A significant turning point in the utilization of Cu in catalysis occurred with the discovery of organocopper species, famously known as Stryker's reagent.28,29 This breakthrough expanded the range of applications for copper-based materials, enabling the construction of useful chemical bonds beyond their purely reductive capabilities. From that moment on, organocopper species garnered significant attention in the scientific community due to copper's economic advantages as a catalyst. Researchers recognized the potential of copper as a superior catalyst, which spurred further exploration and experimentation. The development of Cu NC-based catalysts has a pronounced effect on this field. There are few studies now available in the literature where Cu NCs have already been utilized in various catalytic reactions; however, the examples are still limited and in most of the cases, the mechanism of the catalyst is not properly explored.16,20,30–32 Thus, it would be useful if a precise structural architecture could be developed in such a way that it overcomes the challenges associated with the thiolate Cu NCs to be utilized as an active catalyst.
Carbon–nitrogen (C–N) bonds are pervasive in natural products, agrochemicals, and pharmaceutical compounds, underscoring their immense importance.33,34 Recognizing the significance of C–N bonds, there is a growing emphasis on designing effective catalysts in synthetic chemistry to facilitate the convenient formation of these bonds.35 Traditionally, Cu catalysts, coordinated with ligands, have been the primary choice for such reactions. One illustrative example of this is the synthesis of organic carbamates through carbonylation, where N–H bond cleavage and C–N bond formation occur simultaneously under the influence of Cu catalysts at elevated temperatures and with the use of various toxic chemicals.36,37 Over time, several approaches have been explored, including the substitution of Cu with other transition metal atoms. However, the ultimate objective of synthesizing carbamates through simpler and more convenient methods has remained elusive. Hence, the quest for alternative methods remains a desirable pursuit. In this context, a noteworthy development by Usman et al. involves the use of dialkyl azodicarboxylates as the carbonyl source in the carbonylation of aniline.38 This innovative approach employs the Cu(OAc)2 salt as a catalyst, and notably, the reaction occurs at room temperature, offering a promising alternative for the synthesis of carbamates. However, the use of dialkyl azodicarboxylates in copper-catalyzed carbonylation reactions is still limited.39 While homogeneous M–H-based molecular catalysts have made strides in organic synthesis, the potential of emerging M–H-based metal NCs as homogeneous catalysts in organic reactions has yet to be fully explored. Recently, Lee et al. have reported the synthesis of a Cu32 NC that exhibited catalytic effectiveness in a carbonylation reaction of aniline using diisopropyl azodicarboxylate, albeit with a limited substrate scope.15 However, the influence of the precise structural architecture of the Cu32 NC on this specific reaction remains unknown.
Based on the aforementioned facts, a one-pot synthesis strategy is presented for producing a [Cu29(StBu)12(PPh3)4Cl6H10][BF4] (Cu29) NC with a core–shell architecture. This precise core–shell configuration not only imparts an appealing structure but also exerts an influence on the optical and catalytic properties. The computational investigation constructs the electronic structure of the as-synthesized Cu29 NC where it is found that the occupied orbitals are mostly contributed by the metal–ligand motif shell; however, the unoccupied orbitals are predominantly contributed by the inner core. Due to this unique arrangement, this NC can take part efficiently in the carbonylation reaction. The efficiency under milder conditions makes it highly valuable for C–N bond-forming reactions due to the precise availability of the active catalytic sites which stabilize the radical intermediates.
Results and discussion
In a typical synthesis of Cu NCs, a Cu(I) salt, i.e., Cu(CH3CN)4BF4 in acetonitrile/chloroform 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) was treated with HStBu and PPh3, followed by the addition of methanolic NaBH4 solution at room temperature (Fig. 1a). After a visible color change, the resulting orange precipitate was purified, dissolved in a chloroform/hexane mixture, and left to crystallize under ambient conditions. After seven days, we obtained red-colored, plate-like single crystals, as confirmed by microscopic imaging (Fig. S1†). The resulting structure crystallized in a triclinic crystal system with the space group P
:1 (v/v) was treated with HStBu and PPh3, followed by the addition of methanolic NaBH4 solution at room temperature (Fig. 1a). After a visible color change, the resulting orange precipitate was purified, dissolved in a chloroform/hexane mixture, and left to crystallize under ambient conditions. After seven days, we obtained red-colored, plate-like single crystals, as confirmed by microscopic imaging (Fig. S1†). The resulting structure crystallized in a triclinic crystal system with the space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2) (Table S1†). Its overall morphology is tetrahedral, consisting of 29 Cu atoms protected by 12 –StBu, 6 Cl−, 10 hydrides, and 4 PPh3 ligands (Fig. 1b). Single-crystal X-ray diffraction analysis also revealed the presence of one BF4− ion and uncoordinated CHCl3 in the lattice per formula unit. The origin of the Cl− ions in the structure can be attributed to the cleavage of carbon–chlorine (C–Cl) bonds in CHCl3 during the reaction process. Additionally, the presence of hydrides in the structure can be traced back to the addition of sodium borohydride (NaBH4). This observation is substantiated by analyzing 1H NMR peaks of the as-synthesized deuteride analogue (Cu29D NC) of Cu29 NC under similar reaction conditions using NaBD4. The 1H NMR spectrum of the Cu29 NC distinctly shows hydride peaks ranging from 3.7 to 1.9 ppm (Fig. S2†). The deshielding order of these bridged hydrides aligns with our structural classification, as detailed in Table S2.† However, the absence of these hydride peaks in the Cu29D NC spectrum, together with the unchanged appearance of other peaks, indicates the complete replacement of all ten hydrides in the Cu29 NC with deuterium (Fig. S3†). The presence of respective elements is further confirmed through 11B, 19F, and 31P NMR measurements (Fig. S4–S6†).
(no. 2) (Table S1†). Its overall morphology is tetrahedral, consisting of 29 Cu atoms protected by 12 –StBu, 6 Cl−, 10 hydrides, and 4 PPh3 ligands (Fig. 1b). Single-crystal X-ray diffraction analysis also revealed the presence of one BF4− ion and uncoordinated CHCl3 in the lattice per formula unit. The origin of the Cl− ions in the structure can be attributed to the cleavage of carbon–chlorine (C–Cl) bonds in CHCl3 during the reaction process. Additionally, the presence of hydrides in the structure can be traced back to the addition of sodium borohydride (NaBH4). This observation is substantiated by analyzing 1H NMR peaks of the as-synthesized deuteride analogue (Cu29D NC) of Cu29 NC under similar reaction conditions using NaBD4. The 1H NMR spectrum of the Cu29 NC distinctly shows hydride peaks ranging from 3.7 to 1.9 ppm (Fig. S2†). The deshielding order of these bridged hydrides aligns with our structural classification, as detailed in Table S2.† However, the absence of these hydride peaks in the Cu29D NC spectrum, together with the unchanged appearance of other peaks, indicates the complete replacement of all ten hydrides in the Cu29 NC with deuterium (Fig. S3†). The presence of respective elements is further confirmed through 11B, 19F, and 31P NMR measurements (Fig. S4–S6†).
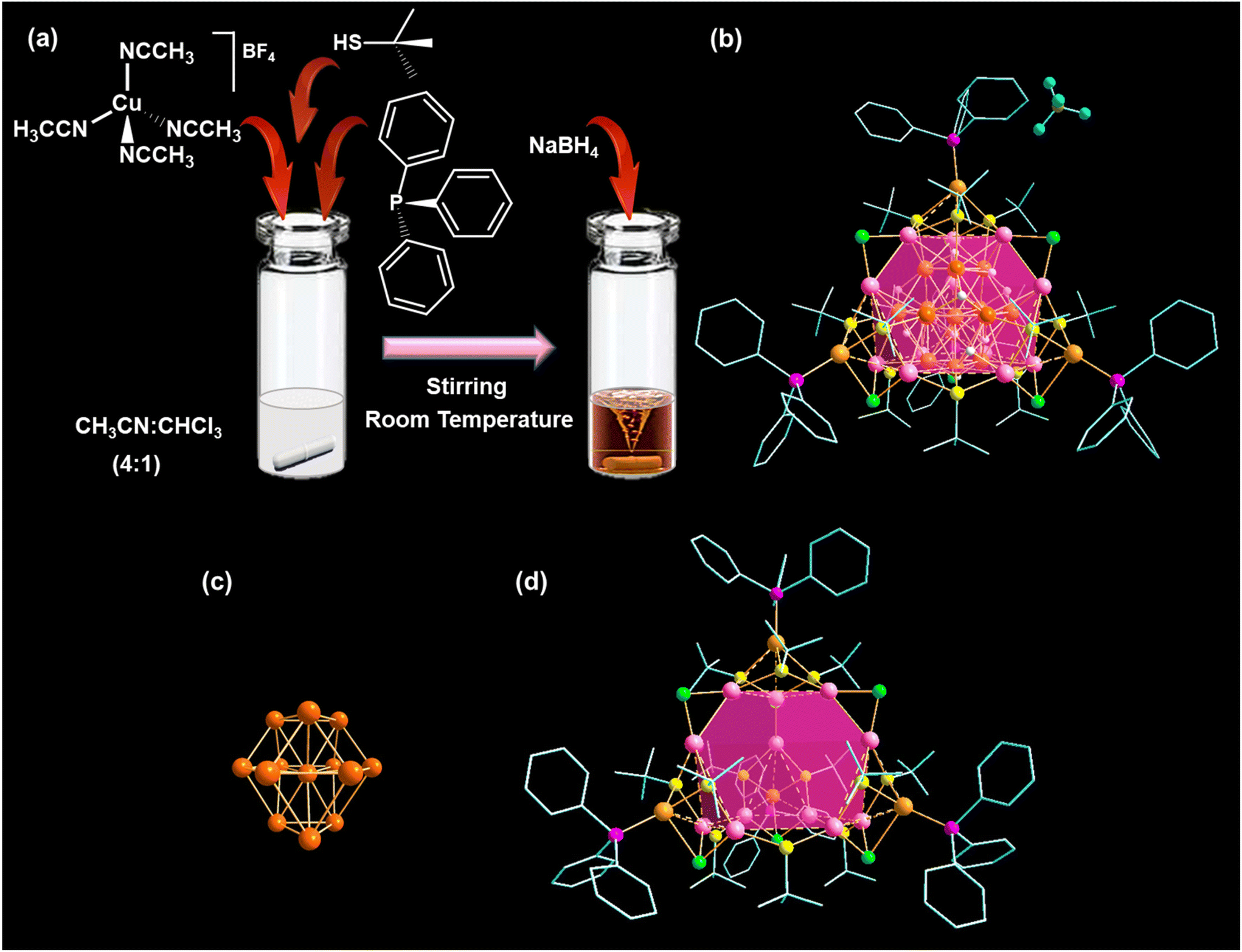 | ||
| Fig. 1 (a) Reaction scheme for the synthesis of the Cu29 NC, (b) obtained crystal structure, (c) distorted triangular orthobicupola-like geometry of the Cu@Cu12 core, and (d) the metal–ligand motif shell of the Cu29 NC. Color legend: Cu(core), deep orange; Cu(Cu12 shell), rose; Cu(Cu4 shell), light orange; S, yellow; P, magenta; Cl, green; C, grey stick; B, dark yellow; F, turquoise; H, white. H atoms and solvent molecules are partially omitted for clarity. | ||
The Cu29 NC exhibits a core–shell architecture, wherein a central Cu-centered Cu12 (Cu@Cu12) core is protected by a Cu16S12P4Cl6 metal–ligand motif shell (Fig. 1c and d). This structural architecture was previously reported by Sun et al.40 In contrast to another reported Cu29 NC (Cu29-Adm NC) with adamantane thiolate (–SAdm), we observe a difference in the connecting chlorides (Fig. S7†).41 This discrepancy in chloride connections is a critical factor that contributes to the structural disparity between these two scenarios.
The existing literature has established a correlation between the incorporation of chloride ligands onto the motif shell and the steric properties of the thiolate ligands utilized in the synthesis.41 However, in our specific case, a noteworthy observation emerges. Despite using a less bulky thiolate ligand (–StBu) in comparison with the adamantane thiolate used in previous studies, we have encountered a higher number of chloride ligands within the metal–ligand motif shell. This intriguing finding indicates that steric effects alone do not determine the addition of smaller ligands to the motifs. To accommodate a greater quantity of these smaller ligands on the outer surface, we intentionally altered the polarity of the reaction mixture. Notably, whereas Bao et al. previously employed a tertiary solvent mixture initially composed of acetonitrile and chloroform in a 1:1 (v/v) ratio, followed by the addition of methanol, our approach utilized a 4:1 (v/v) mixture of acetonitrile and chloroform initially, followed by the addition of methanol.41 This deliberate adjustment in solvent polarity, favouring a higher polar nature, accentuated the addition of chloride ligands. Consequently, it resulted in the formation of the metal–ligand motif shell with a reduced number of thiolate ligands. Therefore, it becomes evident that solvent polarity might play a role in shaping the intricate structure of Cu NCs, further emphasizing the multifaceted factors influencing their composition and morphology.
A positive mode ESI-MS measurement was performed by dissolving the as-synthesized NC in a CHCl3 and methanol mixture (v/v ∼1:1) resulting in a single molecular ion at m/z = 4185.04 with no cluster fragmentation signal, indicating its structural stability (Fig. S8a†). The isotopic distribution of the obtained peak shows its mono-cationic nature and the peak is assigned to the formula [Cu29(StBu)12(PPh3)4Cl6H10]+, which matches the simulated m/z value of 4184.93. The good agreement between experimental and simulated isotopic patterns confirmed its complete molecular formula (inset; Fig. S8a†). However, hydride identification by crystallographic techniques is notoriously challenging due to its low electron density compared to other elements present in the structure. Here, the complete formula of the NC was determined as [Cu29(StBu)12(PPh3)4Cl6H10][BF4], combining SCXRD and ESI-MS data. Additionally, the ESI-MS measurement of the deuteride analogue [Cu29(StBu)12(PPh3)4Cl6D10]+ confirmed the presence of ten hydrides in the NC, as evidenced by a distinct peak at m/z 4195.12 (Fig. S8b†). The valence state and composition of the Cu29 NC were further characterized by XPS and EDS analyses (Fig. S9 and S10†). Peaks corresponding to Cu 2p3/2 and Cu 2p1/2 are observed in the binding energy spectrum of Cu 2p at 931.38 eV and 951.25 eV, respectively (Fig. S11†). The doublet separation is found to be 19.87 eV, and the dominant peak in the Cu LMM Auger spectrum is noticed at 916.3 eV, justifying the +1 oxidation state of all Cu (Fig. S12†). Consequently, it further verifies the obtained crystal structure of the Cu29 NC, where the mono-catatonic nature of the Cu29 NC was identified.
The as-synthesized Cu29 NC in CHCl3 exhibits absorbance peaks at 325, 445, and 527 nm in the UV-vis absorption spectrum (Fig. 2a). These peaks align well with the simulated absorption spectrum obtained through TD-DFT calculations. The highest oscillator strength peaks at 326, 437, and 517 nm correspond to specific transitions: HOMO−10 → LUMO+2, HOMO−8 → LUMO, and HOMO−2 → LUMO, respectively (Fig. 2b and c). These slight deviations can be attributed to the ligand simplifications employed in the theoretical calculations. The Kohn–Sham diagram reveals that the occupied orbitals primarily involve s and p-like states of the ligand and s, p, and d-like states of the Cu atoms in the shell, while the unoccupied orbitals mainly consist of s, p, and d-like states of the Cu atoms in the core. The presence of discernible peaks in the absorption spectrum suggests that these peaks originate from charge transitions occurring between the metal–ligand shell and the inner core of the Cu29 NC. Consequently, any alteration in the architecture of the ligand shell will inevitably influence the dynamics of these charge transition processes. This phenomenon becomes evident when we compare our UV-vis absorbance results with the reported literature concerning Cu29 NCs featuring Cu16S18P4 and Cu16S15P4Cl3 motif shells.41 So, the introduction of chloride ions into the ligand shell is likely to induce changes in the electronic environment of the ligand shell itself. These alterations have a profound impact on the charge transition process, which is apparent from the discrepancies observed in the UV-vis absorption spectrum. This underscores that the sensitivity of the absorption spectrum depends on the ligand shell architecture. Moreover, the absorption spectra of the as-synthesized NC remain largely unchanged even after exposure to the ambient atmosphere for 7 days and sunlight for 4 hours, demonstrating its excellent resistance to photodegradation (Fig. S13†). This structural stability can be attributed to the distinctive structural architecture of the Cu29 NC.42
 | ||
| Fig. 2 (a) UV-vis absorption spectrum of the as-synthesized Cu29 NC in CHCl3; the inset shows the simulated absorption spectrum with oscillator strength, (b) Kohn–Sham diagram and the associated transitions, and (c) FMOs of the associated energy states. Color legend: Cu, orange; S, yellow; P, magenta; Cl, green; C, grey. | ||
Encouraged by the unique characteristics, including the precise atomic structures, exact molecular formula and inherent stability of emerging copper nanoclusters, we wondered if the as-synthesized Cu29 NC would be catalytically active to promote the organic transformations, despite the presence of a sulfur-containing ligand at the outer surface of the NC. We commenced our study by reacting aniline (1a) with diisopropyl azodicarboxylate (2a) in the presence of Cu29 NCs (0.25 mol% Cu) in CDCl3 solvent at room temperature for 3 h, furnishing the desired carbamate product 3aa in a good yield (Fig. 3). The control experiments conducted in the absence of Cu29 NCs or even in the presence of a CuI salt, in lieu of Cu29 NCs, resulted in no 3aa product formation or its formation in traces (Fig. 3). In addition to that, we have performed the controlled reaction of 3aa product formation in the presence of another Cu(I) salt, Cu(CH3CN)4BF4 as a catalyst, which is a precursor salt of the synthesis of the as-synthesized Cu29 NC, resulting in the formation of product 3aa in a trace amount (<5%) (Fig. 3). Furthermore, we explored the same catalytic reaction using an in situ mixture of Cu(CH3CN)4BF4, HS-tBu, PPh3, and NaBH4 as the catalyst precursor. Notably, the yield decreased to 11%. This reduction in yield may be attributed to the potential interference from organic ligands and reducing agents with the product. It is indicative of an imperative role of the Cu29 NC in this catalytic transformation. The synthetic utility of Cu29 NC-catalyzed C–N coupling was evaluated by employing a plethora of distinctly substituted anilines and a variety of dialkyl azodicarboxylates under standard reaction conditions. Gratifyingly, the electron-rich p-methyl and p-tert-butyl anilines (1b and 1c) reacted well with diisopropyl azodicarboxylate (2a) in this transformation, delivering the carbamate products 3ba (69%) and 3ca (68%), respectively, in good yields. Furthermore, 3,5-dimethylaniline (1d) also provided the desired product 3da (69%) in a good yield. Moreover, the electron-poor p-chloro- and p-bromoanilines (1e and 1f) remained as viable substrates, affording carbamates 3ea (65%) and 3fa (62%), respectively. Notably, heteroaryl 2-aminopyridine (1g) was also a competent reaction partner, furnishing product 3ga (47%) in a moderate yield. Delightfully, this method was extendable to different dialkyl azodicarboxylates, such as diethyl azodicarboxylate (2b) and di-tert-butyl azodicarboxylate (2c), delivering carbamates 3ab (78%) and 3ac (65%), respectively, in good yields. Nevertheless, the success of various anilines prompted us to further explore the catalytic activity of the Cu29 NC in the C–N coupling of the benzimidazole N–H bond with dialkyl azodicarboxylates. Indeed, the dialkyl azodicarboxylates (2a–2c) exhibited viability in this transformation, while they were treated with benzimidazole (4), offering the carbamate products 5a–5c (66%, 73%, and 59%, respectively) in good yields. In a comparative study, our Cu29 NC catalyst exhibited superior catalytic activity compared to a previously reported Cu32 NC catalyst for carbonylative C–N bond formation. Following the identical catalytic procedure reported in the literature, the Cu29 catalyst achieved an impressive yield of 59% for the synthesis of 3fa, surpassing the reported yield of 46.7% using the Cu32 catalyst. This significant increase in yield highlights the superior efficiency of the Cu29 catalyst, solidifying its potential as a highly efficient homogeneous catalyst for carbonylative C–N bond formation reactions.
 | ||
| Fig. 3 Cu29 NC-catalyzed carbamate synthesis from anilines and benzimidazole, mechanistic studies, and mechanistic rationale. | ||
To shed light on the mechanistic scenario, radical inhibition experiments were conducted in the presence of radical scavengers. An adduct (7) of the so-formed oxyacyl radical and TEMPO (6) was detected by high-resolution mass spectrometric (HRMS) analysis when the standard reaction was executed in the presence of TEMPO (6) (TEMPO = (2,2,6,6-tetramethylpiperidin-1-yl)oxyl) as a radical scavenger. Furthermore, the standard reaction was inhibited by 1,1-diphenylethene (8), forming an oxyacyl radical-trapped intermediate (9) that was identified by HRMS analysis. These experiments support the involvement of a radical intermediate in the reaction mechanism.
To understand the mechanism more properly, we have plotted the free energy profile diagram which we obtained by theoretical calculations (Fig. 4 and Table S3†). Initially, the addition of diisopropyl azodicarboxylates (Azo) to the reaction medium with the catalyst produces b with an exergonic free energy change (−0.74 eV), which could be due to the H-bonding interaction (2.8–3.1 Å) between the Azo and Cl atoms of the Cu29 NC. Furthermore, complex c with two carbonyl radicals is formed upon two C–N bond cleavage of Azo with a free energy change of 2.58 eV. As the radical generation reaction is too fast, we have considered the concerted pathway. This endergonic reaction might be due to the electronic repulsion between free radicals on carbon atoms and lone pairs of Cl atoms in the Cu29 NC. Next, complex d is formed in the presence of aniline with a reaction free energy change of −0.61 eV. Complex e is formed by C–H bond formation between the carbonyl compound and the aniline radical. A transition state is located in between d and e, which requires an activation barrier of 0.59 eV. The activated e complex undergoes a concerted pathway with the second carbonyl radical and forms a stable C–N complex with a free energy change of −3.7 eV, suggesting a favourable carbamate formation. Hence, the highly exergonic nature of the overall reaction free energy change (−2.95 eV) indicates the thermodynamic favourability of the reaction.
 | ||
| Fig. 4 Mechanistic study of the catalytic reaction from the theoretical investigation. Color legend: Cu, dark orange; S, yellow; P, light orange; Cl, green; N, blue; O, red; C, grey; H, black. H atoms are partially removed for clarity. | ||
Based on experimental and theoretical studies, a proposed mechanism suggests that in the rate-determining step, diisopropyl azodicarboxylates (2a) decompose over Cu29 NCs, generating oxyacyl radicals (2 equiv.) with the extrusion of N2 gas. Hydrogen atom abstraction (HAT) from aniline (1a) by the oxyacyl radical generates an aminyl radical, which then combines with another oxyacyl radical to produce the final carbamate product 3aa. The catalytic activity of the Cu29 NC is influenced by its specific structural architecture, including the arrangement of active sites and the surrounding chemical environment, which contribute to achieving the desired catalytic activity for different substrates.
The key stabilizing interaction identified is the H-bonding interaction (2.8–3.1 Å) between the Azo and Cl atoms of the Cu29 NC. This underscores the pivotal role of the Cl atom within the catalyst, influencing the entire catalytic reaction pathway. In essence, the presence of a Cl atom on the motif shell emerges as a crucial factor shaping the catalytic properties of the system. The involvement of the Cl atom is central to the enhancement of the catalytic activity of the Cu29 NC, particularly when compared to the Cu32 and Cu29-Adm NCs, which contain fewer chloride bridges in the motif (Fig. 3). Indeed, the introduction of a higher quantity of Cl atoms into the Cu29 NC structure significantly amplifies the number of available catalytic sites. These catalytic sites represent critical locations where chemical transformations and reactions can occur efficiently. This augmentation of catalytic sites is indispensable for the Cu29 NC to exhibit and sustain its catalytic activities effectively.
To ensure post-catalytic stability, we conducted several tests. TEM images of the Cu29 NC catalyst before and after the 3fa formation showed consistent cluster size, indicating the stability and uniform distribution of the NC throughout the catalysis reaction (Fig. S14†). Comparable absorption spectra of the catalyst (before and after the catalytic reaction) revealed significant similarity, which also suggests the stability of the NC after the catalytic reaction (Fig. S15†). Additionally, the ESI-MS data of the post-reaction catalyst closely matched those of the as-synthesized catalyst, confirming the intact structure and composition (Fig. S16†). 1H NMR measurements of the post-reaction catalyst showed clear matching with the catalyst before the reaction (Fig. S17†). Notably, consistent peak integration in the hydride region confirmed and quantified the presence of hydrides in the isolated catalyst after the reaction.
Conclusions
In conclusion, our study has successfully engineered a core–shell architecture for atomically precise Cu NCs, showcasing their efficacy as homogeneous catalysts in carbonylative C–N coupling reactions. The designed NC exhibits a distinctive geometry, featuring a central Cu@Cu12 core intricately enveloped by a Cu16S12P4Cl6 metal–ligand motif shell, accompanied by ten hydride ligands. We have systematically investigated the impact of this nanocluster on carbonylative C–N coupling reactions involving aromatic amines and heteroaromatic benzimidazole substrates. Remarkably, these transformations occur under mild reaction conditions, yielding a diverse array of carbamates in high yields through a radical-assisted mechanistic pathway. Our innovative synthetic approach, together with comprehensive structural insights, has unveiled the significant role of the Cl atom within the NC structure. This observation holds the potential to inspire further advancements in the synthesis of novel metal nanoclusters with tailored catalytic properties. The implications of our findings extend beyond the specific catalytic system studied here, contributing valuable knowledge to the broader landscape of materials science and catalysis research.Data availability
The ESI contains experimental details, computational methods, structural refinement parameters, the presence of hydrides in the structure, theoretical insights into the catalysis, SEM micrograph of the as-synthesized crystal, 1H, 11B, 19F, 31P NMR of the Cu29 NC, 1H NMR of the Cu29D NC, structural illustration, XPS and EDS spectra of the Cu29 NC, stability of the Cu29 NC, catalyst characterization after the catalytic reaction, NMR spectra of all products obtained after catalysis, and coordinates of the optimized structure and references.Author contributions
A. K. D., S. B., and A. S. performed the synthesis, characterization and data interpretation. A. P. and B. S. were involved in the catalysis. P. K. M. solved the crystal structure. S. S. M. and B. P. performed the theoretical calculation. S. M. was involved in manuscript preparation. All authors discussed the results and were involved in manuscript writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge funding from the Science and Engineering Research Board (SERB) through the grants CRG/2022/000984. We are also thankful to Ms Saniya Gratious and Ms Sariga Mangalamundackal Vijayan for their help in ESI-MS measurement and initial synthesis, respectively.References
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 CrossRef CAS PubMed.
- S. Biswas, S. Das and Y. Negishi, Coord. Chem. Rev., 2023, 492, 215255 CrossRef CAS.
- S. Sharma, K. K. Chakrahari, J.-Y. Saillard and C. Liu, Acc. Chem. Res., 2018, 51, 2475–2483 CrossRef CAS PubMed.
- X. Liu and D. Astruc, Coord. Chem. Rev., 2018, 359, 112–126 CrossRef CAS.
- A. Baghdasaryan and T. Bürgi, Nanoscale, 2021, 13, 6283–6340 RSC.
- A. K. Das, S. Biswas, V. S. Wani, A. S. Nair, B. Pathak and S. Mandal, Chem. Sci., 2022, 13, 7616–7625 RSC.
- S. Biswas, S. Hossian, T. Kosaka, J. Sakai, D. Arima, Y. Niihori, M. Mitsui, D.-e. Jiang, S. Das, S. Wang and Y. Negishi, Chem. Commun., 2023, 59, 9336–9339 RSC.
- T. Jia, Z.-J. Guan, C. Zhang, X.-Z. Zhu, Y.-X. Chen, Q. Zhang, Y. Yang and D. Sun, J. Am. Chem. Soc., 2023, 145, 10355–10363 CrossRef CAS PubMed.
- B.-L. Han, Z. Liu, L. Feng, Z. Wang, R. K. Gupta, C. M. Aikens, C.-H. Tung and D. Sun, J. Am. Chem. Soc., 2020, 142, 5834–5841 CrossRef CAS PubMed.
- R. S. Dhayal, W. E. van Zyl and C. Liu, Acc. Chem. Res., 2016, 49, 86–95 CrossRef CAS PubMed.
- C. M. Aikens, Acc. Chem. Res., 2018, 51, 3065–3073 CrossRef CAS PubMed.
- R. S. Dhayal, J.-H. Liao, Y.-R. Lin, P.-K. Liao, S. Kahlal, J.-Y. Saillard and C. Liu, J. Am. Chem. Soc., 2013, 135, 4704–4707 CrossRef CAS PubMed.
- S. Nematulloev, A. Sagadevan, B. Alamer, A. Shkurenko, R. Huang, J. Yin, C. Dong, P. Yuan, K. E. Yorov and A. A. Karluk, Angew. Chem., Int. Ed., 2023, 62, e202303572 CrossRef CAS PubMed.
- S. Lee, M. S. Bootharaju, G. Deng, S. Malola, W. Baek, H. Hakkinen, N. Zheng and T. Hyeon, J. Am. Chem. Soc., 2020, 142, 13974–13981 CrossRef CAS PubMed.
- A. W. Cook, Z. R. Jones, G. Wu, S. L. Scott and T. W. Hayton, J. Am. Chem. Soc., 2018, 140, 394–400 CrossRef CAS PubMed.
- Y. Du, H. Sheng, D. Astruc and M. Zhu, Chem. Rev., 2019, 120, 526–622 CrossRef PubMed.
- C. Sun, N. Mammen, S. Kaappa, P. Yuan, G. Deng, C. Zhao, J. Yan, S. Malola, K. Honkala and H. Häkkinen, ACS Nano, 2019, 13, 5975–5986 CrossRef CAS PubMed.
- G.-G. Luo, Z.-H. Pan, B.-L. Han, G.-L. Dong, C.-L. Deng, M. Azam, Y.-W. Tao, J. He, C.-F. Sun and D. Sun, Angew. Chem., Int. Ed., 2023, 62, e202306849 CrossRef CAS PubMed.
- G. Dong, Z. Pan, B. Han, Y. Tao, X. Chen, G. Luo, P. Sun, C. Sun and D. Sun, Angew. Chem., Int. Ed., 2023, 62, e202302595 CrossRef CAS PubMed.
- Q. Tang, Y. Lee, D.-Y. Li, W. Choi, C. W. Liu, D. Lee and D.-e. Jiang, J. Am. Chem. Soc., 2017, 139, 9728–9736 CrossRef CAS PubMed.
- Y.-M. Wang, X.-C. Lin, K.-M. Mo, M. Xie, Y.-L. Huang, G.-H. Ning and D. Li, Angew. Chem., Int. Ed., 2023, 62, e202218369 CrossRef CAS PubMed.
- C. Dong, R.-W. Huang, A. Sagadevan, P. Yuan, L. Gutiérrez-Arzaluz, A. Ghosh, S. Nematulloev, B. Alamer, O. F. Mohammed, I. Hussain, M. Rueping and O. M. Bakr, Angew. Chem., Int. Ed., 2023, 62, e202307140 CrossRef CAS PubMed.
- Y. Liu, J. Yu, Y. Lun, Y. Wang, Y. Wang and S. Song, Adv. Funct. Mater., 2023, 2304184 CrossRef CAS.
- G. V. Goeden and K. G. Caulton, J. Am. Chem. Soc., 1981, 103, 7354–7355 CrossRef CAS.
- S. A. Bezman, M. R. Churchill, J. A. Osborn and J. Wormald, J. Am. Chem. Soc., 1971, 93, 2063–2065 CrossRef.
- M. T. Pirnot, Y. M. Wang and S. L. Buchwald, Angew. Chem., Int. Ed., 2016, 55, 48–57 CrossRef CAS PubMed.
- J.-X. Chen, J. F. Daeuble, D. M. Brestensky and J. M. Stryker, Tetrahedron, 2000, 56, 2153–2166 CrossRef CAS.
- D. M. Brestensky, D. E. Huseland, C. McGettigan and J. M. Stryker, Tetrahedron Lett., 1988, 29, 3749–3752 CrossRef CAS.
- C. Zhang, Z. Wang, W.-D. Si, L. Wang, J.-M. Dou, Z.-Y. Gao, C.-H. Tung and D. Sun, ACS Nano, 2022, 16, 9598–9607 CrossRef CAS PubMed.
- F. Li and Q. Tang, J. Catal., 2020, 387, 95–101 CrossRef CAS.
- S. Biswas, S. Das and Y. Negishi, Nanoscale Horiz., 2023, 8, 1509–1522 RSC.
- A. K. Ghosh and M. Brindisi, J. Med. Chem., 2015, 58, 2895–2940 CrossRef CAS PubMed.
- A. Matošević and A. Bosak, Arch. Ind. Hyg. Toxicol., 2020, 71, 285–299 Search PubMed.
- J. Bariwal and E. Van der Eycken, Chem. Soc. Rev., 2013, 42, 9283–9303 RSC.
- Y. N. Zheng, H. Zheng, T. Li and W. T. Wei, ChemSusChem, 2021, 14, 5340–5358 CrossRef CAS PubMed.
- Z. H. Guan, H. Lei, M. Chen, Z. H. Ren, Y. Bai and Y. Y. Wang, Adv. Synth. Catal., 2012, 354, 489–496 CrossRef CAS.
- M. Usman, Z.-H. Ren, Y.-Y. Wang and Z.-H. Guan, RSC Adv., 2016, 6, 107542–107546 RSC.
- A. J. Jordan, G. Lalic and J. P. Sadighi, Chem. Rev., 2016, 116, 8318–8372 CrossRef CAS PubMed.
- J. Sun, X. Yan, L. Wang, Z. Xie, G. Tian, L. Wang, A. He, S. Li, Q. Guo, C. Chaolumen, J. He and H. Shen, Inorg. Chem., 2023, 62, 9005–9013 CrossRef CAS PubMed.
- Y. Bao, X. Wu, B. Yin, X. Kang, Z. Lin, H. Deng, H. Yu, S. Jin, S. Chen and M. Zhu, Chem. Sci., 2022, 13, 14357–14365 RSC.
- R. Jin, G. Li, S. Sharma, Y. Li and X. Du, Chem. Rev., 2020, 121, 567–648 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2242226. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3nr05912j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |